* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
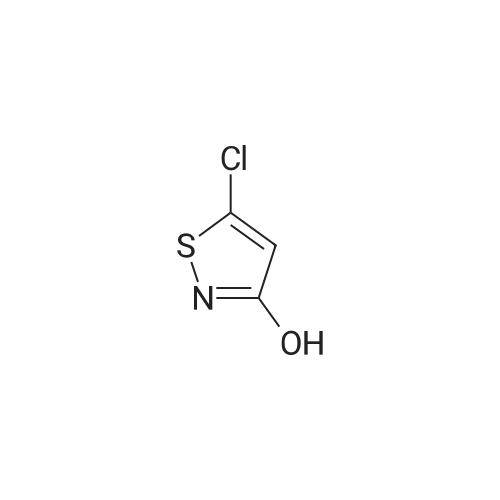
Example 8 Preparation of 2-[3-(5-Chloro-4-isothiazolin-3-one-2 -yl)propionyl]-1,4-benzoquinone (Compound 10) A stirred solution of 2-acryloyl-1,4-benzoquinone (1.0 g, 0.007 mole) and <strong>[25629-58-7]5-chloro-3-hydroxyisothiazole</strong> (0.81 g, 0.006 mole) in 20 ml of toluene was heated at 80 C. for 4 hours. Upon cooling, the precipitate which formed during the reaction period was removed by filtration and recrystallized from ethyl acetate/acetone mixture. A gray/tan solid was obtained; 0.85 g; mp 181-183 C.; IR (KBR) 1635 cm-1 (broad);NMR (acetone-d6) delta7.35 (s,1H); 7.15 (d 1H); 6.85 (d 1H); 6.35 (s,1H); 4.2 (t,2H); 3.52 (t,2H).
Example 2 Preparation of 2-(3-Oxobutyl)-5-chloro-4-isothiazolin-3-one) (Compound 1) A solution of 0.5 g (0.0037 mole) of <strong>[25629-58-7]5-chloro-3-hydroxyisothiazole</strong> and methyl vinyl ketone (1.0 g, 0.014 mole) in 15 ml of toluene was heated at 80 C. for 24 hours. After cooling and removal of toluene and excess reagent by rotary evaporation, the residual oil was purified by column chromatography on silica gel, using diethyl ether/methanol (9/1) as eluant. Compound 1 was obtained as an oil; 0.65 g (85%); IR (neat) 1725,1650 cm-1; NMR (CDCl3) delta6.25 (s,1H); 4.0 (t,2H); 2.9 (t,2H); 2.2 (s,3H).
With hydroquinone; In methanol; diethyl ether; toluene;
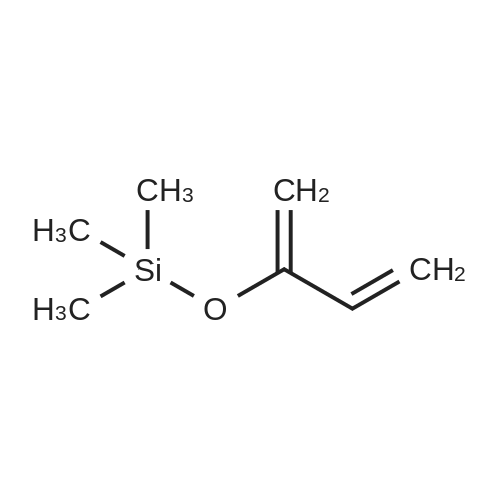
Example 1 Preparation of 2-(3-Oxobutyl)-5-chloro-4-isothiazolin-3-one (Compound 1) A solution of <strong>[25629-58-7]5-chloro-3-hydroxyisothiazole</strong> (1.5 g, 0.011 mole), 2-trimethylsiloxy-1,3-butadiene (5.6 g, 0.04 mole) and 0.5 g of hydroquinone in 20 ml of toluene was heated at 80 C. for 96 hours. After cooling, the mixture was concentrated in vacuo. The residual oil was dissolved in diethyl ether and washed with saturated NaHCO3 solution and then with water. After drying (MgSO4) and concentrating the solution, the residual oil was purified by column chromatography on silica gel, using diethyl ether/methanol (9/1) as eluant. Compound 1 was obtained as an oil, 0.3 g; IR (neat) 1625,1650 cm-1; NMR (CDCl3) delta6.25 (s,1H); 4.0 (t, 2H); 2.9 (t,2H); 2.2 (s, 3H).
With toluene-4-sulfonic acid; In diethyl ether; hexane; toluene;
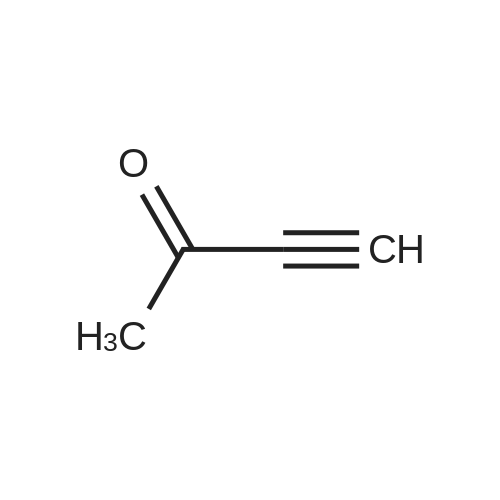
Example 9 Preparation of 2-(2-Oxo-3-buten-4-yl)-5-chloro-4-isothiazolin-3-one (Compound 11) A stirred solution of <strong>[25629-58-7]5-chloro-3-hydroxyisothiazole</strong> (1.5 g, 0.011 mole), 1-butyn-3-one (3.5 g, 0.05 mole) and p-toluenesulfonic acid (0.02 g) in 25 ml of toluene was heated at 80 C. for 24 hours. Upon cooling, the dark brown reaction mixture was concentrated and the residual oil was column chromatographed on silica gel, using diethyl ether/hexane as eluant. Compound 11 was obtained as a yellowish solid; 1.8 g; mp 137-139 C.; IR (KBr) 1675, 1620 cm-1; NMR (CDCl3) delta8.2 (d,1H,J=13.9 Hz); 6.35 (s,1H); 5.85 (d,1H,J=13.9 Hz); 2.35 (s,3H).
5-chloro-2-(N-4-tolunenesulfonylcarbamoyl)3-isothiazolone[ No CAS ]
[ 121946-95-0 ]
Yield
Reaction Conditions
Operation in experiment
85.9%
In toluene;
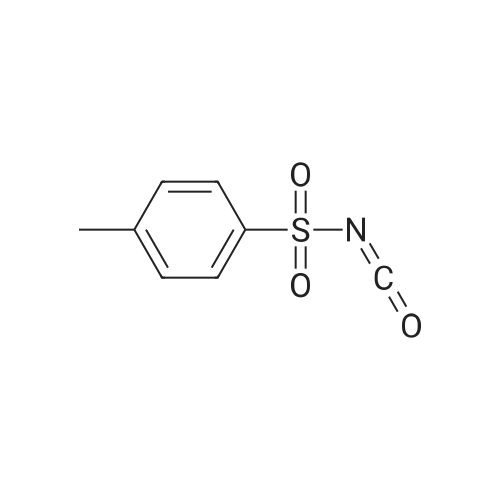
EXAMPLE 1 Preparation of 5-chloro-2-(N-4-tolunenesulfonylcarbamoyl)3-isothiazolone (Compound 1) 4.3 parts (0.032 mol) of <strong>[25629-58-7]5-chloro-3-hydroxyisothiazole</strong> was dissolved in 50 parts of dry toluene and stirred at 25 C. To the resulting solution, 6.3 parts (0.032 mol) of p-toluenesulfonyl isocyanate dissolved in 20 parts of toluene was added. The pale yellow precipitate thus formed increased by continuously stirring. The pre-cipitate was collected by filtration. Thus, 9.14 parts (yield: 85.9%) of 5-chloro-2-(N-4-toluenesulfonylcarbamoyl)-3-isothiazolone (Compound 1) was obtained in the form of a pale yellow solid. m.p.: 125-128 C.
5-chloro-2 (N-2-toluenesulfonylcarbamoyl)3-isothiazolone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
81.2%
In toluene;
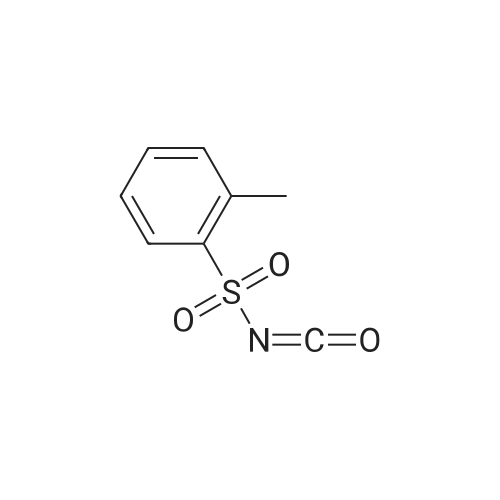
EXAMPLE 2 Preparation of 5-chloro-2 (N-2-toluenesulfonylcarbamoyl)3-isothiazolone (Compound 2) 4.3 parts (0.032 mol) of <strong>[25629-58-7]5-chloro-3-hydroxyisothiazole</strong> was dissolved in 50 parts of dry toluene and stirred at 25 C. To the resulting solution, 6.3 parts (0.032 mol) of o-toluenesulfonyl isocyanate dissolved in 20 parts of toluene was added. Then, the procedure of Example 1 was repeated. Thus, 8.62 parts (yield: 81.2%) of 5-chloro-2-(N-2-toluenesulfonylcarbamoyl)-3-isothiazolone (Compound 2) was obtained in the form of a brown solid. m.p.: 82 to 84 C.
With 3-chloro-benzenecarboperoxoic acid; In dichloromethane; at 20℃; for 15.0h;cooling with ice;
<Step 5> Synthesis of <strong>[25629-58-7]5-chloro-isothiazol-3-ol</strong> 1-oxide To a suspension of <strong>[25629-58-7]5-chloro-isothiazol-3-ol</strong> (31.8 g) in dichloromethane (640 mL), m-chloroperbenzoic acid (content: 65%) (60.7 g) was added under ice-cooling and the resultant reaction mixture was stirred at room temperature for 15 hours. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure. To the resultant residue, dichloromethane was added and the resulting precipitates were filtered out. The filtrate was concentrated under reduced pressure and the resultant residue was purified by silica gel chromatography (eluate; n-hexane:ethyl acetate=67:33 to 60:40) to obtain the subject compound (26.0 g) as a white solid.
26.0 g
With 3-chloro-benzenecarboperoxoic acid; In dichloromethane; at 20℃; for 15.0h;Cooling with ice;
To a suspension of <strong>[25629-58-7]5-chloroisothiazol-3-ol</strong> (31.8 g) in dichloromethane (640 mL), m-chloroperbenzoic acid (content: 65%) (60.7 g) was added under ice-cooling and the resultant reaction mixture was stirred at room temperature for 15 hours. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure. To the resultant residue, dichloromethane was added and precipitates were filtered off. The filtrate was concentrated under reduced pressure and the resultant residue was purified by silica gel chromatography (eluate; n-hexane:ethyl acetate=67:33 to 60:40) to obtain the subject compound (26.0 g) as a white solid.
26.0 g
With 3-chloro-benzenecarboperoxoic acid; In dichloromethane; at 20℃;Cooling with ice;
<Step 4> Synthesis of <strong>[25629-58-7]5-chloro-isothiazol-3-ol</strong> 1-oxide To a suspension of <strong>[25629-58-7]5-chloro-isothiazol-3-ol</strong> (31.8 g) in dichloromethane (640 mL), m-chloroperbenzoic acid (content 65%) (60.7 g) was added under ice-cooling and the resultant reaction mixture was stirred at room temperature for 15 hours. The reaction mixture was filtered and then the filtrate was concentrated under reduced pressure. To the residue, dichloromethane was added and the precipitate was filtered off. The filtrate was concentrated under reduced pressure and the resultant residue was purified by silica gel chromatography (eluent; n-hexane:ethyl acetate=67:33 to 60:40) to give the title compound (26.0 g) as a white solid.
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In tetrahydrofuran; at 0 - 20℃; for 16.0h;
j00451j To a solution of <strong>[25629-58-7]5-chloroisothiazol-3-ol</strong> (4.03 g, 29.73 mmol) and DBU (7.07 g, 46.44 mmol) in THF (80 mL) at 0C, a solution of chloro(methoxy)methane (5.74 g, 71.3 mmol) in THF (40 mL) was added dropwise. The mixture was warmed to rt and stirred for 16 h. EtOAc (300 mL) was added, and the solution was washed with water (100 mL). The organic layer was dried over Na2SO4, filtered, concentrated, and purified by chromatography on silica gel (petroleum ether/EtOAc = 100/1) to give 5-chloro-3-(methoxymethoxy)isothiazole (701.5 mg, 3.91 mmol, 52.92% yield) as yellow oil. ?H NIVIR (400 MHz, CDC13): 6.59 (s, 1 H), 5.43 (s, 2 H), 3.52 (s, 3 H); MS: 180.1 [M+H].
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In tetrahydrofuran; at 10 - 20℃;
<Step 1> Synthesis of 5-chloro-3-(methoxymethoxy)isothiazole To a solution of <strong>[25629-58-7]5-chloro-isothiazol-3-ol</strong> (5.00 g) in tetrahydrofuran (50 mL), 1,8-diazabicyclo[5.4.0]undeca-7-ene (8.42 g) was added, and to the resultant reaction mixture, a solution of chloromethyl methyl ether (4.45 g) in tetrahydrofuran (25 mL) was dropped under ice-cooling at an inside temperature of 10 C. or less. After the completion of dropping, the inside temperature was elevated to room temperature and the reaction mixture was stirred for 10 minutes. To the reaction mixture, water was added and the resultant reaction mixture was extracted with ethyl acetate. The organic phase was washed with saturated saline and was dried over anhydrous sodium sulfate. From the organic phase, the solvent was distilled off under reduced pressure to obtain the subject compound (6.15 g) as a brown oil.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping