There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
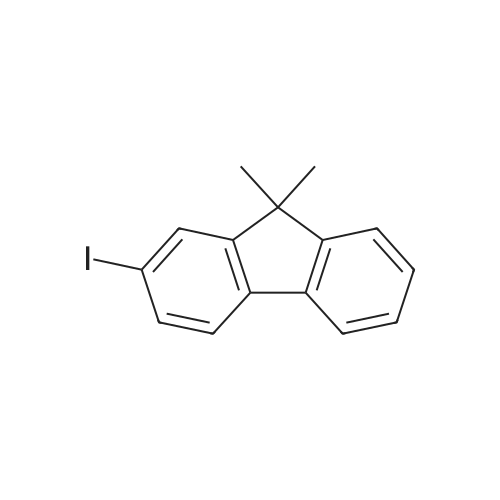
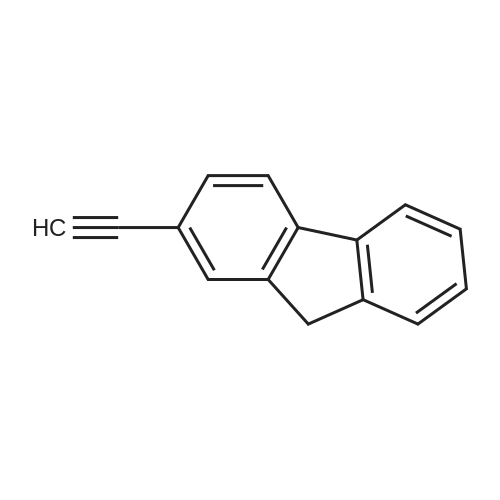
Structure of 2523-42-4 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With sodium chlorate; sulfuric acid; iodine; sodium thiosulfate In methanol; water; acetic acid
Example 1 Synthesis of 2-iodofluorene In a 300-mi four-necked flask were charged 90 ml of glacial acetic acid and 90 ml of water, followed by the addition of 41.6 g (0.25 mole) of fluorene, 12.7 g (0.10 mole) of iodine, 1.93 g (0.018 mole) of sodium chlorate and 4.5 ml of concentrated sulfuric acid under vigorous stirring. The resulting mixture was refluxed under heat at 85 to 90° C. for 30 minutes and then at 95 to 100° C. for 30 minutes. After completion of the reaction, the reaction mixture was extracted with 55 ml of toluene. The organic layer was then washed with a 5percent aqueous solution of sodium thiosulfate. After the organic layer was washed further with 50 ml of a 20percent saline solution, 216 ml of methanol was added. The resulting mixture was crystallized for 2 hours at an internal temperature of 25 to 30° C. and the resulting crystals were collected by filtration. The crude crystals thus obtained were washed with 76 ml of methanol and then dried, whereby 24.8 g (yield: 84.8percent) of the target compound was obtained as white crystals. As a result of HPLC analysis (column: YMC-A-312, detection UV: 254 nm, flow rate: 1.0 ml/min, eluent: methanol/water=9/1 (by volume), buffer: triethylamine and acetic acid, each 0.1percent), the compound was found to be composed of 1.3percent of fluorene, 98.4percent of 2-iodofluorene and 0.02percent of 2, 7-diiodofluorene.
72%
With iodine; potassium carbonate; periodic acid In sulfuric acid; water; acetic acid at 65℃; for 4 h;
2-1: Synthesis of 2-iodofluorene: Fluorene (30.0 g, 180 mmol) was dissolved in a boiling solvent (acetic acid: water: sulfuric acid / 100:20:3(v/v/v))to prepare a first mixed solution, and then periodic acid dehydrate (8.0 g, 45 mmol) and iodine (23.0 g, 91.0 mmol) wereadded to the first mixed solution to prepare a second mixed solution. Then, the second mixed solution was stirred at65h for 4 hours to obtain a precipitate. The precipitate was filtered and then washed with a 2N aqueous sodium carbonatesolution and water to obtain crystals. The crystals were recrystallized by hexane. The yield ofthe product was 72percent.1H NMR(300 MHz, CDCl3) δ(TMS, ppm): 3.81(2H, s, -CH2), 7.31(2H, m, Ar-H), 7.44(2H, m, Ar-H), 7.66(1H, d, Ar-H),7.73(1H, d, Ar-H), 7.85(1H, s, Ar-H)
65%
With iodic acid In water
Fluorene (5.0 g, 0.30 mol) was treated with iodic acid. Themixture was poured into water, and the product was filtered off,washed with water, and dried (8.1 g). A portion (1.02 g) waschromatographed on silica gel in cyclohexane, giving almost colourless 2-iodofluorene (0.73 g, 65percent), m.p. 127-128 [14].
60%
With iodine; acetic acid; periodic acid In water at 80℃; for 4 h; Inert atmosphere
2-Iodofluorene was synthesized by iodination of fluorene (1 g, 6.02 mmol) with iodine (0.8 g, 3.2 mmol) in the presence of ortho-periodinic acid (H5IO6) (20 g, 0.88 mmol) in 80percent acetic acid aqueous solution (20 mL) at 80 °C for 4 h under nitrogen atmosphere.After cooling, the solvent was removed by decantation, and a brown solid was obtained. This was dissolved in toluene and washed with 5percent NaHSO3aqueous solution to remove the remaining iodine. Then, the resulting solid was purified by alumina column chromatography using toluene as an eluent, to obtain 8(the chemical structure in Scheme 2).Yield: 1.05 g (60percent).1H NMR (CDCl3, 400 MHz) δH7.89 (s,1H), 7.76 (d, J= 8.4 Hz, 2H), 7.70 (d, J= 8.4 Hz, 2H), 7.73(d, J= 8.4 Hz, 2H), 7.38-7.35 (m, 2H), 3.87 (s, 2H), MS(GC-mass): m/z292.0 [M+]; C13H9I (292.1).
Reference:
[1] Bulletin of the Chemical Society of Japan, 1989, vol. 62, # 2, p. 439 - 443
[2] New Journal of Chemistry, 2015, vol. 39, # 5, p. 4086 - 4092
[3] Patent: US6437203, 2002, B1,
[4] Organic Electronics: physics, materials, applications, 2014, vol. 15, # 7, p. 1324 - 1337
[5] Journal of Materials Chemistry, 2012, vol. 22, # 12, p. 5319 - 5329
[6] Doklady Chemistry, 2016, vol. 468, # 2, p. 174 - 178[7] Dokl. Akad. Nauk, 2016, vol. 468, # 5, p. 525 - 529,5
[8] Organic Letters, 2001, vol. 3, # 13, p. 2005 - 2007
[9] Nucleosides, Nucleotides and Nucleic Acids, 2007, vol. 26, # 8-9, p. 1199 - 1202
[10] Patent: EP2711359, 2014, A1, . Location in patent: Paragraph 0035; 0068; 0069
[11] Chemical Communications, 2014, vol. 50, # 88, p. 13477 - 13480
[12] Russian Journal of Organic Chemistry, 2005, vol. 41, # 12, p. 1750 - 1751
[13] Chinese Journal of Chemistry, 2010, vol. 28, # 5, p. 699 - 704
[14] Dyes and Pigments, 2017, vol. 147, p. 385 - 392
[15] Bulletin of the Korean Chemical Society, 2014, vol. 35, # 10, p. 3052 - 3058
[16] Journal of Chemical Research, Miniprint, 1997, # 12, p. 2701 - 2733
[17] Chemistry - A European Journal, 2011, vol. 17, # 8, p. 2479 - 2491
[18] Dyes and Pigments, 2017, vol. 146, p. 331 - 343
[19] Inorganica Chimica Acta, 2012, vol. 388, p. 140 - 147
[20] European Journal of Inorganic Chemistry, 2013, # 27, p. 4732 - 4742
[21] Patent: EP1170273, 2002, A1, . Location in patent: Example 1
[22] Journal of the American Chemical Society, 2012, vol. 134, # 7, p. 3542 - 3548
[23] Journal of the American Chemical Society, 2000, vol. 122, # 44, p. 11021 - 11022
[24] Patent: EP1990373, 2008, A1, . Location in patent: Page/Page column 51
[25] Chemistry of Materials, 2010, vol. 22, # 11, p. 3472 - 3481
[26] Organic Electronics: physics, materials, applications, 2014, vol. 15, # 11, p. 3316 - 3326
[27] Dyes and Pigments, 2018, vol. 159, p. 590 - 599
2
[ 153-78-6 ]
[ 2523-42-4 ]
Yield
Reaction Conditions
Operation in experiment
43%
Stage #1: With hydrogenchloride; sodium nitrite In water at 0℃; for 1 h; Stage #2: With sodium iodide In water at 20℃; for 4 h;
General procedure: To a solution of corresponding amine (1 eq) in 18percent HCl (12 mL)was added NaNO2 (1 M in H2O, 1.5 eq) at 0 C. The mixture wasstirred at 0 C for 1 h, and then NaI (2 M in H2O, 2 eq) was added.After the mixture was stirred at room temperature for 4 h, Na2SO3(4 eq) was added. The suspension was filtered to give the desiredproducts 10-12. 6.1.4.1 2-Iodo-9H-fluorene (10) Yellow solid (43percent); 1H NMR (400 MHz, CDCl3) δ 7.89 (s, 1H), 7.76 (d, J = 7.6 Hz, 1H), 7.70 (d, J = 8.0 Hz, 1H), 7.54 (d, J = 8.0 Hz, 2H), 7.41-7.31 (m, 2H), 3.88 (s, 2H).
Reference:
[1] Organic Letters, 2014, vol. 16, # 9, p. 2306 - 2309
[2] European Journal of Medicinal Chemistry, 2016, vol. 108, p. 154 - 165
3
[ 86-73-7 ]
[ 16218-28-3 ]
[ 2523-42-4 ]
Reference:
[1] Organic Process Research and Development, 2008, vol. 12, # 6, p. 1130 - 1136
[2] Russian Journal of Organic Chemistry, 2001, vol. 37, # 8, p. 1130 - 1133
[3] Patent: WO2006/73124, 2006, A1, . Location in patent: Page/Page column 18-19; 24-25
[4] Patent: EP1837324, 2007, A1,
[5] Patent: EP1837324, 2007, A1,
4
[ 2523-42-4 ]
[ 123348-27-6 ]
Reference:
[1] Chemistry - A European Journal, 2011, vol. 17, # 8, p. 2479 - 2491
5
[ 2523-42-4 ]
[ 74-88-4 ]
[ 144981-85-1 ]
Yield
Reaction Conditions
Operation in experiment
91%
Stage #1: With potassium <i>tert</i>-butylate In tetrahydrofuran at 0℃; for 0.166667 h; Stage #2: at 0 - 20℃;
2-Iodofluorene (3 g; 10.27 mmol, 1 equiv) was dissolved in 50 mL of anhydrous tetrahydrofuran. Thesolution was cooled to 0°C and 2.6 g of potassium tert-butoxide (23.16 mmol, 3equiv) was added. During the process, the transparent solution became red.After the solution had been stirred for 10 min, 1.44 g of iodomethane(23.16 mmol, 3 equiv) was added, turning the red solution milky. The solution wasreturned to room temperature and stirred for another 12 hours. After 100 mLdeionized water was added, the solution was extracted with ethyl acetate (100mL X 3). The combined organic solution was then dried over magnesium sulfate,and filtered off. The filtrate was condensed under vacuum to obtain a viscousliquid. Further purification by column chromatography (all n-hexane) on silicagel afforded 3.0 g of product 1 in a yield of 91percent. 1H NMR (300 MHz,CDCl3, δ) 1.47(s, 6H), 7.34-7.36(m, 2H), 7.40-7.45(m, 1H) 7.48(s,1H), 7.64-70(m, 2H), 7.75-7.76(m, 1H)
90%
Stage #1: With sodium hydroxide; benzyltrimethylammonium chloride In water; dimethyl sulfoxide at 20℃; for 3 h; Stage #2: at 20℃; for 3 h;
[Step 1] Synthesis of 2-iodo-9,9-dimethylfluorene A synthesis method of 2-iodo-9,9-dimethylfluorene is described. A synthesis scheme of 2-iodo-9,9-dimethylfluorene is shown in (D-1). 25 g (86 mmol) of 2-iodofluorene and 1.5 L of dimethylsulfoxide (abbreviation: DMSO) were put into a 2.0 L three-neck flask, and 1.0 g (5.4 mmol) of benzyltrimethylammonium chloride and 24 mL of 50percent sodium hydroxide aqueous solution were added to the mixture. This mixture was stirred at room temperature for 3 hours. Thereafter, 19 g (130 mmol) of iodomethane was added to this reaction mixture and stirred at room temperature for 3 hours. After completion of the reaction, the reaction solution was washed with 1.0 mol/L hydrochloric acid, a water layer was extracted with ethyl acetate, combined with an organic layer and washed with saturated saline, and then dried with magnesium sulfate. After drying, the mixture was subjected to suction filtration, and a filtrate was concentrated. An obtained residue was purified by silica gel column chromatography (developing solvent: hexane), and an obtained solution was concentrated. When an obtained solid was recrystallized with a mixed solvent of chloroform and hexane, 24.7 g of a white, powdery solid of 2-iodo-9,9-dimethylfluorene, which was a target matter, was obtained with the yield of 90percent.
70%
Stage #1: With potassium <i>tert</i>-butylate In tetrahydrofuran at 20℃; for 1.5 h; Stage #2: for 2 h;
2-2: Synthesis of 9,9-dimethyl-2-iodofluorene: Potassium tert-butoxide (21.8 g, 0.19 mol) was added to cooled anhydrous tetrahydrofuran, in which 2-iodofluorene(25.0 g, 85.6 mmol) was dissolved, and then stirred at room temperature for 1.5 hours to prepare a first mixedsolution. Subsequently, methyl iodide (28.2 g, 0.19 mol) was added to the first mixed solution to prepare a second mixedsolution, and then the second mixed solution was stirred for 2 hours to obtain potassium iodide. Subsequently, theobtained potassium iodide was filtered, a solvent was removed under reduced pressure, and a reaction product wasseparated by silica column chromatography (hexane). The yield of the reaction product was 70percent.1H NMR(300 MHz, CDCl3) δ(TMS, ppm): 1.47(6H, s, -CH3), 7.31(2H, m, Ar-H)), 7.45(2H, m, Ar-H), 7.66(1H, d, Ar-H),7.73(1H, d, Ar-H), 7.85(1H, s, Ar-H)
64%
Stage #1: With benzyltrimethylammonium chloride; sodium hydroxide In water; dimethyl sulfoxide at 20℃; for 3 h; Stage #2: at 20℃; for 3 h;
1 g (3.44mmol) of 2-iodofluorene and 100 mL of dimethylsulfoxidewere put into a 500 mL three-neck round-bottom flask.Subsequently, 0.04 g (0.22 mmol) of benzyltrimethylammonium chloride and 1 mL of 50percent sodium hydroxide aqueous solution were added to the mixture. This mixture was stirred at RT for 3 h. Thereafter, 0.76 g (5.2 mmol) of iodomethane was added into the reaction mixture, which was stirred at RT for an additional 3 h. After completion of the reaction, the reaction solution was washed with 1.0 mol/L hydrochloric acid; a water layer was extracted three times with ethyl acetate. The collected organic layer was washed with saturated saline, and then dried with magnesium sulfate.After drying, the mixture was subjected to suction filtration,and the filtrate was concentrated. An obtained residue was purified by silica gel column chromatography (hexane aseluent), and an obtained solution was concentrated. The residue solid was recrystallized with a mixed solvent of chloroform and hexane to yield 0.7 g of a white, powdery solid of 2-iodo-9,9-dimethylfluorene (64percent yield) (the chemical structure in Scheme 2).1H NMR (CDCl3, 400MHz) δH7.92 (s, 1H), 7.70 (d, J= 8.4 Hz, 1H), 7.66 (d, J =8.4 Hz 1H), 7.60 (d, J= 8.4 Hz, 1H), 7.47 (d, J= 8.4 Hz,1H), 1.37 (s, 6H), MS (GC-mass): m/z320.0 [M+]; C15H13I(320.1).
Reference:
[1] Organic Electronics: physics, materials, applications, 2014, vol. 15, # 11, p. 3316 - 3326
[2] Patent: US2008/91012, 2008, A1, . Location in patent: Page/Page column 34
[3] Patent: EP2711359, 2014, A1, . Location in patent: Paragraph 0035; 0070; 0071
[4] Bulletin of the Korean Chemical Society, 2014, vol. 35, # 10, p. 3052 - 3058
[5] Journal of the American Chemical Society, 2000, vol. 122, # 44, p. 11021 - 11022
[6] Patent: EP1990373, 2008, A1, . Location in patent: Page/Page column 62
[7] Organic Electronics: physics, materials, applications, 2014, vol. 15, # 7, p. 1324 - 1337
[8] New Journal of Chemistry, 2015, vol. 39, # 5, p. 4086 - 4092
Synthesis Example 10 20 parts of <strong>[2523-42-4]2-iodofluorene</strong> was dissolved in a mixed solution of 114 parts of dimethyl sulfoxide (DMSO) and 23 parts of tetrahydrofuran (THF), and the thus obtained mixture was then stirred at 25C for 10 minutes. Thereafter, while stirring, 8.7 parts of potassium-tert-butoxide was added to the mixture. Twenty minutes later, 19.2 parts of n-octyl iodide was added to the mixture, and further twenty minutes later, 8.7 parts of potassium-tert-butoxide was added thereto. Further, twenty minutes later, 19.2 parts of n-octyl iodide was added thereto, and the obtained mixture was then stirred at 25C for 2 hours. After completion of the stirring, THF was distilled away from the reaction solution, and it was then extracted with toluene-water. The toluene phase was dried over magnesium sulfate, and the toluene was then distilled away, thereby obtaining a brown tarry solid. This brown tarry solid was separated and purified by column chromatography (hexane-ethyl acetate) to obtain 30 parts of 9,9-di-n-octyl-<strong>[2523-42-4]2-iodofluorene</strong> in the form of a colorless crystal.
With potassium tert-butylate; In tetrahydrofuran; at 50℃; for 12h;Inert atmosphere;
Example 12 Synthesis of 9,9-dibutyl-2-iodo-9H- fluorene (32) Under N2 atmosphere, 0,58 parts of 2-iodo-9H- fluorene, 1.10 parts of 1-iodobutane, and 0.67 parts of potassium tert-butoxide were added into 15 parts of dry tetrahydrofuran, followed by stirring and mixing. Then, the reaction mixture was heated to 50 C. and reacted for 12 hours. After the reaction mixture was cooled, poured the water into the reaction mixture for quenching the reaction, using the diethyl ether to extract the product, and magnesium sulfate was used for dehydration. After removing the solvent, the residual was purified with column chromatography method by using hexane as an eluent, to obtain a compound (32) of the present example. This compound was in a form of a light yellow solid, and the yield of this compound was 94%.
Synthesis Example 2 20 parts of <strong>[2523-42-4]2-iodofluorene</strong> was dissolved in a mixed solution of 114 parts of dimethyl sulfoxide (DMSO) and 23 parts of tetrahydrofuran (THF), and the thus obtained mixture was then stirred at 25C for 10 minutes. Thereafter, while stirring, 8.6 parts of potassium-tert-butoxide was added to the mixture. Twenty minutes later, 14.6 parts of butyl iodide was added to the mixture, and further twenty minutes later, 8.6 parts of potassium-tert-butoxide was added thereto. Further, twenty minutes later, 14.6 parts of butyl iodide was added thereto, and the obtained mixture was then stirred at 25C for 2 hours. After completion of the stirring, THF was distilled away from the reaction solution, and it was then extracted with toluene-water. The toluene phase was dried over magnesium sulfate, and toluene was then distilled away, thereby obtaining a brown tarry solid. This brown tarry solid was separated and purified by column chromatography (hexane-ethyl acetate) to obtain 25 parts of 9,9-dibutyl-<strong>[2523-42-4]2-iodofluorene</strong> in the form of a colorless crystal.
With potassium tert-butylate; In tetrahydrofuran;
EXAMPLE 12 Synthesis of 9,9-dibutyl-<strong>[2523-42-4]2-iodo-9H-fluorene</strong> (32) Under N2 atmosphere, 0.58 parts of <strong>[2523-42-4]2-iodo-9H-fluorene</strong>, 1.10 parts of 1-iodobutane, and 0.67 parts of potassium tert-butoxide were added into 15 parts of dry tetrahydrofuran, followed by stirring and mixing. Then, the reaction mixture was heated to 50 C. and reacted for 12 hours. After the reaction mixture was cooled, poured the water into the reaction mixture for quenching the reaction, using the diethyl ether to extract the product, and magnesium sulfate was used for dehydration. After removing the solvent, the residual was purified with column chromatography method by using hexane as an eluent, to obtain a compound (32) of the present example. This compound was in a form of a light yellow solid, and the yield of this compound was 94%.
With N-butylammonium bromide; potassium hydroxide; In water; toluene; at 65℃; for 2h;
General procedure: N-butylammonium bromide (2.9 mmol, 0.9 g), potassium hydroxide (143.8 mmol,8.1 g) was dissolved in 8 mL of water and heated to 65 C in an oil bath. 2-Iodo-9H-indole (14.4 mmol, 4.2 g) and 1-bromo alkane (86.1 mmol) were dissolved in 15 mL of toluene, and then a toluene solution was added to the above aqueous solution and reacted at 65 C for 2 hours. After the completion of the reaction, the mixture was extracted with methylene chloride. The organic phase was dried over anhydrous sodium sulfate, and concentrated under reduced pressure on a rotary evaporator, and the residue was purified by column chromatography (petroleum ether).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping