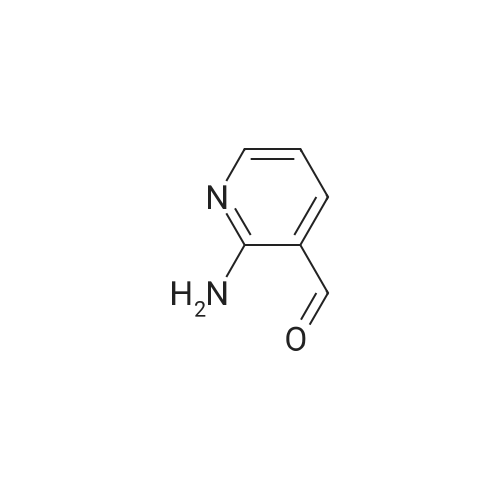
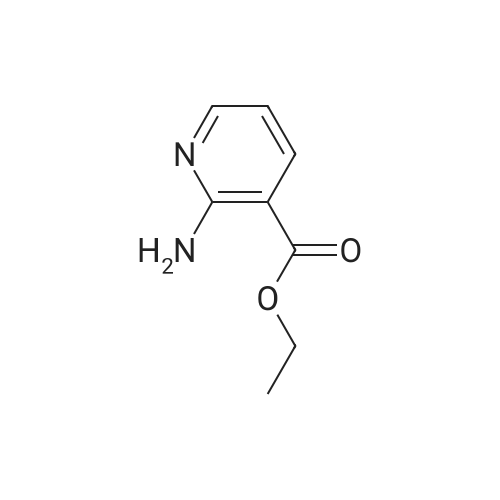
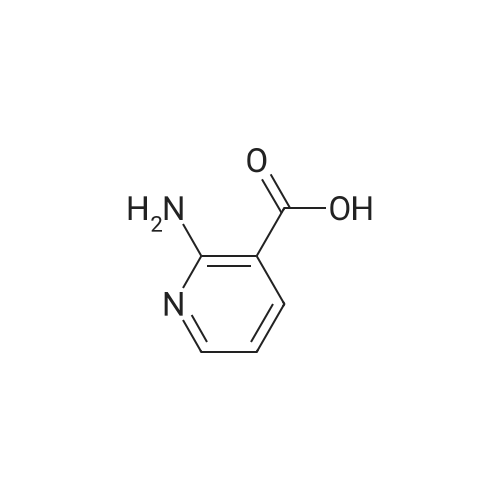
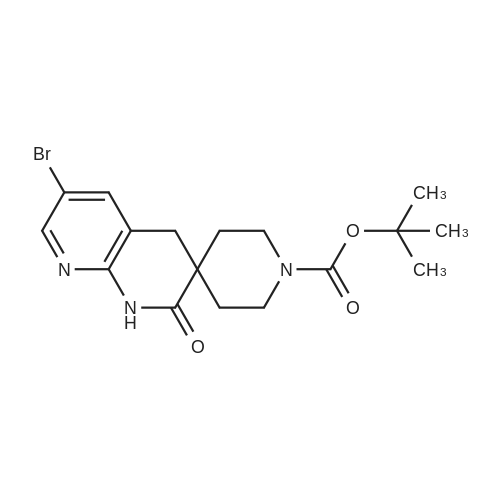
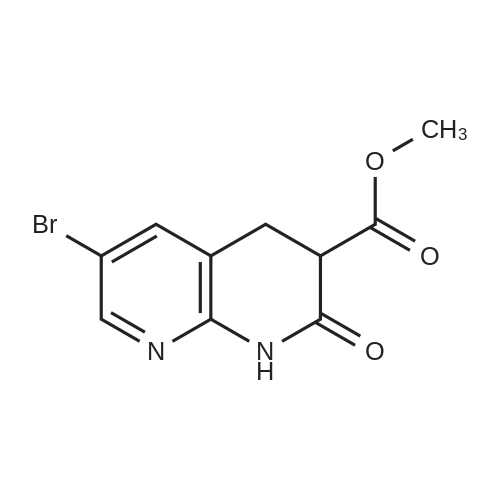
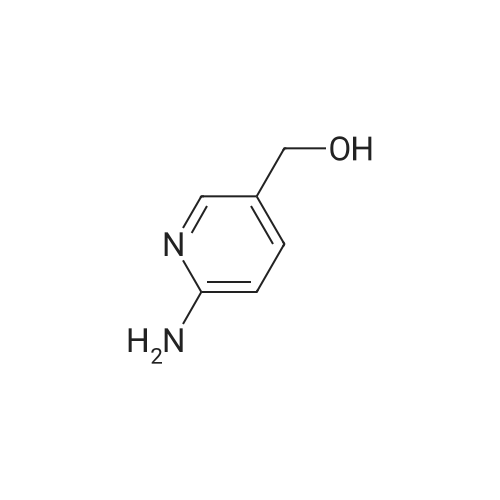
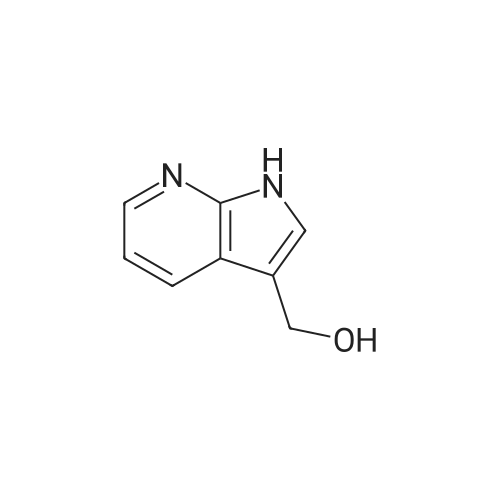
| 95% |
With sodium hydroxide; LiAlH4; In tetrahydrofuran; water; |
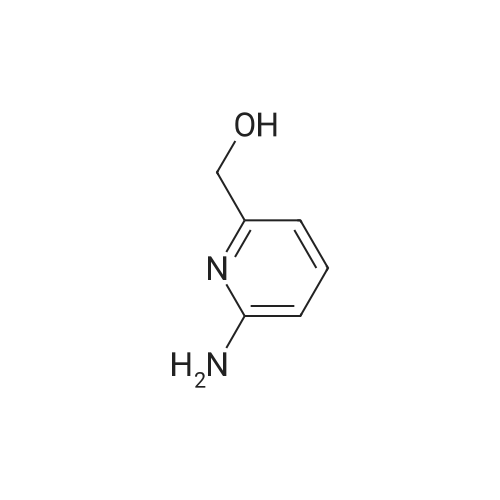
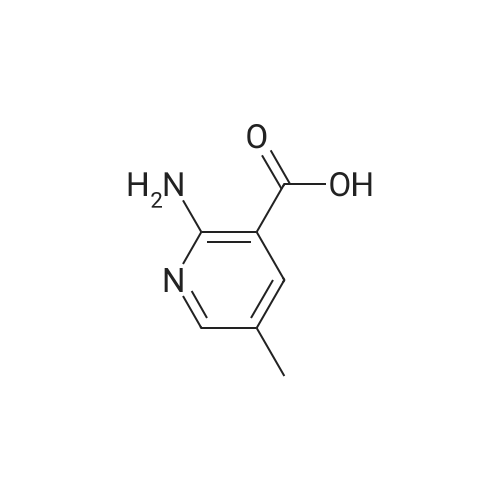
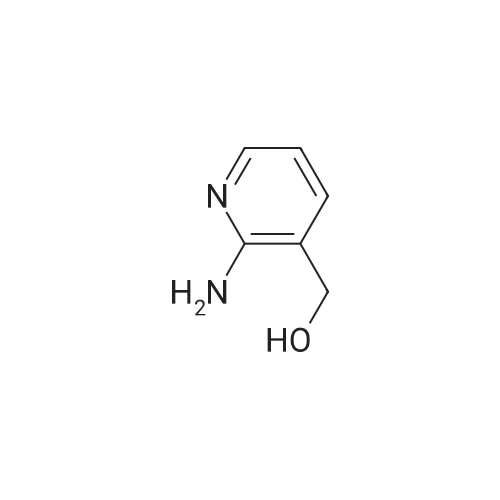
a) 2-Amino-3-(hydroxymethyl)pyridine To a stirred solution of 2-aminonicotinic acid (20 g, 145 mmole) in dry THF (200 mL) under argon was added 1.0 M LiAlH4 in THF (300 mL, 300 mmole) carefully, portionwise, through a reflux condenser, over 4 hr. The reaction became exothermic and refluxed without external heating. After the addition was complete, the reaction was heated at reflux for an additional 16 hr, then was cooled to 0 C and carefully quenched by sequential addition of H2O (12 mL), 15% NaOH in H2O (12 mL), and H2O (35 mL). The resulting thick suspension was stirred for 1 hr, then was filtered through a pad of celite. The filter pad was rinsed with THF (300 mL), and the filtrate was concentrated to dryness to give the title compound (17.04 g, 95%) as a pale yellow waxy solid: LCMS (ES) m/e 125.1 (M + H)+; 1H NMR (400 MHz, DMSO-d6) delta 7.84 (dd, 1 H), 7.37 (m, 1 H), 6.53 (dd, 1 H), 5.65 (br s, 2 H), 5.16 (t, 1 H), 4.34 (d, J = 4.6 Hz, 2 H). |
| 90% |
|
Step 1: 2-Amino-3-(hydroxymethyl)pyridineLithium aluminum hydride (12.4 g, 326.7 mmol) was portionwisely added to a suspension of 2-amino-3-carboxypyridine (30.0 g, 217.2 mmol) in THF (350 mL) at 0C. Once the addition was completed, the reaction mixture was stirred at room temperature for 15 minutes and then at reflux overnight. The mixture was then cooled to 0C and hydrolyzed by the successive addition of water (18 mL), a solution of sodium hydroxyde (18 mL) and water (30 mL) again. The resulting white suspension was filtered on Clarcel and the cake was washed with THF (200 mL) and a mixture of CHCI3/MeOH (250 mL, 9 : 1). After concentration to dryness of the filtrate, the title product was obtained as a yellow solid (24.2 g, 90%).*H NMR (DMSO-c/6, 400 MHz) : delta (ppm) : 7.84 (d, J = 4Hz, 1H), 7.37 (d, J = 7.2 Hz, 1H), 6.55-6.52 (m, 1H), 5.64 (br s, NH2), 4.34 (s, 2H). |
| 83% |
With sodium hydroxide; In tetrahydrofuran; methanol; chloroform; water; |
a) 2-Amino-3-(hydroxymethyl)pyridine To a solution of 2-aminonicotinic acid (20.5 g, 148.1 mmole) in THF was added lithium aluminum hydride (300 mL, 1.0 M in THF) over 30 minutes. The reaction solution was heated to reflux for 18 hrs and then was cooled to room temperature. The reaction was quenched by the sequential dropwise addition of H2O (11.5 mL), 15% NaOH (11.5 mL), and H2O (34.5 mL). The mixture was stirred for 15 min, then was filtered through celite, and the filter pad was washed thoroughly with THF followed by 5% CH3OH/CHCl3. The filtrate was concentrated to give the title compound (15.24 g, 83%) as a waxy light yellow solid: MS (ES) m/e 125 (M + H)+. |
| 83% |
With sodium hydroxide; In tetrahydrofuran; methanol; chloroform; water; |
a 2-Amino-3-(hydroxymethyl)pyridine To a solution of 2-aminonicotinic acid (20.5 g, 148.1 mmole) in THF was added lithium aluminum hydride (300 mL, 1.0 M in THF) over 30 minutes. The reaction solution was heated to reflux for 18 hrs and then was cooled to room temperature. The reaction was quenched by the sequential dropwise addition of H2O (11.5 mL), 15% NaOH (11.5 mL), and H2O (34.5 mL). The mixture was stirred for 15 min, then was filtered through celite, and the filter pad was washed thoroughly with THF followed by 5% CH3OH/CHCl3. The filtrate was concentrated to give the title compound (15.24 g, 83%) as a waxy light yellow solid: MS (ES) m/e 125 (M+H)+. |
|
|
To 100 mL of tetrahydrofuran solution containing 5.7 g of lithium aluminum hydride, 20 mL of tetrahydrofuran solution containing 13.8 g of 2-aminonicotinic acid was added, and heated for 2 hours under reflux. After cooling the reaction mixture back to room temperature, sodium sulfate decahydrate was slowly added until there are no bubbles, and stirred for 2 hours at room temperature. The insolubles were filtered through celite, the filtrate was concentrated under reduced pressure, and 10.7 g of (2-aminopyridin-3-yl)methanol [147-1] was obtained as a white solid. |
|
With lithium aluminium tetrahydride; In tetrahydrofuran; for 16h;Reflux; |
(i) LAH, Dry THF, reflux, 16h; (ii) Br2, HOAc, 20-35C, 3h: (iii) 48% HBr.refhix, dimethyl malonate, CH3OH, 20-35C, 16h; (v) NaOH, CH3OH, reflux, 4h, then HC1, reflux, 16h; (vi) tert-butyl acrylate, Pd(OAc)2, P(o- tolyl)3, DIEA, DMF.Propionitrile, 90C, 16h; (vii) TFA, CH2C12 , 20-35T, 4h, then 4% dioxane. HC1, 20-35C, 2h. (Reference for Step-(i): WO 2005095391 and Step-(ii-vii): J. Med. Chem. 2003, 46, 1627-1635) |
| 0.74 g |
With lithium aluminium tetrahydride; In tetrahydrofuran; diethyl ether; at 0 - 20℃; for 48h;Inert atmosphere; |
To a suspension of 2-aminonicotinic acid (5-Si, 1.58 g, 11.45 mmol) in THF (80 mL), LAH (1M in ether, 22.9 mL, 22.9 mmol) was added slowly at 0 C under argon with stirring. After completion of the addition, the ice bath was removed and the mixture was stirred at room temperature. Additional LAH (1M in ether, 11.4 mL) was added after 48 hours and the reaction was again cooled with an ice bath before saturated NH4C1 aqueous solution (20 mL) was added with stirring to form slurry. The organic layer was separated by decantation and the slurry was washed with EtOAc. The combined organic layer was washed with iN NaOH aqueous solution, brine, and dried over anhydrous Na2SO4. Solvent was removed under reduced pressure to afford (2-aminopyridin-3-yl)methanol (5-S2, 0.74 g) as an off-white solid. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping