|
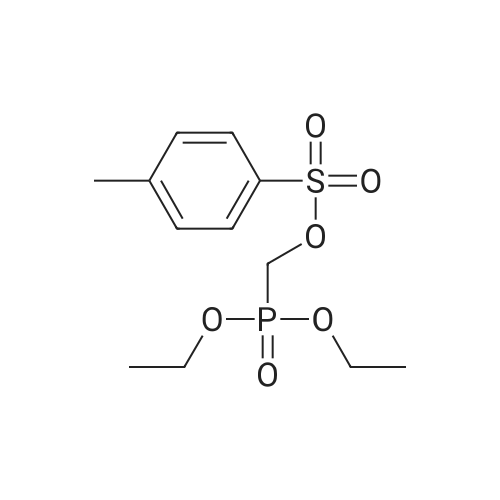
With 2,4,6-triisopropylphenylsulfonyl chloride;1-methyl-1H-imidazole; In dichloromethane; at 20℃; |
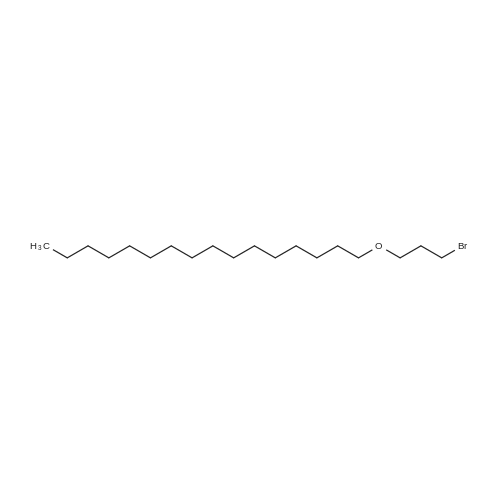
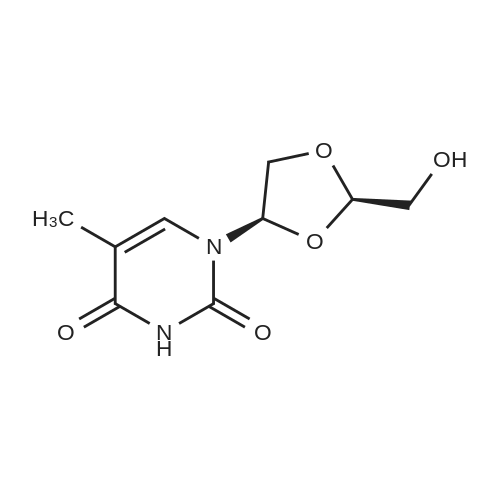
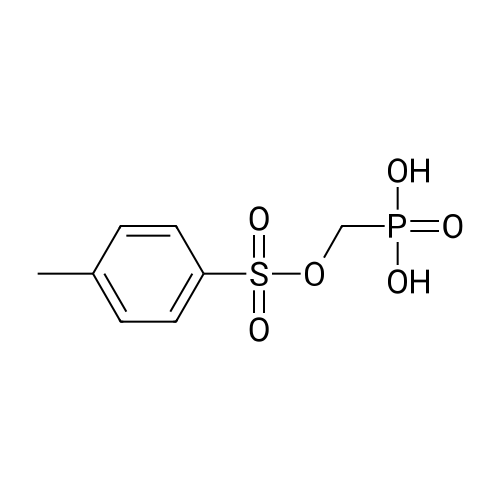
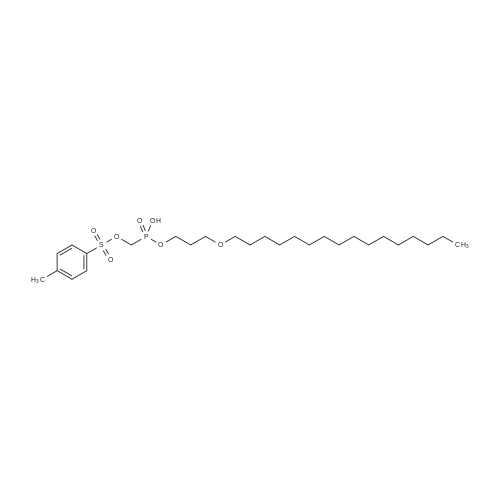
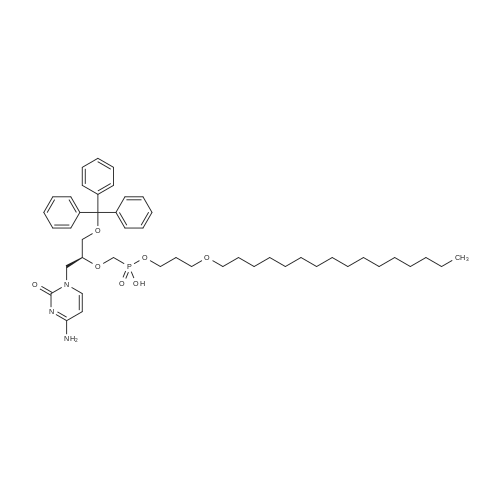
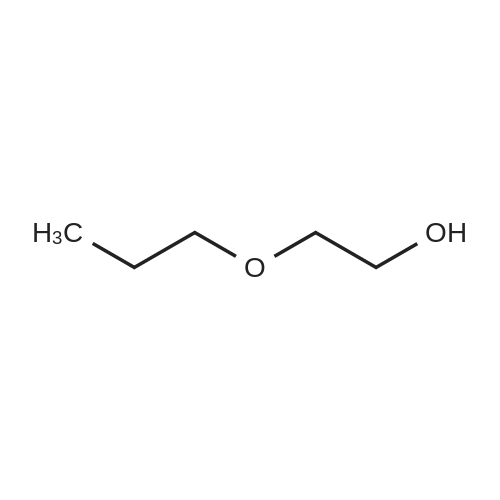
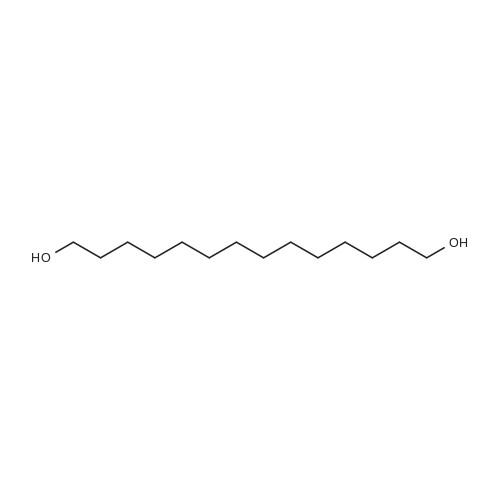
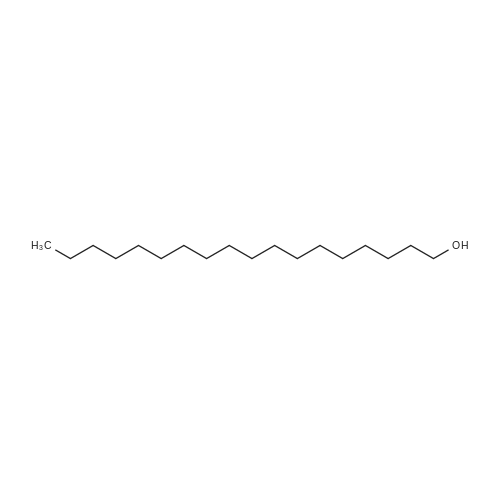
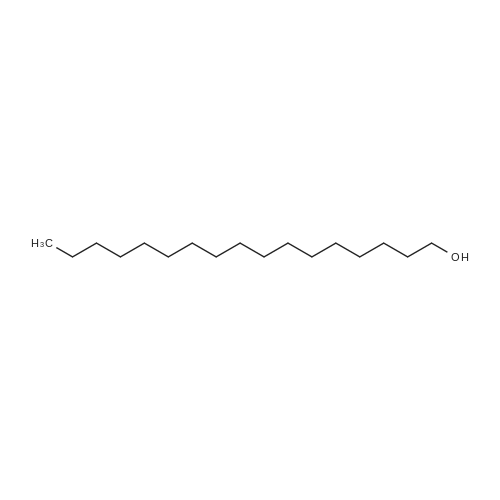
Example 8 Hexadecyloxypropyl 4-N-Benzoyl-2',3'-isopropylidenecytidin-5'-yl vinylphosphonate TPSCl (1.24 g, 4.08 mmol) is added to the mixture of vinylphosphonic acid from the example 7 (0.65 g, 1.36 mmol), hexadecyloxypropanol (0.81 g, 2.7 mmol), and 1-methylimidazole (1.45 ml, 18.3 mmol) in DCM (60 ml). The reaction mixture is stirred overnight at rt, then extracted/washed with saturated solution of NaHCO3 (2 x 100 ml), 3% aqueous citric acid (2 x 100 ml) and dried over Na2SO4. Organic phase is concentrated in vacuo and product is obtained by flash chromatography on silica gel using linear gradient of ethanol in chloroform in 78% yield (0.81 g, 1.06 mmol) in the form of colorless wax. A mixture of diastereoisomers ~ 1:1 1H NMR (600.1 MHz, CD3OD): 0.895 (m, 6H, CH3(CH2)15); 1.24-1.35 (m, 52H, CH3(CH2)13CH2CH2O); 1.368, 1.371 (2 * q, 2 * 3H, 4J = 0.6, (CH3)2C); 1.51 (m, 4H, CH3(CH2)13CH2CH2O); 1.57 (s, 6H, (CH3)2C); 1.89, 1.91 (2 * p, 2 * 2H, Jvic = 6.1, OCH2CH2CH2OCl6H33); 3.38, 3.39 (2 * t, 2 * 2H, Jvic = 6.7, CH3(CH2)13CH2CH2O); 3.47 (td, 2H, Jvic = 6.1, JH,P = 1.2, OCH2CH2CH2OC16H33); 3.49 (t, 2H, Jvic = 6.1, OCH2CH2CH2OC16H33); 4.09-4.16 (m, 4H, OCH2CH2CH2OC16H33); 4.27-4.36 (m, 4H, H-5'); 4.48 (m, 2H, H-4'); 4.91 (dd, 2H, J3',2' = 6.2, J3',4' = 3.6, H-3'); 5.06, 507 (2 * dd, 2 * 1H, J2',3' = 6.2, J2',1' = 1.8, H-2'); 5.87, 5.88 (2 * d, 2 * 1H, J1',2' = 1.8, H-1'); 6.145, 6.17 (2 * ddd, 2 * 1H, JH,P = 24.0, Jtrans = 18.4, Jcis = 12.8, =CHP); 6.257, 6.261 (2 * ddd, 2 * 1H, JH,P = 52.0, Jcis = 12.8, Jgem = 2.1, CHcisHtrans=CHP); 6.30, 6.31 (2 * ddd, 2 * 1H, JH,P = 26.0, Jtrans = 18.4, Jgem = 2.1, CHcisHtrans=CHP); 7.54 (m, 4H, H-m-Bz); 7.59 (d, 2H, J5,6 = 7.7, H-5); 7.64 (m, 2H, H-p-Bz); 7.99 (m, 4H, H-o-Bz); 8.16, 8.17 (2 * d, 2 * 1H, J6,5 = 7.7, H-5). 13C NMR (150.9 MHz, CD3OD): 14.48 (CH3(CH2)15); 23.75 (CH3(CH2)13CH2CH2O); 25.46, 25.48 ((CH3)2C); 27.27 (CH3(CH2)14CH2O); 27.43 ((CH3)2C); 30.49, 30.61, 30.78, 30.80, 30.81 (CH3(CH2)14CH2O); 31.70 (d, JC,P = 6.5, OCH2CH2CH2OC16H33); 33.09 (CH3(CH2)13CH2CH2O); 64.96, 64.98 (d, JC,P = 5.7, OCH2CH2CH2OC16H33); 66.97, 67.04 (d, JC,P = 5.6, CH2-5'); 67.42, 67.43 (OCH2CH2CH2OC16H33); 72.10 (CH3(CH2)14CH2O); 82.64, 82.635, 82.644 (CH-3'); 86.57, 86.58 (CH-2'); 88.23, 88.30 (d, JC,P = 7.2, CH-4'); 97.60, 97.71 (CH-1'); 98.51, 98.54 (CH-5); 115.10, 115.11 (C(CH3)2); 125.63, 125.67 (d, JC,P = 183.9, =CHP); 129.25 (CH-o-Bz); 129.83 (cH-m-Bz); 134.14 (CH-p-Bz); 134.70 (C-i-Bz); 138.01, 138.04 (d, JC,P = 1.9, CH2=CHP); 148.26, 148.34 (CH-6); 157.58, 157.60 (C-2); 165.58 (C-4); 169.19 (CO-Bz). 31P NMR (202.3 MHz, CD3OD): 19.62, 19.72. IR vmax(CHCl3) 3406 (w), 2928 (s), 2856 (m), 1703 (m), 1670 (s), 1628 (m), 1603 (w), 1554 (m), 1582 (w), 1497 (m, sh), 1480 (vs), 1455 (w, sh), 1400 (m), 1385 (m), 1377 (m), 1301 (m), 1248 (s, sh), 1186 (w), 1158 (m), 1121 (m), 1110 (m), 1078 (s), 1070 (s), 1028 (m), 1003 (m, sh), 989 (m), 909 (w), 865 (w), 842 (vw), 708 (w), 687 (w), 512 (w) cm-1. HR-EI C40H62N3O9P (M+H)+ calcd 759.4224; found 759.4225 |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping