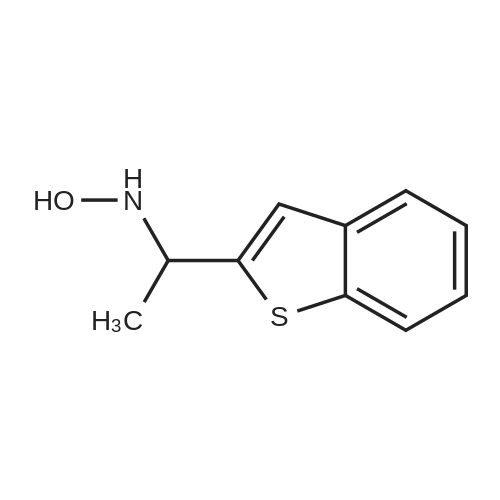
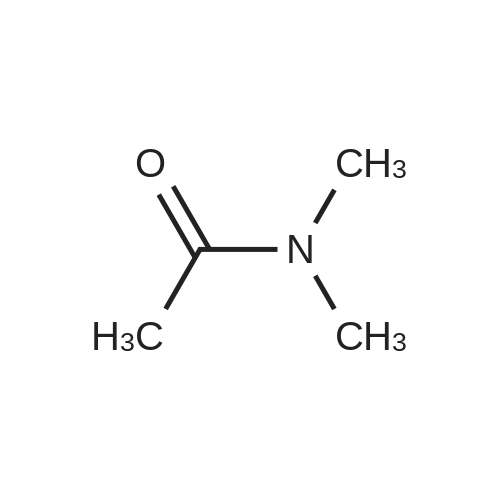
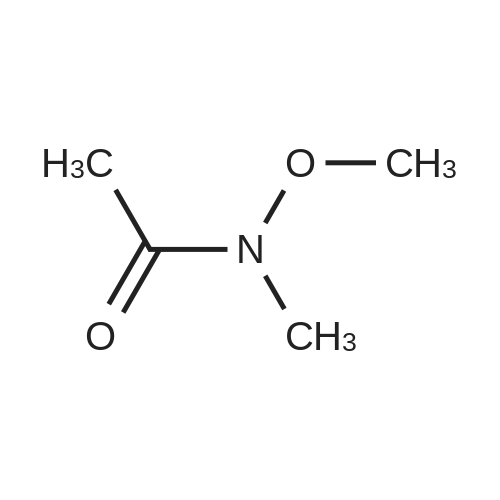
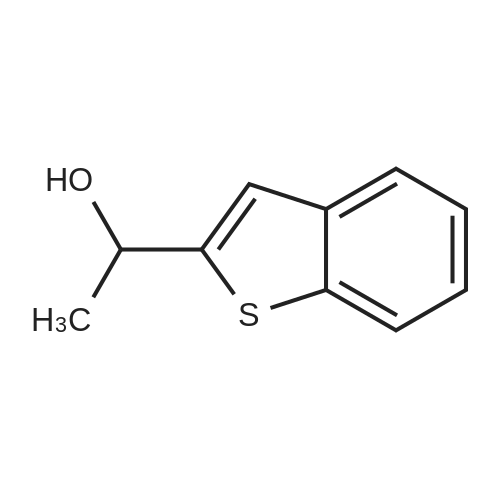
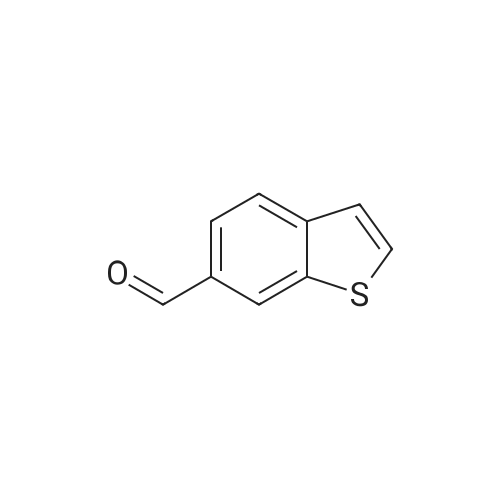
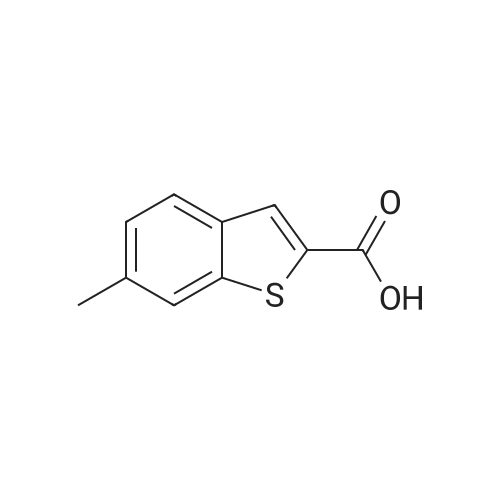
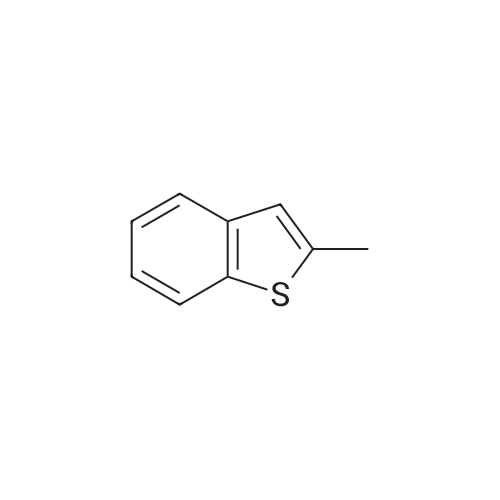
| 100% |
With tetra-N-butylammonium tribromide; In methanol; dichloromethane; at 20℃; for 15h;Product distribution / selectivity; |
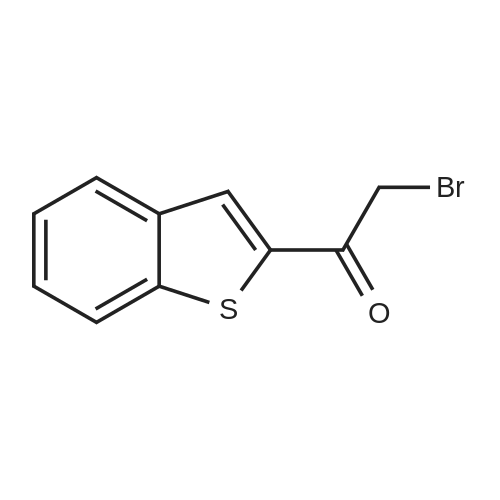
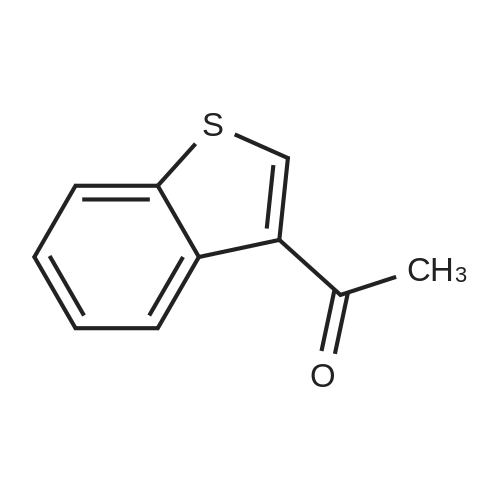
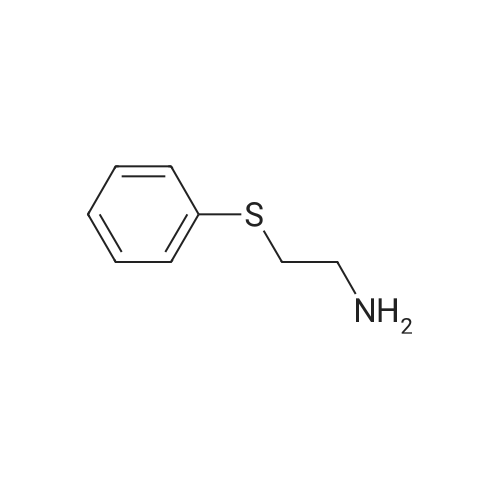
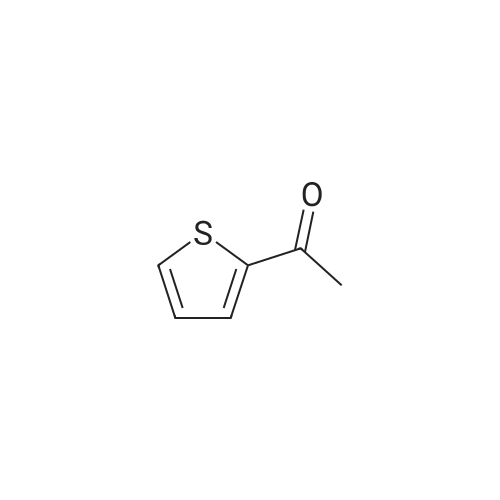
Step A: A solution of tetrabutylammonium tribromide (15.0 g, 312 mmol) in dichloromethane (80 ml) was added dropwise to a solution of 2-acetylbenzothiophene (5.0 g, 28 mmol) in dichloromethane (20 ml) and methanol (20 ml) at room temperature. At completion of the addition, the resulting red-orange solution was stirred at room temperature for 15 hours. The mixture was concentrated under vacuum and the residue was taken into ethyl acetate and water. The layers were separated and the aqueous phase was extracted with ethyl acetate. The combined organic extracts were washed with saturated aqueous sodium bicarbonate and brine and dried over anhydrous sodium sulfate to give the desired bromo compound (7.3 g, 100percent): 1HNMR (300 MHz, CDCl3) delta 8.07 (s, 1H), 7.91 (dd, J=12.0, 8.1 Hz, 2 H), 7.42-7.53 (m, 2H), 4.47 (s, 2H). |
| 89% |
With copper(ll) bromide; In methanol; chloroform; at 60℃; for 15h; |
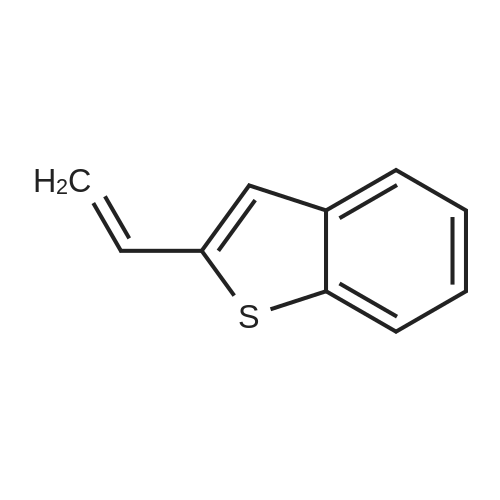
A mixture consisting of 10 mL of CHCl3, 2 mL of MeOH, 1.50 g (8.52 mmol, 1.00 mol eq) of acebenzothiophene (7) and 3.76 g (17.0 mmol, 2.00 mol eq) of CuBr2 was heated at 60 °C within 15 h. After cooling to rt the mixture was filtered and organic solution extracted by 30 mL NaHCO3 (10 percent aq solution) and brine (2 x 30 mL). Organic layer was dried over Na2SO4, filtered and evaporated by RVO and HV to yield 1.93 g (7.58 mmol, 89 percent) of product 8 |
| 84% |
With N,N,N-trimethylanilinium bromide; acetic acid; at 20 - 50℃; for 1.5h;Reflux; |
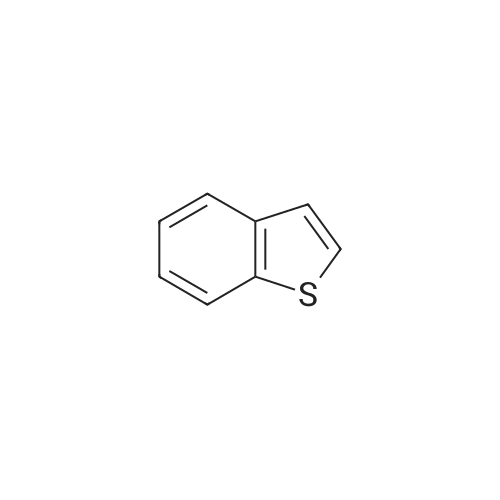
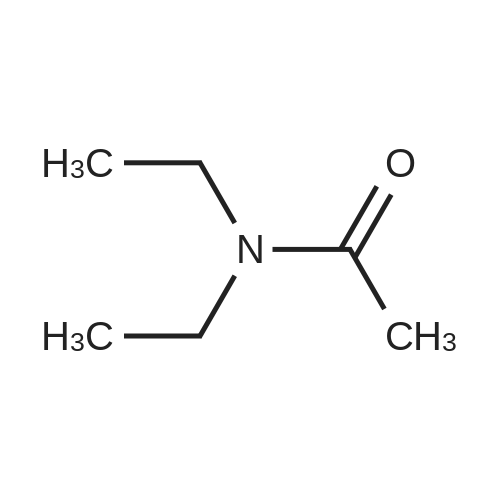
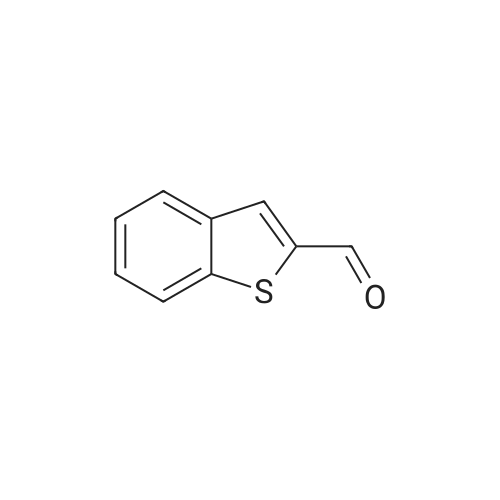
4.2.1 1-(Benzo[b]thiophen-2-yl)-2-bromoethanone (1) N-Butyl lithium (2.5 M in hexane) (4.5 ml, 11.2 mmol, 1.5 equiv) was added dropwise to a solution of benzo[b]thiophene (1 g, 7.45 mmol, 1 equiv) in anhydrous Et2O (5 ml) at -78 °C. The mixture was stirred at -78 °C for 30 min then refluxed for 1 h. A solution of DMA (7.8 mmol, 0.7 ml, 1.05 equiv) in anhydrous Et2O (5 ml) was added dropwise to the mixture. The mixture was refluxed for 2 h. After addition of aqueous HCl 1 N (20 ml), the aqueous layer was extracted with Et2O (3 * 10 ml). The combined organic layers were dried over anhydrous MgSO4, filtered and concentrated in vacuum to give a crude product which was purified by flash chromatography on silica gel (cyclohexane/EtOAc: 90:10) to provide intermediate 2-acetylbenzo[b]thiophene (1.03 g, 78percent) as a crystalline yellow solid. Trimethylphenylammonium tribromide (677 mg, 1.8 mmol, 1.2 equiv) in small portions was added into the solution of 2-acetylbenzo[b]thiophene (270 mg, 1.5 mmol, 1 equiv) in acetic acid (3 ml). The mixture was stirred at 50 °C for 30 min and then at room temperature for 1 h. The mixture was evaporated in vacuum. The solid was dissolved in CH2Cl2 (20 ml), washed with a saturated sodium bicarbonate solution (3 * 5 ml) and dried over anhydrous MgSO4. The solvent was removed under reduced pressure and the product was purified by flash chromatography on silica gel (cyclohexane/EtOAc: 95:5) to provide 1 (300 mg, 84percent) as a crystalline yellow solid. Mp = 114-116 °C (Et2O); 1H NMR (300 MHz, CDCl3) delta (ppm) 8.07 (s, 1H, H3), 7.94-7.88 (m, 2H, Harom), 7.53-7.41 (m, 2H, Harom), 4.46 (s, 2H, CH2); 13C NMR and MS were conform to the literature values (25). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping