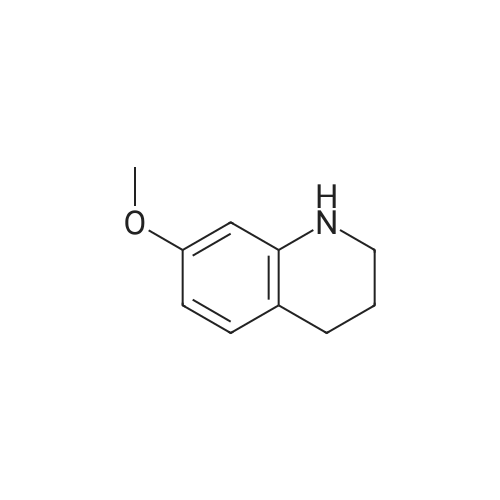
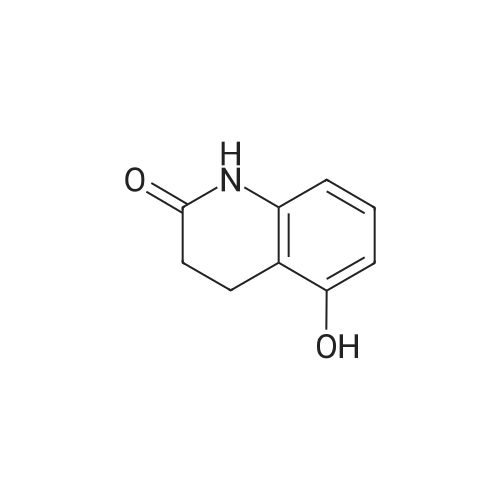
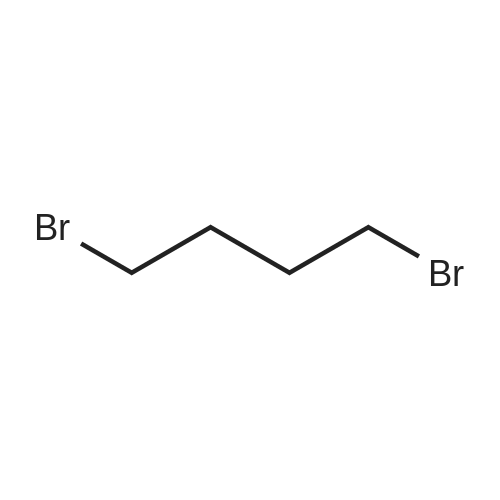
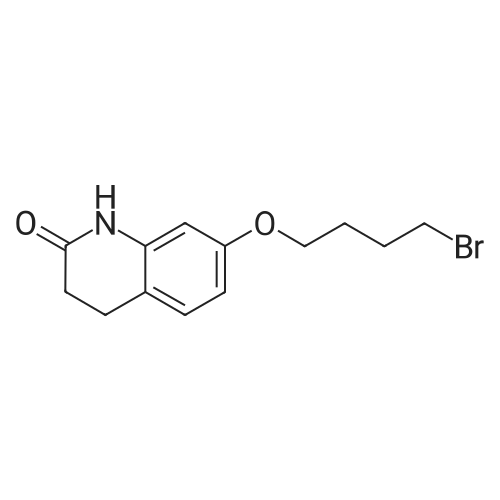
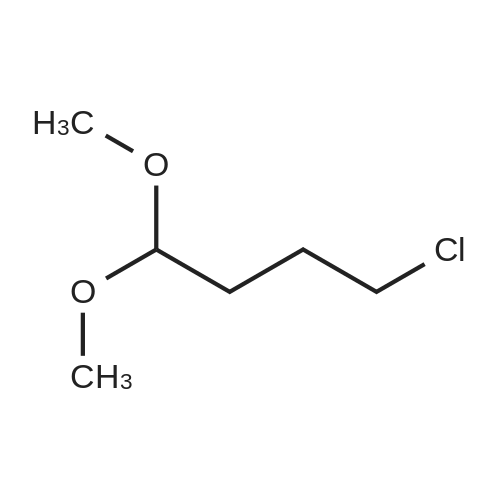
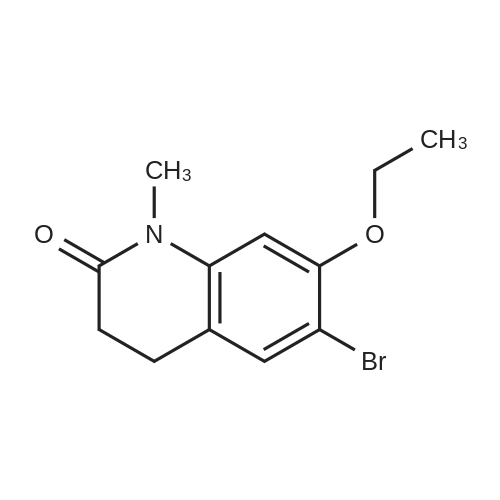
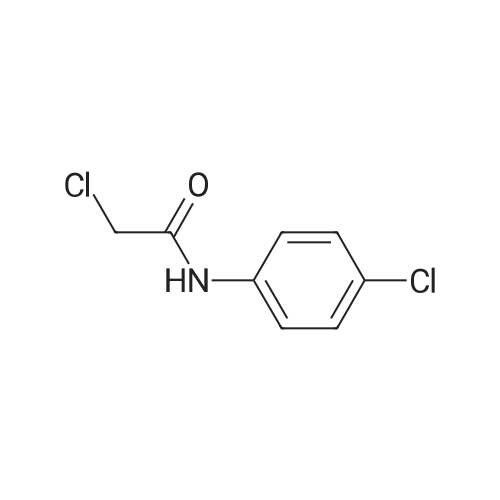
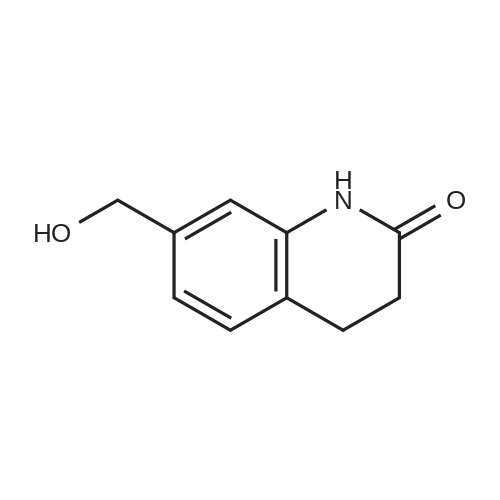
| 74% |
With potassium carbonate; In ethanol;Reflux; |
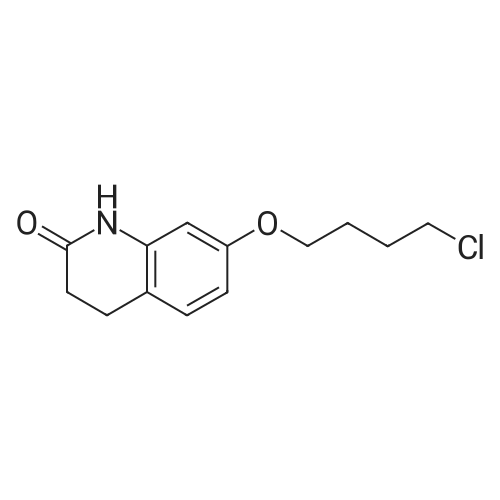
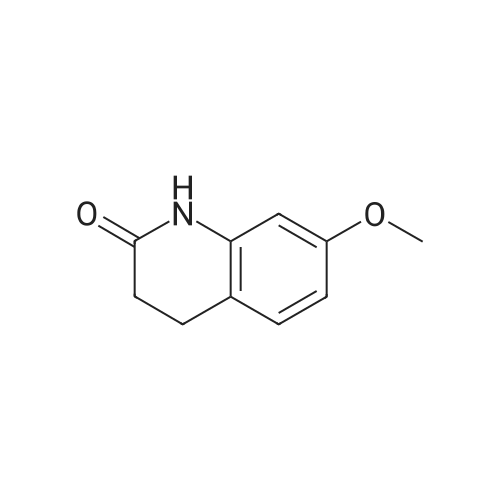
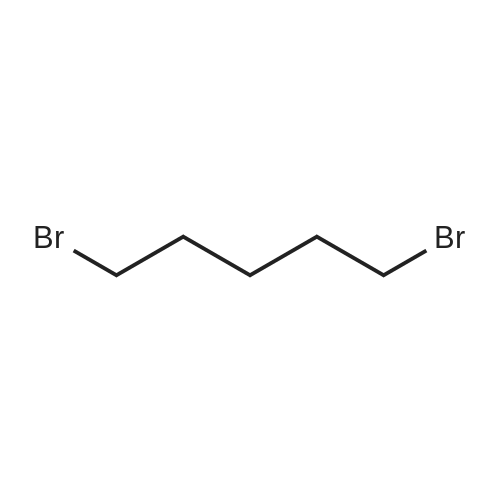
Intermediate 4:[0118] 7-hydroxy-3,4-dihydroquinolin-2(lH)-one (163 mg, 1.0 mmol), 1,4- dibromobutane (0.36 mL, 3.0 mmol) and anhydrous K2C03 (138 mg, 1.0 mmol) were dissolved in EtOH and the solution was heated to reflux overnight. The solution was diluted with water and extracted with EtOAc. The combined organic layers were washed with saturated NaHC03, brine, dried over anhydrous Na2S04, concentrated in vacuo and purified by flash chromatography on silica gel column (elution with PE EtOAc = 2: 1) to give 7-(4-bromobutoxy)-3,4-dihydroquinolin- 2(lH)-one (intermediate 4) (220 mg, 74percent) as a white solid, mp: 106-109°C. |
| 73.3% |
With potassium carbonate; In water; acetonitrile; for 4h;Reflux; |
40 g of acetonitrile was added to 82 g (0.5 mol) of 7-hydroxy-3,4-dihydrocarbostyril,to this,After adding a potassium carbonate aqueous solution (a solution obtained by dissolving 69 g (0.5 mol) of potassium carbonate in 204 g of water)further,324 g (1.5 mol) of 1,4-dibromobutane was added,Refluxed for 6 hours.This reaction solution was concentrated under reduced pressure,408 g of 2-propanol was added to the residue,After stirring at 10 ° C. or less for 1 hour,204 g of water was added,The mixture was stirred at 10 ° C. or less for 1 hour for crystallization and filtration.The obtained crystals were dried at 80 ° C.,109 g (yield: 73.3percent) of 7- (4-bromobutoxy) -3,4-dihydrocarbostyril was obtained. |
| 72% |
With potassium carbonate; sodium sulfate;Aliquat 336; In toluene; for 2h;Heating / reflux;Product distribution / selectivity; |
Example 11; Preparation of 7-(4-bromobutoxy)-3,4-dihydro-2(1H)-quinolinone (7-BBQ) by reaction of 7-hydroxy-3,4-dihydro-2(1H)-quinolinone with 1,4-dibromobutane in the Presence of Phase Transfer Catalyst; A reactor was charged with 7-hydroxy-3,4-dihydro-2(1H)-quinolinone (50 g, 0.307 mole), 1,4-dibromobutane (98percent purity, 110.8 ml, 198.7 g, 0.92 mol, 3 eq.), potassium carbonate (55.1 g, 0.399 mole, 1.3 eq.), tricaprylylmethylammonium chloride (Aliquat.(R). 336) (5 g), sodium sulfate (30 g), water (250 ml) and toluene (75 ml) and the mixture was heated under reflux for 2 hours (the reaction mixture contained 12.1percent of BQB after reaction completion). The organic phase was collected and the solvent and an excess of 1,4-dibromobutane were removed from to dryness by evaporation in vacuum. 2-Propanol (270 ml) was added to the residue thus obtained and the mixture was stirred at 5-10° C. for 5 hours. A precipitate was then collected by filtration, washed with cold 2-propanol (2.x.35 ml) and dried at 50° C. overnight to give crude 7-BBQ (82.3 g, 90.0percent yield, containing 14.3percent of BQB). The crude 7-CBQ (82.3 g) was slurried in ethyl acetate (1150 ml) at room temperature for 8 hours. A precipitate (BQB) was collected by filtration and washed with ethyl acetate (2.x.100 ml). The ethyl acetate was removed from the filtrate to dryness by evaporation under reduced pressure and the colorless solid thus obtained was dried at 60° C. overnight to give 7-BBQ (65.9 g, 72.0percent total yield, containing 1.9percent of BQB). Melting point=110-111° C. |
| 72% |
With potassium carbonate; In N,N-dimethyl-formamide; at 60℃; for 12h; |
Example 1; 7-(4-Bromobutoxy)-3,4-dihydroquinolinone-2(1H)-one (3) (Scheme 1); To a stirred suspension of anhydrous potassium carbonate (K2CO3) (10.50 g, 0.075 mol) in 100 mL of anhydrous N,N-dimethylformamide (DMF) was added 7-hydroxyquinolinone 1 (10.00 g, 0.06 mol) followed by 1,4-dibromobutane 2 (25.91 g, 0.12 mol). The resulting mixture was heated at 60° C. for 12 h. The progress of the reaction was monitored by thin layer chromatography (TLC) technique. After cooling to room temperature, the reaction mixture was filtered through a sintered funnel and the precipitate was washed with ethyl acetate (25 mL.x.2). The combined filtrate was concentrated on a rotavapor. The residue was diluted with 250 mL of ethyl acetate, washed with water (100 mL.x.2), dried over anhydrous magnesium sulfate and evaporated the solvent. The residue was triturated with hexane and then filtered the precipitate to give the pure title compound 3. White solid, 12.88 g (72percent). 1H NMR (400 MHz, CDCl3): delta 1.92-1.97 (m, 2H); 2.05-2.10 (m, 2H); 2.64 (t, J=9.2 Hz, 2H); 2.92 (t, J=9.2 Hz, 2H); 3.50 (t, J=7.6 Hz, 2H); 3.98 (t, J=7.6 Hz, 2H); 6.37 (d, J=3.2 Hz, 1H); 6.50-6.54 (m, 1H); 7.04 (d, J=11.2 Hz, 1H), 8.61 (broad s, 1H). |
| 68.7% |
With potassium carbonate; In acetonitrile; at 60 - 65℃;Inert atmosphere; |
General procedure: The appropriate dibromoalkane derivative 2a?2d (4.4 mmol)was added to a mixture of the starting material 7-hydroxy-3,4-dihydro-2(1H)-quinoline (1) (2.0 mmol), anhydrous K2CO3(290 mg, 2.1 mmol) in CH3CN (8 mL). The reaction mixture washeated to 60?65 C and stirred for 8?10 h under an argon atmosphere.After complete reaction, the solvent was evaporated underreduced pressure. Water (30 mL) was added to the residue and themixture was extracted with dichloromethane (30 mL 3). Thecombined organic phases were washed with saturated aqueoussodium chloride, dried over sodium sulfate, and filtered. The solventwas evaporated to dryness under reduced pressure. The residuewas purified on a silica gel chromatography usingdichloromethane/acetone (50:1) as eluent to give the intermediates3a?3d. |
| 55% |
With potassium hydroxide; In isopropyl alcohol; for 16h;Reflux; |
Potassium hydroxide (0.120 g, 2.139 mmol) was added to a solution of 7-hydroxy-3,4-dihydro-2(1H)-quinolinone (0.200 g, 1.226 mmol) in 2-propanol (3 mL). After a clear solution was obtained, 1,4-dibromobutane (0.44 mL, 3.685 mmol) was added and the mixture was refluxed for 16 h. It was then cooled, diluted with ethyl acetate and filtered. The filtrate was concentrated to provide a crude residue which was purified by chromatography on neutral alumina (20percent acetone in dichloromethane) to give the title compound as a white solid (0.200 g, 55percent). 1H NMR (400 MHz, CDCl3) delta 1.87-1.98 (m, 2H), 2.00-2.10 (m, 2H), 2.62 (t, J=7.7 Hz, 2H), 2.90 (t, J=7.5 Hz, 2H), 3.49 (t, J=6.8 Hz, 2H), 3.97 (t, J=6.0 Hz, 2H), 6.29 (d, J=2.1 Hz, 1H), 6.52 (dd, J=8.3, 2.5 Hz, 1H), 7.05 (d, J=8.3 Hz, 1H), 7.69 (br, exchangeable with D2O, 1H); IR (KBr) upsilon 2928, 1677, 1631, 1594, 1383 cm-1; MS 298, 300 [(M+1), (M+3)]. |
| 32% |
With potassium carbonate; In N,N-dimethyl-formamide; at 50℃; |
To a mixture of 7-hydroxy-3,4-dihydroquinolin-2(lH)-one (23-1) (14.0 g, 85.9 mmol, 1.0 eq) and potassium carbonate (17.8 g, 129 mmol, 1.5 eq) in N,N-dimethylformamide (200 mL) was added 1,4-dibromobutane (46.0 g, 215 mmol, 2.5 eq). The mixture was stirred at 50 °C overnight. The reaction mixture was poured into 500 mL of water, extracted with ethyl acetate (200 mL x 2), dried over sodium sulfate and concentrated in vacuo to give the crude product, which was purified by silica gel column chromatography (petroleum ether : ethyl acetate = 5: 1) to give compound 23-2 as a white solid (8.2 g, yield: 32 percent). MS (ESI): m/z 298, 300 [M+H]+. |
|
With potassium carbonate; In water; |
Reference Example 6 To a solution of 4.06 g of potassium carbonate with 400 ml of water was added 40 g of 7-hydroxy-3,4-dihydrocarbostyril and 158 g of 1,4-dibromobutane, then the mixture was refluxed for 3 hours. The reaction mixture thus obtained was extracted with dichloromethane, dried with anhydrous magnesium sulfate, then the solvent was removed by evaporation. The residue thus obtained was purified by means of a silica gel column chromatography (eluent: dichloromethane), and recrystallized from n-hexane-ethanol to yield 50 g of 7-(4-bromobutoxy)-3,4-dihydrocarbostyril. |
|
With potassium carbonate; In acetone; at 60 - 65℃; for 16h; |
Example-1 Preparation of 7-(4-bromobutoxy)-3,4-dihydrocarbostyryl The solution of 7-(4-hydroxy)-3,4-dihydrocarbostyryl (125 g), potassium carbonate (158.75 g) and 1,4-dibromobutane (662.5 g) was prepared in acetone (1375 mL) and refluxed at 60° C.-65° C. for 16 hours. After the completion of the reaction, the reaction mixture was cooled to 15° C.-20° C. and stirred for 1.0 h. Isolated solid was filtered and washed with chilled acetone (250 mL). The filtrate was distilled under vacuum at 60° C. to remove access of acetone and at 120° C. to remove traces of 1,4-dibromobutane. The oily residue were cooled at 40° C.-45° C. and treated with methylene dichloride and stirred for 15-20 minutes till clear solution was obtained. The reaction mixture was treated with 5percent NaOH and process water at 25° C.-35° C. and the organic layer was separated. The organic layer was further charcoalized and filtered through cilete bed. The filtrate was subjected to distillation under vacuum to remove excess of methylene dichloride. The resulting mass was treated with ethyl acetate (125 mL) and cyclohexane (500 mL) at 25° C.-35° C. and stirred for 1 h. The solid thus obtained was filtered, washed with cyclohexane and suck dried. The product was dried in hot air oven at 55° C.-60° C. |
|
|
Example-1Step A:Preparation of Pure 7-(4-Bromobutoxy)-3,4-Dihydro-2(1H)-Quinolinone; (IV):7-Hydroxy-3,4-dihydro-2(1H)-quinolinone (20 g, 0.153 mol) was added to a solution of potassium carbonate (25 g, 0.181 mol) in DM water (400 ml) at 25-35° C. and the contents were heated to 80-90° C. to obtain a clear solution. 1,4-Dibromobutane (200 ml) was added to the reaction mass, heated to reflux temperature (100-105° C.) and stirred for 7 hours at the same temperature. After completion of reaction, reaction mass was cooled to ambient temperature and organic layer was separated. Organic layer containing 7-(4-bromobutoxy)-3,4-dihydro-2(1H)-quinolinone having 5percent 1,4-bis[3,4-dihydro-2(1H)quinolinon-7-oxy]butane (by HPLC, by area normalization) was washed with precooled 5percent w/v aqueous sodium hydroxide (75 ml) at 15-20° C., to remove unreacted 7-hydroxy-3,4-dihydro-2(1H)-quinolinone. Organic layer was concentrated at 90-105° C. under reduced pressure varying from 50 to 5 mm of Hg to dryness. The concentrated mass was diluted with toluene (2000 ml) and heated to 60° C. Silica gel (50 g) was added and stirred for 30 min at 60-70° C. Silica gel was removed by filtration and the filtrate was concentrated at 60-80° C. under reduced pressure varying from 200 to 10 mm of Hg. The resulting concentrated mass having 0.3percent of 1,4-bis[3,4-dihydro-2(1H)quinolinon-7-oxy]butane (by HPLC, by area normalization) was diluted with hexanes (50 ml) and stirred for 30 minutes to crystallize the product. Product was filtered and washed with hexanes.Yield: 18 gChromatographic purity: 97.89percent (by HPLC, by area normalization)1,4-Bis[3,4-dihydro-2(1H)quinolinone-7-oxy]butane content: 0.33percent |
|
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In acetonitrile; at 40 - 45℃; for 24h;Product distribution / selectivity; |
Example 1Preparation of 7-(4-bromobutoxy)-3,4-dihvdrocarbostyril7-Hydroxy-3,4-dihydrocarbostyril (60gm) and potassium carbonate (76.3 gm) were taken in acetonitrile (1200ml) at room temperature. To this tetra butyl ammonium iodide (13.7 gm) and 1 ,4-dibromobutane (238.5gm) were added and heated at 40- 45°C for 24 hours. Reaction mass was cooled upto room temperature and was filtered off. The resulting filtrate was distilled off under vacuum. The resultant mass was cooled to 25-30°C and cyclohexane (300 ml) was added under stirring. The resulting solid was filtered off and was dried. The resulting solid was taken in water and was stirred for few minutes. The solid was filtered and dried under vacuum at 55-60°C for 20 hours to obtain title compound.Yield: 73-75percent; Purity: 93-95percent |
| 93 g |
With potassium carbonate; In water; N,N-dimethyl-formamide; at 25 - 30℃; for 19h; |
A mixture of 7-hydroxy-3,4-dihydroquinolin-2(1H)-one (75 gm), 1 ,4-dibromobutane (196.6 gm), potassium carbonate (90 gm), dimethylformamide (375 ml) and water (37.5 ml) was stirred for 19 hrs at 25-30°C. Dichloromethane was added to the reaction mixture at 25- 30°C and stirred for 30 min at the same temperature. Filtered the unwanted solid and extracted the desired compound trapper in the unwanted solid by using dichloromethane. Combined the obtained dichloromethane solution with the above obtained filtrate. Water was added to the reaction mixture at 25-30°C and stirred for 15 min at the same temperature. Both the organic and aqueous layers were separated and extracted the aqueous layer with dichloromethane. Combined the organic layers and washed with water. Distilled off the solvent completely from the organic layer and co-distilled with cyclohexane. Cyclohexane (75 ml) was added to the obtained compound at 25-30°C and stirred the reaction mixture for 2 hrs at the same temperature. Filtered the solid, washed with cyclohexane and dried the material to get the title compound. Yield: 93.0 gm. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping