* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: at 5 - 10℃; Stage #2: at 5 - 25℃; for 6 h;
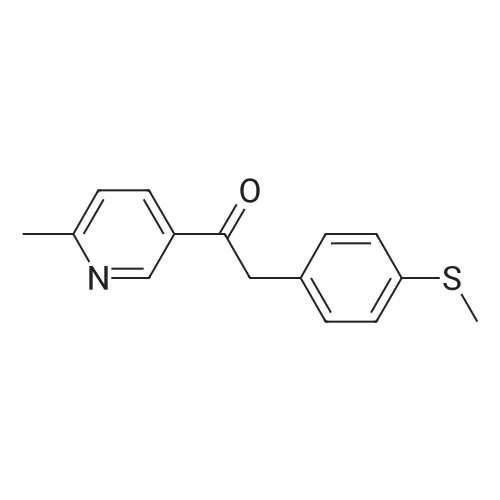
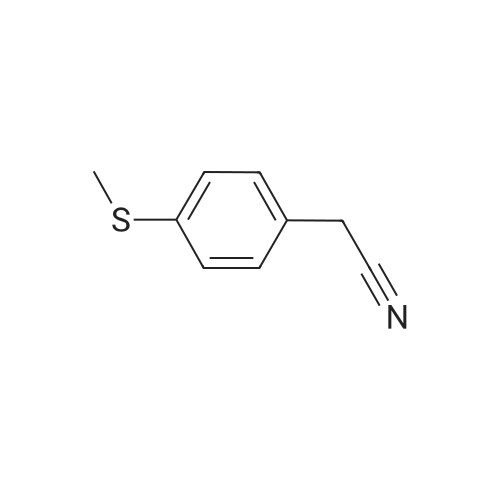
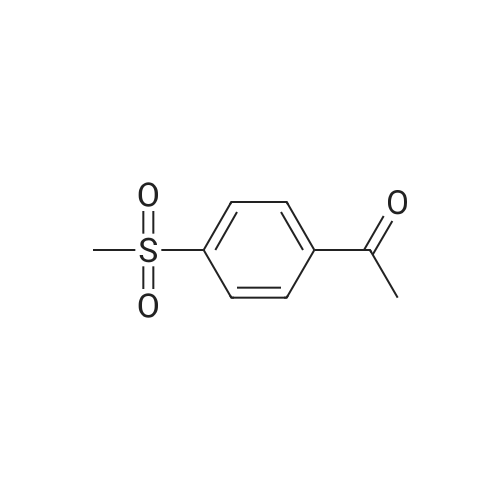
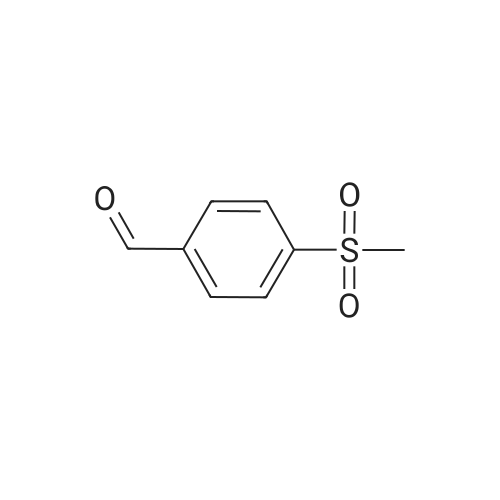
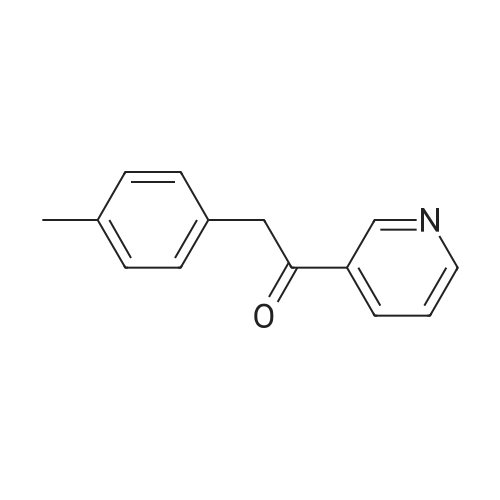
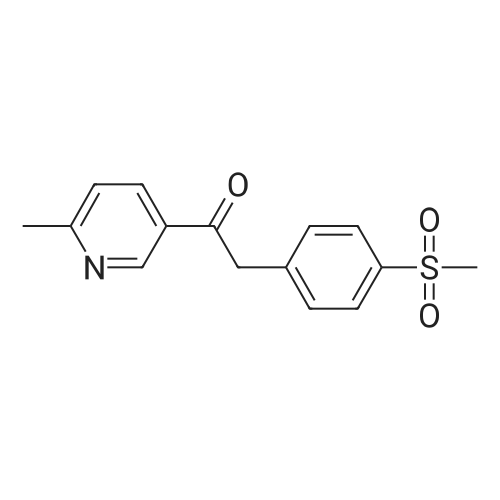
100 g 1-(6-methylpyridin-3-yl)-2-[4-(methylsulfanyl)phenyl]ethanone of formula (VI) (1.0 equiv.), 150 mL ice-cold acetic acid (1.5 vol) and 30 mL methanesulfonic acid (1.2 equiv, 0.3 vol-96.11 g/mol; 1.481 g/ml) are introduced into a 3000 ml flask provided with high-temperature thermometer, coolant and dropping funnel.The reaction mixture is quenched to 5-10 ° C.120 mL hydrogen peroxide 30percent w/w (3 equiv, 1.20 vol; 34.02 g/mol: 1.13 g/ml) are added under stirring at 5-10° C. The reaction mixture is stirred at 20-25° C. for at least 6 hours. At the end of the reaction, it is quenched to 0-5° C. and a solution consisting of 300 g sodium thiosulfate (3 wt) and water (10 vol) is added in portions.Approximately 180-260 ml of an aqueous solution of 30percent sodium hydroxide are added up to reach a pH of about 5.5-6.5.It is stirred at 20-25° C. for 2 h and the suspension is filtered. The solid is washed with 2x400 mL water, then the solid is dried at 40° C. in a vacuum for at least 12 h. 105.7 g product are obtained, for a molar yield of 94percent HPLC purity, equal to 97.5percent (A percent).
80.4%
With sodium tungstate; sulfuric acid; dihydrogen peroxide In methanol at 55℃; for 1 h;
To the reaction flask was added C50g, 2N sulfuric acid 3.7ml and 500ml methanol, 1. Lg sodium tungstate, heated to 55 degrees, droppingAfter stirring, the mixture was stirred for 1 hour, 500 ml of water was added, cooled to room temperature, filtered, and the filter cake was washed with 500 ml of water and dried to obtain 45.2 g, yield: 80.4percent purity: 98.1percent.
72%
With sodium tungstate; dihydrogen peroxide In ethyl acetate at 30℃; for 1 h; Flow reactor
In a continuous flow reactor, enter the first material input port with 1-(6-methylpyridin-3-yl)-2-(4-methanesulfonylphenyl)ethanone in ethyl acetate (1 mol /L concentration, 1mL/s flow rate), The second input port was fed with an aqueous solution of sodium tungstate (0.1 mol/L concentration, 0.1 mL/s flow rate) and heated to 30 degrees via a heat exchanger. Enter a 10.5percent hydrogen peroxide solution (1 mL/s flow rate, 3 equivalents) at the third material input port and mix with the solution shown in the first inlet. After mixing the two solutions, pass 20-30 degrees in 60 minutes. Cooler, Then, a sodium thiosulfate solution (2 mol/L concentration, 1 mL/s flow rate) was input from the fourth material input port, and the upper organic phase was collected and output in a continuous flow dispenser. The organic phase was collected into a container and, after reaching 20 liters, a post-treatment of concentration and crystallization was carried out. The yield of the two-step reaction was 72percent and the purity was 98.5percent.
21 g
With sodium tungstate; sulfuric acid; dihydrogen peroxide; Aliquat 336 In dichloromethane; water at 18 - 20℃;
Product (wet) mass of 1-(6-methyl-3-pyridinyl)-2-[(4-Methylthio)phenyl]ethanone (V) was dissolved in dichloromethane (185 ml), stirred allowed to settle and aqueous layer was separated. Dichloromethane solution of the product was directly forwarded to the subsequent synthetic stage of oxidation with a mixture of concentrate Sulphuric acid (4.10 gm.)+water (4.50 ml) and stirred for 15-20 minutes. A solution of sodium tungstate (0.74 gm) in water (14.0 ml) was also charged and reaction mass was stirred for 10 minutes followed by addition of mixture of methyl-tri-n-octyl chloride (0.80 gm) in dichloromethane (8.0 ml). The reaction mixture was stirred for 10 minutes, and cooled to 18°-20° C. Mixture of 50percent Hydrogen peroxide (19.50 gm) and water (12.50 ml) was gradually added within 45 minutes at 18°-20° C. temp. Reaction mass was maintained for several hours and monitored by TLC examination, till the spot corresponding to 1-(6-methyl-3-pyridinyl)-2-[(4-methylthio)phenyl]ethanone (V) was practically absent by TLC. Reaction mass pH was set between 6.95 to 7.10 using mixture of diluted ammonia solution under string within 30 minutes at 25°-30° C. It was stirred for 10 minutes, aqueous layer separated and extracted with dichloromethane (130 ml). Combined dichloromethane layer containing product was washed with water (110 ml), dried over anhydrous sodium sulphate and filtered. Dichloromethane at atmospheric pressure to obtained. Vacuum was finally applied and the mass was degassed Then isopropyl alcohol (96 ml) was added, stirred mass and cooled to 25°-30° C. followed by chilling to 0°-5° C. temp, maintained for an hour, filtered, washed with chilled isopropyl alcohol and dried at 50°-60° C. (yield 21 gms, m.p. 176°-180° C., purity by HPLC-92.50percent).
Reference:
[1] Patent: US2012/232281, 2012, A1, . Location in patent: Page/Page column 7
[2] Journal of Organic Chemistry, 2000, vol. 65, # 25, p. 8415 - 8420
[3] Patent: US6369275, 2002, B1, . Location in patent: Page column 17
[4] Patent: CN104045596, 2017, B, . Location in patent: Paragraph 0056-0057
[5] Patent: CN108689917, 2018, A, . Location in patent: Paragraph 0036; 0044; 0051-0054; 0062-0065
[6] Patent: WO2012/66570, 2012, A2, . Location in patent: Page/Page column 21
[7] Patent: EP2497767, 2012, A1, . Location in patent: Page/Page column 100-11
[8] Patent: US2013/245272, 2013, A1, . Location in patent: Paragraph 0100
2
[ 36357-38-7 ]
[ 3466-32-8 ]
[ 221615-75-4 ]
Yield
Reaction Conditions
Operation in experiment
91%
With potassium phosphate In 1-methyl-pyrrolidin-2-one at 100℃; for 18 h; Inert atmosphere
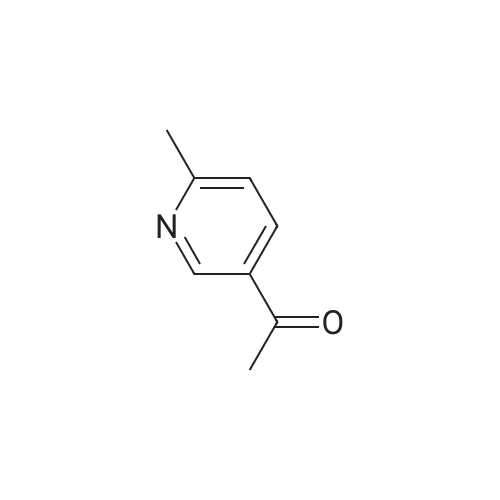
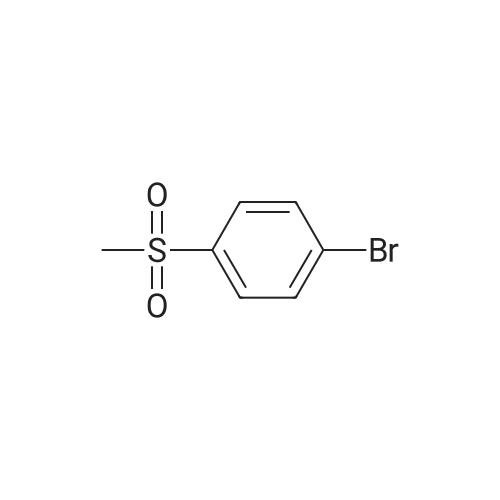
Pd(acac)2 (6.1 mg, 0.02 mmol, 0.5 mol percent) and Xantphos (23.2 mg, 0.04 mmol, 1 mol o) are introduced into a flared flask provided with coolant. 4-bromophenylmethylsulfone of formula (III, XBr) (1.17 g, 5 mmol), acetylpicoline of formula (II) (541 mg, 4 mmol) and K3PO4 (2.55 g, 12.0 mmol, 3 eq) are added thereto. Once the argon atmosphere has been stabilized with vacuum-argon cycles, anhydrous and degassed NMP (15 ml) is added with a syringe. The mixture is then kept stirred under stirring in an argon atmosphere for 18 h at 100° C. The conversion is quantitative. The reaction mixture is diluted with a saturated solution of NaHCO3 (50 mL) and extracted with AcOEt (4.x.50 mL). The combined organic phases were washed with an aqueous solution saturated with NaHCO3 (30 mL), anhydrified on MgSO4 and concentrated in a vacuum. The residue was purified by silica gel chromatography using AcOEt/cyclohexane as eluent in a gradient from 5:5 to 10:0. 1.05 g product were obtained, for a molar yield of 91percent as a white crystalline solid.
75%
With tris-(dibenzylideneacetone)dipalladium(0); [5-(diphenylphosphanyl)-9,9-dimethyl-9H-xanthen-4-yl]diphenylphosphane; sodium t-butanolate In toluene for 5 h; Inert atmosphere; Reflux
Xantphos 0.00267 g (0.0046 mmol) and Pd2(dba)3 0.00177 g (0.0031 mmol) in 15 ml of anhydrous toluene were charged in a reactor under inert atmosphere. 4-bromophenyl methyl sulfone 0.724 g (3.078 mmol) and 3-acetyl-6-methyl pyridine 0.416 g (3.079 mmol) were then added. The mixture was heated to reflux and a suspension of t-BuONa 0.71 g in 15 ml of anhydrous toluene was added dropwise over about 4 h. After about 1 h from completion of the addition, the reaction mixture was cooled to 20 °C and a solution of diluted hydrochloric acid to acidic pH was added. The aqueous phase was separated and added dropwise over 1 h to a mixture of water 8.3 g, ethyl acetate 15.3 g and sodium bicarbonate 2.1 g at 60 °C. At addition completed and after maintaining the temperature at 60 °C for 1 h, it was checked that the pH was between 4 and 7, the mixture was cooled to 20 °C filtered and dried under vacuum at 50 °C. 0.67 g of the compound of formula 1 was obtained with a yield of 75 percent.
75%
With tris-(dibenzylideneacetone)dipalladium(0); [5-(diphenylphosphanyl)-9,9-dimethyl-9H-xanthen-4-yl]diphenylphosphane; sodium t-butanolate In toluene for 4 h; Inert atmosphere; Reflux
Xantphos 0.00267 g (0.0046 mmol) and Pd2(dba)3 0.00177 g (0.0031 mmol) in 15 ml of anhydrous toluene were charged in a reactor under inert atmosphere. 4-bromophenyl methyl sulfone 0.724 g (3.078 mmol) and 3-acetyl-6-methyl pyridine 0.416 g (3.079 mmol) were then added. The mixture was heated to reflux and a suspension of t-BuONa 0.71 g in 15 ml of anhydrous toluene was added dropwise over about 4 h. After about 1 h from completion of the addition, the reaction mixture was cooled to 20°C and a solution of diluted hydrochloric acid to acidic pH was added. The aqueous phase was separated and added dropwise over 1 h to a mixture of water 8.3 g, ethyl acetate 15.3 g and sodium bicarbonate 2.1 g at 60°C. At addition completed and after maintaining the temperature at 60°C for 1 h, it was checked that the pH was between 4 and 7, the mixture was cooled to 20°C filtered and dried under vacuum at 50 °C. 0.67 g of the compound of formula 1 was obtained with a yield of 75 percent.
72%
With tris-(dibenzylideneacetone)dipalladium(0); [5-(diphenylphosphanyl)-9,9-dimethyl-9H-xanthen-4-yl]diphenylphosphane; sodium t-butanolate In toluene for 5 h; Inert atmosphere; Reflux
EXAMPLE 1 [0068] Synthesis of 1-(6-methylpyridin-3-yl)-2-[(4-methylsulfonyl)-phenyl]-ethanone. [0069] Xantphos 0.027 g (0.0477 mmol) and Pd2(dba)3 0.0182 g (0.0198 mmol) in 100 ml of anhydrous toluene were charged in a reactor under inert atmosphere. 4-bromophenyl methyl sulfone 9.3 g (39.7 mmol) and 3-acetyl-6-methyl pyridine 5.4 g (39.7 mmol) were then added. The mixture was heated to reflux and a suspension of t-BuONa 8.4 g in 100 ml of anhydrous toluene was added dropwise over about 4 h. After about 1 h from completion of the addition, the reaction mixture was cooled to 20° C. and a solution of diluted hydrochloric acid to acidic pH was added. The aqueous phase was separated and added dropwise over 1 h to a mixture of water 83.3 g, ethyl acetate 153 g and sodium bicarbonate 20.1 g at 60° C. At addition completed and after maintaining the temperature at 60° C. for 1 h, it was checked that the pH was between 4 and 7, the mixture was cooled to 20° C., filtered, and dried under vacuum at 50° C. 8.3 g of the compound of formula 1 were obtained with a yield of 72percent.
With tert-butylmagnesium chloride In tetrahydrofuran at 65 - 70℃; for 1.5 h;
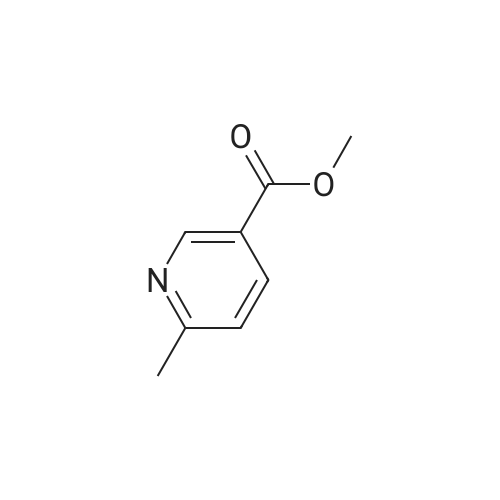
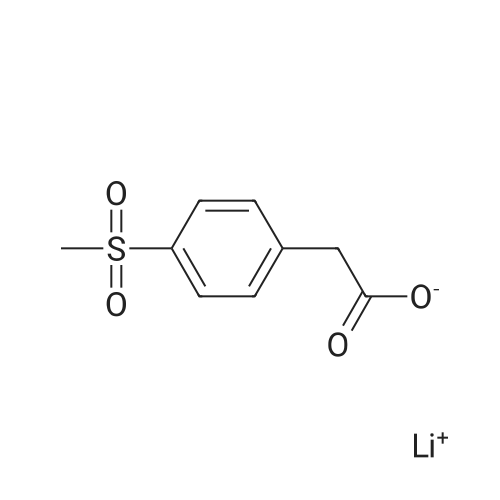
In a 4-neck anhydrous flask there are introduced at 20-25°C, 10.2 g of Lithium (4-methylsulfonyl)phenyl acetate of formula (II- M=Li) (1.0 mol. equiv), 200 mL of Anhydrous THF and the mixture is heated to 65-70°C (up to reflux). Maintaining at T=65-70 °C there are simultaneously dosed in the mixture in about 1 hour: a) 66.0 g of t-BuMgCl 1.0 M solution in THF (about 74.2 mL) (1.6 mol. equiv.) andb) a solution of 4.64 g of the methyl ester of the 6-methylpyridine-3-carboxylic acid of formula (III - R=Me) (0.65 mol. equiv.) in 15 mL of Anhydrous THF. After completing the addition, the mixture is stirred at 65-70 °C for 30 minutes. The reaction is controlled through HPLC then it is cooled to 20-25°C and the reaction mixture is diluted under vigorous stirring with 100 mL of water. Maintaining the temperature comprised in the range between 20°C and 25°C the mixture is brought to pH comprised between 1 and 0 by adding about 15 mL of 32percent hydrochloric acid. Stirring is carried out at 20-25°C for 30 minutes controlling the pH. The phases are separated by putting aside the organic phase useful for recovering the lithium (4-methylsulfonyl)phenyl acetate. The aqueous phase is washed using 2x50 mL of MTBE and the organic phases are recombined and used for recovering the (4-methylsulfonyl)phenyl acetate lithium. The aqueous phase is distilled at low pressure for removing the residue organic solvent. The product is precipitated under heat (40-45°C) by adding 7.5 mL of 30percent aqueous NaOH followed by cooling to room temperature. The product is filtered and dried in an oven under vacuum at 50°C for 8 hours. There are obtained 6.9 g of product as a white solid corresponding to a 78percent molar yield. HPLC purity 96.9percent (Apercent). Impurity '408' = 0.21percent (HPLC percent) (Figure 3). The organic phases obtained as described above are recombined to bring to a pH comprised between 11 and 12 by adding about 1 g of LiOH monohydrate solid. The mixture is concentrated to residue at low pressure at a maximum temperature of 40°C. The residue is suspended at room temperature in 2 volumes of Methanol. Stirring is carried out at room temperature for at least 3 hours. The suspension is filtered and the solid is washed using cold methanol then dried at low pressure at 90°C for 8 hours, to obtain 2.5 g of (4-methylsulfonyl)phenyl acetate lithium.
With tert-butylmagnesium chloride In tetrahydrofuran at 70 - 80℃; Large scale
4-methanesulfonylphenylacetic acid (3.0 kg) was added tetrahydrofuran (6 L),A solution of 1 M t-butylmagnesium chloride in tetrahydrofuran (40 L) was added dropwise,Heating 70 ~ 80 ,A solution of methyl 6-methylnicotinate (1.7 kg) in tetrahydrofuran (5 L) was slowly added dropwise,About 2 to 3 hours dripping.After refluxing for 1 hour.Cooling to 20 ~ 25 ,add water,The pH of the sodium hydroxide aqueous solution (the mass concentration refers to the mass of sodium hydroxide as a percentage of the total mass of the aqueous sodium hydroxide solution) is adjusted to pH = 7 to 8,Precipitate a lot of solid.Centrifugal,The filter cake was rinsed with water and dried in vacuo at 50 ° C for 16 hours,About 3.6 kg of a yellow solid was obtained.Recrystallization from dichloromethane (20 L) gave 1.8 kg of etoposide intermediate II.
Reference:
[1] Journal of Organic Chemistry, 2000, vol. 65, # 25, p. 8415 - 8420
[2] Patent: EP2551265, 2013, A1, . Location in patent: Paragraph 0062-0066
[3] Patent: CN106632003, 2017, A, . Location in patent: Paragraph 0046; 0047; 0048
2-(N-phthalomidoyl)-1,3-bis(dimethylamino)trimethinium hexafluorophosphate[ No CAS ]
2-[2-Dimethylamino-1-[1-dimethylamino-meth-(Z)-ylidene]-3-(4-methanesulfonyl-phenyl)-4-(6-methyl-pyridin-3-yl)-4-oxo-butyl]-isoindole-1,3-dione[ No CAS ]
With tert-butylmagnesium chloride; In tetrahydrofuran; at 65 - 70℃; for 1.5h;
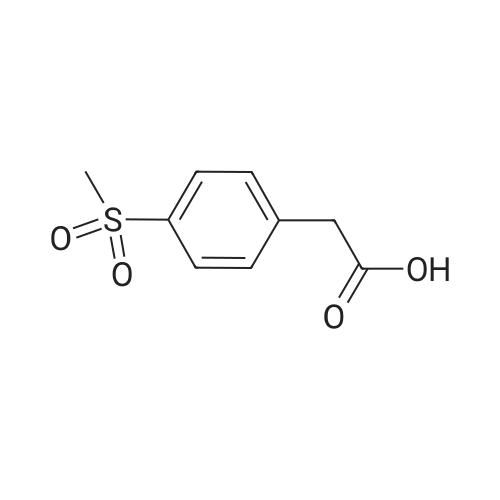
In a 4-neck anhydrous flask there are introduced at 20-25C 5.0 g of (4-methylsulfonyl)phenyl acetic acid of formula (IV) (1.0 mol. equiv.), 100 mL of Anhydrous THF and the mixture is heated to 65-70C (up to reflux). Maintaining at T=65-70 C there are simultaneously dosed in the mixture in about 1 hour: a) 62.0 g of t-BuMgCl 1.0 M solution in THF (about 70.0 mL) (3.0 mol. equiv.) andb) a solution of 2.3 g of methyl ester of 6-methylpyridine-3-carboxylic acid of formula (III - R=Me) (0.65 mol. equiv.) in 7.5 mL of Anhydrous THF. After completing addition, maintain at 65-70 C for 30 minutes. The reaction is controlled through HPLC: HPLC purity 72.05% (A%) and Impurity '408' = 0.26% (A%).
1.8 kg
With tert-butylmagnesium chloride; In tetrahydrofuran; at 70 - 80℃;Large scale;
4-methanesulfonylphenylacetic acid (3.0 kg) was added tetrahydrofuran (6 L),A solution of 1 M t-butylmagnesium chloride in tetrahydrofuran (40 L) was added dropwise,Heating 70 ~ 80 ,A solution of methyl 6-methylnicotinate (1.7 kg) in tetrahydrofuran (5 L) was slowly added dropwise,About 2 to 3 hours dripping.After refluxing for 1 hour.Cooling to 20 ~ 25 ,add water,The pH of the sodium hydroxide aqueous solution (the mass concentration refers to the mass of sodium hydroxide as a percentage of the total mass of the aqueous sodium hydroxide solution) is adjusted to pH = 7 to 8,Precipitate a lot of solid.Centrifugal,The filter cake was rinsed with water and dried in vacuo at 50 C for 16 hours,About 3.6 kg of a yellow solid was obtained.Recrystallization from dichloromethane (20 L) gave 1.8 kg of etoposide intermediate II.
100 g 1-(6-methylpyridin-3-yl)-2-[4-(methylsulfanyl)phenyl]ethanone of formula (VI) (1.0 equiv.), 150 mL ice-cold acetic acid (1.5 vol) and 30 mL methanesulfonic acid (1.2 equiv, 0.3 vol-96.11 g/mol; 1.481 g/ml) are introduced into a 3000 ml flask provided with high-temperature thermometer, coolant and dropping funnel.The reaction mixture is quenched to 5-10 C.120 mL hydrogen peroxide 30% w/w (3 equiv, 1.20 vol; 34.02 g/mol: 1.13 g/ml) are added under stirring at 5-10 C. The reaction mixture is stirred at 20-25 C. for at least 6 hours. At the end of the reaction, it is quenched to 0-5 C. and a solution consisting of 300 g sodium thiosulfate (3 wt) and water (10 vol) is added in portions.Approximately 180-260 ml of an aqueous solution of 30% sodium hydroxide are added up to reach a pH of about 5.5-6.5.It is stirred at 20-25 C. for 2 h and the suspension is filtered. The solid is washed with 2x400 mL water, then the solid is dried at 40 C. in a vacuum for at least 12 h. 105.7 g product are obtained, for a molar yield of 94% HPLC purity, equal to 97.5% (A %).
92%
With sodium tungstate; dihydrogen peroxide; In methanol; at 40 - 50℃; for 2h;
1.1) A compound AYTKX17 (800.0 g, 3.109 mol), methanol (7200 ml) and sodium tungstate (20.51 g, 0.062 mol) were added to a 20 L reactor, and stirred to obtain a yellow suspension. Slowly add 10% hydrogen peroxide (2325.9g, 6.840mol), and control the temperature below 50 C. After the completion of the dropwise addition, the reaction was carried out at a temperature of 40-50 C for 2 h. TLC showed that the reaction was almost complete. A solution of sodium sulfite (78.4 g, 0.622 mol) dissolved in 800 ml of water was added, stirred for 20 min, and then cooled to 5-10 C for 1 h. After suction filtration, the filter cake was washed with purified water (500 ml × 2), and dried at 45-55 C to constant weight to give a pale yellow solid, 827.1 g, yield 92.0%. The pale yellow solid (825.0 g) obtained in the step 1.1) was added to a 10 L three-necked flask, and acetonitrile (3300 ml) and purified water (2475 ml) were added thereto, and the mixture was heated to reflux under stirring for 0.5 h, and the solution was suspended and crystallized. The temperature was controlled at 5-10 C for 1 h, suction filtration, and the filter cake was washed with a mixture of 900 ml of acetonitrile/water (1:2 (v/v)), and dried at 45-55 C to constant weight to obtain 706.0 g of a pale yellow solid. That is, the compound AYTKX01 has a yield of 78.5%.
80.4%
With sodium tungstate; sulfuric acid; dihydrogen peroxide; In methanol; at 55℃; for 1h;
To the reaction flask was added C50g, 2N sulfuric acid 3.7ml and 500ml methanol, 1. Lg sodium tungstate, heated to 55 degrees, droppingAfter stirring, the mixture was stirred for 1 hour, 500 ml of water was added, cooled to room temperature, filtered, and the filter cake was washed with 500 ml of water and dried to obtain 45.2 g, yield: 80.4% purity: 98.1%.
72%
With sodium tungstate; dihydrogen peroxide; In ethyl acetate; at 30℃; for 1h;Flow reactor;
In a continuous flow reactor, enter the first material input port with 1-(6-methylpyridin-3-yl)-2-(4-methanesulfonylphenyl)ethanone in ethyl acetate (1 mol /L concentration, 1mL/s flow rate), The second input port was fed with an aqueous solution of sodium tungstate (0.1 mol/L concentration, 0.1 mL/s flow rate) and heated to 30 degrees via a heat exchanger. Enter a 10.5% hydrogen peroxide solution (1 mL/s flow rate, 3 equivalents) at the third material input port and mix with the solution shown in the first inlet. After mixing the two solutions, pass 20-30 degrees in 60 minutes. Cooler, Then, a sodium thiosulfate solution (2 mol/L concentration, 1 mL/s flow rate) was input from the fourth material input port, and the upper organic phase was collected and output in a continuous flow dispenser. The organic phase was collected into a container and, after reaching 20 liters, a post-treatment of concentration and crystallization was carried out. The yield of the two-step reaction was 72% and the purity was 98.5%.
Product (wet) mass of l-(6-methyl-3-pyridinyl)-2-[(4-Methylthio) phenyl] ethanone (V) was dissolved in dichloromethane (185 ml), stirred allowed to settle and aqueous layer was separated. Dichloromethane solution of the product was directly forwarded to the subsequent synthetic stage of oxidation with a mixture of concentrate Sulphuric acid (4.10 gm.) + water (4.50 ml) and stirred for 15-20 minutes. A solution of sodium tungstate (0.74 gm) in water (14.0 ml) was also charged and reaction mass was stirred for 10 minutes followed by addition of mixture of methyl-tri-n-octyl chloride (0.80 gm) in dichloromethane (8.0 ml).. The reaction mixture was stirred for 10 minutes, and cooled to 18-20C Mixture of 50% Hydrogen peroxide (19.50 gm) and water (12.50 ml) was gradually added within 45 minutes at 18-20C temp. Reaction mass was maintained for several hours and monitored by TLC examination, till the spot corresponding to l-(6- methyl-3-pyridinyl)-2-[(4-methylthio) phenyl] ethanone (V) was practically absent by TLC. Reaction mass pH was set between 6.95 to 7.10 using mixture of diluted ammonia solution under string within 30 minutes at 25-30C It was stirred for 10 minutes, aqueous layer separated and extracted with dichloromethane (130 ml). Combined dichloromethane layer containing product was washed with water (1 10 ml), dried over anhydrous sodium sulphate and filtered. Dichloromethane at atmospheric pressure to obtained. Vacuum was finally applied and the mass was degassed Then isopropyl alcohol (96 ml) was added, stirred mass and cooled to 25-30C followed by chilling to 0-5C temp, maintained for an hour, filtered, washed with chilled isopropyl alcohol and dried at 50-60C(yield 21 gms, m.p. 176o-180C,purity by HPLC-92.50%)
Example 8 - Synthesis of 1-(6-methylpyridin-3-yl)-2-[4-(methylsulfonyl)phenyl]ethanone of formula (I) - Exemplary variation of the invention 100 g 1-(6-methylpyridin-3-yl)-2-[4-(methylsulfanyl)phenyl]ethanone of formula (VI) (1.0 equiv.), 150 mL ice-cold acetic acid (1.5 vol) and 30 mL methanesulfonic acid (1.2 equiv, 0.3 vol - 96.11 g/mol; 1.481 g/ml) are introduced into a 3000 ml flask provided with high-temperature thermometer, coolant and dropping funnel. The reaction mixture is quenched to 5-10 C. 120 mL hydrogen peroxide 30% w/w (3 equiv, 1.20 vol; 34.02 g/mol: 1.13 g/ml) are added under stirring at 5-10C. The reaction mixture is stirred at 20-25 C for at least 6 hours. At the end of the reaction, it is quenched to 0-5C and a solution consisting of 300 g sodium thiosulfate (3 wt) and water (10 vol) is added in portions. Approximately 180-260 ml of an aqueous solution of 30% sodium hydroxide are added up to reach a pH of about 5.5-6.5. It is stirred at 20-25C for 2 h and the suspension is filtered. The solid is washed with 2 x 400 mL water, then the solid is dried at 40C in a vacuum for at least 12 h. 105.7 g product are obtained, for a molar yield of 94% HPLC purity, equal to 97.5% (A%).
21 g
With sodium tungstate; sulfuric acid; dihydrogen peroxide; Aliquat 336; In dichloromethane; water; at 18 - 20℃;pH 6.95-7.10;
Product (wet) mass of 1-(6-methyl-3-pyridinyl)-2-[(4-Methylthio)phenyl]ethanone (V) was dissolved in dichloromethane (185 ml), stirred allowed to settle and aqueous layer was separated. Dichloromethane solution of the product was directly forwarded to the subsequent synthetic stage of oxidation with a mixture of concentrate Sulphuric acid (4.10 gm.)+water (4.50 ml) and stirred for 15-20 minutes. A solution of sodium tungstate (0.74 gm) in water (14.0 ml) was also charged and reaction mass was stirred for 10 minutes followed by addition of mixture of methyl-tri-n-octyl chloride (0.80 gm) in dichloromethane (8.0 ml). The reaction mixture was stirred for 10 minutes, and cooled to 18-20 C. Mixture of 50% Hydrogen peroxide (19.50 gm) and water (12.50 ml) was gradually added within 45 minutes at 18-20 C. temp. Reaction mass was maintained for several hours and monitored by TLC examination, till the spot corresponding to 1-(6-methyl-3-pyridinyl)-2-[(4-methylthio)phenyl]ethanone (V) was practically absent by TLC. Reaction mass pH was set between 6.95 to 7.10 using mixture of diluted ammonia solution under string within 30 minutes at 25-30 C. It was stirred for 10 minutes, aqueous layer separated and extracted with dichloromethane (130 ml). Combined dichloromethane layer containing product was washed with water (110 ml), dried over anhydrous sodium sulphate and filtered. Dichloromethane at atmospheric pressure to obtained. Vacuum was finally applied and the mass was degassed Then isopropyl alcohol (96 ml) was added, stirred mass and cooled to 25-30 C. followed by chilling to 0-5 C. temp, maintained for an hour, filtered, washed with chilled isopropyl alcohol and dried at 50-60 C. (yield 21 gms, m.p. 176-180 C., purity by HPLC-92.50%).
2-chloro-1,3-(bis-morpholinyl)trimethinium hexafluoro phosphate[ No CAS ]
[ 221615-75-4 ]
[ 202409-33-4 ]
Yield
Reaction Conditions
Operation in experiment
Example-7; Preparation of Etoricoxib; Potassium tert. butoxide (1.4g) was dissolved in THF (7ml) and the solution was maintained at 25-30 0C for 10 minutes and then cooled to 10 0C. Subsequently, solution of l-(6-methylpyridin-3-yl)-2[4-(methylsulfonyl)rhohenyl] ethanone (5g) in THF was added and kept the mixture for lhr at 25-30 0C. Subsequently, 2-chloro 1,3 -(bis morpholinyl) trimethinium hexafluoro phosphate (4g) followed by THF (20ml) was added and the reaction mixture was maintained at 25-30 0C for 2 hrs. The reaction mixture was dumped in to the mixture of trifluoroacetic acid (0.6g), acetic acid (4.4g) and THF (10ml) and maintained the reaction mixture at 25-30 0C for 2 hrs. The reaction mixture was stirred at 55-60 0C for 3 hrs and aq. ammonia solution (30ml), and ammonium acetate (0.8g) was added and further stirred at 55-60 0C for 20 hrs. The reaction mixture was cooled to 25-30 0C then after water was added and extract with ethyl acetate (50ml). Organic layer was separated and washed with 10 % sodium bicarbonate solution, dried and concentrated to dryness <; 500C to obtain Etoricoxib.Yield = 3.1 g, (12.82 %), HPLC purity = 15.36% (Etoricoxib), (unreacted 64.15 %-sulfone compound).
2-chloro-1,3-(bis-piperidinyl)trimethinium hexafluorophosphate[ No CAS ]
[ 221615-75-4 ]
[ 202409-33-4 ]
Yield
Reaction Conditions
Operation in experiment
75.9%
Example-5; Preparation of Etoricoxib; Potassium tert. butoxide (2.Ig) was dissolved in THF (20ml) and the solution was maintained at 5-25 0C for 10 minutes. Subsequently, a solution of l-(6-methylpyridin-3- yl)-2[4-(rnethylsulfonyl)phenyl] ethanone (5g) in THF was added and kept the mixture for lhr at 25-30 0C. 2-chloro l,3-(bis piperidyl) trimethinium hexafluoro phosphate salt of formula (HIc) (8g), obtained in Example 4 above, in THF, was added to the previously prepared solution of ketosulfone and the reaction mixture was heated at 30-55 0C for 2-24 hrs. The reaction mixture was dumped into a mixture of triflorbacetic acid (Ig), acetic acid (7.5g) and THF. The reaction mixture was further heated to 55-60 0C for 6-20 hrs, after adding ammonia solution (125 ml). Then cooled the reaction mixture to room temperature. The reaction mixture was dumped into water and extracted with ethyl acetate and washed with 10% aqueous sodium bicarbonate solution. The organic layer was separated and concentrated to dryness at less than 50 0C to give Etoricoxib 8.6 g. HPLC purity- 91.28 % (Etoricoxib). Crude mass was purified to obtain Etoricoxib (4.7g), yield = 75.9 %; HPLC purity = 96.31 %.
2-chloro-1,3-(bis-pyrrolidinyl)trimethinium hexafluorophosphate[ No CAS ]
[ 221615-75-4 ]
[ 202409-33-4 ]
Yield
Reaction Conditions
Operation in experiment
ExampIe-9; Preparation of Etoricoxib; Potassium tert. butoxide (0.426g) was dissolved in THF (40ml) and the solution was maintained at 25-30 0C for 10 minutes and then cooled to 10-15 0C. Subsequently, solution of l-(6-methylpyridin-3-yl)-2[4-(methylsulfonyl)phenyl] ethanone (Ig) in THF was added and kept the mixture for lhr at 10-15 0C. Subsequently, 2-chloro l,3-(bis pyrolidinyl) trimethinium hexafluoro phosphate salt (1.3g), in THF (8ml) was added and maintained the reaction mixture for 2 hrs at 25-30 0C. The reaction mixture was dumped in to the mixture of trifloroaceticacid (0.2g), acetic acid (1.5g), and THF (10ml) and maintained for 1 hour at 25-30 0C. Further the reaction mixture was heated at 60-70 0C for 15 hrs after aq. ammonia solution (25ml) was added and the reaction mixture was cooled to 25-30 0C. After then ethyl acetate and water was added, organic layer was separated and washed with water and 10 % sodium bicarbonate solution. Organic layer was separated, dried and concentrated to dryness <; 50 0C to give crude Etoricoxib. Yield = 0.9 g (40.84 %), % HPLC purity = 55.82 % (Etoricoxib), (unreacted sulfone compound = 34.44 %).
Toluene (40 ml), propionic acid (25 ml), l-(6-methylpyridin-3-yl)-2-[4- (methylsulfonyl)phenyl]ethanone (5 g), 3-amino-2-chloroacrolein (5.4 g) and methanesulfonic acid (2.5 ml) were taken into a reaction flask, and the resulting mixture was heated at 100-110C, followed by maintaining for 15 hours. Upon completion of the reaction, the reaction mass was cooled to 20-25C, followed by the addition of water (25 ml) and ethyl acetate (50 ml). The pH of the reaction mass was adjusted to 7 to 7.5 using sodium bicarbonate solution (25%, 25 ml). The reaction mass was stirred and extracted with ethyl acetate (50 ml), followed by separation of the organic layer. The organic layer was washed with water (50 ml) twice, followed by carbon treatment [Carbon grade-Darco(D-60)]. The organic layer was heated at 50C and filtered over a hyflo bed at 50C. Ethyl acetate was distilled out under vacuum at 45-50C to produce 5.5 g of crude etoricoxib as an oily mass (Purity by HPLC : 83.10%).
Example 2 Synthesis of 1-(6-methylpyridin-3-yl)-2-[4-(methylsulfonyl)phenyl]ethanone of Formula (I) To a four-neck round-bottom 250 ml flask equipped with mechanical stirring and condenser were added at 20-25 C. and under nitrogen atmosphere Xantphos, palladium acetate, tripotassium phosphate trihydrate, PVP, 4-bromophenyl methyl sulfone, 5-acetyl-2-methyl pyridine, water (50.00 ml). A vacuum/nitrogen cycle was repeated for at least three times at 20-25 C. The resulting reaction mixture was heated up to 85-90 C. and stirred for at least 24 h. The reaction mixture was transferred in an other recipient then cooled down to 40-50 C. and diluted with water (400 ml). The resulting mixture was stirred at 40-45 C. for 15 min and cooled down to -5-0 C. The resulting reaction mixture was stirred at -5-0 C. for at least 2 h, then it was filtered and the cake was washed with water (3*100 ml) and dried at 65-70 C. under reduced pressure to afford a crude product of formula (I) as a yellow solid (48.3 g). yield: 90.1%, Purity: 75.7% by HPLC A %.
With potassium phosphate;palladium(II) acetylacetonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1-methyl-pyrrolidin-2-one; at 100℃; for 18h;Inert atmosphere;Product distribution / selectivity;
Pd(acac)2 (6.1 mg, 0.02 mmol, 0.5 mol %) and Xantphos (23.2 mg, 0.04 mmol, 1 mol o) are introduced into a flared flask provided with coolant. 4-bromophenylmethylsulfone of formula (III, XBr) (1.17 g, 5 mmol), acetylpicoline of formula (II) (541 mg, 4 mmol) and K3PO4 (2.55 g, 12.0 mmol, 3 eq) are added thereto. Once the argon atmosphere has been stabilized with vacuum-argon cycles, anhydrous and degassed NMP (15 ml) is added with a syringe. The mixture is then kept stirred under stirring in an argon atmosphere for 18 h at 100 C. The conversion is quantitative. The reaction mixture is diluted with a saturated solution of NaHCO3 (50 mL) and extracted with AcOEt (4×50 mL). The combined organic phases were washed with an aqueous solution saturated with NaHCO3 (30 mL), anhydrified on MgSO4 and concentrated in a vacuum. The residue was purified by silica gel chromatography using AcOEt/cyclohexane as eluent in a gradient from 5:5 to 10:0. 1.05 g product were obtained, for a molar yield of 91% as a white crystalline solid.
75%
With tris-(dibenzylideneacetone)dipalladium(0); 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; sodium t-butanolate; In toluene; for 5h;Inert atmosphere; Reflux;
Xantphos 0.00267 g (0.0046 mmol) and Pd2(dba)3 0.00177 g (0.0031 mmol) in 15 ml of anhydrous toluene were charged in a reactor under inert atmosphere. 4-bromophenyl methyl sulfone 0.724 g (3.078 mmol) and 3-acetyl-6-methyl pyridine 0.416 g (3.079 mmol) were then added. The mixture was heated to reflux and a suspension of t-BuONa 0.71 g in 15 ml of anhydrous toluene was added dropwise over about 4 h. After about 1 h from completion of the addition, the reaction mixture was cooled to 20 C and a solution of diluted hydrochloric acid to acidic pH was added. The aqueous phase was separated and added dropwise over 1 h to a mixture of water 8.3 g, ethyl acetate 15.3 g and sodium bicarbonate 2.1 g at 60 C. At addition completed and after maintaining the temperature at 60 C for 1 h, it was checked that the pH was between 4 and 7, the mixture was cooled to 20 C filtered and dried under vacuum at 50 C. 0.67 g of the compound of formula 1 was obtained with a yield of 75 %.
75%
With tris-(dibenzylideneacetone)dipalladium(0); 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; sodium t-butanolate; In toluene; for 4h;Inert atmosphere; Reflux;
Xantphos 0.00267 g (0.0046 mmol) and Pd2(dba)3 0.00177 g (0.0031 mmol) in 15 ml of anhydrous toluene were charged in a reactor under inert atmosphere. 4-bromophenyl methyl sulfone 0.724 g (3.078 mmol) and 3-acetyl-6-methyl pyridine 0.416 g (3.079 mmol) were then added. The mixture was heated to reflux and a suspension of t-BuONa 0.71 g in 15 ml of anhydrous toluene was added dropwise over about 4 h. After about 1 h from completion of the addition, the reaction mixture was cooled to 20C and a solution of diluted hydrochloric acid to acidic pH was added. The aqueous phase was separated and added dropwise over 1 h to a mixture of water 8.3 g, ethyl acetate 15.3 g and sodium bicarbonate 2.1 g at 60C. At addition completed and after maintaining the temperature at 60C for 1 h, it was checked that the pH was between 4 and 7, the mixture was cooled to 20C filtered and dried under vacuum at 50 C. 0.67 g of the compound of formula 1 was obtained with a yield of 75 %.
72%
With tris-(dibenzylideneacetone)dipalladium(0); 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; sodium t-butanolate; In toluene; for 5h;Inert atmosphere; Reflux;
EXAMPLE 1 [0068] Synthesis of 1-(6-methylpyridin-3-yl)-2-[(4-methylsulfonyl)-phenyl]-ethanone. [0069] Xantphos 0.027 g (0.0477 mmol) and Pd2(dba)3 0.0182 g (0.0198 mmol) in 100 ml of anhydrous toluene were charged in a reactor under inert atmosphere. 4-bromophenyl methyl sulfone 9.3 g (39.7 mmol) and 3-acetyl-6-methyl pyridine 5.4 g (39.7 mmol) were then added. The mixture was heated to reflux and a suspension of t-BuONa 8.4 g in 100 ml of anhydrous toluene was added dropwise over about 4 h. After about 1 h from completion of the addition, the reaction mixture was cooled to 20 C. and a solution of diluted hydrochloric acid to acidic pH was added. The aqueous phase was separated and added dropwise over 1 h to a mixture of water 83.3 g, ethyl acetate 153 g and sodium bicarbonate 20.1 g at 60 C. At addition completed and after maintaining the temperature at 60 C. for 1 h, it was checked that the pH was between 4 and 7, the mixture was cooled to 20 C., filtered, and dried under vacuum at 50 C. 8.3 g of the compound of formula 1 were obtained with a yield of 72%.
With potassium phosphate tribasic trihydrate; triphenylphosphine-3,3',3"-trisulfonic acid, trisodium salt; palladium diacetate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In water; at 80 - 90℃; for 20h;Inert atmosphere;
Example 1 - Synthesis of 1-(6-methylpyridin-3-yl)-2-[4-(methylsulfonyl) phenyl]ethanone of formula (I). Wherein BMS = 4-bromophenylmethylsulfone. [0051] Experimental Procedure [0052] To a four-neck round-bottom 100 ml flask equipped with mechanical stirring and condenser were added at 20-25 C and under nitrogen atmosphere Xantphos, TPPTS, palladium acetate, PVP, tripotassium phosphate trihydrate, BMS, 5-acetyl-2-methylpyridine and water according to the above amounts. A vacuum/nitrogen cycle was repeated for at least three times at 20-25 C. The resulting reaction mixture was heated up to 80-90 C and stirred for at least 20 h. The reaction mixture was then cooled down to 40-50C and diluted with water (200 ml). The resulting mixture was stirred at 40-45C for 15 min. and cooled down to -5-0C. The resulting reaction mixture was stirred at -5-0C for at least 2 h, then it was filtered and the cake was washed with water (3 x 50 ml) and dried at 60-65C under reduced pressure to afford crude product of formula (I) as a yellow solid (11.2g). Molar yield: 83.6%, Purity: 88.4% by HPLC A%.
In a 4-neck anhydrous flask there are introduced at 20-25C 11 g of (4-methylsulfonyl)phenyl acetate sodium of formula (II- M=Na), 200 mL of Anhydrous THF and the mixture is heated to 65-70C. Maintaining at T=65-70C there are rapidly added 21 g (0.5 equiv.) of t-BuMgCl 1.0 M solution in THF (about 23 mL). The mixture is left under stirring for 1 hr at 65-70 C then there is rapidly added, still at 65-70C, a solution of 3.6 g of methyl ester of the 6-methylpyridine-3-carboxylic acid of formula (III - R=Me) (0.5 equiv.) in 10 mL of Anhydrous THF. After completing addition the mixture is maintained at 65-70 C for 1 hr. Maintaining at T=65-70 C there are rapidly added 21 g (0.5 equiv.) of t-BuMgCl 1.0 M solution in THF (about 23 mL). It is left under stirring for 1 hr at 65-70C then there is rapidly added, still at 65-70C, a solution of 520 mg of methyl ester of the 6-methylpyridine-3-carboxylic acid of formula (III - R=Me) (0.075 equiv.) in 2 mL of anhydrous THF. After completing addition, the mixture is maintained at 65-70C for 1 hr. Maintaining at T=65-70 C, there are rapidly added 33.6 g (0.8 equiv.) of t-BuMgCl 1.0 M solution in THF (about 37 mL). The mixture is left under stirring for 1 hr at 65-70C then there is rapidly added, still at 65-70C, a solution of 520 mg of methyl ester of the 6-methylpyridine-3-carboxylic acid of formula (III - R=Me) (0.075 equiv.) in 2 mL of Anhydrous THF. The mixture is left under stirring for 1 hr at 65-70C then it is cooled to 20-25C. The molar yield in solution is equivalent to 90%. After completing the reaction cooling is carried out at 20-25C and the reaction mixture is diluted under vigorous stirring using 100 mL of Water. Maintaining at T= 20-25 C the mixture is brought to pH=1-0 by adding about 25 mL of 32% Hydrochloric acid. Stirring is carried out at 20-25C for 0.5 hrs controlling the pH. The organic phase is put aside (recovery of the sodium salt of the acidic sulfone). The aqueous phase is washed using 2x50 mL of MTBE. The organic phases are combined to the previous ones and they are put aside (recovery of the sodium salt of the acidic sulfon). The process continues with the aqueous phase containing the product. The aqueous phase is distilled to remove the residual organic solvent. The pH of the aqueous phase is corrected up to about pH=6.5-7.5 using 25 mL of 30% NaOH. There is observed the precipitation of the product. Stirring is carried out for at least one hour at 20-25C. Filtration is carried out by washing the solid using 20 mL of purified water and 20 mL of THF. The raw product is dried in an oven at 50C for 8 hrs to obtain 7.7 g of product for a molar yield equivalent to 87%. HPLC purity 85.9% (A%) and Impurity '408'= 3.85% (A%). The (4-methylsulfonyl)phenyl acetate of formula (II-M=Na) used in excess is recovered as follows. The organic phase obtained as described above is brought to pH=11-12 by dosing a solution of 1.25 g of NaOH in form of pearls or scales in 10 mL of Methanol. The mixture is concentrated to residue at low pressure at Tmax=40C. The residue is suspended at room temperature using 10 mL of methanol. Stirring is carried out for at least 3 hrs at room temperature. Filtration is carried out by washing using 5 mL of methanol. The raw product is dried in an oven at 50C for 8 hrs to afford 3.5 g of recovered product
In an anhydrified 4-neck flask there were introduced at 20-25C 5.0 g of (4-methylsulfonyl)phenyl acetic acid of formula (IV) and 100 mL of anhydrous THF and stirring is carried out at 20-25C. 1.0 g of sodium hydride dispersion in mineral oil (60% w/w, MW: 24.00; 1.1 molar equivalents) is added in portions in about 5 minutes, ensuring that the temperature does not exceed 45-50C. After completing the addition the mixture is heated to 65-70C for 1 hr. Maintaining at T=65-70C there are rapidly added 21 g (1.0 equiv.) of t-BuMgCl 1.0 M solution in THF (PM116.87) (23 mL). The mixture is left under stirring for 1 hr at 65-70C then is rapidly added, still at 65-70C, a solution of 1.78 g (0.5 equiv.) of methyl ester of the 6-methylpyridine-3-carboxylic acid of formula (III - R=Me) in 5 ml of Anhydrous THF. After completing the addition the mixture is maintained at 65-70C for 1 hr. Maintaining at T=65-70C there are rapidly added 10.5 g (0.5 equiv.) of t-BuMgCl 1.0 M solution in THF (PM 116.87) (11.5 mL). The mixture is left under stirring for 1 hr at 65-70C then there is rapidly added, still at 65-70C, a solution of 260 mg (0.075 equiv.) of methyl ester of 6-methylpyridine-3-carboxylic acid of formula (III - R=Me) in 1 mL of anhydrous THF. After completing addition the mixture is maintained at 65-70 C for 1 hr. Maintaining at T=65-70 C there are rapidly added 10.5 g (0.5 equiv.) of t-BuMgCl 1.0 M solution in THF (PM 116.87) (11.5 mL). The mixture is left under stirring for 1 hr at 65-70 C then there is rapidly added, still at 65-70 C, a solution of 260 mg (0.075 equiv.) of methyl ester of the 6-methylpyridine-3-carboxylic acid of formula (III - R=Me) in 1 mL of Anhydrous THF. After completing addition the mixture is maintained at 65-70 C for 1 hr. Cooling is carried out to 20-25C. The yield is calculated in solution through titration: 80%. The product is isolated according to the preceding example. It is obtained a molar yield equivalent to 73% and with 94.4% HPLC purity (A%) and amount of impurity '408' = 2.02% (A%) (see figure 4).
With tert-butylmagnesium chloride; In tetrahydrofuran; at 65 - 70℃; for 1.5h;
In a 4-neck anhydrous flask there are introduced at 20-25C, 10.2 g of Lithium (4-methylsulfonyl)phenyl acetate of formula (II- M=Li) (1.0 mol. equiv), 200 mL of Anhydrous THF and the mixture is heated to 65-70C (up to reflux). Maintaining at T=65-70 C there are simultaneously dosed in the mixture in about 1 hour: a) 66.0 g of t-BuMgCl 1.0 M solution in THF (about 74.2 mL) (1.6 mol. equiv.) andb) a solution of 4.64 g of the methyl ester of the 6-methylpyridine-3-carboxylic acid of formula (III - R=Me) (0.65 mol. equiv.) in 15 mL of Anhydrous THF. After completing the addition, the mixture is stirred at 65-70 C for 30 minutes. The reaction is controlled through HPLC then it is cooled to 20-25C and the reaction mixture is diluted under vigorous stirring with 100 mL of water. Maintaining the temperature comprised in the range between 20C and 25C the mixture is brought to pH comprised between 1 and 0 by adding about 15 mL of 32% hydrochloric acid. Stirring is carried out at 20-25C for 30 minutes controlling the pH. The phases are separated by putting aside the organic phase useful for recovering the lithium (4-methylsulfonyl)phenyl acetate. The aqueous phase is washed using 2x50 mL of MTBE and the organic phases are recombined and used for recovering the (4-methylsulfonyl)phenyl acetate lithium. The aqueous phase is distilled at low pressure for removing the residue organic solvent. The product is precipitated under heat (40-45C) by adding 7.5 mL of 30% aqueous NaOH followed by cooling to room temperature. The product is filtered and dried in an oven under vacuum at 50C for 8 hours. There are obtained 6.9 g of product as a white solid corresponding to a 78% molar yield. HPLC purity 96.9% (A%). Impurity '408' = 0.21% (HPLC %) (Figure 3). The organic phases obtained as described above are recombined to bring to a pH comprised between 11 and 12 by adding about 1 g of LiOH monohydrate solid. The mixture is concentrated to residue at low pressure at a maximum temperature of 40C. The residue is suspended at room temperature in 2 volumes of Methanol. Stirring is carried out at room temperature for at least 3 hours. The suspension is filtered and the solid is washed using cold methanol then dried at low pressure at 90C for 8 hours, to obtain 2.5 g of (4-methylsulfonyl)phenyl acetate lithium.
3-[2-(4-(methylthio)phenyl)-2-cyanoacetyl](6-methyl)pyridine[ No CAS ]
[ 221615-75-4 ]
Yield
Reaction Conditions
Operation in experiment
DM water (1.78 L) and concentrated sulfuric acid (3.4 kg) were stirred and heated in RBF at 25-30 C. The solution was heated to 60-65C and 3-[2-(4- (methylthio)phenyl)-2-cyanoacetyl](6-methyl)pyridine (1.25kg) was added into it in lots. The reaction mass was further heated to 95-100C and maintain till the completion of reaction. After completion of the reaction the mass was cooled to 20-25C. Acetic acid (1.96 kg) was added and cooled' to 6- 10C. 48% hydrogen peroxide (0.258kg) was added drop wise and stirred at 10-12C till the completion of reaction. After completion of the reaction, DM water (3.75 L) was added at 5- 10C and the excess hydrogen peroxide was neutralized using sodium thiosulpahte solution (0.12kg of sodium thiosulphate in 0.36 L of DM water) at 5-10C. Filter the reaction mass at 10-15 C. The filtration bed was washed with DM water (0.62 L) and the clear filtrate was taken in another RBF at 5-10C, pH was adjusted to 7.5-8.0 using ammonia solution (8.5 kg) at below 15C and stirred. The reaction mass was filtered, dried under vacuum. The wet ketosulfone (~ 2.04 kg) in 6.2 L water was heated to 50-55C and stirred. The reaction mass was cooled to 25- 30C, filtered under vacuum, washed with DM water (0.62L) and dried under vacuum at 25-30C. The product was washed under vacuum at 65-70C till moisture content comes less than 5%.
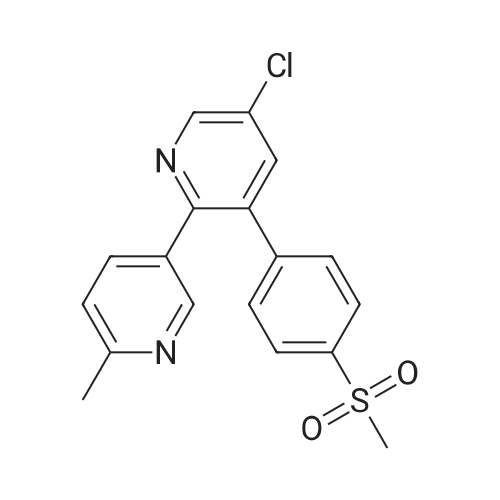
5-chloro-3-(4-methanesulfonylphenyl)-6'-methyl-[2,3']bipyridinyl hydrochloride[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Potassium tertiary butoxide (0.456 Kg), l-(6-methylpyridin-3-yl)-2[4- (methylsulfonyl)- phenyl]ethanone (1.0 Kg) and 2-chloro-l,3-(bis morpholinyl) trimethinium hexafluoro phosphate of (Formula III) (1.612 Kg) were dissolved in THF (10.0 L) and the solution was heated at 30-55C for 2-24 hrs. The reaction mixture was treated with acetic acid (1.45 Kg) and basified with ammonia gas (1.2 Kg) followed by the addition of ammonium acetate (0.277 Kg). The solvent was removed by distillation under vacuum to obtain a residue. The residue was treated with toluene and water mixture followed by the removal of toluene. The residue was crystallized in isopropanol to obtain etoricoxib. The etoricoxib thus obtained was converted to its hydrochloride by treating with methanolic HCl. The etoricoxib hydrochloride was recrystallized in methanol to obtain pure etoricoxib hydrochloride. HPLC purity > 99.0%.
N-(2-chloro-3-(dimethylamino)allylidene)-N-methylmethanaminium hexafluorophosphate(V)[ No CAS ]
5-chloro-3-(4-methanesulfonylphenyl)-6'-methyl-[2,3']bipyridinyl hydrochloride[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Sodium hydride (15 g) and l-(6-methylpyridin-3-yl)-2[4-(methylsulfonyl)- phenyl]ethanone (100 g) were taken in dimethylformamide (200 mL) at 10C. Toluene (500 mL) and compound of (Formula PiIota-Alpha) ( 1 12.32 g) were added to the reaction mixture and stirred for 3 hours at 10C. The reaction mixture was treated with acetic acid (145.3 g) and basified with ammonia gas (277 g) followed by the addition of ammonium acetate (28.5 g). The solvent was removed by distillation under vacuum to obtain a residue. The reaction mixture was heated at 60C for 6 hours and cooled to 40C. Formaldehyde (21.6 g) was added and the reaction mixture was heated at 60C for 1 hour. The separated aqueous layer was treated with tetrahydrofuran (5 mL) and toluene (300 mL) and solvents were removed to obtain a residue. The residue was treated with toluene and water mixture followed by the removal of toluene. The residue was crystallized in isopropanol to obtain etoricoxib in the presence of seed crystals of Form-I.
N-(2-chloro-3-(dimethylamino)allylidene)-N-methylmethanaminium hexafluorophosphate(V)[ No CAS ]
[ 202409-33-4 ]
Yield
Reaction Conditions
Operation in experiment
84.4%
Add compound AYTKX01 (2.0 g, 6.91 mmol, purchased from Chengdu Claymont Pharmaceutical Technology Co., Ltd., batch number 20160401) and ultra-dry tetrahydrofuran (THF, 15 ml) to a 100 mL three-necked flask, and stir under nitrogen atmosphere, temperature control 0-10 C, Potassium tert-butoxide (0.85 g, 7.60 mmol) was added in portions. The temperature was controlled at 10-20 C for 15 min, then the compound AYTKX02 (2.23 g, 7.26 mmol, purchased from Chengdu Claymont Pharmaceutical Technology Co., Ltd., batch number 20160301) was added in one time. Add 5 ml of ultra-dry tetrahydrofuran, control at 20-25 C for 1 h, pour the reaction into acetic acid (2.90 g, 48.37 mmol), and incubate at 20-25 C for 1.5 h. Concentrated ammonia water (2.66g, 76.01mmol) was added dropwise, and after heating, it was heated to 60-65 C for 2-3 h. TLC showed that the basic reaction was complete, and the mixture was allowed to stand for separation. The organic phase was added dropwise with 120 ml of saturated aqueous sodium hydrogencarbonate solution. The solid was precipitated, stirred under temperature, and stirred at 5-10 C for about 1 h. Filtration, washing with 5 ml of THF/H2O (1:2 (v/v)) mixed solution, vacuum drying to constant weight at 40-45 C, to obtain 2.09 g of a yellow solid, which is etoric (compound AYTKX05), yield 84.4%. After testing, the content of impurity M in AYTKX05 was 0.50%, and the purity was 96.36%.
With methanesulfonic acid; ammonium acetate; propionic acid; In toluene; at 25 - 75℃; for 14 - 16h;Reflux;
n-Propionic acid (30 ml) and methanesulfonic acid (5.7gm) was added to 2-(4-methanesulfonyl- phenyl)-1-(6-methyl-pyridin-3-yl)-ethanone(10 gm) in toluene (100 ml) at 25 to 35C. The resulting mixture was heated to 70-75C to get clear solution. Vinamidinium hexaflurophophate (15.9 gm) was added, followed by ammonium acetate (18.7gm). Reaction mass was refluxed for 14-16hrs. After completion, the reaction mixture was cooled to room temperature. Ethyl acetate and water was added to the reaction mass, adjusted pH to 7.5 -8.5 with ammonia solution. Salts were removed through hyflo bed and the filtrate was washed with 30% aqueous sodium chloride solution, followed by water. Ethyl acetate layer was concentrated to residue. The residue was dissolved in isopropyl alcohol and a solution of p-toluenesulfonic acid (6.55 gm) in isopropyl alcohol was added. Stirred the solution for 3-4 hrs at room temperature, followed by reflux for 1 hr. Reaction mass was slowly cooled to room temperature, the obtained solid was filtered and washed with isopropyl alcohol to gave 12.4 gm of PTSA salt of etoricoxib. PTSA salt of Etoricoxib (12.4 gm) was taken in a mixture of ethyl acetate and water, to this mixture 18-20% of aqueous ammonia (8.0 ml) solution was added. The organic layer was separated and the ethyl acetate layer was washed with 30% sodium chloride solution. Ethyl acetate layer was concentrated to get residue, isopropyl alcohol was added and then heated to 65-70C for 1hr. The reaction mixture was cooled 5-10C and the obtained solid was filtered to give pure Etoricoxib.
To a suspension of 1- (6-methylpyridin-3-yl)-2- (4- (methylsulfonyl)phenyl)ethanone(1.74 g) in THF (50 ml), stirring at --5C, potassium 2-methylpropan-2-olate (1.2 g) wasadded. The reaction mixture was stirred at ambient temperature for 30 mm. and cooled (icewater) again, before addition of methyl 2-chloro-3-(tosyloxy)acrylate (1.9 g) in portions (in 5 mm). The mixture was further stirred at 5C for 30 mm.After stirring for another 30 mm., 70 ml of ethyl ether was added. The formedsuspension was stirred for 20 mm. and the solid was filtered off (washed with 20 ml ether). The obtained solid was treated with 50 ml water and 10 ml dichloromethane (DCM) at ambient temperature for 2 hrs. Then, 50 ml of DCM was added and the separated water layer was extracted again with 25 ml of DCM. The combined DCM layers were dried and concentrated to give a crude product (1.65 g). The crude product was purified bychromatography to give a foam-like solid (1.10 g).
With methanesulfonic acid; In 1,2-dichloro-ethane; at 80 - 100℃; for 24h;
A mixture of 2-chloro-3-hydroxyacrylaldehyde (2 g), l-(6-methylpyridin-3-yl)-2-(4- (methylsulfonyl)-phenyl)ethanone (2 g), formamide (4 ml), methanesulfonic acid (0.5 ml) and 1 ,2-dichloroethane (2 ml) was stirred (80C heating block) for 20 hrs and further stirred at (100C heating block) for 4 hrs. After cooling down to ambient temperature, water (10 ml) was added, followed by addition of ethyl acetate (25 ml) and ether (25 ml). The mixture was stirred for 15 min, and layers were separated. The water layer was extracted with ethyl acetate (50 ml). Combined organic layers were washed with water (10 ml), dried and concentrated to give a crude product (3 g). The above material was re-dissolved in ethyl acetate (30 ml) and treated with HC1 1M (10 ml) for 10 min. Separated ethyl acetate layer was extracted again with 1M HC1 (5 ml). Combined water layers were basified to pH 7 by addition of NaOH solution (2M), and extracted with ethyl acetate (50 ml). The combined ethyl acetate layers were washed with water, brine, dried and concentrated to give desired product (1.81 g, solid), yield 73%.
ethyl (2-chloro-3-oxoprop-1-en-1-yl)carbamate[ No CAS ]
[ 51-79-6 ]
[ 221615-75-4 ]
diethyl ((1Z,3E)-2-chloro-4-(4-(methylsulfonyl)phenyl)-5-oxo-5-(6-methylpyridin-3-yl)penta-1,3-diene-1,3-diyl)dicarbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With propionic acid; In chloroform; at 110 - 140℃;
A mixture of ethyl 2-chloro-3-oxoprop-l-enylcarbamate (200 mg), l-(6-methylpyridin-3- yl)-2-(4-(methylsulfonyl)phenyl)ethanone (200 mg), ethyl carbamate (2 g) and propionic acid (0.1 ml) in CHC13 (1 ml) was stirred at 140C for 2 hrs and 20 min. CHCI3 (0.5 ml) was added, and the mixture was further stirred at 110C over night. The reaction mixture was diluted in DCM, washed twice with water , base (5 NaOH), water, dried and concentrated to give a dark mixture. The crude product was partly purified by chromatography. It was further isolated by prep. HPLC and the structure was proved by MS.
N-(2-chloro-3-(dimethylamino)allylidene)-N-methylmethanaminium hexafluorophosphate(V)[ No CAS ]
3-methyl-8-hydroxy-7-(4-methanesulfonylphenyl)isoquinolin-5-formaldehyde[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
42%
Adding Formula I (1g) to the reaction flask at 25 C,Add 12 ml of isopropyl acetate,Add sodium tert-butoxide (0.58g) for 2h, add 2-chloro-1,3-bis(dimethylamino)trimethylene hexafluorophosphate (formula II, 1.27g) for 4h.The system was transferred to a solution of isopropyl acetate (3 ml) in acetic acid (0.84 g) at ambient temperature for 4 h, filtered.5 ml of water was added to the filter cake and reacted at 35 to 45 C for 5 h, and then the column chromatography method of Example 1 was used.420 mg of the compound of formula IV are obtained in a yield of 42%, HPLC purity 99.1%.
With potassium tert-butylate; In tetrahydrofuran; for 3h;Reflux;
1.74 g (6·0 mmol) of compound C, 1.36 g (9·0 mmol) of methyl 6-methylnicotinate, 1.01 g (9.0 mmol) of potassium t-butoxide were mixed with 21 mL of tetrahydrofuran, and heated to reflux. After the reaction was completed by 3h. TLC, 30 mL of water, 40 mL of dichloromethane was slowly added, and the liquid phase was separated, and the aqueous phase was extracted with dichloromethane (40 mL of X 2 ), dichloromethane was combined, washed twice with saturated brine, and the organic phase was separated. Drying over anhydrous sodium sulfate, the solvent was evaporated under reduced pressure, and purified by column chromatography to yield 1.56 g of pale yellow solid, Compound D, yield 63.8%
N-(2-chloro-3-(dimethylamino)allylidene)-N-methylmethanaminium hexafluorophosphate(V)[ No CAS ]
(2Z,4Z)-4-Chloro-5-dimethylamino-2-(4-methanesulfonyl-phenyl)-1-(6-methyl-pyridin-3-yl)-penta-2,4-dien-1-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
1.2) Add a compound AYTKX01 (703.0g, 2.430mol) and ultra-dry tetrahydrofuran (THF, 6300ml) to a 20L reactor, stir under nitrogen atmosphere, control temperature 10-15 C, add potassium tert-butoxide (313.5g) , 2.794mol), temperature control 15-20 C reaction for 15min, Then, the compound AYTKX02 (782.2g, 2.551mol) was added in one portion, and the reaction was carried out at a temperature of 30-35 C for 1 h. The reaction solution was poured into acetic acid (1021.3 g, 17.010 mol), and the reaction was carried out at 30-40 C for 1 h, and a yellow solid precipitated. Add n-heptane (2109ml), cool to about 5-10 C for 30min, suction filtration, wash the filter cake with 1500ml THF / n-heptane (1:1 (v / v)) mixed solution to obtain a yellow solid compound AYTKX04 And go directly to the next step.
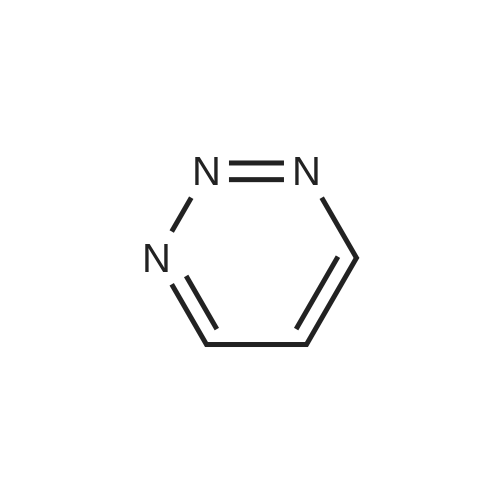
With potassium hydroxide; In acetonitrile; at 80℃; for 8h;Inert atmosphere;
In a 10 mL vacuum tube, add 5-chloro-1,2,3-triazine 2a (11.5 mg, 0.10 mmol), 2- (4-methanesulfonylphenyl) -1- (6-methylpyridine-3 -Yl) -ethanone 6b (43.4 mg, 0.15 mmol) and KOH (2.8 mg, 0.05 mmol). Nitrogen was replaced three times, then 0.5mL MeCN was added, and the reaction tube was placed at 80 C for 8h. Follow the reaction by TLC, after terminating the reaction, add CH2Cl2 (3x10mL) for extraction, separate the organic phase, dry with Na2SO4, concentrate the organic phase in vacuo, and then obtain the target product etoricoxib 1d by column chromatography, yield (30.8mg, 89%).
6'-methyl-3-[4-(methylsulfonyl)phenyl]-2,3'-bipyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
With potassium hydroxide; In acetonitrile; at 80℃; for 8h;Inert atmosphere;
In a 10 mL vacuum tube, add 1,2,3-triazine 2a (8.1 mg, 0.10 mmol), 2- (4-methanesulfonylphenyl) -1- (6-methylpyridin-3-yl)- Ethanone 6b (29 mg, 0.1 mmol) and KOH (2.8 mg, 0.05 mmol). Nitrogen was replaced three times, then 0.5mL MeCN was added, and the reaction tube was placed at 80 C for 8h. The reaction was followed by TLC. After the reaction was terminated, CH2Cl2 (3x10mL) was added for extraction, the organic phase was separated, dried over Na2SO4, and the organic phase was concentrated in vacuo, and then subjected to column chromatography to obtain the target product 2d, yield (26.9mg, 83%) .






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping