* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With potassium iodide In N,N-dimethyl-formamide at 120℃; for 30 h;
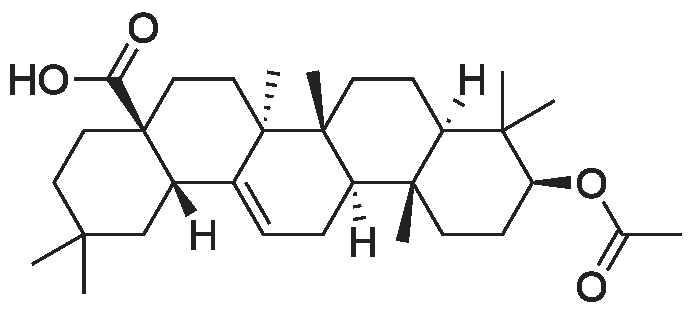
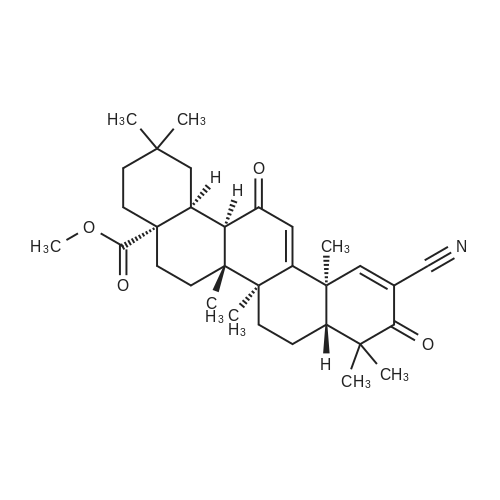
5.58 g of compound V (10 mmol) were dissolved in 80 ml of anhydrous DMF, 1.1648 g of cuprous cyanide (13 mmol) and0.332 g of potassium iodide (2 mmol) was added, and a balloon was added to prevent the water in the air from entering the reaction system and reacting at 120 ° C for 30 hours. reactionAfter the reaction was cooled to room temperature, the reaction solution was poured into 200ml of water to quench, add 500ml of ethyl acetate, stir, filterThe insolubles were removed and the organic layer was washed with saturated sodium bicarbonate (200 ml x 2) and saturated sodium chloride (200 ml), respectively,Drying, suction filtration, rotary drying and column chromatography (PE / EA = 5: 1) gave 4.22 g of yellow powder with a yield of 83.6percent.
73%
With potassium iodide In N,N-dimethyl-formamide at 120℃;
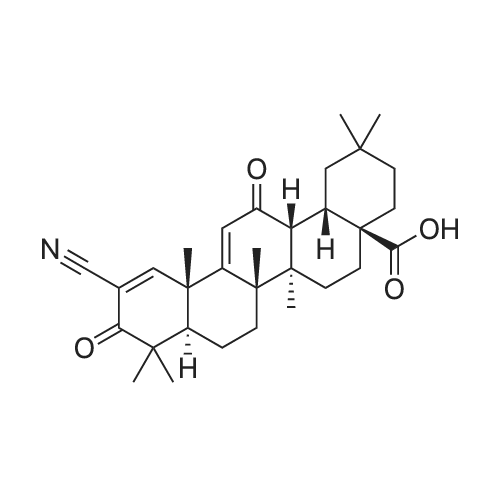
As shown in Scheme 4, the natural triterpenoid oleanolic acid (1) was used as the starting material in the synthesis of CDDO-Me. The improved method commences with methylation of the carboxylic acid of oleanolic acid (1) to afford methyl ester 2 in quantitative yield. With ester 2, activation of the A-ring is fulfilled by 2-iodoxybenzoic acid-mediated two-fold oxidation to give enone 3. Epoxidation with meta-chloroperoxybenzoic acid, followed by direct C-ring enolization and A-ring enone bromination with bromine and hydrobromic acid, affords key intermediate 4. With bromide 4 in hand, a cross-coupling reaction with copper cyanide provides CDDO-Me (5) in only five steps (four operations) from oleanolic acid (Scheme 4). This is an improvement over conventional methods, which require 10 steps. Moreover, intermediate 4 was prepared in high yield and few overall steps, thereby providing a base compound for development of additional analogs and derivatives.
73%
With potassium iodide In N,N-dimethyl-formamide at 120℃;
As a specific example of using this synthetic method in the synthesis of triterpenoids, CDDO-Me was synthesized from oleanolic acid. As shown in Scheme 2, the natural triterpenoid oleanolic acid (1) was used as the starting material in the synthesis of CDDO-Me. The method commences with methylation of the carboxylic acid of oleanolic acid (1) to afford methyl ester 2 in quantitative yield. With ester 2, activation of the A-ring is fulfilled by 2-iodoxybenzoic acid-mediated two-fold oxidation to give enone 3. Epoxidation with meta-chloroperoxybenzoic acid, followed by direct C-ring enolization and A-ring enone bromination with bromine and hydrobromic acid, affords key intermediate 4. With bromide 4 in hand, a cross-coupling reaction with copper cyanide provides CDDO-Me (5) (Scheme 2). Intermediate 4 was prepared in high yield and few overall steps, thereby providing a base compound for development of the analogs and derivatives described herein.
Reference:
[1] Patent: CN106632576, 2017, A, . Location in patent: Paragraph 0058; 0059; 0060; 0061
[2] Patent: US2013/303797, 2013, A1, . Location in patent: Paragraph 0106
[3] Patent: US2013/303607, 2013, A1, . Location in patent: Paragraph 0103
[4] Organic Letters, 2013, vol. 15, # 7, p. 1622 - 1625
methyl 2-cyano-3,12-dioxoolean-9(11)-en-28-oate[ No CAS ]
[ 218600-53-4 ]
Yield
Reaction Conditions
Operation in experiment
40%
Compound 10 was prepared by formylation of OA (Compound 9) (Simonsen and Ross, 1957) with ethyl formate in the presence of sodium methoxide in THE (Clinton et al., 1961). Compound 7 was obtained by introduction of a double bond at C-1 of Compound 10 with phenylselenenyl chloride in ethyl acetate and sequential addition of 30% hydrogen peroxide (Sharpless et al., 1973). Compound II was synthesized from Compound 10 by addition of hydroxylamine in aqueous ethanol; cleavage of Compound 11 with sodium methoxide gave Compound 12 (Johnson and Shelberg, 1945). Compound 14 was prepared from Compound 13 (Picard et al., 1939) by alkali hydrolysis followed by Jones oxidation. Compound 15 was prepared by formylation of Compound 14 with ethyl formate in the presence of sodium methoxide in benzene. Compound 16 was synthesized from Compound 15 by addition of hydroxylamine. Nitrile 17 was obtained by cleavage of isoxazole 16 with sodium methoxide (yield, 100%), followed by introduction of a double bond at C-1 with PhSeCl-H2O2 (yield, 40%). CDDO (6) was prepared in 71% yield by halogenolysis of 17 with lithium iodide in DMF (Dean, P. D. G., 1965).
Compound 10 was prepared by formylation of OA (Compound 9) (Simonsen and Ross, 1957) with ethyl formate in the presence of sodium methoxide in THE (Clinton et al., 1961). Compound 7 was obtained by introduction of a double bond at C-1 of Compound 10 with phenylselenenyl chloride in ethyl acetate and sequential addition of 30% hydrogen peroxide (Sharpless et al., 1973). Compound II was synthesized from Compound 10 by addition of hydroxylamine in aqueous ethanol; cleavage of Compound 11 with sodium methoxide gave Compound 12 (Johnson and Shelberg, 1945). Compound 14 was prepared from Compound 13 (Picard et al., 1939) by alkali hydrolysis followed by Jones oxidation. Compound 15 was prepared by formylation of Compound 14 with ethyl formate in the presence of sodium methoxide in benzene. Compound 16 was synthesized from Compound 15 by addition of hydroxylamine. Nitrile 17 was obtained by cleavage of isoxazole 16 with sodium methoxide (yield, 100%), followed by introduction of a double bond at C-1 with PhSeCl-H2O2 (yield, 40%). CDDO (6) was prepared in 71% yield by halogenolysis of 17 with lithium iodide in DMF (Dean, P. D. G., 1965).
70.3%
With lithium iodide; In N,N-dimethyl-formamide; at 153℃; for 12.0h;Inert atmosphere;
Dissolve 2 g of CDDO-Me in 80 ml of anhydrous DMF, add 12 g of anhydrous lithium iodide, protect with nitrogen and heat to reflux (153 C) for 12 hours. After the reaction was completed, the reaction solution was cooled to room temperature, the reaction solution was diluted with 200 ml of ethyl acetate, 200 ml of 5%Dilute hydrochloric acid, partition, aqueous layer was washed with ethyl acetate (120ml × 2), the organic layers were combined and washed with saturated sodium chloride(200ml), dried over anhydrous sodium sulfate, suction filtered and dried. residue was purified by column chromatography (PE / EA = 3: 1) to give 1.37g of white powder. Yield 70.3%.
methyl 2-cyano-3,12-dioxoleana-1,9(11)-dien-28-oate hemibenzenate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In acetone; for 0.666667h;Ultrasonic processor;
Various experiments, replicating the last recovery step from the synthesis of CDDO methyl ester, were performed. Honda et al., 2000. The objective was the isolation of crystalline material from a solution mixture of (10:1) benzene/acetone.Approximately 100 mg of CDDO-methyl ester was dissolved in 300 muL of benzene/acetone (10:1) and filtered through a 0.2-mum nylon filter. The solution was then sonicated using an ultrasonic processor for 10 minutes and allowed to evaporate at room temperature in an uncapped vial overnight. A clear gel formed and 100 muL of benzene/acetone (10:1) was added. The solution was submitted to sonication on an ultrasonic processor for approximately 30 minutes. A white precipitate formed. The solids were allowed to air dry.In other experiments, approximately 200 mg of CDDO-methyl ester was dissolved in 0.8 mL of benzene/acetone (10:1) and filtered through a 0.2-mum nylon filter. The solution was then evenly divided into two 1-dram vials. Samples A and B were then allowed to fast evaporate at room temperature for a few hours. Sample A was capped and placed in a freezer. After the sample froze, the sample was allowed to thaw at room temperature. A small scratch was introduced using a spatula and the sample was allowed to evaporate at room temperature. White solids formed and were allowed to air dry. Sample B was capped, left at room temperature and was a clear solution after sitting at room temperature overnight. A small scratch was introduced using a spatula and the sample was allowed to evaporate at room temperature. White solids formed and were allowed to air dry.Crystalline material, determined to be a hemibenzenate, was obtained from several of these experiments. As described above, minor disturbances, such as sonication or merely introducing a small scratch within the recovery vessel, will facilitate the crystallization of the benzene solvate (Table 12).Characterization data of the hemibenzenate are summarized in Table 13. Characteristic peaks for the hemibenzenate XRPD pattern are provided in Table 19. The DSC curve exhibits a broad endotherm near 133 C., associated with 7.0% of weight loss in the TG thermograph FIG. 23. The weight loss is likely due to the volatilization of benzene (see NMR discussion below), and corresponds to 0.5 moles of benzene for each mole of CDDO-methyl ester. The DSC endotherm observed near 223 C. most likely results from the melt of desolvated material.These data distinguish previously isolated forms of CDDO-methyl ester from the present invention.
methyl 2-cyano-3,12-dioxoleana-1,9(11)-dien-28-oate dimethanol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
at -10 - 60℃;
A CDDO-methyl ester dimethanol solvate was prepared according to the below procedure. Approximately 500 mg of CDDO-methyl ester was dissolved in 20 mL of methanol at 60 C. The solution was then slowly added to 20 mL of cold methanol at -10 C. with agitation. White solids were collected by vacuum filtration and then stored in a freezer.Characterization data are summarized in Table 14. Characteristic peaks for the dimethanolate XRPD pattern are provided in Table 18.The DSC curve shows a broad endotherm near 102 C., associated with 11% of weight loss in the TG thermograph (FIG. 24). The TGIR data confirms the weight loss is due to volatilization of 2.0 moles of methanol (FIG. 25). The resulting material from the TGIR experiment was recovered and was amorphous by XRPD (FIG. 26). A baseline shift at approximately 130 C., a broad exotherm near 203 C. followed by a sharp endotherm (onset: 223 C.) are also observed in the DSC curve. These events are most likely indicative of the Tg of the amorphous material (Form B) obtained through the desolvation of the dimethanol solvate followed by crystallization of the amorphous material to Form A and melting of that crystalline material.The solution proton NMR spectrum was obtained. The chemical assignments were not performed; however it appears consistent with the chemical structure of CDDO-methyl ester. The peaks at 3.51 ppm are assigned to methanol and correspond to 1.7 moles. This result is consistent with the thermal data above.
at -15 - 60℃;Industry scale;Product distribution / selectivity;
One kilogram of Form A CDDO-Me was dissolved in 60+/-5 C. methanol to afford a complete solution. The resulting hot solution of CDDO-Me was added to a vessel containing cold ?5 C. to ?15 C. methanol, while maintaining agitation and a temperature of ?5 C. to ?15 C. throughout the addition. The resulting suspension of crystalline dimethanol solvate of CDDO-Me was filtered. The resulting solids, which displayed an XRPD pattern consistent with that presented in FIG. 26 (prior to TGIR analysis), were dried in an oven at 70+/-5 C. Drying was continued until the XRPD profile displayed no reflections characteristic of crystalline substance. The resulting XRPD amorphous CDDO-Me solids were passed through a sieve and packaged. Product recovery ranges from 65-95%.
With sodium periodate; (x)ClH*Cl3Ru; In water; ethyl acetate; acetonitrile; at 0℃; for 0.0833333h;
A solution of 23 (100 mg, 0.20 mmol) in MeCN (1.0 mL) and EtOAc (1.0 mL) was added to a solution of RuClyxHCl (11 mg, 0.053 mmol) and NaIO4 (80 mg, 0.37 mmol) in water (0.5 mL) at 0 C. After stirring at 0 C for 5 min, the reaction mixture was poured onto 10% Na2SO3(aq) solution (20 mL). After stirring at ambient temperature for 2 min, the reaction mixture was transferred to a separatory funnel, which was extracted with EtOAc. The combined organic extracts were filtered through a pad of celite, which was washed with additional EtOAc. The combined filtrate and washes were dried over MgSO4, filtered, and concentrated. The crude product was purified by column chromatography (silica gel, 0% to 25% EtOAc in hexanes) to give product 63316 (76 mg, 71% yield) as a white foam solid: 1H NMR (400 MHz, CDCl3) delta 5.80 (s, 1H), 4.98 (s, 1H), 4.54 (bs, 1H), 3.70 (s, 3H), 3.03 (m, 1H), 2.97 (d, 1H, J = 1.6 Hz), 2.90 (d, 1H, J = 4.8 Hz), 2.02 (m, 1H), 1.78-1.94 (m, 3H), 1.74 (s, 3H), 1.44 (s, 3H), 1.36 (s, 3H), 1.29 (s, 3H), 1.10- 1.78 (m, HH), 1.03 (s, 3H), 0.99 (s, 3H), 0.88 (s, 3H); m/z 540.3 (M+l).
With potassium iodide; In N,N-dimethyl-formamide; at 120℃; for 30h;
5.58 g of compound V (10 mmol) were dissolved in 80 ml of anhydrous DMF, 1.1648 g of cuprous cyanide (13 mmol) and0.332 g of potassium iodide (2 mmol) was added, and a balloon was added to prevent the water in the air from entering the reaction system and reacting at 120 C for 30 hours. reactionAfter the reaction was cooled to room temperature, the reaction solution was poured into 200ml of water to quench, add 500ml of ethyl acetate, stir, filterThe insolubles were removed and the organic layer was washed with saturated sodium bicarbonate (200 ml x 2) and saturated sodium chloride (200 ml), respectively,Drying, suction filtration, rotary drying and column chromatography (PE / EA = 5: 1) gave 4.22 g of yellow powder with a yield of 83.6%.
73%
With potassium iodide; In N,N-dimethyl-formamide; at 120℃;
As shown in Scheme 4, the natural triterpenoid oleanolic acid (1) was used as the starting material in the synthesis of CDDO-Me. The improved method commences with methylation of the carboxylic acid of oleanolic acid (1) to afford methyl ester 2 in quantitative yield. With ester 2, activation of the A-ring is fulfilled by 2-iodoxybenzoic acid-mediated two-fold oxidation to give enone 3. Epoxidation with meta-chloroperoxybenzoic acid, followed by direct C-ring enolization and A-ring enone bromination with bromine and hydrobromic acid, affords key intermediate 4. With bromide 4 in hand, a cross-coupling reaction with copper cyanide provides CDDO-Me (5) in only five steps (four operations) from oleanolic acid (Scheme 4). This is an improvement over conventional methods, which require 10 steps. Moreover, intermediate 4 was prepared in high yield and few overall steps, thereby providing a base compound for development of additional analogs and derivatives.
73%
With potassium iodide; In N,N-dimethyl-formamide; at 120℃;
As a specific example of using this synthetic method in the synthesis of triterpenoids, CDDO-Me was synthesized from oleanolic acid. As shown in Scheme 2, the natural triterpenoid oleanolic acid (1) was used as the starting material in the synthesis of CDDO-Me. The method commences with methylation of the carboxylic acid of oleanolic acid (1) to afford methyl ester 2 in quantitative yield. With ester 2, activation of the A-ring is fulfilled by 2-iodoxybenzoic acid-mediated two-fold oxidation to give enone 3. Epoxidation with meta-chloroperoxybenzoic acid, followed by direct C-ring enolization and A-ring enone bromination with bromine and hydrobromic acid, affords key intermediate 4. With bromide 4 in hand, a cross-coupling reaction with copper cyanide provides CDDO-Me (5) (Scheme 2). Intermediate 4 was prepared in high yield and few overall steps, thereby providing a base compound for development of the analogs and derivatives described herein.
Compound TX63212: At room temperature RTA 402 (200 mg, 0.4 mmol) and Nbromosaccharin (156 mg, 0.6 mmol) were mixed in acetonitrile (3 mL) and water (1 mL). The mixture was stirred at room temperature for 2 h. After dilution with dichloromethane, the mixture was washed with water. The organic phase was dried over MgSO4 and concentrated. The residue was purified by flash chromatography (silica gel, 33% EtOAc in hexanes) to give a mixture of two 1 ,2-bromohydrin products 1 and 2.The obtained mixture of Compounds 1 and 2 was dissolved in benzene (10 mL), and triethylamine (84 jiL, 0.6 mmol) was added. The mixture was stirred at room temperature for 1 hour. The reaction mixture was diluted with EtOAc and was then washed with water, dried over MgSO4 and concentrated. The residue was purified by flash chromatography (silica gel, 40% to 50% EtOAc in hexanes) to give the desired product TX63212 (10.2 mg, 4.9%) as a minor product. ?H NMR (400 MHz, CDC13) oe 5.95 (s, 1H), 4.09 (s, 1H), 3.67 (s, 3H), 3.08- 3.04 (m, 1H), 2.97 (d, 1H, 4.8 Hz), 2.00-1.40 (m, 13H), 1.36-1.20 (m, 2H), 1.30 (s, 3H), 1.26 (s, 3H), 1.17 (s, 3H), 1.12 (s, 3H), 1.10 (s, 3H), 1.01 (s, 3H), 0.92 (s, 3H); mlz = 522.3 (M+1).
With N-bromosaccharin; water; In acetonitrile; at 20℃; for 2h;
At room temperature RTA 402 (200 mg, 0.4 mmol) and Nbromosaccharin (156 mg, 0.6 mmol) were mixed in acetonitrile (3 mL) and water (1 mL). The mixture was stirred at room temperature for 2 h. After dilution with dichloromethane, the mixture was washed with water. The organic phase was dried over MgSO4 and concentrated. The residue was purified by flash chromatography (silica gel, 33% EtOAc in hexanes) to give a mixture of two 1 ,2-bromohydrin products 1 and 2.
methyl 2β-cyano-3,12-dioxo-1,2-epoxyolean-9(11)-en-28-oate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
99%
With 3-chloro-benzenecarboperoxoic acid; In dichloromethane; at 20℃; for 3h;Cooling with ice;
CDDO-Me (8.00 g, 15.8 mmol) was dissolved in dichloromethane (40 ml). To this solution, 3-chloroperbenzoic acid (>65%) (5.04 g, 19.0 mmol) was added under ice cooling, and the mixture was stirred at room temperature for 3 hours. To the reaction mixture, a saturated aqueous solution of sodium bicarbonate (40 ml) was added, and the mixture was stirred at the same temperature as above for 15 minutes. To this mixture, a 10% aqueous sodium thiosulfate solution (40 ml) was further added, and the resulting mixture was stirred at the same temperature as above for 5 minutes, followed by extraction with ethyl acetate. The separated organic layer was washed with saturated saline and dried over magnesium sulfate, and the solvent was evaporated off under reduced pressure. The residue was purified by silica gel column chromatography (n-hexane:ethyl acetate=4:1, v/v) to obtain substrate compound (2) (8.16 g, 99%) as a white amorphous solid.
92%
With dihydrogen peroxide; In water; acetonitrile; at 20℃; for 48h;
Compound TX63098: At room temperature RTA 402 (25.0 g, 49.4 mmol) wasdissolved in acetonitrile (250 mL), to which H202 solution (30% in water, 34 mL, 300 mmol)was added in one portion. The mixture was stirred for 48 h at room temperature. The reactionmixture was concentrated, and the crude product was partitioned between 250 mL EtOAc and100 mL water. The organic phase was separated and washed with water and brine, then driedover MgSO4 and concentrated. The residue was purified by flash chromatography (silica gel,33% EtOAc in hexanes) to give the desired product TX63098 (23.8 g, 92%) as white solid.?H NMR (400 MHz, CDC13) oe 6.07 (s, 1H), 4.34 (s, 1H), 3.70 (s, 3H), 3.06-3.00 (m, 1H),2.94 (d, 1H, J=4.8 Hz), 2.00-1.80 (m, 3H), 1.75-1.60 (m, 6H), 1.56-1.44 (m, 2H), 1.37-1.21(m, 4H), 1.27 (s, 6H), 1.19 (s, 3H), 1.12 (s, 3H), 1.08 (s, 3H), 1.01 (s, 3H), 0.91 (s, 3H); mlz= 522.3 (M+1).
Compound TX63212: At room temperature RTA 402 (200 mg, 0.4 mmol) and Nbromosaccharin (156 mg, 0.6 mmol) were mixed in acetonitrile (3 mL) and water (1 mL). The mixture was stirred at room temperature for 2 h. After dilution with dichloromethane, the mixture was washed with water. The organic phase was dried over MgSO4 and concentrated. The residue was purified by flash chromatography (silica gel, 33% EtOAc in hexanes) to give a mixture of two 1 ,2-bromohydrin products 1 and 2.The obtained mixture of Compounds 1 and 2 was dissolved in benzene (10 mL), and triethylamine (84 jiL, 0.6 mmol) was added. The mixture was stirred at room temperature for 1 hour. The reaction mixture was diluted with EtOAc and was then washed with water, dried over MgSO4 and concentrated. The residue was purified by flash chromatography (silica gel, 40% to 50% EtOAc in hexanes) to give the desired product TX63212 (10.2 mg, 4.9%) as a minor product. ?H NMR (400 MHz, CDC13) oe 5.95 (s, 1H), 4.09 (s, 1H), 3.67 (s, 3H), 3.08- 3.04 (m, 1H), 2.97 (d, 1H, 4.8 Hz), 2.00-1.40 (m, 13H), 1.36-1.20 (m, 2H), 1.30 (s, 3H), 1.26 (s, 3H), 1.17 (s, 3H), 1.12 (s, 3H), 1.10 (s, 3H), 1.01 (s, 3H), 0.92 (s, 3H); mlz = 522.3 (M+1).Compound TX63315: At room temperature the mixture of Compounds 1 and 2 (25 mg, 0.04 1 mmol) was dissolved in KOH solution (0.83 mL) (5 g/mL in 20:1 EtOH/ water). The mixture was stirred at room temperature for 1.5 h. After dilution with dichloromethane, the reaction mixture was washed with 1 N HC1(aq) and water. The organic phase was dried over MgSO4 and concentrated. The residue was purified by flash chromatography (silica gel, 0% to 40% EtOAc in hexanes) to give the desired product TX63315 (6.6 mg, 30%) as a white solid. ?H NMR (400 MHz, CDC13) oe 6.13 (s, 1H), 4.78 (br s, 2H), 4.16 (s, 1H), 3.68 (s, 3H), 3.04-3.01 (m, 1H), 2.90 (d, 1H, J=4.8 Hz), 2.06-2.01 (m, 1H), 1.91-1.80 (m, 3H), 1.72-1.27 (m, 11H), 1.25 (s, 3H), 1.24 (s, 3H), 1.19 (s, 3H), 1.10 (s, 3H), 1.07 (s, 3H), 0.99 (s, 3H),0.89 (s, 3H); mlz = 540.3 (M+1).
methyl N<SUP>2</SUP>-(((9H-fluoren-9-yl)methoxy)carbonyl)-N<SUP>5</SUP>-(4-(bromomethyl)phenyl)-L-glutaminate[ No CAS ]
[ 218600-53-4 ]
C61H73N3O10[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
38%
Referring to the synthesis method of VII1,CDDO-Me (298 mg, 0.59 mmol)Was dissolved in 10 ml of DMF,Anhydrous methanol (188 mg, 5.9 mmol) was added,K2CO3 (162.8 mg, 1.18 mmol) was stirred at room temperature overnight,Compound V (325 mg, 0.59 mol) was added and stirring was continued for 0.5 h,The reaction was stopped by adding 5 ml of 1N diluted hydrochloric acid.The reaction solution was diluted with an appropriate amount of dichloromethane (50 mL)Saturated sodium hydrogencarbonate solution and saturated brine.The organic layer was dried over anhydrous sodium sulfate,Filtered, and the filtrate was concentrated to a pale yellow solid,Purification by flash silica gel column chromatography gave white solid VII2 (269 mg, 38%).
methyl N<SUP>2</SUP>-(((9H-fluoren-9-yl)methoxy)carbonyl)-N<SUP>5</SUP>-(4-(bromomethyl)phenyl)-L-glutaminate[ No CAS ]
[ 218600-53-4 ]
C62H75N3O10[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
39%
Referring to the synthesis method of VII1,CDDO-Me (298 mg, 0.59 mmol) was dissolved in 10 ml of DMF,Anhydrous ethanol (271.4 mg, 5.9 mmol) was added,K2CO3 (162.8 mg, 1.18 mmol) was stirred at room temperature overnight,Compound V (325 mg, 0.59 mol) was added and stirring was continued for 0.5 h,The reaction was stopped by adding 5 ml of 1N diluted hydrochloric acid.The reaction solution was diluted with an appropriate amount of dichloromethane (50 mL)Saturated sodium hydrogencarbonate solution and saturated brine.The organic layer was dried over anhydrous sodium sulfate,Filtered, and the filtrate was concentrated to a pale yellow solid,Purification by flash silica gel column chromatography gave white solid VII3 (295 mg, 39%).
methyl N<SUP>2</SUP>-(((9H-fluoren-9-yl)methoxy)carbonyl)-N<SUP>5</SUP>-(4-(bromomethyl)phenyl)-L-glutaminate[ No CAS ]
[ 218600-53-4 ]
1-hydroxyl-2-cyano-3-(4-(methyl-L-glutaminate-N<SUP>2</SUP>-(((9H-fluoren-9-yl)methoxy)carbonyl)-N<SUP>5</SUP>-yl)-benzyloxy)-12-oxooleana-2(3),9(11)-dien-28-oic acid methyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
53%
CDDO-Me (298 mg, 0.59 mmol) was dissolved in 10 ml of DMF,Pure water (106 mg, 5.9 mmol) was added,K2CO3 (162.8 mg, 1.18 mmol) was stirred at room temperature overnight,Compound V (325 mg, 0.59 mol) was added and stirring was continued for 0.5 h,The reaction was stopped by adding 5 ml of 1N diluted hydrochloric acid.The reaction solution was diluted with an appropriate amount of dichloromethane (50 mL)Saturated sodium hydrogencarbonate solution and saturated brine.The organic layer was dried over anhydrous sodium sulfate,Filtered, and the filtrate was concentrated to a pale yellow solid,Flash silica gel column chromatography gave white solid VII (305 mg, 53%).
1-isosorbide mononitrate 2-cyano-3-acetoxy-12-oxoolean-2(3),9(11)-diene-28-carboxylic acid methyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
35%
CDDO-Me (100 mg, 0.2 mmol) was dissolved in 2.5 ml of dry DMF,Isosorbide mononitrate (57.3 mg, 0.3 mmol) was added,K2CO3 (41 mg, 0.3 mmol) was stirred at room temperature overnight,The next day, compound V (23.6 mg, 0.3 mmol) was added and stirring was continued for 6 h,The reaction solution was diluted with an appropriate amount of dichloromethane (50 mL)Saturated sodium bicarbonate solution and saturated brine were washed three times.The organic layer was dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated to a white solid which was rapidly passed Silica gel column chromatography gave white solid I1 (35 mg, 35%).
methyl (1α,2α,21β)-2-cyano-21-hydroxy-3,12-dioxo-1,2-epoxyolean-9(11)-en-28-oate[ No CAS ]
C32H43NO5[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
2.19 g; 192 mg
With SANK 11867; In dimethyl sulfoxide; at 23℃; for 144h;Enzymatic reaction;
80 ml of a main culture medium having the composition shown in Table 2 of Example 1 was placed in each of 583 500-ml Erlenmeyer flasks, sterilized at 121 C. for 20 minutes, and then cooled to room temperature. 4 ml of the seed culture solution of SANK 11867 was aseptically inoculated to the medium in each flask and cultured for 2 days under conditions of 23 C. and 210 rpm in a rotating shaker. 0.8 ml of a solution of CDDO-Me dissolved at a concentration of 30 mg/ml in dimethyl sulfoxide was added to this culture solution in each flask, and the mixture was cultured again at 210 rpm at 23 C. for 6 days in a rotating shaker. (2) Isolation of Terpenoid Derivative (I) and Terpenoid Derivative (III) (0386) The behaviors of terpenoid derivative (I) and terpenoid derivative (III) in this Example were monitored by HPLC under conditions given below. column: Unison UK-C18 (0387) (4.6 mm in diameter×75 mm in length; manufactured by Imtakt Corp.) solvent: A: 10 mM aqueous ammonium formate solution containing 0.01% formic acid (0388) B: acetonitrile containing 0.01% formic acid (0389) A/B=equilibration with 5/5 followed by linear gradient for 7 minutes up to A/B=1/9 (0390) flow rate: 1.0 ml/min (0391) detection: ultraviolet absorption lambda=230 nm (0392) retention time: 4.3 minutes (terpenoid derivative (I)) 4.9 minutes (terpenoid derivative (III)). (0393) To 42.6 L of the culture solution obtained in paragraph (1), an equal amount of acetone was added, and the mixture was well stirred and then allowed to stand overnight at room temperature. Then, the mycelia was removed by filtration under reduced pressure to obtain 78 L of a filtrate. This filtrate was applied to a Sepabeads SP207 (2 L; manufactured by Mitsubishi Chemical Corp.) column washed in advance with acetone and then buffer-replaced with water. The column was washed with 6 L of a mixed solvent of acetone:water=5:5 and subsequently with 6 L of a mixed solvent of acetone:water=6:4, followed by elution with 6 L of a mixed solvent of acetone:water=7:3. To this eluate containing the compounds of interest, 1.2 L of water was added, and further 2.6 L of ethyl acetate was added for liquid-liquid distribution to separate an organic layer containing the compounds of interest. The organic layer was washed with saturated saline, then dried over anhydrous sodium sulfate, and then concentrated under reduced pressure to obtain 6.92 g of extracts containing terpenoid derivative (I) and terpenoid derivative (III). A 1.5 g aliquot of the extracts was dissolved in 5 ml of methanol. This solution was adsorbed onto ODS-A (7.5 g; manufactured by YMC Co., Ltd.), and a mini-column was packed with the resulting carrier and then applied to an ODS-A (100 g; manufactured by YMC Co., Ltd.) column equilibrated in advance with a mixed solvent of acetonitrile:water containing 0.01% formic acid=55:45, followed by elution at a flow rate of 15 ml/min with the same mixed solvent as above. The ultraviolet absorption of the compounds of interest was detected at a wavelength lambda=230 nm. A peak appearing at a retention time of 42.0 minutes (terpenoid derivative (I)) and a peak appearing at a retention time of 50.0 minutes (terpenoid derivative (III)) were each separated 5 times. The fraction solutions of each compound were combined and concentrated under reduced pressure to obtain 192 mg of terpenoid derivative (I) as a colorless powder and 2.45 g of a pale yellow powder containing terpenoid derivative (III). 2.45 g of the pale yellow powder containing terpenoid derivative (III) was dissolved in 2 ml of acetonitrile. This solution was applied to a Sepabeads SP207SS (500 ml; manufactured by Mitsubishi Chemical Corp.) column buffer-replaced in advance with a mixed solvent of acetonitrile:water=55:45. The column was washed with 1,750 ml of a mixed solvent of acetonitrile:water=60:40, followed by elution with 2,340 ml of a mixed solvent of acetonitrile:water=65:35. A 1,200 ml aliquot, of the eluates, containing terpenoid derivative (III) with high purity was concentrated under reduced pressure and subsequently lyophilized to obtain 2.19 g of terpenoid derivative (III).
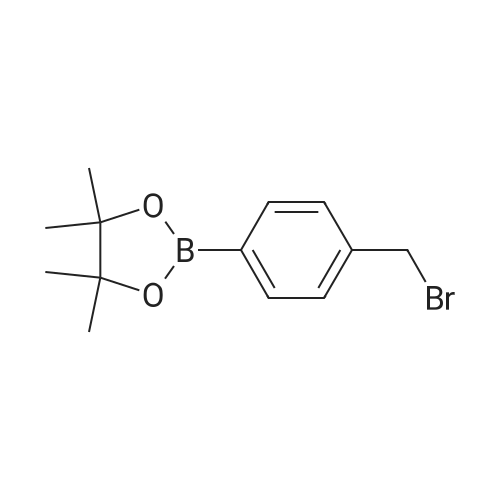
CDDO-Me (298 mg, 0. 59 mmol) was dissolved in 10 ml of DMF and anhydrous methanol (188 mg, 5.9 mmol) was added.K2CO3 (162.8 mg, 1.18 mmol) was stirred at room temperature overnight, bromomethylbenzeneboronic acid pinacol (349.3 mg, 1.18 mmol) was added and stirring was continued for 0.5 h, and the reaction was quenched by adding 5 ml of diluted hydrochloric acid of IN. The reaction solution was diluted with an appropriate amount of dichloromethane (50 mL), and then washed three times with saturated sodium hydrogen carbonate and brine. The organic layer was dried over anhydrous sodium sulfate.Filtration and concentration of the filtrate gave a pale yellow solid.A white solid I2 (297 mg, 67%).
CDDO-Me (298 mg, 0.59 mmol) was dissolved in 10 ml of DMF and anhydrous ethanol (271 mg, 5.9 mmol) was added.K2CO3 (162.8 mg, 1.18 mmol) was stirred at room temperature overnight, bromomethylbenzeneboronic acid pinacol (349.3 mg, 1.18 mmol) was added and stirring was continued for 0.5 h, and the reaction was quenched by adding 5 ml of diluted hydrochloric acid of IN. The reaction solution was diluted with an appropriate amount of dichloromethane (50 mL), and then washed three times with saturated sodium hydrogen carbonate and brine. The organic layer was dried over anhydrous sodium sulfate and filtered.The filtrate was concentrated to give a pale yellow solid.A white solid I3 (221 mg, 49%) was obtained.
CDDO-Me (298 mg, 0 · 59 mmol) was dissolved in 10 ml of DMF, purified water (106 mg, 5·9 mmol), K2CO3 (162.8 mg, 1.18 mmol), stirred at room temperature overnight, bromomethylbenzene Boric acid pinacol (349.3 mg, 1.18 mmol) was stirred for 0.5 h, and the reaction was quenched by adding 5 ml of diluted hydrochloric acid of IN. The reaction solution was diluted with an appropriate amount of dichloromethane (50 mL), and then washed three times with saturated sodium hydrogen carbonate and brine. The organic layer was dried over anhydrous sodium sulfate and filtered.The filtrate was concentrated to give a pale yellow solid.The white solid I1 (305 mg, 53%) was obtained by flash chromatography.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping