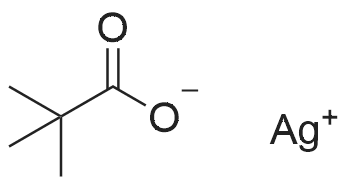
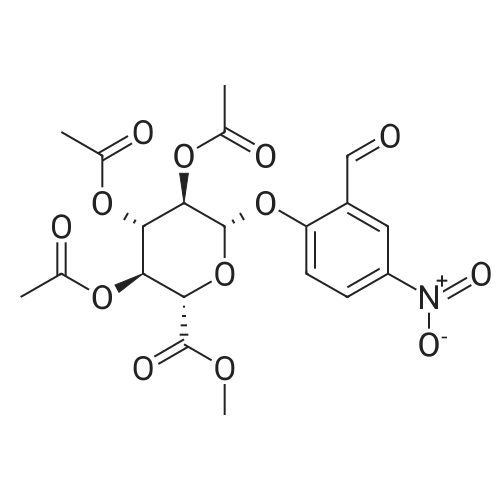
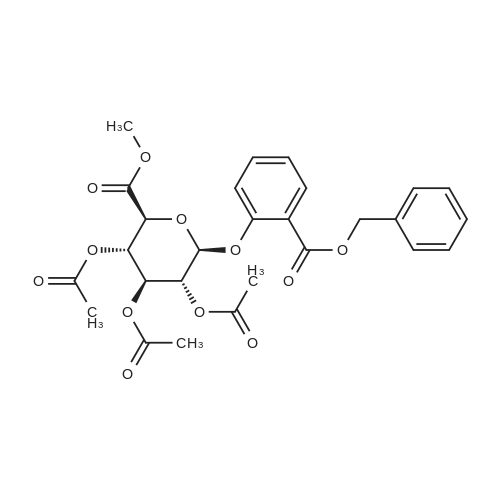
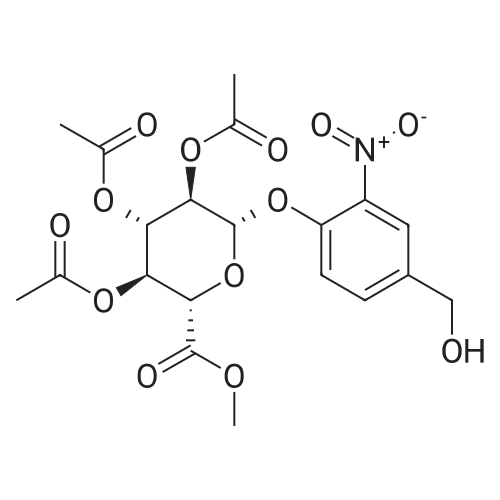
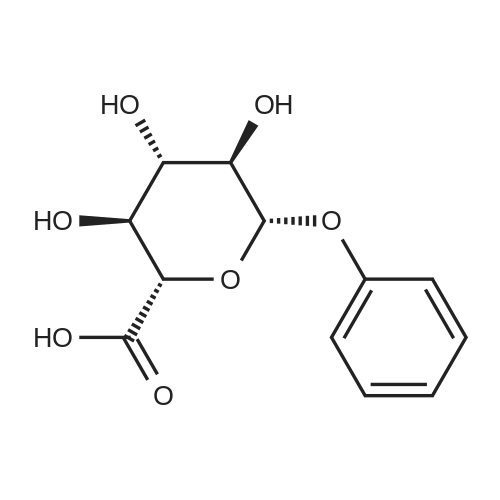
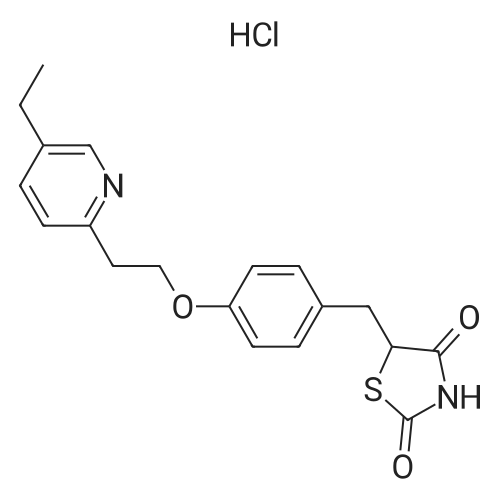
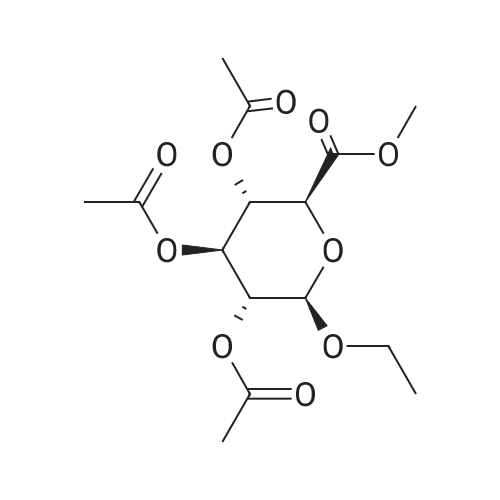
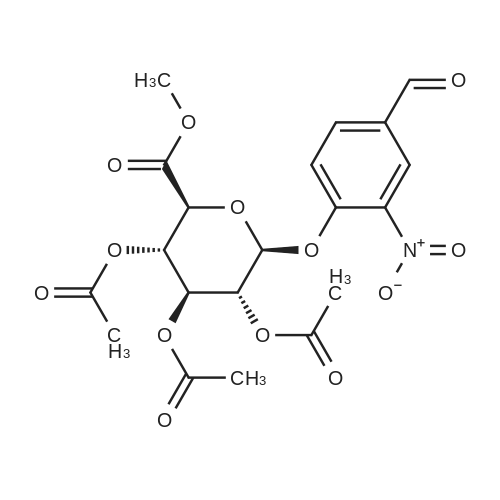
| 92% |
With sulfuric acid; sodium bromide; In tert-butyl methyl ether; at 45 - 50℃; for 24h; |
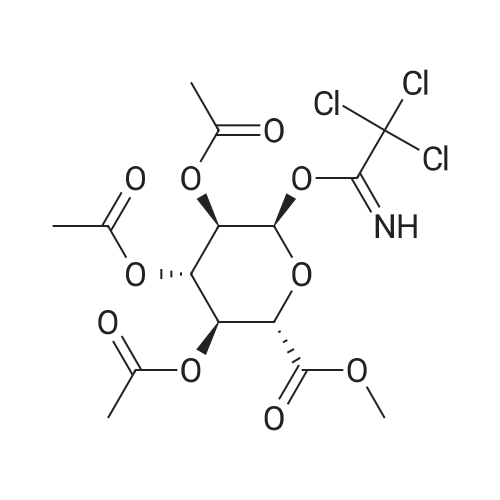
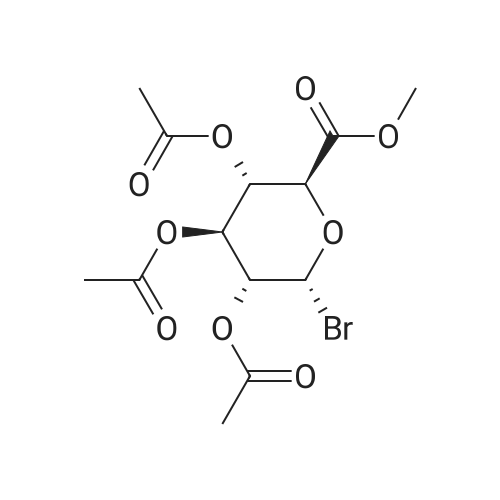
(1) Add 1000g to 10L MTBE solvent while stirring bBeta-tetra-O-acetylglucuronic acid methyl ester,After the system was stirred and dissolved, 273 g of sodium bromide was added. After the feeding, the system was heated to 45 C.Then, 266 g of concentrated sulfuric acid was added dropwise to the system with stirring,Control the temperature of the system in the range of 40 C to 50 C during the dropping process;After the dropwise addition, the system is kept at 45 ± 5 for reaction,HPLC tracks the reaction,The material disappears after about 24 hours,After the reaction is completed, the bodyThe temperature was lowered to 25 , and the system was filtered to obtain sodium sulfate crystals; (2) The filtrate obtained in step (1) is concentrated to about 3 L at normal pressure,Collect the distilled methyl tertiary ether and continue to use in the next batch of reactions.Then the temperature was lowered to 0 C with stirring for 3 hours, and the product and mother liquor were obtained after filtration;The mother liquor can be directly used in the next batch of reactions without concentration.Product alpha-tri-O-acetyl methyl glucuronide bromide,After drying, the mass was 971 g, the yield was 92%, and the purity was 99.5% by HPLC. |
| 90% |
With hydrogen bromide; acetic acid; at 10 - 20℃; for 2h; |
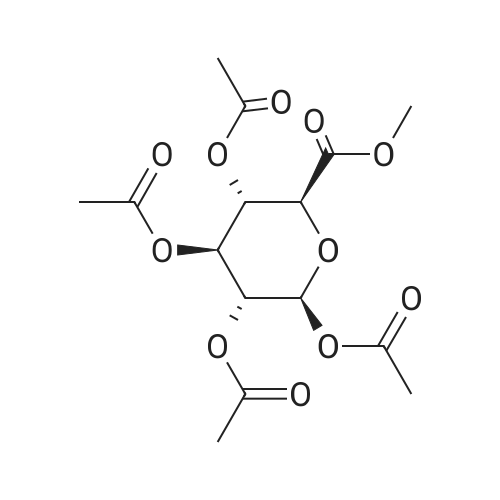
Add 1,2,3,4-tetra-O-acetyl-D-glucuronic acid methyl ester (3) (15.0 g, 4 mmol) to a solution of hydrogen bromide in acetic acid (% by mass of hydrogen bromide) 33%) (60 mL) remained stable no more than 10 C, then naturally warmed to room temperature and stirred for 2 hours. The reaction was concentrated to dryness and then purified with ethyl acetate ethyl acetate. The solid was filtered to give acetobromo-alpha-D-glucuronic acid methyl ester(4) (yield: 14.3 g, yield: 90%). |
| 89% |
With bromine; In ethyl acetate; at 30℃; for 19h;Irradiation; Green chemistry; |
General procedure: Bromine (1.5 mmol, 0.08 mL) was added slowly to a magnetic stirring barand perfluorohexanes (4.0 mL) in a test tube (14 mmphi x 105 mm) with a septum and then 1-O-acetylsugar 1a (1 mmol, 392 mg) in ethyl acetate (2.0 mL) wasadded slowly, forming three layers. The test tube was stirring upon irradiationwith 15 W black light (at 352 nm, TOSHIBA EFD15BLB-T) at 30 C. The light source wasplaced away from the test tube. After 23 hours, the bromine layer disappearedand the fluorous layer recovered transparency. The ethyl acetate layer wastaken up with a pipette. Then, additional ethyl acetate (2 mL x 4) was placedon the residual FC-72 layer, followed by decanting off. The combined ethylacetate layer was washed with water (15 mL), aqueous sat. NaHCO3 (20mL), brine (20 mL) and, dried over Na2SO4, andconcentrated. Purification by chromatography on silica gel with hexane/AcOEt = 2/1gave glycosyl bromide 2a (0.91 mmol,374 mg) in 91% yield |
| 83.9% |
With hydrogen bromide; acetic acid; In dichloromethane; at 0℃; for 2h;Inert atmosphere; |
25.6 g of methyl 1,2,3,4-tetra-O-acetyl-beta-D-glucuronide (4) (68.0 mmol) was dissolved in 120 ml of dichloromethane under nitrogen and cooled to At 0 C, 150 mL of 33% HBr in acetic acid was added dropwise. Stirring was continued for 2 hours at this temperature. TLC was applied. After completion, it was diluted with water and extracted with dichloromethane. The organic layer was washed with saturated sodium hydrogen carbonate and brine. The aqueous solution was dried over sodium sulfate, filtered, evaporated, evaporated22.7 g of a white solid were obtained in a yield of 83.9%. |
| 82% |
With hydrogen bromide; acetic acid; In dichloromethane; at 0 - 20℃; for 0.666667h; |
(2S,3R,4S,5S,6S)-6-(methoxycarbonyl)tetrahydro-2H-pyran-2, 3,4,5- tetrayl tetraacetate 18 (2.0 g, 5.31 mmol) was dissolved in CH2CI2 (20 mL) and cooled to 0 C. To this solution was added a solution of 33% HBr in AcOH (10 mL) dropwise over 10 minutes. The mixture was stirred at 0 C for 30 minutes and then allowed to warm to room temperature. The reaction mixture was diluted with CH2CI2 and washed with sodium bicarbonate solution, followed by brine. The organic layer was dried over MgS04, filtered and evaporated to give an orange syrup. Toluene was added and the residue evaporated to dryness three more times. The same azeotroping process was repeated with ether to give a light beige solid in 1.75 g for a 82% yield. |
| 14% |
With hydrogen bromide; In dichloromethane; water; at 20℃; for 4h;Cooling with ice; |
Methyl-1,2,3,4-tetra-O-acetyl-beta-D-glucuronate (1.5 g, 4.50 mmol) was dissolved in 20 mL of DCM, and hydrogen bromide (2.34 mL, 3.0 eq.) was added thereto in an ice bath. After the dropwise addition, the ice bath was removed, and the mixture was stirred at room temperature for 4 hours. After the completion of reaction, H2O was added, and the mixture was subjected to extraction with CHCl3. The organic layer was washed with brine, dried by adding anhydrous sodium sulfate, and then distilled under reduced pressure. Then, the residue was purified by column chromatography (hexane:EtOAc=4:1) to isolate and obtain a compound 45 (0.22 g, 14%) as a white crystal. |
|
With hydrogen bromide; In acetic acid; for 3.5h; |
Methyl yl (2,3,4-tri-O-acetyl-ss-D-glucopyranosyl azide) uronate (2) Methyl (2,3, 4-tri-O-acetyl- (3-D-glucopyranosyl azide) uronate was prepared by reference to the methodology of Tropper, F. D. et al., Synthesis, 1992,618-620. Methyl 1, 2,3, 4-tetra-O-acetyl- P-D-glucopyranuronate (Bollenback, G. N. et al. J. Am. Chem. Soc., 1955,77, 3310-3315) (1.23 g, 3.26 mmol) was dissolved in HBr/AcOH solution (20 mL, 33% HBr in AcOH) and the solution was stirred. After 3.5 hours the mixture was diluted with CH2Cl2 (100 mL), washed with ice-cold water (2 x 100 mL), saturated sodium bicarbonate (3 x 100 mL), and brine (100 mL). The organic layer was dried (MgS04), filtered and the filtrate evaporated in vacuo to afford a pale yellow oil. This oil was dissolved in CH2CI2 (50 mL), and saturated sodium bicarbonate solution (50 mL), sodium azide (1.1 g, 17 mmol) and tetrabutylammonium hydrogensulfate (1. 1 g, 3.2 mmol) were added. The mixture was stirred vigorously for 16 hours and then CH2Cl2 (150 mL) was added and the solution was washed with saturated sodium bicarbonate (3 x 200 mL), brine (1 x 100 mL), then dried (MgS04), filtered and the solvent evaporated in vacuo to afford Methyl (2,3, 4-tri-O-acetyl-p- D-glucopyranosyl azide) uronate as a white crystalline solid (1.00 g, 85%), m. p. 135-137 C [lit. m. p. 153 C (Gyorgydeak, Z. and Thiem, J. Carb. Res., 1995, 268, 85-92)].'H NMR (500 MHz, CDCl3) 6 5.23 (t, 1H, J= 9.1 Hz, H3); 5. 18 (t, 1H, J= 9.5 Hz, H4); 4.91 (t, 1H, J= 8.8 Hz, H2); 4.68 (d, 1H, J = 8.7 Hz, H1) ; 4.09 (d, 1H, J = 9.6 Hz, H5) ; 3.73 (s, 3H, COOCH3) ; 2.03 (s, 3H, COCH3) ; 1.98 (s, 3H, COCH3) ; 1.97 (s, 3H, COCH3). 13C NMR (125 MHz, CDCl3) 8 169.9 (C=O), 169.2 (C=O), 169.0 (C=O), 166.5 (C=O), 88.0 (Cl), 74.2 (C5), 71.8 (C3), 70.4 (C2), 69.0 (C4), 53. 0 (COOCH3), 20.4 (COCH3), 20.34 (COCH3). |
|
With titanium(IV) bromide; In dichloromethane; at 20℃; for 24h; |
Methyl l-broino-l-deoxy-2,3,4-tri-0- cetyl-o.-D- glucopyr nuronate, compound 10, scheme I.[0045] A solution of 9 ( 1 g, 2.66 mmol) and TiBr4 ( 1 g, 2.72 mmol) in CH2CI2 (30 ml) was stirred at room temperature for 24 h. The mixture was washed with ice water (30 ml) and saturated aqueous NaHCC>3 solution (30 ml), dried over Na2S04, and evaporated to dryness to give 10 (0.95 g, 90%), which was used directly in the next step without further purification. |
| 15 g |
With hydrogen bromide; acetic acid;Cooling; |
Methyl tetra-O-acetyl-beta-D-glucopyranuronate produced according to Example 5, 20 g, is dissolved into 80 mL of 30% hydrogen bromide in acetic acid. The mixture, after solution, is refrigerated overnight. Chloroform, 75 mL, is added to the solution and with moderate stirring a saturated solution of sodium bicarbonate in water is slowly added until the acid wad neutralized. The chloroform is then extracted and dried with sodium sulfate. The chloroform is then removed by rotary evaporation. Absolute ethanol, 65 mL, is then added to the remaining syrup from which crystals began to separate. The mixture is allowed to stand in the refrigerator overnight. Colorless crystals of methyl 2,3,4-tri-O-acetyl-1-bromo-1-deoxy-alpha glucopyranuronate are obtained in a yield of 15 g. The reaction is shown below in Formula (6). |
|
With hydrogen bromide; acetic anhydride; acetic acid; In dichloromethane; at 0℃; |
General procedure: The per-acetylated sugar (1 mmol) was dissolved in DCM (1 mL),acetic anhydride (0.1 mL) and HBr/AcOH (0.8 mL, 30% w/w), and stirred overnight at 0 C. The reaction mixture was diluted with DCM (10 mL), washed with cold water (2 × 5 mL), cold NaHCO3 (2 × 5 mL) and cold brine (5 mL), and dried over MgSO4. After evaporation, thealpha-D-glycosyl bromide was obtained |
|
With hydrogen bromide; acetic acid; In dichloromethane; for 2h;Cooling with ice; |
Under ice-bath was added dropwise to the above methylene chloride solution of hydrogen bromide in acetic acid solution 70mL, reaction 2h, water was added 200mL stirred 20min, extracted three times with methylene chloride, the organic layer was washed with water three times, saturated sodium bicarbonate adjusted to pH to 7, washed twice with saturated brine, dried over anhydrous sodium sulfate 8h.Column chromatography (petroleum ether: ethyl acetate = 5: 1) to give the intermediate 4. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping