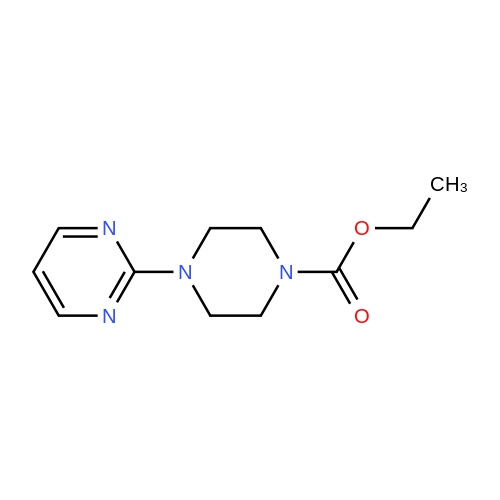
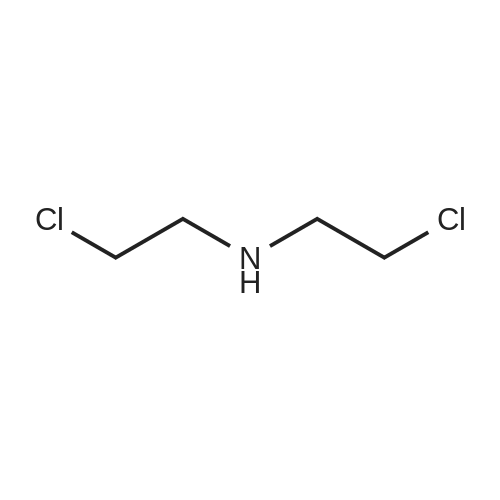
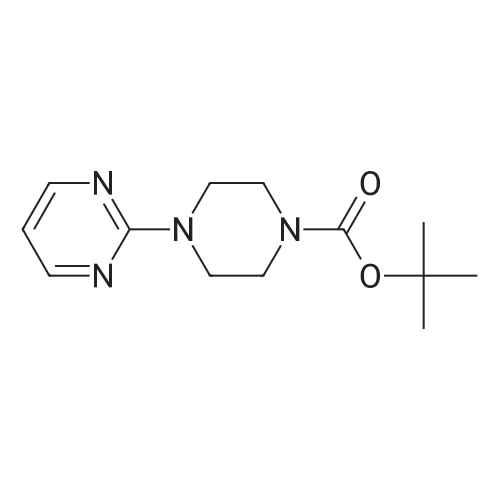
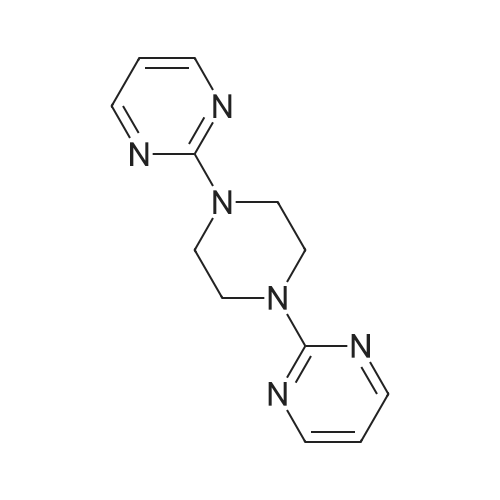
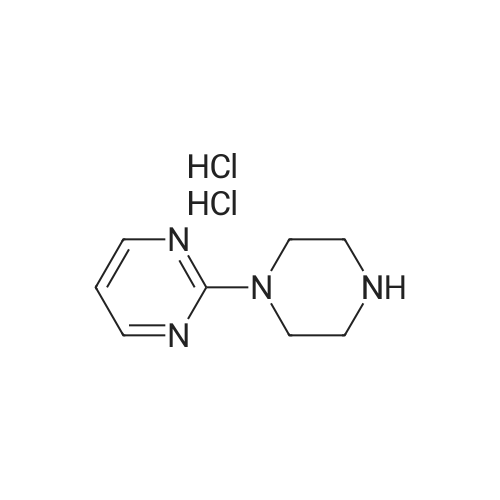
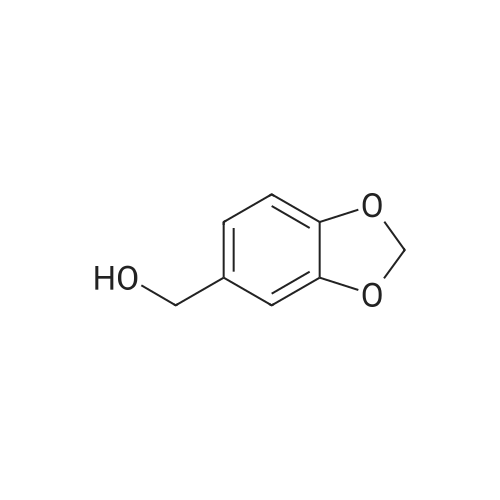
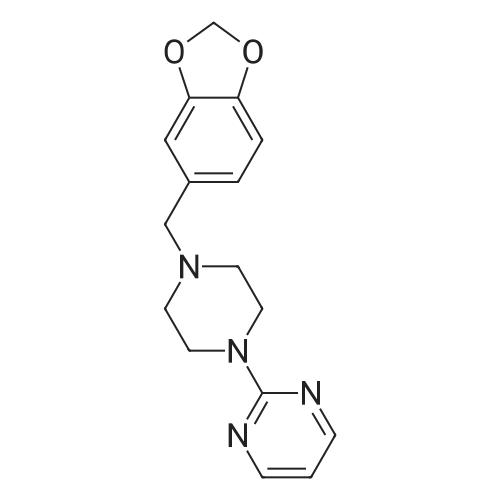
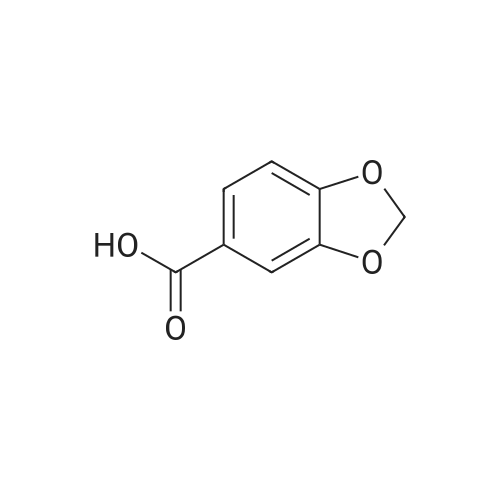
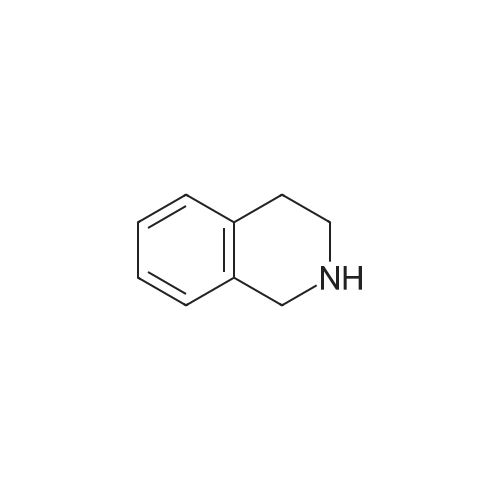
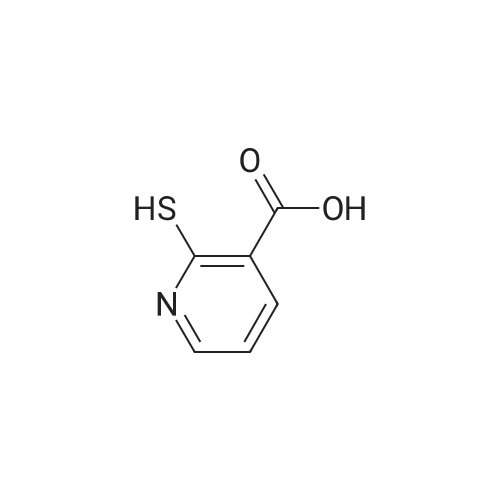
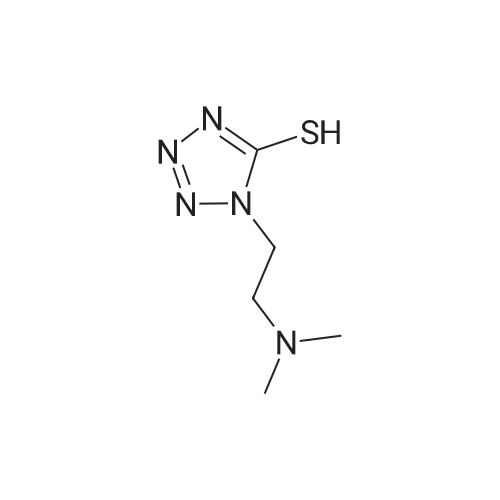
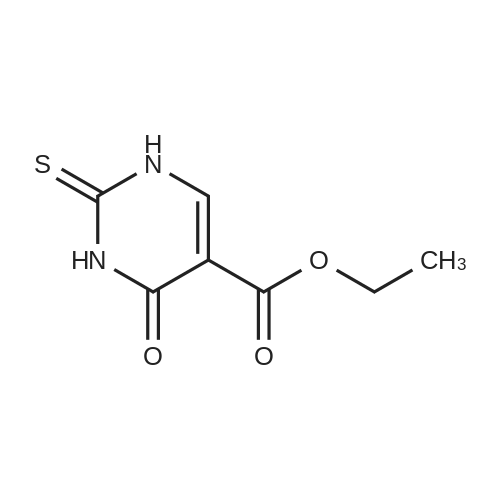
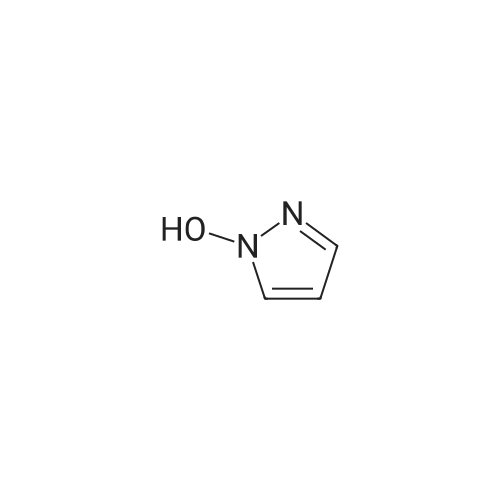
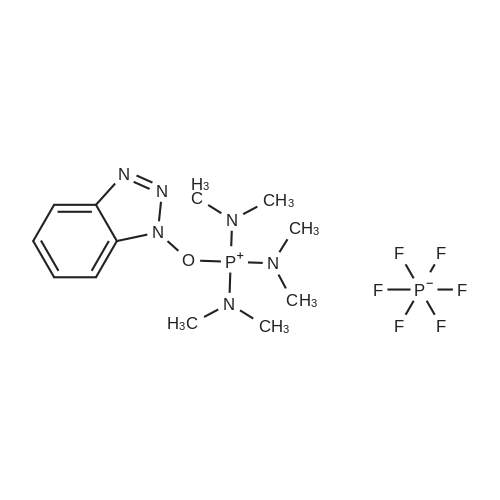
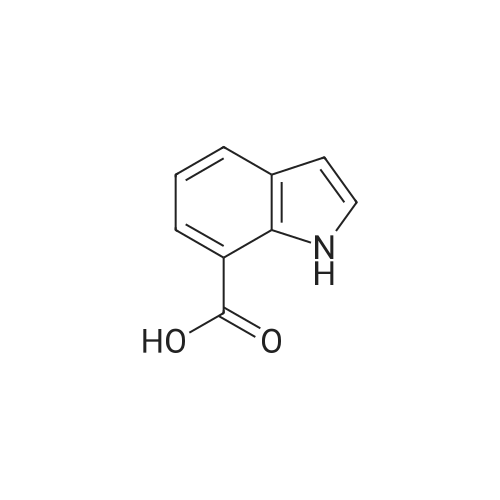
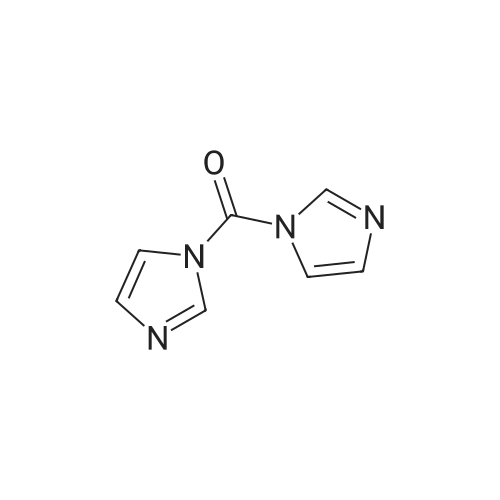
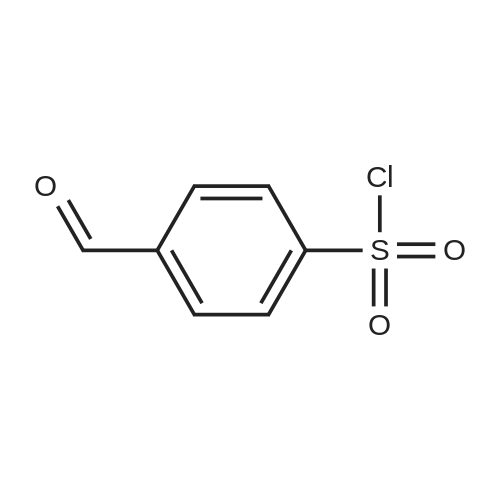
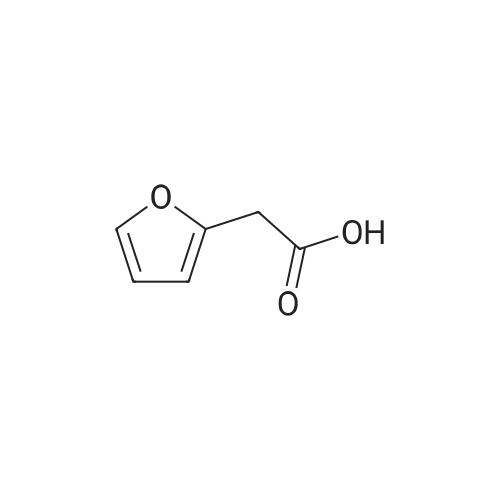
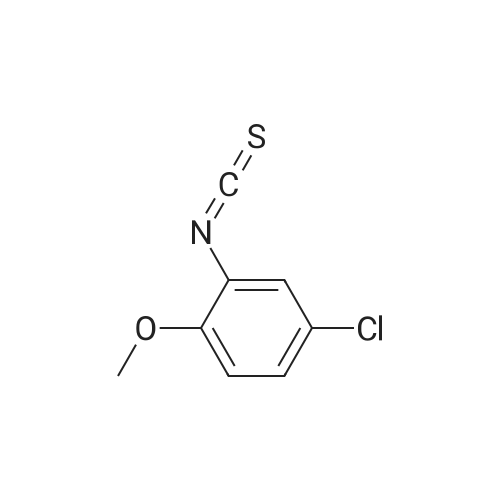
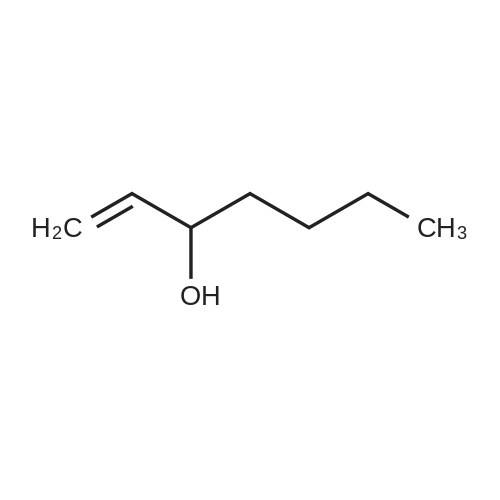
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
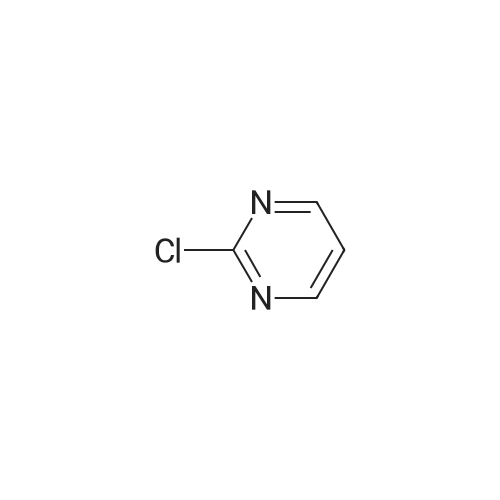
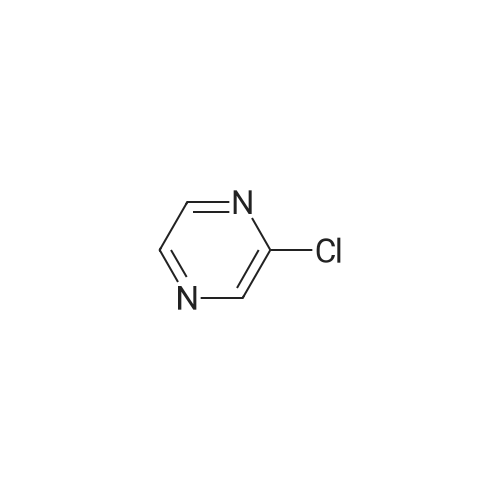
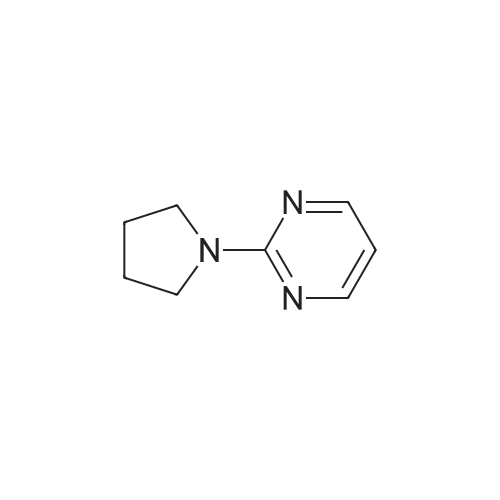
General procedure: 2-Chloro-4,6-disubstituted-pyrimidines 17 were prepared bythe reaction of the diazoniumsalts of 4,6-disubstituted-pyrimidin-2-amines (16) with concentrated hydrochloric acid and ZnCl2 [35].Compound 18 was prepared according to literature [32], and themethod was improved. To a stirred solution of piperazine(45 mmol) and K2CO3 (16.5 mmol) in water (20 mL) was addedchloropyrimidine 17 (18 mmol) in small portions at 50e65 C. Themixture was stirred for 1 h at 60e65 C and cooled to 35 C. Theyellowsolid, 1,4-bispyrimidylpiperazine byproduct, was filtered off,and the filtrate was then extracted three times with chloroform,dried over Na2SO4, and evaporated in vacuum to give compound 18,which was used for the following reactions without further purification. 18a: yellow oil, yield 88percent.
83.8%
With ammonia In water at 98℃; for 3 h;
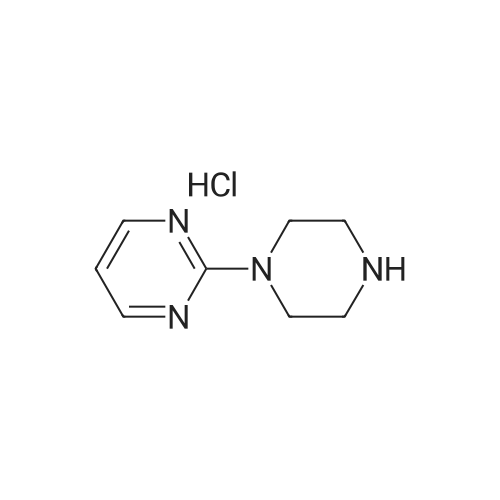
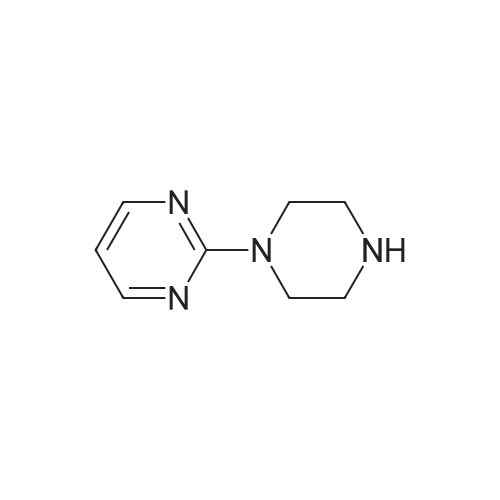
The 24 g anhydrous piperazine, 100 ml water and 8 ml ammonia water into the 250 ml three port into reaction, stirring and heating to 98 °C, dropwise 10 g of 2 - chloro pyrimidine and 90 ml water mixed solution, in 3 hours paused, 98 °C lower heat insulating 0.5 hours. To turns on lathe the distillation, removing about 120 ml water of, cooling to room temperature, dichloromethane is used for extraction 3 times, each time the 30 ml. The combined dichloromethane, adding 4 g anhydrous calcium chloride drying 1 hour, filtering, in the 40 °C lower steaming and removing dichloromethane, shall be 15.6 g product, liquid phase purity 90.04percent, disubstituted by-product (the second piperazine pyrimidine) 11percent. GC analysis of the residual piperazine 0.21percent, yield 83.8percent.
80.7%
Stage #1: With ammonium hydroxide In water at 95 - 100℃; Stage #2: at 95 - 100℃; for 4 h;
100 g of anhydrous piperazine, 360 ml of water and 32 ml of aqueous ammonia were poured into a 1000 ml three-necked reaction flask and heated to 95 ° C to 100 ° C with stirring. A mixed solution of 44 g of dichloropyrimidine and 360 ml of water was added dropwise, and the mixture was dropped at about 3 hours and incubated at 95 ° C to 100 ° C for 1 hour. Spin distillation, remove about 480ml of water, cooling to room temperature, extracted with dichloromethane 3 times, each 120m. The combined methylene chloride was added with 16 g of anhydrous calcium chloride for 1 hour, filtered and the dichloromethane was removed by steaming at 40 ° C to give 57 g of product, liquid purity of 89.74percent, disubstituted by-product (dipyrimidinyl piperazine) 10percent. GC analysis of piperazine residue 0.19percent, the yield of 80.7percent.
Reference:
[1] Chinese Journal of Chemistry, 2015, vol. 33, # 10, p. 1124 - 1134
[2] European Journal of Medicinal Chemistry, 2016, vol. 117, p. 167 - 178
[3] Organic Letters, 2016, vol. 18, # 20, p. 5272 - 5275
[4] Journal of the American Chemical Society, 2017, vol. 139, # 33, p. 11357 - 11360
[5] Patent: CN107216318, 2017, A, . Location in patent: Paragraph 0021; 0024; 0027
[6] Patent: CN106432212, 2017, A, . Location in patent: Paragraph 0010; 0018
[7] Journal of Organic Chemistry, 1953, vol. 18, p. 1484,1487
[8] Farmaco, 2005, vol. 60, # 5, p. 439 - 443
[9] Journal of Chemical Research, 2006, # 12, p. 809 - 811
[10] Patent: US35053, 1995, E1,
[11] Patent: US5099019, 1992, A,
[12] Patent: US4598078, 1986, A,
[13] Patent: US4668687, 1987, A,
[14] Patent: US4355031, 1982, A,
[15] Patent: EP263213, 1988, A1,
[16] Molecules, 2008, vol. 13, # 10, p. 2426 - 2441
[17] Patent: US2005/234046, 2005, A1, . Location in patent: Page/Page column 57
[18] Bioorganic and Medicinal Chemistry Letters, 2014, vol. 24, # 16, p. 3886 - 3890
[19] Patent: CN107266429, 2017, A, . Location in patent: Paragraph 0021; 0024; 0027
2
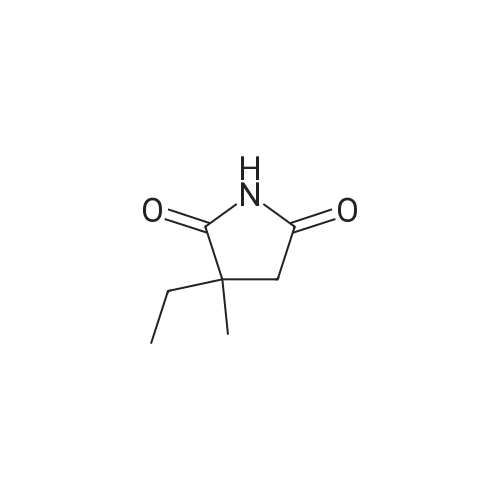
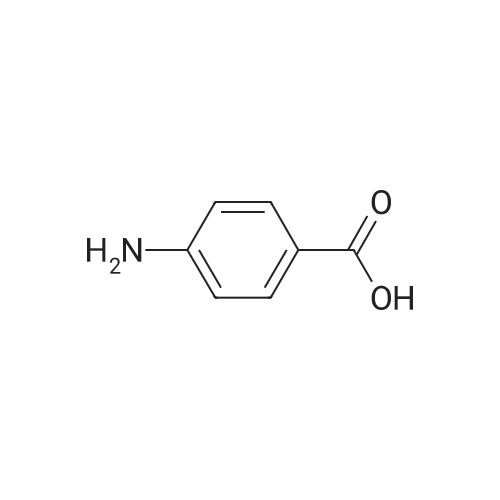
[ 127264-76-0 ]
[ 75-09-2 ]
[ 7732-18-5 ]
[ 20980-22-7 ]
Reference:
[1] Patent: US5204465, 1993, A,
3
[ 109-12-6 ]
[ 821-48-7 ]
[ 20980-22-7 ]
Reference:
[1] European Journal of Pharmaceutical Sciences, 2016, vol. 88, p. 166 - 177
4
[ 78069-54-2 ]
[ 20980-22-7 ]
Reference:
[1] Archiv der Pharmazie, 2003, vol. 336, # 4-5, p. 208 - 215
5
[ 99931-83-6 ]
[ 20980-22-7 ]
Reference:
[1] Collection of Czechoslovak Chemical Communications, 1990, vol. 55, # 9, p. 2304 - 2316
6
[ 109-12-6 ]
[ 334-22-5 ]
[ 20980-22-7 ]
Reference:
[1] Research on Chemical Intermediates, 2011, vol. 37, # 8, p. 1041 - 1045
With Raney nickel In 5,5-dimethyl-1,3-cyclohexadiene for 24 h; Reflux
General procedure: The prepared grades of R-Ni were weighed in water after considering its specific gravity. The residual water was removed using dean stark apparatus. (0043) All the reactions were carried out in a 2-neck round bottom flask, attached with a condenser. Typically, reaction was carried out by stirring and refluxing the reaction mixture of amine and alcohol with pretreated R-Ni in 20ml solvent. After reaction completion, reaction mixture was cooled and filtered using Whatman filter paper 40. The solvent was removed in vacuo. The mixture thus obtained was purified using column chromatography. The purified compounds obtained were characterized by IR, NMR, LC–MS and melting or boiling point. The analytical data obtained of the known compounds are in agreement to the reported literature.
Reference:
[1] Chemistry - A European Journal, 2015, vol. 21, # 27, p. 9656 - 9661
[2] ChemCatChem, 2014, vol. 6, # 3, p. 808 - 814
[3] Chemistry - A European Journal, 2013, vol. 19, # 11, p. 3665 - 3675
[4] Journal of Organic Chemistry, 2011, vol. 76, # 7, p. 2328 - 2331
[5] Tetrahedron Letters, 2007, vol. 48, # 47, p. 8263 - 8265
[6] Journal of the American Chemical Society, 2009, vol. 131, p. 1766 - 1774
[7] Applied Catalysis A: General, 2014, vol. 478, p. 241 - 251
[8] Journal of Organic Chemistry, 2017, vol. 82, # 13, p. 6604 - 6614
[9] Angewandte Chemie - International Edition, 2017, vol. 56, # 46, p. 14702 - 14706[10] Angew. Chem., 2017, vol. 129, # 46, p. 14894 - 14898,5
[11] Green Chemistry, 2012, vol. 14, # 1, p. 226 - 232
11
[ 20850-43-5 ]
[ 20980-22-7 ]
[ 3605-01-4 ]
Yield
Reaction Conditions
Operation in experiment
92%
With triethylamine In isopropyl alcohol at 20 - 50℃;
50 g of 2-piperazin-1-ylpyrimidine, 60 g of triethylamine and 170 ml of isopropyl alcohol were put into a 100 ml three-necked reaction flask, and 58 g of piperonyl chloride was added dropwise with stirring at room temperature, and the mixture was dropped in 30 minutes, heated to 50 °C, stirring to cool to room temperature, filtration, recovery of mother liquor, filter cake by adding 100ml water beating, pumping, plus 50ml water washing cake. Dried at 50 °C, 71.5 g of piribedil crude and 99.1percent by HPLC. Yield 92percent.
52%
With potassium carbonate In 5,5-dimethyl-1,3-cyclohexadiene at 130℃; for 9 h;
16.4 g of piperazinylpyrimidine was dissolved in 300 ml of xylene, 28 g of potassium carbonate and 18 g of piperonyl chloride were added and the suspension was incubated at reflux temperature (130C) for 9 hours, cooled, extracted with 10percent hydrochloric acid several times,The separated hydrochloric acid solution is washed with ether, the acid solution is transferred to alkaline by potassium carbonate, the organic phase is separated, the aqueous phase is extracted with chloroform, the combined organic phase is dried with potassium carbonate, filtered and the solvent is removed by distillation,20 g of the residual oily organics was obtained and recrystallized from ethanol to give 15 g of product, piribedil, yield 52percent.
14.76 g
With triethylamine In isopropyl alcohol at 20 - 50℃; for 2.5 h;
ake 10g about 89.7percent content of piperazine pyrimidine, 12g triethylamine, 34ml isopropanol into 100ml three reaction flask, dropping peppermint chloride at room temperature under stirring, dropping over 30 minutes. Heated to 50 , incubated for 2 hours, cooled to room temperature with stirring, filtered, the mother liquor was recovered, the filter cake was added 20ml water beating, suction filtration, plus 10ml water washing cake. 50 drying, get piribedil crude 16.6g, HPLC analysis content of 99.2percent. Yield 92percent; 16g of crude piribedil (99.2percent), 0.3g of activated charcoal and 42ml of absolute ethanol were added into a 100ml single-necked flask and heated to the reflux temperature for 0.5 hour. The activated carbon was removed by hot filtration and the filtrate was cooled and crystallized under stirring to obtain a white Crystalline solid. Filtered, rinsed with a small amount of anhydrous ethanol, and dried to obtain 14.76g of the first crystal product of piribedil, the content of which was 99.90percent by HPLC and the refined yield was 90.33percent.
Reference:
[1] Patent: CN106432212, 2017, A, . Location in patent: Paragraph 0010; 0019
[2] Patent: CN106432212, 2017, A, . Location in patent: Paragraph 0016
[3] Patent: CN107216318, 2017, A, . Location in patent: Paragraph 0022; 0025; 0028
[4] Patent: CN107266429, 2017, A, . Location in patent: Paragraph 0022; 0025; 0028
12
[ 94-53-1 ]
[ 20980-22-7 ]
[ 3605-01-4 ]
Yield
Reaction Conditions
Operation in experiment
85%
With tris(pentafluorophenyl)borate; phenylsilane In dibutyl ether at 140℃; for 24 h; Schlenk technique; Inert atmosphere; Green chemistry
In the same procedure as in Example 1, The reaction mixture was then heated at 140 ° C for 24 h and cooled to room temperature. The resulting white crystals were isolated by the same post-treatment to yield 85percent yield.The obtained product was subjected to nuclear magnetic resonance spectroscopy and nuclear magnetic resonance spectroscopy to confirm that the obtained product was 2- [4- (1,3-benzodioxol-5-ylmethyl) _ Base] pyrimidine, that ie piribedil. [0050] In a 10 mL Schlenk reaction tube (Beijing Xinwei Er Glass Instrument Co., Ltd., F891410 reaction tube, Capacity 10mL, Grinding mouth 14/20) was added 0.005 mmol of tris (pentafluorophenyl) boron, The inside of the tube is replaced with argon, Then, 1.5 mL of n-butyl ether and 2.0 mmol of phenyl silane were added under argon atmosphere and stirred (using an IKA magnetic stirrer, RCT basic, Stirring speed of 500 rpm). Followed by the addition of 0.5 mmol of 1- (2-pyrimidinyl) piperazine and 1.0 mmol ofd piperic acid. After heating at 100 ° C for 20 h, the mixture was cooled to room temperature. Quenched with sodium hydroxide solution (3M; 3 mL) Ethyl acetate (3 mL) was added, After stirring at room temperature for 3 h, Ethyl acetate extraction (2 mL x 3), The organic phase was dried over anhydrous sodium sulfate, Filtration, organic phase through the rotary evaporator (Swiss step Qi Co., Ltd., BUCHI Rotary Evaporator R-3) Concentrated, And then through the column (Beijing Xin Wei Er Glass Instrument Co., Ltd., C383040C with sand plate storage column chromatography column, 35/20, φ30mm, effective length: 500ml) Chromatography to obtain white crystal products, Yield 41percent. The resulting product was subjected to 1H-NMR (400 MHz, CDCl3) and nuclear magnetic resonance spectroscopy 13C-NMR (101 MHz, CDCl3) analysis, The resulting spectra are shown in Figure 1-2. The resulting product was confirmed to be 2- [4- (1,3-benzodioxol-5-ylmethyl) piperazin-1-yl] pyrimidine, i.e piribedil
With trifuran-2-yl-phosphane; palladacycle; lithium hydroxide In neat (no solvent) at 120℃; for 24 h; Inert atmosphere; Molecular sieve
General procedure: An oven dried Schlenk tube was charged with LiOH (1.5 mmol), pre-catalyst (1.5*10-2 mmol), P(2-Fur)3 (3*10-2 mmol) and activated 4 Å MS (100 mg). The tube was connected to a vacuum line under argon and purged three times. To the reaction mixture, sulfanilamide (3.0 mmol) and arylalcohol (6.0 mmol) were added. The Schlenk tube was closed with PTFE stopper and the reaction mixture was stirred at 120 °C for 24 h. At the end of the reaction time, the reaction mixture was cooled to room temperature, diluted with methanol (5 mL), and the tube was washed with methanol three more times (3*2 mL). The methanol solution was concentrated under vacuum and the crude was subjected to flash column chromatography on silica gel using ethyl acetate and n-hexane mixtures to afford the N-alkylated product.
With bromine; calcium carbonate In water at 50 - 55℃; for 2h;
62%
With hydrogenchloride; bromine at 0 - 100℃;
With bromine In dichloromethane at 0℃;
With sodium hydroxide; bromine In hydrogenchloride
3 5-Bromo-2-(1-piperazinyl)pyrimidine
Example 3 5-Bromo-2-(1-piperazinyl)pyrimidine To an ice-cooled solution of 1-(2-pyrimidinyl) piperazine (16.4 g, 0.1 mole) in 1N HCl (100 mL) was added dropwise bromine (15.98 g, 0.1 mole). After stirring at 0° for 0.5 h. the mixture was heated to 100° C. until dissipation of the red color had occurred. The mixture was filtered, cooled, made alkaline with 50% NaOH and extracted with Et2 O. The dried extract (MgSO4) was concentrated in vacuo to provide 14.5 g (62%) of product, m.p. 73°-75° C. By appropriate modification of this procedure the 5-chloro intermediate and the 5-iodo intermediate may be prepared.
14.5 g (62%)
With sodium hydroxide; bromine In hydrogenchloride
3 5-Bromo-2-(1-piperazinyl)pyrimidine (IIc)
EXAMPLE 3 5-Bromo-2-(1-piperazinyl)pyrimidine (IIc) This example serves to illustrate procedure B of Scheme 2. To an ice-cooled solution of 1-(2-pyrimidinyl)piperazine (16.4 g, 0.1 mole) in 1N HCl (100 mL) was added dropwise bromine (15.98 g, 0.1 mole). After stirring at 0° for 0.5 hr, the mixture was heated to 100° until dissipation of the red color had occurred. The mixture is filtered, cooled, made alkaline with 50% NaOH and extracted with ether. The dried extract (MgSO4) was concentrated in vacuo to provide 14.5 g (62%) of product, m.p. 73°-75°. By appropriate modification of this procedure, the 5-chloro intermediate, IIb, and the 5-iodo intermediate, IId, may be prepared.
2-[(1-methoxycarbonylmethyl-1<i>H</i>-tetrazol-5-yl)-(4-pyrimidin-2-yl-piperazin-1-yl)-methyl]-pyrrolidine-1-carboxylic acid <i>tert</i>-butyl ester[ No CAS ]
Preferably, the Piribedil product having a Piribedil purity of 98 wt % or lower is prepared from a method comprising reacting 1-(2-pyrimidinyl)piperazine and piperonal in the presence of formic acid as a reducing agent at a temperature of 100-140 C
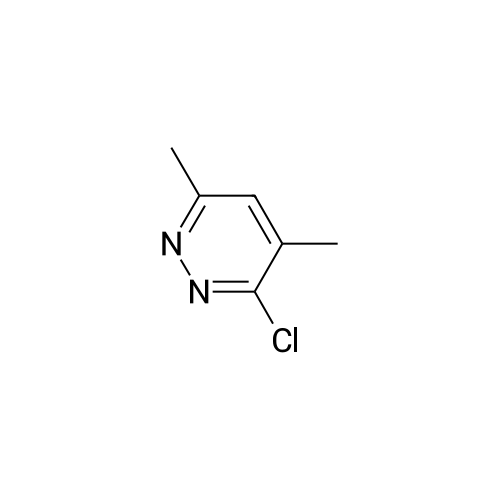
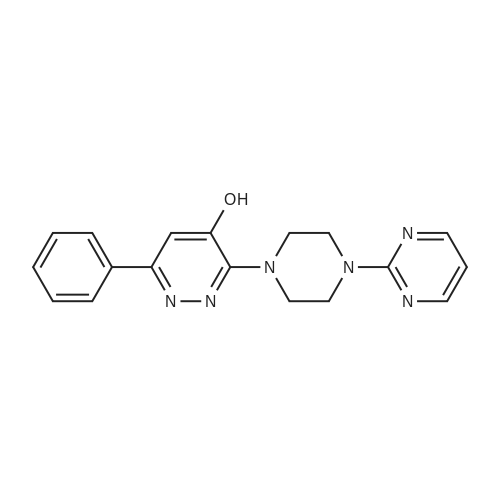
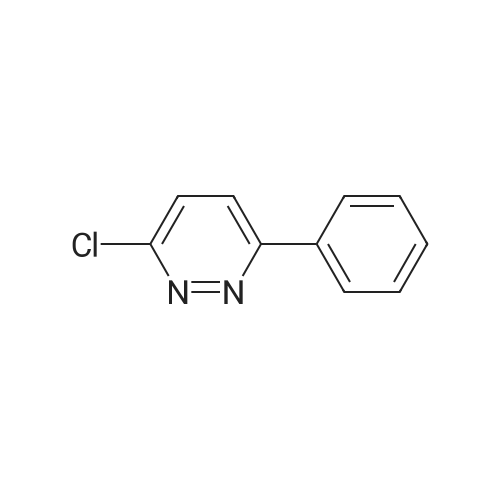
A mixture of 3-chloro-4,6-diphenylpyridazine (267mg, l.Ommol), l-(2-pyrimidyl)piperazine <n="93"/>(656mg, 4.0mmol) in 3ml of 1-BuOH was heated with stirring at 1300C for 3 days. The solvent was removed by evaporation in vacuo the residue was treated with water to give a suspension. The solid was then filtered off, washed with water, dried over filter funnel in vacuo to give light pink solid yielding white solid (22.1g, 66mmol, 97.3%). ESI-MS: m/z 335.2 (M+H+). 1H NMR (DMSO): 1H NMR (DMSO): d 8.406 (d, J=6.5, 2H), 7.740 (d, J=4.0, 2H), 7.558 (s, 3H), 6.686 (t, J=4.8, J=4.4, 1H), 6.841 (s, 1H), 3.881 (s, 4H), 3.620 (s, 4H), 3.776 (S9 4H).
97.3%
In butan-1-ol; at 130.0℃; for 72.0h;Product distribution / selectivity;
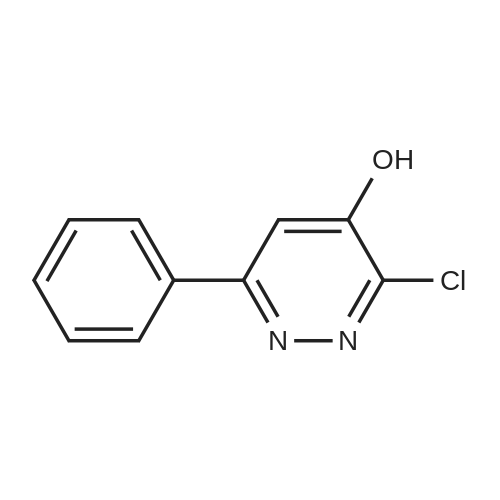
The compound was prepared from <strong>[89868-13-3]3-chloro-4-hydroxy-6-phenylpyridazine</strong> (14g, 68mmol). A mixture of 3-chloro-4,6-diphenylpyridazine (267mg, l.Ommol), l-(2-pyrimidyl)piperazine (656mg, 4.0mmol) in 3ml of 1-BuOH was heated with stirring at 1300C for 3 days. The solvent was removed by evaporation in vacuo the residue was treated with water to give a suspension. The solid was then filtered off, washed with water, dried over filter funnel in vacuo to give light pink solid yielding white solid (22.1g, 66mmol, 97.3%). ESI-MS: m/z 335.2 (M+H+). 1H NMR (DMSO): 1H NMR (DMSO): d 8.406 (d, 1=6.5, 2H), 7.740 (d, J=4.0, 2H), 7.558 (s, 3H)3 6.686 (t, J=4.8, J=4.4, 1H)5 6.841 (s, 1H), 3.881 (s, 4H), 3.620 (s, 4H), 3.776 (s, 4H).
97.3%
In butan-1-ol; at 130.0℃; for 72.0h;Product distribution / selectivity;
6-phenyl-3-(4-(pyrimidin-2-yl)piperazin-1-yl)pyridazin-4-ol (MW01-7-121WH) The compound was prepared from <strong>[89868-13-3]3-chloro-4-hydroxy-6-phenylpyridazine</strong> (14 g, 68 mmol). A mixture of 3-chloro-4,6-diphenylpyridazine (267 mg, 1.0 mmol), 1-(2-pyrimidyl)piperazine (656 mg, 4.0 mmol) in 3 ml of 1-BuOH was heated with stirring at 130 C. for 3 days. The solvent was removed by evaporation in vacuo the residue was treated with water to give a suspension. The solid was then filtered off, washed with water, dried over filter funnel in vacuo to give light pink solid yielding white solid (22.1 g, 66 mmol, 97.3%). ESI-MS: m/z 335.2 (M+H+). 1H NMR (DMSO): 1H NMR (DMSO): d 8.406 (d, J=6.5, 2H), 7.740 (d, J=4.0, 2H), 7.558 (s, 3H), 6.686 (t, J=4.8, J=4.4, 1H), 6.841 (s, 1H), 3.881 (s, 4H), 3.620 (s, 4H), 3.776 (s, 4H).
97.3%
In butan-1-ol; at 130.0℃; for 72.0h;Product distribution / selectivity;
This compound was prepared from <strong>[89868-13-3]3-chloro-4-hydroxy-6-phenylpyridazine</strong> (14g, 68mmol) in the same manner as described below, yielding white solid (22.1g, 66mmol, 97.3%). ESI-MS: m/z 335.2 (M+H+). 1H NMR (DMSO): 1H NMR (DMSO): d 8.406 (d, J=6.5, 2H), 7.740 (d, J=4.0, 2H), 7.558 (s, 3H), 6.686 (t, J=4.8, J=4.4, 1H), 6.841 (s, 1H), 3.881 (s, 4H), 3.620 (s, 4H), 3.776 (s, 4H).; 6-phenyl-3-(4-(pyrimidin-2-yl)piperazin-l-v?pyridazin-4-ol flVlW01-7-121WID The compound was prepared from <strong>[89868-13-3]3-chloro-4-hydroxy-6-phenylpyridazine</strong> (14g, 68mmol). A mixture of 3-chloro-4,6-diphenylpyridazine (267mg, l.Ommol), l-(2-pyrimidyl)piperazine (656mg, 4.0mmol) in 3ml of 1-BuOH was heated with stirring at 13O0C for 3 days. The solvent was removed by evaporation in vacuo the residue was treated with water to give a suspension. The solid was then filtered off, washed with water, dried over filter funnel in vacuo to give light pink solid yielding white solid (22.1g, 66mmol, 97.3%). ESI-MS: m/z 335.2 (M+H+). 1H NMR (DMSO): 1H NMR (DMSO): d 8.406 (d, J=6.5, 2H), 7.740 (d, J=4.0, 2H), 7.558 (s, 3H), 6.686 (t, J=4.8, J=4.4, 1H), 6.841 (s, 1H), 3.881 (s, 4H), 3.620 (s, 4H), 3.776 (s, 4H).;This compound was prepared from <strong>[89868-13-3]3-chloro-4-hydroxy-6-phenylpyridazine</strong> (14g, 68mmol) A mixture of 3-chloro-4,6-diphenylpyridazine (267mg, l.Ommol), l-(2-pyrimidyl)pperazine (656mg, 4.0mmol) in 3ml of 1-BuOH was heated with stirring at 1300C for 3 days. The solvent was removed by evaporation in vacuo the residue was treated with water to give a suspension. The solid was then filtered off, washed with water, dried over filter funnel in vacuo to give light pink solid, yielding white solid (22.1g, 66mmol, 97.3%). ESI-MS: m/z 335.2 (M+H+). 1H NMR (DMSO): 1H NMR (DMSO): d 8.406 (d, J=6.5, 2H), 7.740 (d, J=4.0, <n="97"/>2H), 7.558 (s, 3H), 6.686 (t, J=4.8, J=4.4, 1H), 6.841 (s, 1H), 3.881 (s, 4H), 3.620 (s, 4H), 3.776 (S5 4H).
With triethylamine In N,N-dimethyl-formamide at 20℃;
1
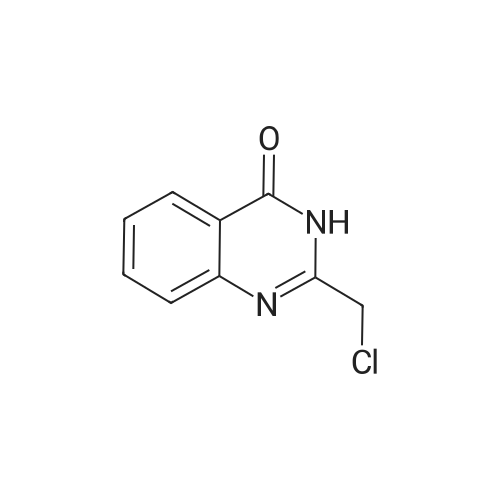
In a typical example, 2-((4-(pyrimidin-2-yl)piperazin-l-yl)methyl)quinazolin-4-one (Compound 123, see, Table 1) was prepared as follows:2-((4-(pyrimidin-2-yl)piperazin- 1 -yl)methy l)quinazolin-4( 1 H)-one300 mg of 2-(chloromethyl)quinazolin-4(3H)-one and 300 mg 2-(piperazin-l- yl)pyrimidine were dissolved in 5 ml of DMF at room temperature. After a few seconds, 0.5 ml of triethylamine was added, and the reaction mixture was stirred at ambient temperature, while being monitored by TLC. Once the reaction was completed (overnight), additional 0.2 ml of triethylamine was added, followed by 3 ml of water, and the mixture was cooled to 0 °C. The solid precipitate was thereafter collected by vacuum filtration, washed with cold water and dried for 2 hours at 50 0C, to give 370 mg (75 % yield) of the product.1H-NMR (CDCl3): δ = 10.05 (bs, IH, NH), 8.32-6.48 (m, 7H, Har), 3.93-3.88 (t, 4H), 3.62 (s, 2H), 2.68-2.63(t, 4H) ppm.
3-phenyl-6-(4-pyrimidin-2-yl-piperazin-1-yl)pyridazine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
96.4%
With ammonium chloride; In butan-1-ol; at 130℃; for 48h;
Figure 1 depicts a synthetic scheme for the preparation of 2-(4-(6-phenylpyridazin-3- yl)piperazin- 1 -yl)pyrimidine (MWO 1-3-183WH). Reagent and condition: (a) 1 -BuOH, NH4Cl, and2-( piperazin-l-yl)pyrimidine. A typical reaction mixture of comprised about 0.01 mol of 3- chloro-6-phenylpyridazine by 2-(piperazin-l- yl)pyrimidine, about 0.05 mol of 2-(piperazin-l- yl)pyrimidine and about 0.01 mol of ammonium hydrochloride was prepared in about 15ml of 1-BuOH. The mixture was stirred at about 13O0C for about 48h, and then the solvent was removed under reduced pressure. The remaining residue was then extracted with ethyl acetate, washed with water and brine, dried over anhydrous Na2SO4. Removal of solvent followed by recrystallization from 95% ethanol yielded light yellow crystals, yield 96.4%; HPLC: 97.4% purity; HRMS calculated 318.1587, found 318.1579; 1H NMR (CDCl3): delta 8.356 (d, J=4.5, 2H), 8.01 l(d,J=7.5, 11 1H), 7.692 (d, J=9.5, 1H), 7.468 (t, J=6.0, 2H), 7.417 (d, J=7.5, 1H), 7.047 (d, J=9.5, 1H), 6.546 (t, J=4.5, 1H), 4.013 (t, J=5.0, 4H), 3.826 (t5 J=5.0, 4H).
96.4%
With ammonium chloride; In butan-1-ol; at 130℃; for 48h;
Figure 1 depicts a synthetic scheme for the preparation of 2-(4-(6-phenylpyridazin-3- yl)piperazin-l-yl)pyrimidine (MW01-3-183WH). Reagent and condition: (a) 1-BuOH, NH4Cl, and 2-( piperazin-l-yl)pyrimidine. A typical reaction mixture of comprised about 0.01 mol of 3- chloro-6-phenylpyridazine by 2-(piperazin-l- yl)pyrimidine, about 0.05 mol of 2-(piperazin-l- yl)pyrimidine and about 0.01 mol of ammonium hydrochloride was prepared in about 15ml of 1- BuOH. The mixture was stirred at about 13O0C for about 48h, and then the solvent was removed under reduced pressure. The remaining residue was then extracted with ethyl acetate, washed with water and brine, dried over anhydrous Na2SO4. Removal of solvent followed by recrystallization from 95% ethanol yielded light yellow crystals, yield 96.4%; HPLC: 97.4% purity; HRMS calculated 318.1587, found 318.1579; 1H NMR (CDCl3): delta 8.356 (d, J=4.5, 2H), 8.01 l(d, J=7.5, 11 2H), 7.692 (d, J=9.5, 1H), 7.468 (t, J=6.0, 2H), 7.417 (d, J=7.5, 1H), 7.047 (d, J=9.5, 1H), 6.546 (t, J=4.5, 1H), 4.013 (t, J=5.0, 4H), 3.826 (t, J=5.0, 4H).
96.4%
With ammonium chloride; In butan-1-ol; at 130℃; for 48h;
FIG. 2 depicts a synthetic scheme for the preparation of 2-(4-(6-phenylpyridazin-3-yl)piperazin-1-yl)pyrimidine (MW01-3-183WH). Reagents and conditions: (a) 1-BuOH, NH4Cl, and 2-(piperazin-1-yl)pyrimidine. A typical reaction mixture comprising about 0.01 mol of <strong>[20375-65-9]3-chloro-6-phenylpyridazine</strong> by 2-(piperazin-1-yl)pyrimidine, about 0.05 mol of 2-(piperazin-1-yl)pyrimidine and about 0.01 mol of ammonium hydrochloride was prepared in about 15 ml of 1-BuOH. The mixture was stirred at about 130 C. for about 48 h, and then the solvent was removed under reduced pressure. The remaining residue was then extracted with ethyl acetate, washed with water and brine, dried over anhydrous Na2SO4. Removal of solvent followed by recrystallization from 95% ethanol yielded light yellow crystals, yield 96.4%; HPLC: 97.4% purity; HRMS calculated 318.1587, found 318.1579; 1H NMR (CDCl3): d 8.356 (d, J=4.5, 2H), 8.011 (d, J=7.5, 11 2H), 7.692 (d, J=9.5, 1H), 7.468 (t, J=6.0, 2H), 7.417 (d, J=7.5, 1H), 7.047 (d, J=9.5, 1H), 6.546 (t, J=4.5, 1H), 4.013 (t, J=5.0, 4H), 3.826 (t, J=5.0, 4H).
96.4%
With ammonium chloride; In butan-1-ol; at 130℃; for 48h;
Reagent and condition: (a) 1-BuOH, NH4Cl, and 2-( piperazin-l-yl)pyrimidine. A typical reaction mixture of comprised about 0.01 mol of 3- chloro-6-phenylpyridazine by 2-(piperazin-l- yl)pyrimidine, about 0.05 mol of 2-(piperazin-l- yl)pyrimidine and about 0.01 mol of ammonium hydrochloride was prepared in about 15ml of 1- BuOH. The mixture was stirred at about 1300C for about 48h, and then the solvent was removed under reduced pressure. The remaining residue was then extracted with ethyl acetate, washed with water and brine, dried over anhydrous Na2SO4. Removal of solvent followed by recrystallization from 95% ethanol yielded light yellow crystals, yield 96.4%; HPLC: 97.4% purity; HRMS <n="87"/>calculated 318.1587, found 318.1579; 1H NMR (CDCl3): delta 8.356 (d, J=4.5, 2H), 8.01 l(d, J=7.5, 11 2H), 7.692 (d, J=9.5, 1H), 7.468 (t, J=6.0, 2H), 7.417 (d, J=7.5, 1H), 7.047 (d, J=9.5, 1H), 6.546 (t, J=4.5, 1H), 4.013 (t, J=5.0, 4H), 3.826 (t, J=5.0, 4H).
In o-xylene; at 150℃; for 30.0h;Inert atmosphere; Sealed tube;
General procedure: Dried pressure tube was charged with magnetic stir bar and50 mg of PdSiO2 catalysts (1 mol% with respect to amine). Then,1.0 mL o-xylene was added, followed by the addition of 0.5 mmolof amine and 1 mmol of benzyl alcohol. The pressure tube wasflushed with argon was closed with screw cap. Then it was placedin the preheated aluminum block and reaction was allowed to progressfor 30 h at 150 C. After completion of the reaction, pressuretube was removed from aluminum block and cooled down to roomtemperature. The catalyst was filtered out by ciliate and reactionproducts were analyzed by GC-MS and the corresponding amineswere purified by column chromatography. The yields of selectedamines were determined by GC analysis using n-hexadecane asstandard. For this purpose, after completion of the reaction, nhexadecane(100 mL) as standard was added to the reaction pressuretube and the reaction products were diluted with ethyl acetate followed by filtration using plug of silica and then subjected GCanalysis.
85%
With Raney nickel; In 5,5-dimethyl-1,3-cyclohexadiene; for 24.0h;Reflux;
General procedure: The prepared grades of R-Ni were weighed in water after considering its specific gravity. The residual water was removed using dean stark apparatus. (0043) All the reactions were carried out in a 2-neck round bottom flask, attached with a condenser. Typically, reaction was carried out by stirring and refluxing the reaction mixture of amine and alcohol with pretreated R-Ni in 20ml solvent. After reaction completion, reaction mixture was cooled and filtered using Whatman filter paper 40. The solvent was removed in vacuo. The mixture thus obtained was purified using column chromatography. The purified compounds obtained were characterized by IR, NMR, LC-MS and melting or boiling point. The analytical data obtained of the known compounds are in agreement to the reported literature.
With sodium tris(acetoxy)borohydride; acetic acid; In 1,2-dichloro-ethane; at 20℃; for 16h;
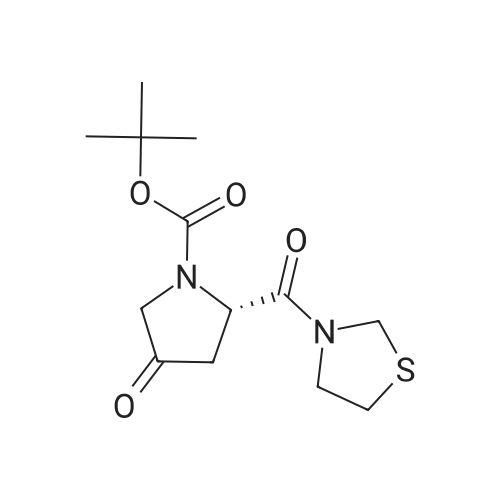
(1) The title compound (1.50 g) of Reference Example 3 and 2-(1-piperazinyl)pyrimidine (0.903 g) were dissolved in 1,2-dichloroethane (25 mL), and acetic acid (0.29 mL) and triacetoxysodium borohydride (2.12 g) were added. The mixture was stirred at room temperature for 16 hr. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction solution and the mixture was extracted with chloroform. The extract solution was washed with saturated brine, dried and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give 3-{(2S,4S)-1-tert-butoxycarbonyl-4-[4-(2-pyrimidinyl)-1-piperazinyl]-2-pyrrolizinylcarbonyl}-1,3-thiazolidine (2.12 g) as a white solid.(2) The above-mentioned compound (2.12 g) was dissolved in a 5.6 mol/L hydrochloric acid-ethanol solution (10 mL) and the mixture was stirred at room temperature for 22 hr. The reaction solution was concentrated under reduced pressure to give the title compound (2.05 g) as a white solid.1H-NMR(DMSO-d6)delta 2.33(1H,m), 2.92-4.33(15H,m), 4.47-4.77(5H,m), 6.79(1H,t,J=4.8Hz), 8.46(2H,d,J=4.8Hz), 9.14(1H,brs), 11.01 (1H,brs).
2-hydroxy-7-[4-(2-pyrimidyl)piperazinomethyl]-2,4,6-cycloheptatrien-1-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
18%
With acetic acid In methanol; water at 20℃; for 72h;
7
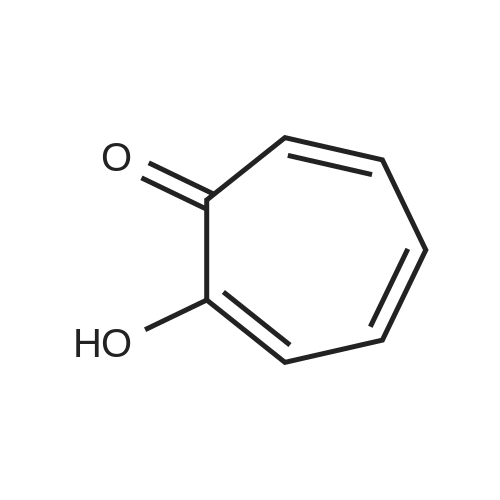
Commercially available tropolone (2.4 g, 20 mmol), 1-(2-pyrimidyl) piperazine (3.3 g, 20 mmol) and acetic acid (1.1 mL, 20 mmol) were mixed with 10 mL methanol and then 37% aqueous formalin (1.6 mL, 20 mmol) was added. After stirring at room temperature for 3 days, the reaction mixture was concentrated under reduced pressure and the residue was purified by silica gel column chromatography (eluent; methylene chloride:methanol=25:1). After suspending the purified fraction in methanol, 2-hydroxy-7-[4-(2-pyrimidyl)piperazinomethyl]-2,4,6-cycloheptatrien-1-one (1.1 g, 3.6 mmol, 18%) was obtained by filtration.
With acetic acid; In methanol; water; at 20℃; for 15h;
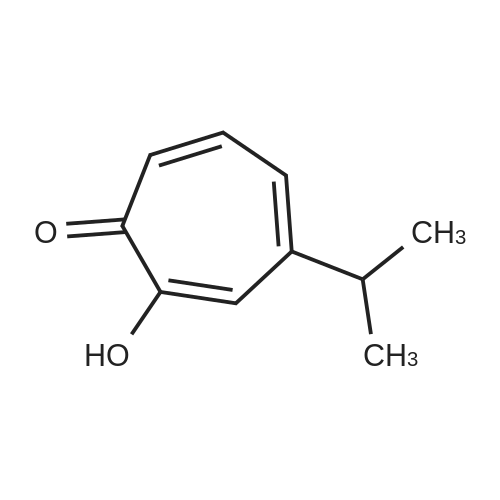
Hinokitiol (3.3 g, 20 mmol), 1-(2-pyrimidyl)piperazine (3.3 g, 20 mmol) and acetic acid (1.1 mL, 20 mmol) were mixed with 10 mL of methanol and then added with 37% aqueous formalin (1.6 mL, 20 mmol). After stirring at room temperature for about 15 hours, 2-hydroxy-4-isopropyl-7-[4-(2-pyrimidyl)piperazinomethyl]-2,4,6-cycloheptatrien-1-one (3.6 g, 10.5 mmol, 53%) was obtained by collecting the precipitated crystals by filtration.
With N-ethyl-N,N-diisopropylamine; at 20.0℃; for 40.0h;Combinatorial reaction / High throughput screening (HTS);
Second Step: N-Alkylation Reaction [0434] 1.2 ml of a corresponding piperazine solution (1.2 mmol, 10 eq) previously prepared is distributed into each reactor. The piperazines used and the amounts added to the reaction are listed below: [TABLE-US-00008] Structure MW Quantity (g) [CHEMMOL-00071] 162.24 0.417 [CHEMMOL-00072] 192.26 0.494 [CHEMMOL-00073] 192.26 0.494 [CHEMMOL-00074] 192.26 0.494 [CHEMMOL-00075] 196.68 0.506 [CHEMMOL-00076] 196.68 0.506 [CHEMMOL-00077] 196.68 0.506 [CHEMMOL-00078] 176.26 0.453 [CHEMMOL-00079] 176.26 0.453 [CHEMMOL-00080] 176.26 0.453 [CHEMMOL-00081] 180.23 0.463 [CHEMMOL-00082] 180.23 0.463 [CHEMMOL-00083] 230.23 0.592 [CHEMMOL-00084] 163.22 0.420 [CHEMMOL-00085] 163.22 0.420 [CHEMMOL-00086] 176.26 0.453 [CHEMMOL-00087] 164.21 0.422 [CHEMMOL-00088] 164.21 0.422 [CHEMMOL-00089] 164.21 0.422 [CHEMMOL-00090] 198.66 0.511 [CHEMMOL-00091] 198.66 0.511 [CHEMMOL-00092] 190.29 0.489 [0435] Next, 75 mul (3.5 eq) of DIEA is added to each well and shaken at room temperature for 40 hours. The resins are filtered and rinsed three times each with three successive solvents: DMF, MeOH, CH2Cl2 (4 ml/reactor per rinse). The resin is allowed to dry in air. [0436] Cleavage [0437] Cleavage is carried out in 2 ml of TFA per reactor at room temperature for 2 hours. The resins are filtered and rinsed with 2×2 ml of CH2Cl2, then the filtrates are concentrated. Each filtrate is taken up in 1 ml of MeOH, the pH is adjusted to 9 with an aqueous 1 M NaOH solution. The mixtures are then purified on cation exchange resins according to the protocol described hereinabove. The 44 filtrates collected into previously tared tubes are vacuum concentrated (Genevac) and analyzed by HPLC/MS before being weighed.
With N-ethyl-N,N-diisopropylamine; at 20.0℃; for 40.0h;Combinatorial reaction / High throughput screening (HTS);
Second Step: N-Alkylation Reaction [0434] 1.2 ml of a corresponding piperazine solution (1.2 mmol, 10 eq) previously prepared is distributed into each reactor. The piperazines used and the amounts added to the reaction are listed below: [TABLE-US-00008] Structure MW Quantity (g) [CHEMMOL-00071] 162.24 0.417 [CHEMMOL-00072] 192.26 0.494 [CHEMMOL-00073] 192.26 0.494 [CHEMMOL-00074] 192.26 0.494 [CHEMMOL-00075] 196.68 0.506 [CHEMMOL-00076] 196.68 0.506 [CHEMMOL-00077] 196.68 0.506 [CHEMMOL-00078] 176.26 0.453 [CHEMMOL-00079] 176.26 0.453 [CHEMMOL-00080] 176.26 0.453 [CHEMMOL-00081] 180.23 0.463 [CHEMMOL-00082] 180.23 0.463 [CHEMMOL-00083] 230.23 0.592 [CHEMMOL-00084] 163.22 0.420 [CHEMMOL-00085] 163.22 0.420 [CHEMMOL-00086] 176.26 0.453 [CHEMMOL-00087] 164.21 0.422 [CHEMMOL-00088] 164.21 0.422 [CHEMMOL-00089] 164.21 0.422 [CHEMMOL-00090] 198.66 0.511 [CHEMMOL-00091] 198.66 0.511 [CHEMMOL-00092] 190.29 0.489 [0435] Next, 75 mul (3.5 eq) of DIEA is added to each well and shaken at room temperature for 40 hours. The resins are filtered and rinsed three times each with three successive solvents: DMF, MeOH, CH2Cl2 (4 ml/reactor per rinse). The resin is allowed to dry in air. [0436] Cleavage [0437] Cleavage is carried out in 2 ml of TFA per reactor at room temperature for 2 hours. The resins are filtered and rinsed with 2×2 ml of CH2Cl2, then the filtrates are concentrated. Each filtrate is taken up in 1 ml of MeOH, the pH is adjusted to 9 with an aqueous 1 M NaOH solution. The mixtures are then purified on cation exchange resins according to the protocol described hereinabove. The 44 filtrates collected into previously tared tubes are vacuum concentrated (Genevac) and analyzed by HPLC/MS before being weighed.
With N-ethyl-N,N-diisopropylamine; In dichloromethane; at 20.0℃; for 40.0h;Combinatorial reaction / High throughput screening (HTS);
Second Step: N-alkylation Reaction [0155] 1.5 ml of a corresponding piperazine solution (1.6 mmol, 10 eq) previously prepared by dissolving the quantity to be weighed in 3.3 ml of CH2Cl2 is distributed into each reactor. A slight excess of each solution is prepared. The piperazines used and the amounts added in the reaction are listed below: [TABLE-US-00007] Structure MW Quantity (g) [CHEMMOL-00012] 162.24 0.578 [CHEMMOL-00013] 192.26 0.685 [CHEMMOL-00014] 192.26 0.685 [CHEMMOL-00015] 192.26 0.685 [CHEMMOL-00016] 196.68 0.700 [CHEMMOL-00017] 196.68 0.700 [CHEMMOL-00018] 196.68 0.700 [CHEMMOL-00019] 176.26 0.628 [CHEMMOL-00020] 176.26 0.628 [CHEMMOL-00021] 176.26 0.628 [CHEMMOL-00022] 180.23 0.642 [CHEMMOL-00023] 180.23 0.642 [CHEMMOL-00024] 230.23 0.820 [CHEMMOL-00025] 163.22 0.581 [CHEMMOL-00026] 163.22 0.581 [CHEMMOL-00027] 176.26 0.628 [CHEMMOL-00028] 164.21 0.585 [CHEMMOL-00029] 164.21 0.585 [CHEMMOL-00030] 164.21 0.585 [CHEMMOL-00031] 164.21 0.585 [CHEMMOL-00032] 198.66 0.708 [CHEMMOL-00033] 198.66 0.708 [CHEMMOL-00034] 190.29 0.678 [CHEMMOL-00035] 178.24 0.635 [0156] Next, 125 mul (4.5 eq) of DIEA to each reactor and shake the mixture at room temperature for 40 hours is added. The resins are filtered and rinsed three times each with three successive solvents: DMF, MeOH, CH2Cl2 (4 ml/reactor per rinse). The resin is allowed to dry in air. [0157] Cleavage [0158] Cleavage is carried out in 2 ml of TFA per reactor at room temperature for 2 hours. The resins are filtered and rinsed with 2×2 ml of CH2Cl2, and the filtrates are concentrated under vacuum. [0159] Extraction in Alkaline Medium [0160] The 48 mixtures are individually taken up in 8 ml of a 1:1 mixture of CH2Cl2/H2O in Whatman 12 ml cartridges equipped with a PTFE filter. The organic phase (=Organic phase A) is filtered, the aqueous phase is washed with 4 ml of CH2Cl2. The pH of the aqueous phase is adjusted to alkaline pH range by adding a saturated aqueous Na2CO3 solution. Then the aqueous phase is extracted with CH2Cl2 (1×4 ml then 1×2 ml). The 48 filtrates (=48 organic phase B) are vacuum concentrated. [0161] Purification on Cation Exchange Resin [0162] The cation exchange resin is a BCX resin packaged by Bodhan (Mettler-Toledo) in the form of 1 g SPE cartridges. The resin is first washed with 2×3 ml of MeOH (conditioning). The crude mixture, dissolved in 1 ml of MeOH and adjusted to pH 9 with a 1 M aqueous NaOH solution, is then deposited on the small resin column (loading). The column is then washed with 2×3 ml of MeOH (wash). The piperazine derivative is then released by elution with 3 ml of a 2 M solution of NH4OH/MeOH (elution). The filtrate is concentrated under vacuum.
With N-ethyl-N,N-diisopropylamine; In dichloromethane; at 20.0℃; for 40.0h;Combinatorial reaction / High throughput screening (HTS);
Second Step: N-alkylation Reaction [0155] 1.5 ml of a corresponding piperazine solution (1.6 mmol, 10 eq) previously prepared by dissolving the quantity to be weighed in 3.3 ml of CH2Cl2 is distributed into each reactor. A slight excess of each solution is prepared. The piperazines used and the amounts added in the reaction are listed below: [TABLE-US-00007] Structure MW Quantity (g) [CHEMMOL-00012] 162.24 0.578 [CHEMMOL-00013] 192.26 0.685 [CHEMMOL-00014] 192.26 0.685 [CHEMMOL-00015] 192.26 0.685 [CHEMMOL-00016] 196.68 0.700 [CHEMMOL-00017] 196.68 0.700 [CHEMMOL-00018] 196.68 0.700 [CHEMMOL-00019] 176.26 0.628 [CHEMMOL-00020] 176.26 0.628 [CHEMMOL-00021] 176.26 0.628 [CHEMMOL-00022] 180.23 0.642 [CHEMMOL-00023] 180.23 0.642 [CHEMMOL-00024] 230.23 0.820 [CHEMMOL-00025] 163.22 0.581 [CHEMMOL-00026] 163.22 0.581 [CHEMMOL-00027] 176.26 0.628 [CHEMMOL-00028] 164.21 0.585 [CHEMMOL-00029] 164.21 0.585 [CHEMMOL-00030] 164.21 0.585 [CHEMMOL-00031] 164.21 0.585 [CHEMMOL-00032] 198.66 0.708 [CHEMMOL-00033] 198.66 0.708 [CHEMMOL-00034] 190.29 0.678 [CHEMMOL-00035] 178.24 0.635 [0156] Next, 125 mul (4.5 eq) of DIEA to each reactor and shake the mixture at room temperature for 40 hours is added. The resins are filtered and rinsed three times each with three successive solvents: DMF, MeOH, CH2Cl2 (4 ml/reactor per rinse). The resin is allowed to dry in air. [0157] Cleavage [0158] Cleavage is carried out in 2 ml of TFA per reactor at room temperature for 2 hours. The resins are filtered and rinsed with 2×2 ml of CH2Cl2, and the filtrates are concentrated under vacuum. [0159] Extraction in Alkaline Medium [0160] The 48 mixtures are individually taken up in 8 ml of a 1:1 mixture of CH2Cl2/H2O in Whatman 12 ml cartridges equipped with a PTFE filter. The organic phase (=Organic phase A) is filtered, the aqueous phase is washed with 4 ml of CH2Cl2. The pH of the aqueous phase is adjusted to alkaline pH range by adding a saturated aqueous Na2CO3 solution. Then the aqueous phase is extracted with CH2Cl2 (1×4 ml then 1×2 ml). The 48 filtrates (=48 organic phase B) are vacuum concentrated. [0161] Purification on Cation Exchange Resin [0162] The cation exchange resin is a BCX resin packaged by Bodhan (Mettler-Toledo) in the form of 1 g SPE cartridges. The resin is first washed with 2×3 ml of MeOH (conditioning). The crude mixture, dissolved in 1 ml of MeOH and adjusted to pH 9 with a 1 M aqueous NaOH solution, is then deposited on the small resin column (loading). The column is then washed with 2×3 ml of MeOH (wash). The piperazine derivative is then released by elution with 3 ml of a 2 M solution of NH4OH/MeOH (elution). The filtrate is concentrated under vacuum.
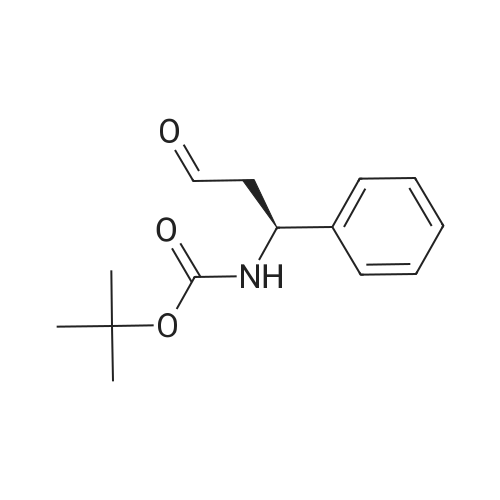
1-(tert-Butoxycarbonyl)-4-oxo-L-proline (1.0 g, 4.4 mmol), 2-piperazin-1-ylpyrimidine (0.73 g, 4.4 mmol), and acetic acid (275 muL, 4.6 mmol) were dissolved in 20 mL of anhydrous 1,2-dichloroethane and sodium triacetoxyborohydride (1.85 g, 8.7 mmol) was added. After agitating at RT for 24 hr, the reaction mixture was quenched with saturated NaHCO3. The pH was adjusted to pH 7 by addition of solid NaHCO3 and concentrated HCl, the mixture was extracted with dichloromethane, dried over MgSO4, filtered, and concentrated to afford 1.0 (61%) of crude material that was sufficiently pure for further use. MS m/z 378 (MH+).
With N-ethyl-N,N-diisopropylamine; In 1-methyl-pyrrolidin-2-one; for 20h;Combinatorial reaction / High throughput screening (HTS);
Each resin from Step 3 was distributed into 24 fritted syringes (Torvig, 50 mg each, 50 mumol), for a total of 96 syringes, and was swelled in NMP (1 mL) for 30 min. The solvent was removed by filtration. Twenty-four solutions of the building blocks listed below (10 mmol each) and DIBA (3.5 mL, 20 mmol) in NMP (10 mL) were prepared. 3 mL of the 24 solutions was added to the 24 syringes for each resin from Step 3, accordingly. The suspensions were then shaken for 20 h on a Titer Plate Shaker. The reaction mixture was filtered and washed 5 times with methylene chloride (5 mL), 3 times with THF (5 mL), 3 times THF/H2O (3/1 v/v, 5 mL), and 3 times with THF (5 mL). The resins were then dried overnight under vacuum.
With N-ethyl-N,N-diisopropylamine; In 1-methyl-pyrrolidin-2-one; for 20h;Combinatorial reaction / High throughput screening (HTS);
Each resin from Step 3 was distributed into 24 fritted syringes (Torvig, 50 mg each, 50 mumol), for a total of 96 syringes, and was swelled in NMP (1 mL) for 30 min. The solvent was removed by filtration. Twenty-four solutions of the building blocks listed below (10 mmol each) and DIBA (3.5 mL, 20 mmol) in NMP (10 mL) were prepared. 3 mL of the 24 solutions was added to the 24 syringes for each resin from Step 3, accordingly. The suspensions were then shaken for 20 h on a Titer Plate Shaker. The reaction mixture was filtered and washed 5 times with methylene chloride (5 mL), 3 times with THF (5 mL), 3 times THF/H2O (3/1 v/v, 5 mL), and 3 times with THF (5 mL). The resins were then dried overnight under vacuum.
With N-ethyl-N,N-diisopropylamine; In 1-methyl-pyrrolidin-2-one; for 20h;Combinatorial reaction / High throughput screening (HTS);
Each resin from Step 3 was distributed into 24 fritted syringes (Torvig, 50 mg each, 50 mumol), for a total of 96 syringes, and was swelled in NMP (1 mL) for 30 min. The solvent was removed by filtration. Twenty-four solutions of the building blocks listed below (10 mmol each) and DIBA (3.5 mL, 20 mmol) in NMP (10 mL) were prepared. 3 mL of the 24 solutions was added to the 24 syringes for each resin from Step 3, accordingly. The suspensions were then shaken for 20 h on a Titer Plate Shaker. The reaction mixture was filtered and washed 5 times with methylene chloride (5 mL), 3 times with THF (5 mL), 3 times THF/H2O (3/1 v/v, 5 mL), and 3 times with THF (5 mL). The resins were then dried overnight under vacuum.
With N-ethyl-N,N-diisopropylamine; In 1-methyl-pyrrolidin-2-one; for 20h;Combinatorial reaction / High throughput screening (HTS);
Each resin from Step 3 was distributed into 24 fritted syringes (Torvig, 50 mg each, 50 mumol), for a total of 96 syringes, and was swelled in NMP (1 mL) for 30 min. The solvent was removed by filtration. Twenty-four solutions of the building blocks listed below (10 mmol each) and DIBA (3.5 mL, 20 mmol) in NMP (10 mL) were prepared. 3 mL of the 24 solutions was added to the 24 syringes for each resin from Step 3, accordingly. The suspensions were then shaken for 20 h on a Titer Plate Shaker. The reaction mixture was filtered and washed 5 times with methylene chloride (5 mL), 3 times with THF (5 mL), 3 times THF/H2O (3/1 v/v, 5 mL), and 3 times with THF (5 mL). The resins were then dried overnight under vacuum.
With 4-methyl-morpholine; In N,N-dimethyl-formamide;
C. 4-(1,1-Dimethylethyl)-N-[5-[[[3-[[4-(2-pyrimidinyl)piperazin-1-yl]carbonyl]-4-methylphenyl]thio]methyl]thiazol-2-yl]benzamide A mixture of the product of Example 32, part B (0.250 g, 0.567 mmol), 1-(2-pyrimidyl) piperazine (0.121 g, 0.737 mmol), benzotriazol-1-yloxytris(dimethylamino)phosphoniumhexafluorophosphate (0.476 g, 1.08 mmol), 4-methylmorpholine (0.31 mL, 2.82 mmol) in DMF (6 mL) was heated at 65 C. for 5 h. The mixture was then diluted with ethyl acetate, washed with water, 1N NaOH solution, water, and brine. The solution was dried over anhydrous MgSO4 and concentrated under vacuum. The residue was purified using flash column chromatography (ethyl acetate) to provide the desired product (0.210 g, 63%) as a white solid: LC/MS RT=3.94 min; mass spectrum (M+H)+=587.42.
1-(1H-indol-7-ylcarbonyl)-4-(2-pyrimidinyl)piperazine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In N-methyl-acetamide;
IX. 1-(1H-Indol-7-ylcarbonyl)-4-(2-pyrimidinyl)piperazine To a chilled solution of <strong>[1670-83-3]indole-7-carboxylic acid</strong> (3.22 g; 0.02 mole) in 30ml dimethylformamide was added 1,1'-carbonyldiimidazole (3.24 g; 0.02 mole). This was stirred at ice bath temperature for 40 minutes after whicha solution of 1-(2-pyrimidyl)piperazine (4.1 g; 0.025 moles) was added. This was stirred at ambient temperature for 3 hours and the solvent was concentrated in vacuo. The resulting oil was triturated with water to give5.7 g (93%) of a solid, m.p. 194-197 C. The solid was recrystallized from methanol/water to give 4.7 g (76%) of 1-(1H-Indol-7-ylcarbonyl)-4-(2-pyrimidinyl)piperazine, m.p. 200-202 C. ANALYSIS: Calculated for C17 H17 N5 O: 66.43% C 5.58% H 22.79% N Found: 66.05% C 5.84% H 22.93% N
In N-methyl-acetamide; hydrogenchloride; chloroform;
EXAMPLE Preparation of 2-{4- [4- (4-chloro-1-pyrazolyl)-butyl]-1-piperazinyl}pyrimidine. To a mixture of 2- (1-piperazinyl)pyrimidine (32.8 g; 0.2 mol), 1,4-dibromobutane (47.5 g; 0.22 mol) and K2 CO3 (69 g; 0.5 tool) in 400 ml of dimethylformamide is added 4 -chloropyrazole (20.5 g; 0.2 mol), and the mixture is held at the reflux for 17 hours. The reaction mixture is filtered hot and evaporated to dryness. The residue is dissolved in HCl, washed with CHCl3, rendered alkaline with dilute NaOH and extracted in basic medium with CHCl3. The organic phase is then dried and subsequently evaporated to dryness, and 61 g (95%) of 2-{4-[4-(4-chloro-1-pyrazolyl)butyl]-1-piperazinyl}pyrimidine are obtained. Spectroscopic data: IR (film); 2843, 1586, 1547, 1358, 983 cm-1. 1 H NMR (delta, CDCl3): 8.25 (d, 2H, J=4.7 Hz); 7.39 (s, 1H); 7.35 (s, 1H); 6.44 (t, 1H, J=4.7 Hz); 4.0 (t, 2H, J=6.8 Hz); 3.80 (m, 4H); 2.43 (m, 6H); 1.90 (m, 2H) 1.52 (m, 2H).
(4-pyrimidin-2-yl-piperazin-1-yl)-[4-(4-chlorophenyl)phenyl]-methanone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
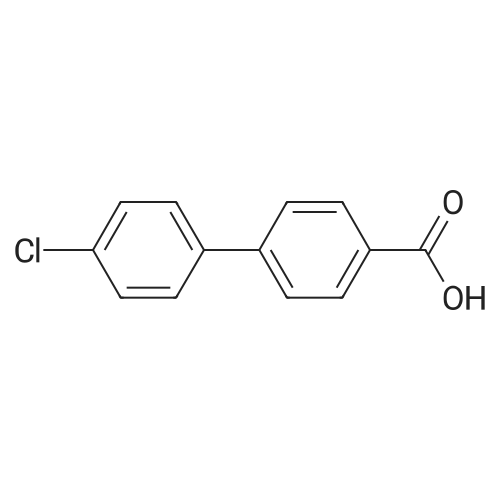
With 1-hydroxy-7-aza-benzotriazole; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; triethylamine; In dichloromethane; for 16h;
6.2. Preparation of (4-Pyrimidin-2-yl-piperazin-l-yl)-r4-(4- chloromethylphenylphenyll-methanoneTo a solution of 4'-chloro-biphenyl-4-carboxylic acid (0.1 g, 0.43 mmol) and l-(2- pyrimidyl)-piperazine (0.07 g, 0.43 mmol) in methylene chloride (3 ml), was added EDCI (0.098 g, 0.43 mmol) and HOAt (0.07 g, 0.43 mmol) triethylamine (0.07 ml, 0.52 mmol). The mixture was stirred for 16 hours and then washed with brine. The layers were separated, and the organic phase was dried over magnesium sulfate and concentrated. The resulting oil was purified by flash chromatography, and a white solid (0.1 Ig) was collected. Spectral data was consistent with structure. MS (M+l) = 379. HPLC (> 95 %). 1H NMR (CDC13) 8.35 (d, 2H), 7.55 ( m, 8H), 6.58 (t, IH), 3.80 (bm, 8H).
With 1-hydroxy-7-aza-benzotriazole; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; triethylamine; In dichloromethane; for 16h;
6.2. Preparation of (4-Pyrimidin-2-yl-piperazin-1-yl)-[4-(4-chloromethylphenylphenyl]-methanone To a solution of 4'-chloro-biphenyl-4-carboxylic acid (0.1 g, 0.43 mmol) and 1-(2-pyrimidyl)-piperazine (0.07 g, 0.43 mmol) in methylene chloride (3 ml), was added EDCI (0.098 g, 0.43 mmol) and HOAt (0.07 g, 0.43 mmol) triethylamine (0.07 ml, 0.52 mmol). The mixture was stirred for 16 hours and then washed with brine. The layers were separated, and the organic phase was dried over magnesium sulfate and concentrated. The resulting oil was purified by flash chromatography, and a white solid (0.11 g) was collected. Spectral data was consistent with structure. MS (M+1)=379. HPLC (>95%). 1H NMR (CDCl3) 8.35 (d, 2H), 7.55 (m, 8H), 6.58 (t, 1H), 3.80 (bm, 8H).
With potassium carbonate In acetone for 24h; Heating / reflux;
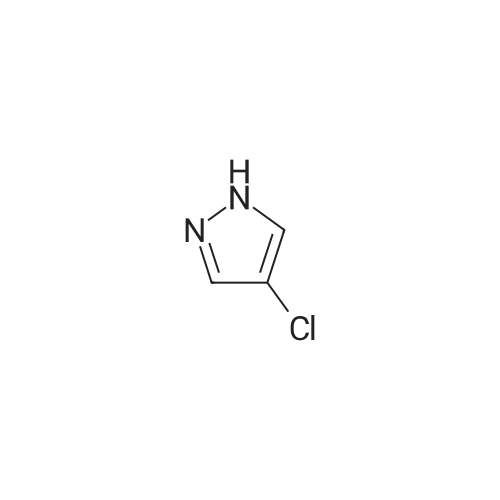
3
Example 3; N1-[4-chloro-2-(2-chlorObenzoyl)phenyl]-2-[4-(2-pyrimidinyl)piperazino] acetamide (6b):; To a compound of λ/1-[4-chloro-2-(2-chlorobenzoyl)phenyl]-2-chloroacetamide (500 mg, 1.46 mmol} in dry acetone (20 mL) was added anhydrous potassium carbonate (1g, 7.30 mmol) and 1-(2-pyrimidinyl)piperazine (239 mg, 1.46 mmol). The reaction mixture was refluxed for 24h and the reaction was monitored by TLC using ethyl acetate-hexane (6:4) as a solvent system. The potassium carbonate was then removed by suction filtration and the solvent was evaporated under vacuum to afford the crude product. This was further purified by column chromatography using ethyl acetate: hexane (6:4) as a solvent system to obtain the pure product 6b (575 mg, 83% yield).1H NMR (CDCI3) δ 2.68-2.72 (m, 4H), 3.40 (s, 2H)1 4.0-4.16 (m, 4H), 6.42-6.44 (d, 1H J= 8.65 Hz), (7.25-7.80 (m, 6H)1 8.22-8.25 (d, 2H J= 9.16 Hz), 8.80-8.92 (d, 2H J= 9.06, Hz)1 12.40 (s, 1H); FABMS: 470 (M+H).
With O-(benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate; N-ethyl-N,N-diisopropylamine In N,N-dimethyl-formamide at 20℃; for 0.5h;
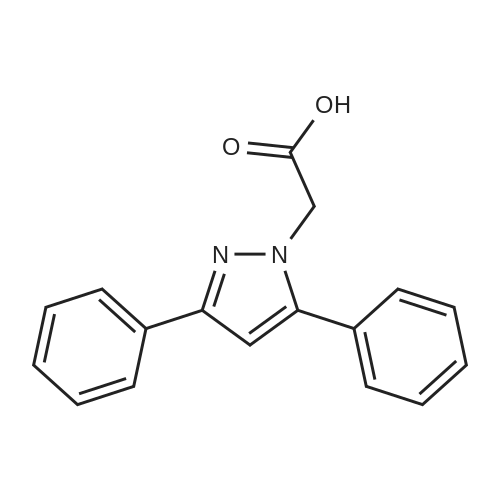
7.02.01 2-(3, 5-di henyl-pyrazol- 1 -yl)- 1 -(4-(2-pyrimidinyl)- 1 -piperazinyl)-ethanon
2-(3, 5-di henyl-pyrazol- 1 -yl)- 1 -(4-(2-pyrimidinyl)- 1 -piperazinyl)-ethanon278 mg (3,5-diphenyl-pyrazol-l-yl)-acetic acid was dissolved in 5 mL DMF. 321 mg TBTU and 172?? DIPEA were added to this solution and the mixture was stirred for a few minutes at RT. 164 mg 2-(l-piperazinyl)-pyrimidine was added and stirring was continued for 30 min. Then 10 mL of a 1M NaOH solution and 50 mL CH2CI2 were added, the organic phase was separated.The solvent was removed and the residue was purified by flash chromatography (silica gel,DCM/MeOH/NH3 0-5%). Diethylether was given to the residue and the precipitate was filltered to yield 310 mg of the desired compound.Rt: 1.48 (method F)(M+H)+: 425
310 mg
Stage #1: 2-(3,5-diphenyl-1H-pyrazol-1-yl)acetic acid With O-(benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate; N-ethyl-N,N-diisopropylamine In N,N-dimethyl-formamide at 20℃;
Stage #2: N-(2-pyridinyl)piperazine With sodium hydroxide In N,N-dimethyl-formamide for 0.5h;
7.02.01. 7.02.01.2-(3,5-diphenyl-pyrazol-1-yl)-1-(4-(2-pyrimidinyl)-1-piperazinyl)-ethanon _
General procedure: 7.02.01. 2-(3,5-diphenyl-pyrazol-1-yl)-1-(4-(2-pyrimidinyl)-1-piperazinyl)-ethanon 278 mg (3,5-diphenyl-pyrazol-1-yl)-acetic acid was dissolved in 5 mL DMF. 321 mg TBTU and 172 μL DIPEA were added to this solution and the mixture was stirred for a few minutes at RT. 164 mg 2-(1-piperazinyl)-pyrimidine was added and stiffing was continued for 30 min. Then 10 mL of a 1M NaOH solution and 50 mL CH2Cl2 were added, the organic phase was separated. The solvent was removed and the residue was purified by flash chromatography (silica gel, DCM/MeOH/NH3 0-5%). Diethylether was given to the residue and the precipitate was filtered to yield 310 mg of the desired compound. Rt: 1.48 (method F) (M+H)+: 425
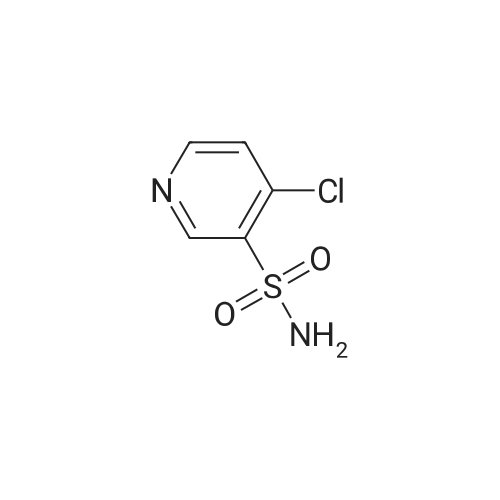
4-[4-(pyrimidin-2-yl)piperazin-1-yl]-3-pyridinesulfonamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
65%
In methanol; at 20℃; for 48h;
General procedure: To a suspension of <strong>[33263-43-3]4-chloro-3-pyridinesulfonamide</strong> 1 (0.77 g, 4 mmol) in dry methanol (10 ml) the appropriate 4-substituted-piperazine (8 mmol) was added and reaction mixture was stirred at room temperature for 48 h. The resulting precipitate was collected by filtration, washed with water (3ml) and dried.
Stage #1: N-(2-pyridinyl)piperazine; 4-nitro-benzoyl chloride With N-ethyl-N,N-diisopropylamine In dichloromethane at 20℃;
Stage #2: With hydrogenchloride; palladium 10% on activated carbon; hydrogen In methanol; water
5; 7
General procedure: General procedure for the preparation of intermediates 8 (Figure 12), 11 (Figure 13), and 15 and 16 (Figure 11): Piperazines 7, 10 and 14 were stirred over night at room temperature in dichloromethane with N,N-Diisopropylethylamine (5 equivalents) and 4-nitrobenzoyl chloride (1 .2 equivalents) or 4-nitrobenzenesulfonyl chloride (1.2 equivalents), respectively. Aqueous workup was followed by dichloromethane extraction and removal of volaliles. Without further purification, the crude intermediates were diluted in methanol (0.05M) and aqueous 6N hydrochloric acid solution (10% by volume to the methanol). Hydrogenation was done overnight under hydrogen atmosphere over Pd/C (10%, 0.1 equivalents). The reaction mixture was filtered over Celite* and removal of volatiles afforded crude intermediates 8, 11, 15 and 16 which were advanced to the next step without further purification.
5-nitro-6-(4-pyrimidin-2-ylpiperazin-1-yl)nicotinic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
38%
In ethyl acetate; at 20℃; for 1h;
General procedure: A solution of compound 4 and different primary and secondary amines were stirred at rt for 1h, followed by extraction with EtOAc. The extract was then washed with 1N HCl, water, and brine, dried over Na2SO4, and evaporated in vacuo. The residue was purified by column chromatography (hexane/EtOAc=2:1) to give product 6 as a solid.4.2.4.5 5-Nitro-6-(4-pyrimidin-2-ylpiperazin-1-yl)nicotinic acid (6e) Procedure A was used with compound 5 (170 mg, 0.84 mmol) and 1-(2-pyrimidyl)piperazine (278 mg, 1.7 mmol) to afford product 6e as a yellow solid (106 mg, 38%). 1H NMR (300 MHz, CDCl3) delta: 8.83 (d, J = 2.1 Hz, 1H), 8.56 (d, J = 2.1 Hz, 1H), 8.41 (d, J = 8.4 Hz, 2H), 6.70 (t, J = 4.8 Hz, 1H), 3.90-3.86 (m, 4H), 3.66-3.62 (m, 4H). ESI-MS: m/z (329, MH-).
With tris(pentafluorophenyl)borate; phenylsilane; In dibutyl ether; at 140℃; for 24.0h;Schlenk technique; Inert atmosphere; Green chemistry;
In the same procedure as in Example 1, The reaction mixture was then heated at 140 C for 24 h and cooled to room temperature. The resulting white crystals were isolated by the same post-treatment to yield 85% yield.The obtained product was subjected to nuclear magnetic resonance spectroscopy and nuclear magnetic resonance spectroscopy to confirm that the obtained product was 2- [4- (1,3-benzodioxol-5-ylmethyl) _ Base] pyrimidine, that ie piribedil. [0050] In a 10 mL Schlenk reaction tube (Beijing Xinwei Er Glass Instrument Co., Ltd., F891410 reaction tube, Capacity 10mL, Grinding mouth 14/20) was added 0.005 mmol of tris (pentafluorophenyl) boron, The inside of the tube is replaced with argon, Then, 1.5 mL of n-butyl ether and 2.0 mmol of phenyl silane were added under argon atmosphere and stirred (using an IKA magnetic stirrer, RCT basic, Stirring speed of 500 rpm). Followed by the addition of 0.5 mmol of 1- (2-pyrimidinyl) piperazine and 1.0 mmol ofd piperic acid. After heating at 100 C for 20 h, the mixture was cooled to room temperature. Quenched with sodium hydroxide solution (3M; 3 mL) Ethyl acetate (3 mL) was added, After stirring at room temperature for 3 h, Ethyl acetate extraction (2 mL x 3), The organic phase was dried over anhydrous sodium sulfate, Filtration, organic phase through the rotary evaporator (Swiss step Qi Co., Ltd., BUCHI Rotary Evaporator R-3) Concentrated, And then through the column (Beijing Xin Wei Er Glass Instrument Co., Ltd., C383040C with sand plate storage column chromatography column, 35/20, φ30mm, effective length: 500ml) Chromatography to obtain white crystal products, Yield 41%. The resulting product was subjected to 1H-NMR (400 MHz, CDCl3) and nuclear magnetic resonance spectroscopy 13C-NMR (101 MHz, CDCl3) analysis, The resulting spectra are shown in Figure 1-2. The resulting product was confirmed to be 2- [4- (1,3-benzodioxol-5-ylmethyl) piperazin-1-yl] pyrimidine, i.e piribedil
With triethylamine; In isopropyl alcohol; at 20 - 50℃;
50 g of 2-piperazin-1-ylpyrimidine, 60 g of triethylamine and 170 ml of isopropyl alcohol were put into a 100 ml three-necked reaction flask, and 58 g of piperonyl chloride was added dropwise with stirring at room temperature, and the mixture was dropped in 30 minutes, heated to 50 C, stirring to cool to room temperature, filtration, recovery of mother liquor, filter cake by adding 100ml water beating, pumping, plus 50ml water washing cake. Dried at 50 C, 71.5 g of piribedil crude and 99.1% by HPLC. Yield 92%.
52%
With potassium carbonate; In 5,5-dimethyl-1,3-cyclohexadiene; at 130℃; for 9.0h;
16.4 g of piperazinylpyrimidine was dissolved in 300 ml of xylene, 28 g of potassium carbonate and 18 g of piperonyl chloride were added and the suspension was incubated at reflux temperature (130C) for 9 hours, cooled, extracted with 10% hydrochloric acid several times,The separated hydrochloric acid solution is washed with ether, the acid solution is transferred to alkaline by potassium carbonate, the organic phase is separated, the aqueous phase is extracted with chloroform, the combined organic phase is dried with potassium carbonate, filtered and the solvent is removed by distillation,20 g of the residual oily organics was obtained and recrystallized from ethanol to give 15 g of product, piribedil, yield 52%.
With triethylamine; In isopropyl alcohol; at 50℃; for 4.5h;
Taking 10 g about 89.7% content piperazine pyrimidine, 12 g triethylamine, 34 ml isopropanol into 100 ml three port into reaction, stirring at the room temperature under the chlorosilane dropping pepper, in 30 minutes in the transfusion. Heating to 50 C, thermal insulation 4 hours, stirring under cooling to room temperature, filter, recovery mother liquor, in the filter cake by adding 20 ml water beating, filtering, adding 10 ml water washes filters cake. 50 C drying, the crude product shall piribedil 16.6 g, HPLC analysis content 99.2%. Yield 92%
14.76 g
With triethylamine; In isopropyl alcohol; at 20 - 50℃; for 2.5h;
ake 10g about 89.7% content of piperazine pyrimidine, 12g triethylamine, 34ml isopropanol into 100ml three reaction flask, dropping peppermint chloride at room temperature under stirring, dropping over 30 minutes. Heated to 50 , incubated for 2 hours, cooled to room temperature with stirring, filtered, the mother liquor was recovered, the filter cake was added 20ml water beating, suction filtration, plus 10ml water washing cake. 50 drying, get piribedil crude 16.6g, HPLC analysis content of 99.2%. Yield 92%; 16g of crude piribedil (99.2%), 0.3g of activated charcoal and 42ml of absolute ethanol were added into a 100ml single-necked flask and heated to the reflux temperature for 0.5 hour. The activated carbon was removed by hot filtration and the filtrate was cooled and crystallized under stirring to obtain a white Crystalline solid. Filtered, rinsed with a small amount of anhydrous ethanol, and dried to obtain 14.76g of the first crystal product of piribedil, the content of which was 99.90% by HPLC and the refined yield was 90.33%.
With trifuran-2-yl-phosphane; palladacycle; lithium hydroxide; In neat (no solvent); at 120℃; for 24.0h;Inert atmosphere; Molecular sieve;
General procedure: An oven dried Schlenk tube was charged with LiOH (1.5 mmol), pre-catalyst (1.5*10-2 mmol), P(2-Fur)3 (3*10-2 mmol) and activated 4 Å MS (100 mg). The tube was connected to a vacuum line under argon and purged three times. To the reaction mixture, sulfanilamide (3.0 mmol) and arylalcohol (6.0 mmol) were added. The Schlenk tube was closed with PTFE stopper and the reaction mixture was stirred at 120 C for 24 h. At the end of the reaction time, the reaction mixture was cooled to room temperature, diluted with methanol (5 mL), and the tube was washed with methanol three more times (3*2 mL). The methanol solution was concentrated under vacuum and the crude was subjected to flash column chromatography on silica gel using ethyl acetate and n-hexane mixtures to afford the N-alkylated product.
2-amino-6-[4-(pyrimidine-2-yl)piperazin-1-yl]-9-(β-D-ribofuranosyl)-9H-purine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
23%
With triethylamine; In ethanol;Reflux;
General procedure: 2-Amino/6-chloro-9-(beta-D-ribofuranosyl)purine (7, 8) were dissolved in 10 ml absolute EtOH, then 1-substituted piperazines and (Et)3N (3 equiv) were added. The mixture was refluxed for 5-10 h. The reaction mixture was concentrated in vacuo and the residue was purified by column chromatography.
2-(4-(2-methyl-4-nitrophenyl)piperazin-1-yl)pyrimidine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
78%
1-(2-pyrimidinyl)piperazine (2.46 g, 15.00 mmol), potassium carbonate (4.14 g, 30.00 mmol) and N,N-dimethylThe carbamide (50 mL) was added to a 100 mL single-necked flask and heated to 65 ° C for 2 hours. Slowly add <strong>[455-88-9]2-fluoro-5-nitrotoluene</strong>(2.79 g, 18.00 mmol) in N,N-dimethylformamide (20 mL). Cool to room temperature and mixThe mixture was poured into ice water (100 mL) and stirred for 30 minutes, a solid precipitated, filtered, and the filter cake was washed with water (100 mL x 3) to giveThe title compound (yellow solid, 3.50 g, yield: 78percent).
(1-(4-methoxybenzyl)-1H-indazol-3-yl)(4-(pyrimidin-2-yl)piperazin-1-yl)methanone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
61.6%
With benzotriazol-1-ol; N-[3-(N,N-dimethylamino)-propyl]-N'-ethyl-carbodiimide hydrochloride; triethylamine In dichloromethane at 20℃; for 6h; Inert atmosphere;
4.1.2. General synthesis of compounds (5a-p)
General procedure: To the solution of 4a-p (1.54 mmol) in CH2Cl2 (20 mL), EDCI (0.59 g,3.08 mmol), HOBt (0.42 g, 3.08 mmol), TEA (0.31 g, 3.08 mmol) and 1-(2- pyrimidin-1-yl)piperazine (0.51 g, 3.08 mmol) were added under N2 atmosphere. The reaction mixture was stirred for 6 h at room temperature.After 4a-p was totally consumed, the mixture was poured intowater and extracted with CH2Cl2 (30 mL × 3). The organic layer waswashed with brine, dried with anhydrous Na2SO4, and concentrated.The residue was purified by silica gel chromatography (PE: EA =10:1-2:1; V/V) to afford the desired product 5a-p
61.6%
With benzotriazol-1-ol; N-[3-(N,N-dimethylamino)-propyl]-N'-ethyl-carbodiimide hydrochloride; triethylamine In dichloromethane at 20℃; for 6h; Inert atmosphere;
4.1.2. General synthesis of compounds (5a-p)
General procedure: To the solution of 4a-p (1.54 mmol) in CH2Cl2 (20 mL), EDCI (0.59 g,3.08 mmol), HOBt (0.42 g, 3.08 mmol), TEA (0.31 g, 3.08 mmol) and 1-(2- pyrimidin-1-yl)piperazine (0.51 g, 3.08 mmol) were added under N2 atmosphere. The reaction mixture was stirred for 6 h at room temperature.After 4a-p was totally consumed, the mixture was poured intowater and extracted with CH2Cl2 (30 mL × 3). The organic layer waswashed with brine, dried with anhydrous Na2SO4, and concentrated.The residue was purified by silica gel chromatography (PE: EA =10:1-2:1; V/V) to afford the desired product 5a-p






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping