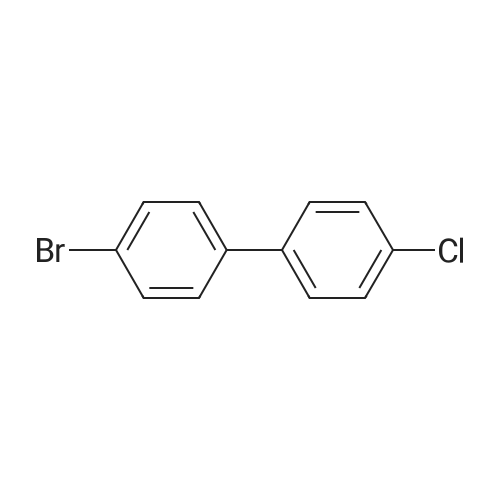
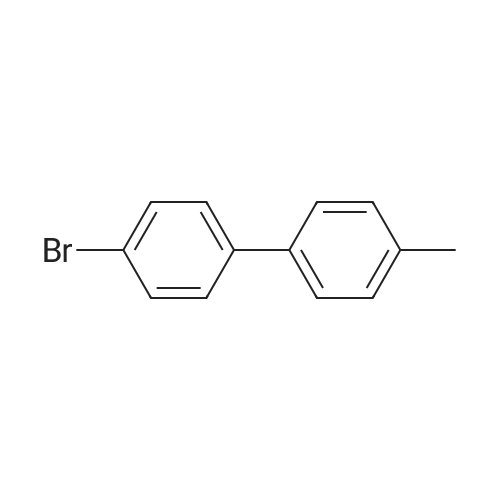
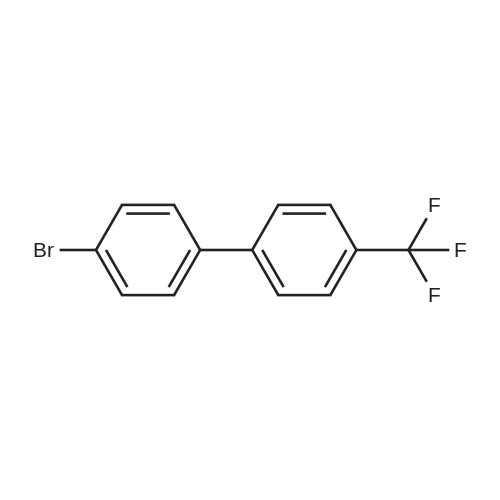
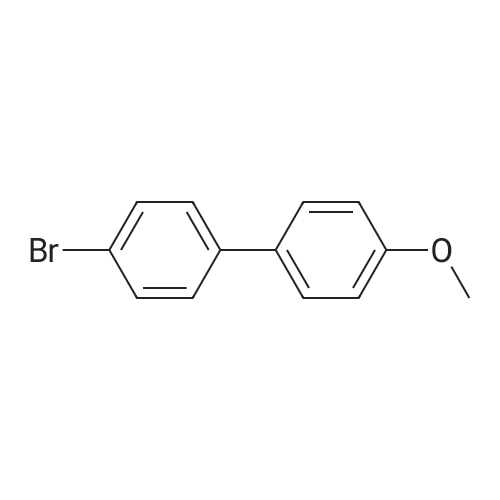
| 54% |
Stage #1: With pyrrolidine; sodium bis(2-methoxyethoxy)aluminium dihydride In tert-butyl methyl ether; toluene at -20 - 25℃; for 1.33333 h;
Stage #2: With potassium <i>tert</i>-butylate In tetrahydrofuran; tert-butyl methyl ether; toluene
Stage #3: at 10℃; for 0.25 h; |
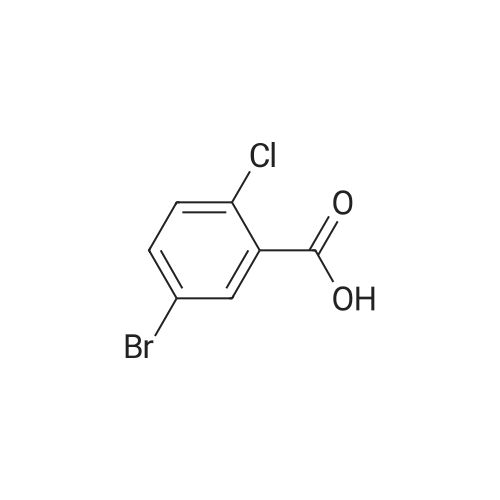
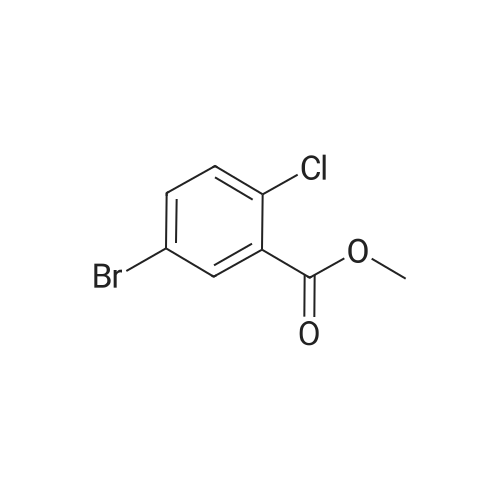
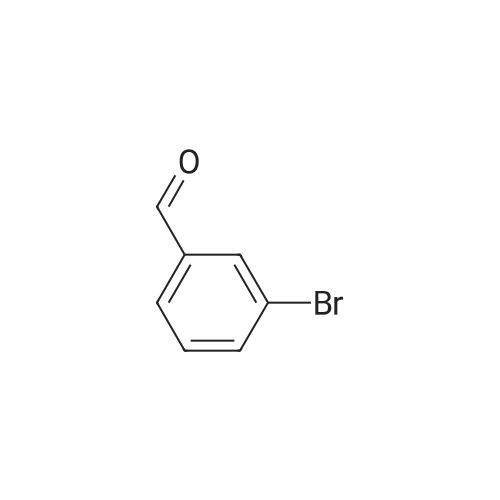
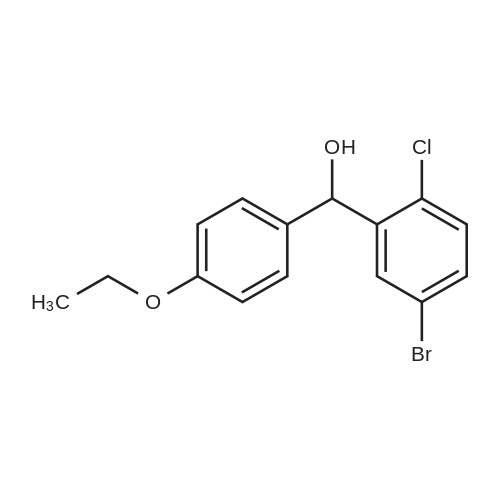
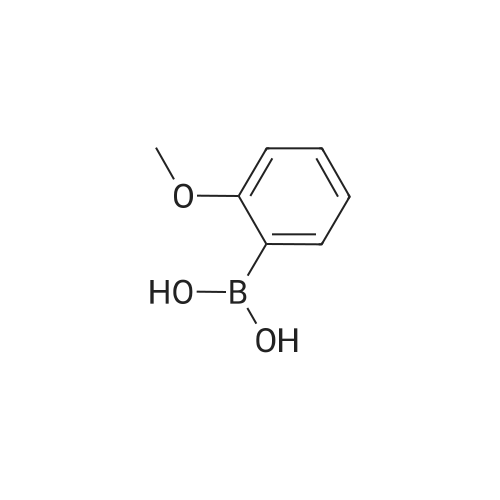
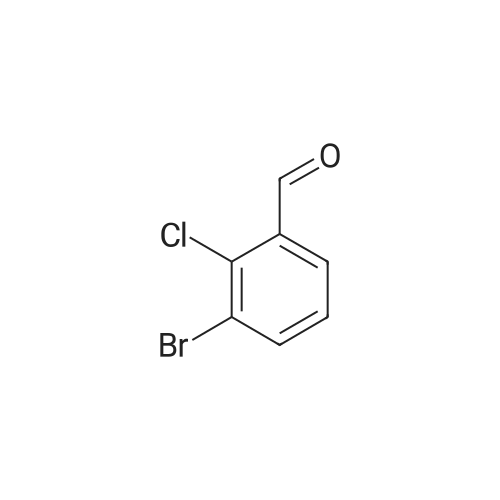
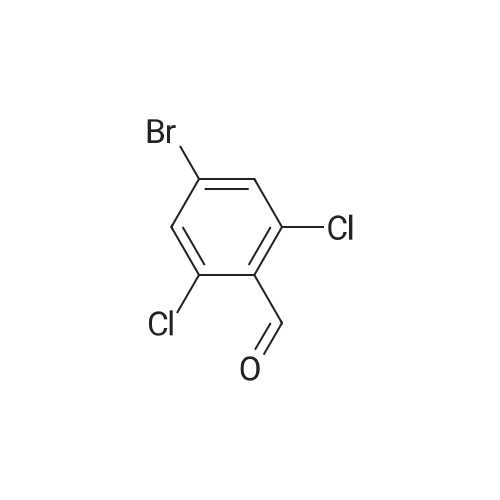
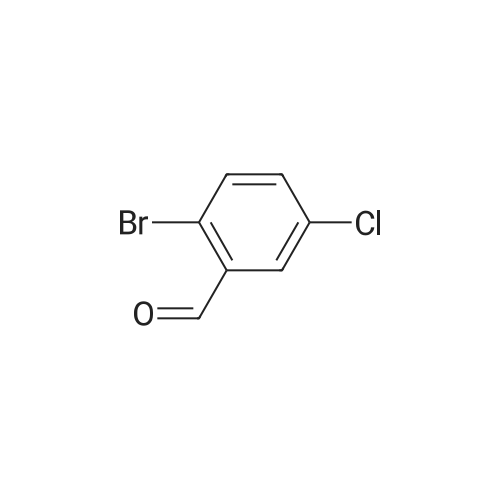
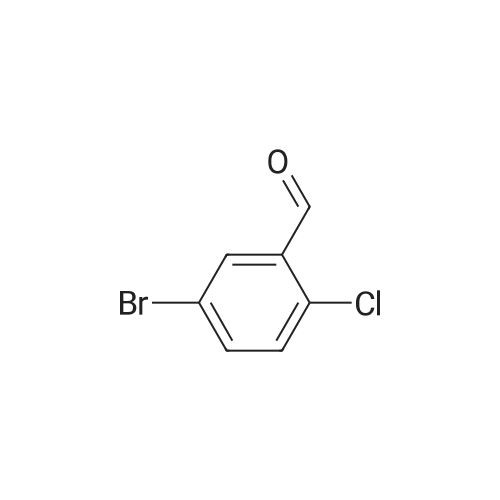
5-Bromo-2-chlorobenzaldehyde (74c). A solution of pyrrolidine (4.00 g, 56.0 mmol) in MTBE (12 mL) was added dropwise over 20 min to a solution of Red-Al.(R). (3.4 M solution in toluene, 16 ml, 54.4 mmol) in MTBE (33 mL) maintained at -20 °C. The mixture was stirred for 1 h at 25 °C. A solution of potassium tert-butoxide (0.60 g, 5.36 mmol) in THF (3 mL) was added. The resulting solution was added dropwise to a solution of 2-chloro-5-bromobenzoic acid methyl ester (72, 6.80 g, 27.3 mmol) in MTBE (15 mL) at 10 °C. After 15 min the mixture was quenched with 2 N HCl (300 mL). Repeated recystallizations (hexanes) of the recovered material gave a crude solid (3.22 g, 54percent, mp 43-46 °C), which was used without further purification in the next step.; The synthesis of benzaldehyde chlorooxime synthons 52 is depicted in Scheme 4, below. Of the eight aldehydes 44e-f and 74a-f, only 44e and 74d were commercially available. The preparation of aldehyde 44f began with the known three-step transformation of 4-methyl-3-nitrobenzonitrile 63 to methoxy compound 64. See Reiner, J. E., et al., Bioorg. Med. Chem. Lett., 12, 1203-1208 (2002). α-Bromination of 64 using one equivalent of N-bromosuccinimide gave little selectivity between the mono- and dibromo adducts, but the analogous reaction using 2.5 equivalents gave dibromide 65 almost exclusively. Silver nitrate oxidation of dibromide 65 gave aldehyde 44f. See Hill. R. A., et al., J. Chem. Soc., Perkin Trans., 1, 2209-2215 (1987). The reaction of the o-nitrotoluene 63 with N,N-dimethylformamide dimethyl acetal in DMF gave the enamine 66, which underwent oxidative cleavage using sodium periodate in THF, see Riesgo, E. C., et al., J. Org. Chem., 61, 3017-3022 (1996), to give aldehyde 74a via a more facile preparation than previously reported. See Dann, O., et al., Liebigs Ann. Chem., 3, 409-425 (1984). Aldehyde 74b also has been prepared previously. See Schultz, E. M., et al., J. Med. Chem., 19(6), 783-787 (1976). A more expedient preparation of 74b began with chlorotoluene 67 undergoing α-bromination to 68, see Gilbert, A. M., et al., J. Med. Chem., 43, 1203-1214 (2000), by a modification of the original procedure. See Liu, P., et al., Synthesis, 14, 2078-2080 (2001). The reaction of 68 with 2-nitropropane and sodium ethoxide in ethanol gave 74b. See Mallory, F. M., et al., Tetrahedron, 57, 3715-3724 (2001). Commercially available aldehyde 69 and aldehyde 71, see Hino, K., et al., Chem. Pharm. Bull., 36(6), 3462-3467 (1988), which had been prepared via a Sandmeyer reaction from commercially available 70, were converted to methyl esters 72 and 73, respectively. The esters were converted to aldehydes 74c and 74e, respectively, using Red-Al.(R).(sodium bis(2-methoxyethoxy)aluminium hydride) (Aldrich Chemical Co., Inc., Milwaukee, Wisconsin, United States of America), pyrrolidine, and potassium tert-butoxide in methyl tert-butyl ether. See Abe, T., et al., Tetrahedron, 57, 2701-2710 (2001). Cyanoaldehyde 74f was prepared by debromocyanation of 44d. See Laali, K. K., et al., J. Org. Chem., 58, 1385-1392 (1993). Aldehydes 44e-44f and 74a-74f were converted to oxime derivatives 75a-h (of which 75a,e were known previously), see Quan, M. L., et al., J. Med. Chem., 42(15), 2752-2759 (1999), using hydroxylamine hydrochloride in either water/ethanol or pyridine/ethanol. The oximes were treated with N-chlorosuccinimide in DMF to give chlorooximes 52a-h, following the procedure reported for 52e. See Liu. K.-C., et al., J. Org. Chem., 45, 3916-3918 (1980). The chlorooximes 52 were reacted with acetylenes 51 without further purification.; Reagents and conditions: (a) H2, 10percent Pd/C, EtOH; (b) NaNO2, aq. H2SO4; (c) CH3l, NaH, DMF; (d) NBS, benzoyl peroxide, CCl4; (e) AgNO3, aq. EtOH; (f) DMFDMA, DMF; (g) NalO4; aq. THF; (h) diethyl phosphite, (i-Pr)2NEt, THF; (j) 2-nitropropane, NaOEt, EtOH; (k) NaNO2, aq. HCl, then CuCN, KCN; (I) MeOH, H2SO4; (m) DCC, DMAP, MeOH, CH2Cl2; (n) Red-Al.(R)., t-BuOK, pyrrolidine, MTBE; (o) NH2OH HCl, H2O/EtOH or Py/EtOH (p) NCS, DMF. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping