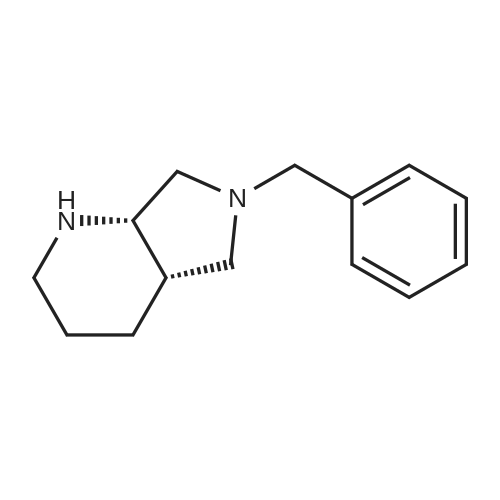
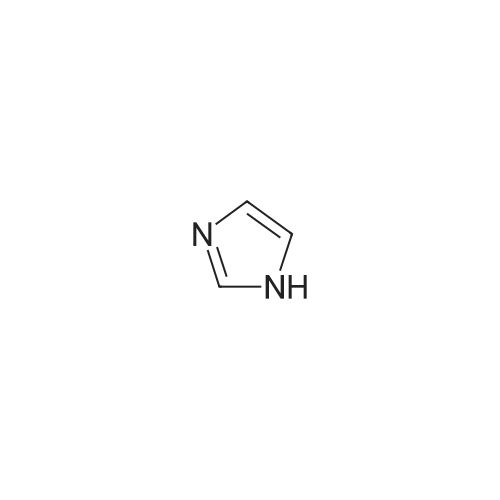
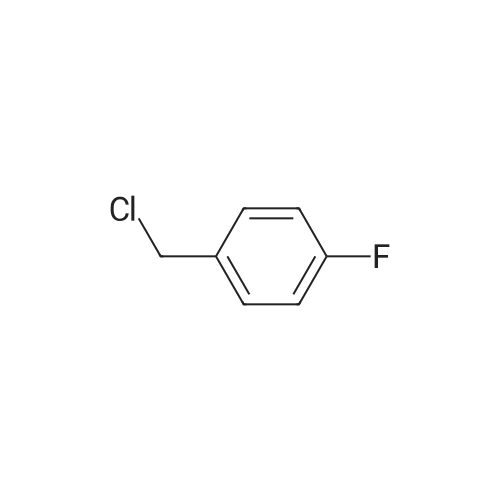
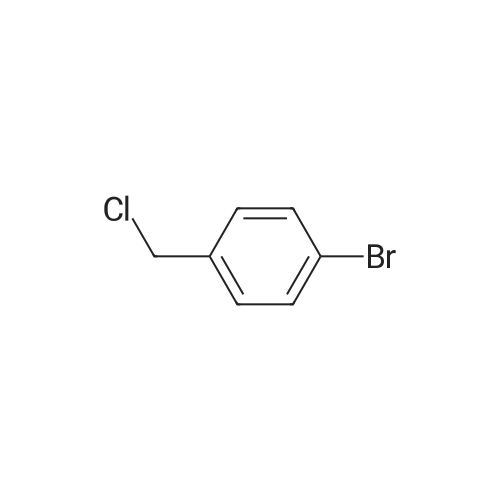
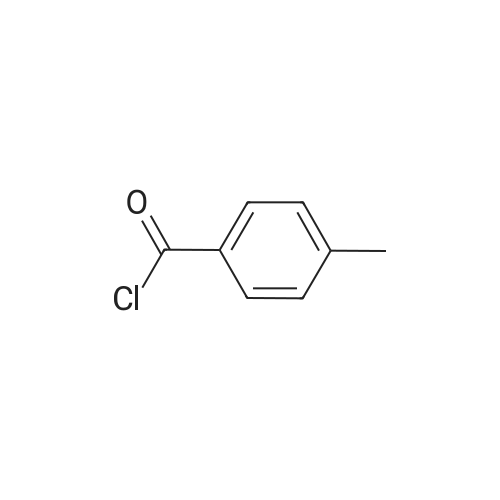
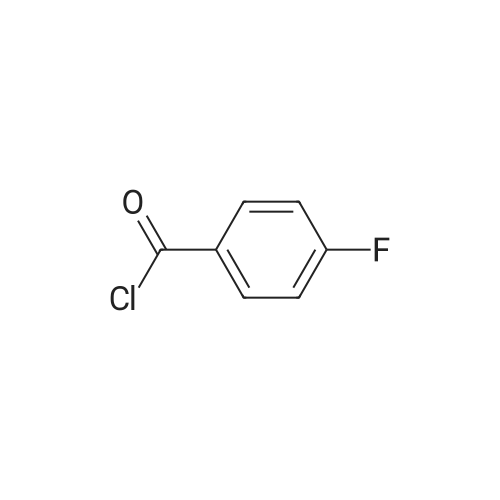
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
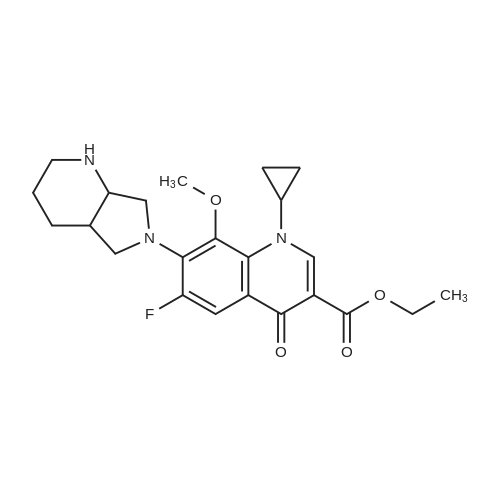
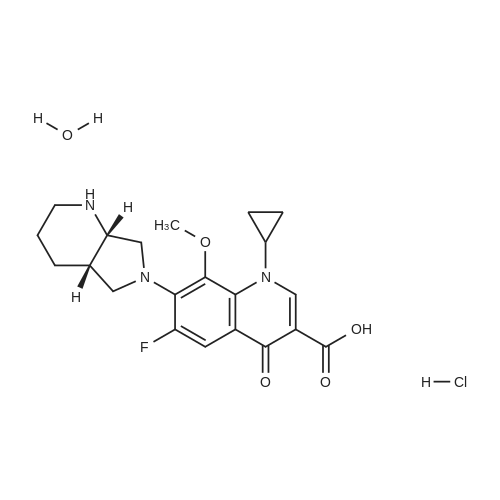
With hydrogenchloride In ethanol; water at 25 - 30℃; for 1 h;
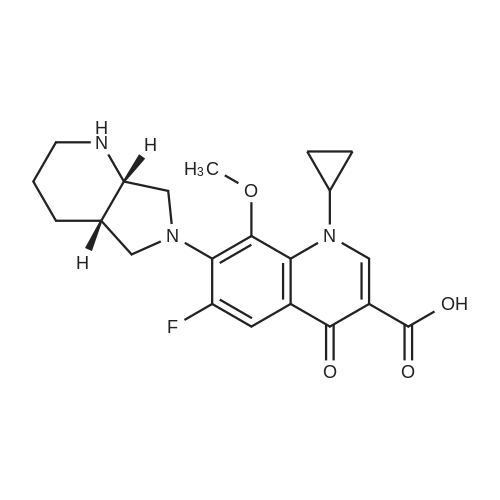
The 50g tartaric acid moxifloxacin is added to250ml water and 250ml of ethanol and stirred, was added 16. 6ml of concentratedhydrochloric acid at 25~30 ° C, the reaction was stirred at this temperaturefor 1 hour, suction filtered, the filter cake was washed with 100ml of ethanol,50~55 ° C for 3 hours in vacuo give moxifloxacin hydrochloride 35. 10g (88.3percent). Yield: 86 61percent isomer content: 0•05percent
Stage #1: at 90 - 100℃; for 3 h; Stage #2: With triethylamine In acetonitrile at 20℃; for 0.5 h;
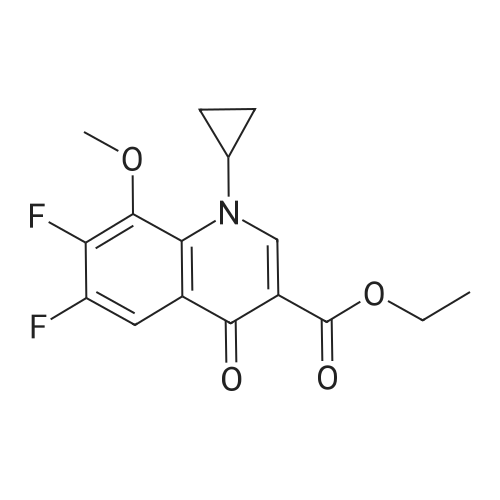
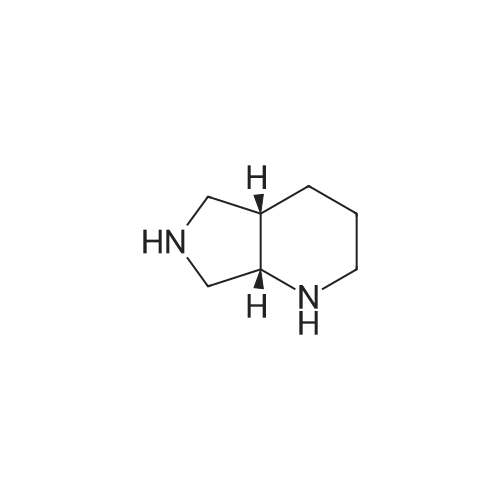
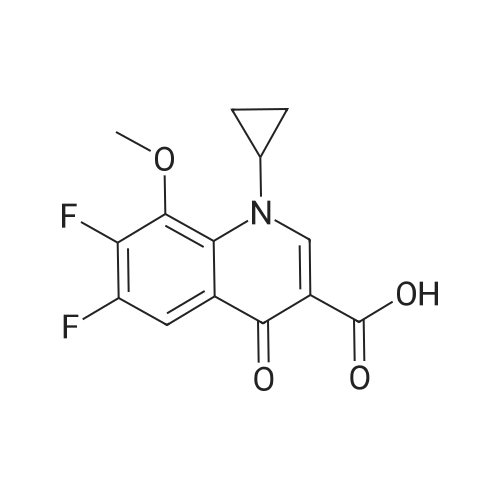
In a 2000 mL three-necked round-bottomed flask, 150.00 g of acetic anhydride was added, stirred, heated to 80 °C,boric acid 28. 00g, stirring evenly, slowly warming to 110 °C, stirring reaction 2 hours. Cooled to 60-70 ° C, ethyl 1-cyclopropyl-6,7-difluoro-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylate 100 was added. And the reaction was continued at 90 to 100 °C for 3 hours. After completion of the reaction, the reaction mixture was cooled to room temperature. 650 mL of acetonitrile and 592 mL of triethylamine were added to the reaction solution, and the mixture was stirred for 30 minutes. To the reaction solution was added 39.39 g of (S, S) -2,8-diazabicyclo [4.3.0] nonane (1.01eq), heating reflux reaction 3 hours, TLC detection reaction is completed, down to room temperature, stirring 30 minutes, ice bath temperature 5 ~ 10 °C 90mL concentrated hydrochloric acid, adjust PH = 1. 2, continue ice bath Stirring and crystallization for 8 hours, filtration, ice-ethanol 50mLX2 washing filter cake, filter cake 50 ~ 60 ° C / _0.095MPa vacuum drying 12 hours to obtain crude moxifloxacin hydrochloride 121. 48g light yellow powder, yield: 89 78percent, HPLC: 99.7percent.
Stage #1: With triethylamine In acetonitrile at 15℃; Stage #2: With hydrogenchloride In water
To the other reactor was added 60 g of (S, S) -2,8-diazabicyclo [4.3.0] nonane,40 (^ acetonitrile,4 (^ triethylamine,120 g of the above-prepared moxifloxacin hydrochloride intermediate was added with stirring,The reaction temperature was 15 ° C,The tracking reaction was monitored by TLC,To (S, S) -2,8-diazabicyclo [4.3.0] nonane;After the reaction is complete,The filtrate was added with 50 g of purified water, Keeping the temperature of the reaction solution at about 20 ° C,Concentrated hydrochloric acid,To a pH of 0.5, Stirring crystallization centrifugal filtration, And the resulting solid was dried at 70 ° C for about 7 hours,Moxifloxacin hydrochloride was crude 114g,The crude yield of moxifloxacin hydrochloride was 91.8percent and the purity of HPCL was 99.45percent.
Stage #1: With triethylamine In acetonitrile at 80 - 90℃; for 5 h; Large scale Stage #2: With hydrogenchloride In methanol at 0 - 20℃; for 3 h; Large scale
(1) The 100L reactor was sequentially charged with 24.49 kg of Im-1 and 60 L of acetonitrile.Further, 8.77 kg of SM2 and 11.71 kg of triethylamine were added, and the mixture was heated to reflux at 80 to 90 ° C with stirring.(2) Reflow reaction for 5 h, TLC monitoring (developing solvent: ethyl acetate).(3) After the reaction is completed, the temperature is lowered to room temperature, and the reaction liquid is transferred to a 500 L reaction vessel.60 L of methanol was added with stirring.(4) Add hydrochloric acid dropwise under stirring to adjust pH ≈0.5, about 18.44L concentrated hydrochloric acid, dark green solid precipitated.(5) After the completion of the dropwise addition, stirring at room temperature for 2 h; cooling to 0 to 5 ° C, stirring for 1 h;(6) Centrifugation, the filter cake is washed with 24 L of methanol cooled to 0-5 ° C.Drying at 50-55 ° C for 20 to 24 hours, to obtain crude product Im-2 19.35kg,Yellow powder solid, yield 76.3percent.
Reference:
[1] Patent: CN108276402, 2018, A, . Location in patent: Paragraph 0075; 0083; 0084; 0085; 0086; 0087; 0088; 0089
6
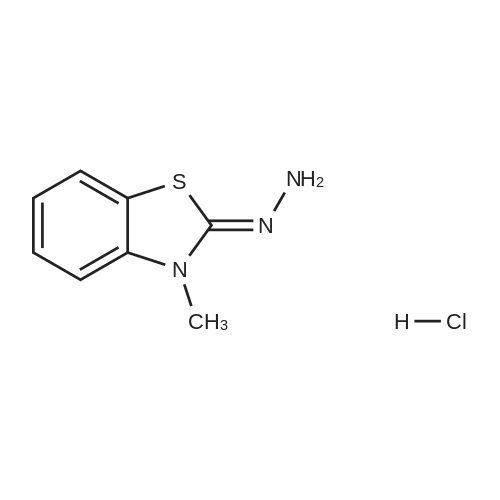
[ 151096-09-2 ]
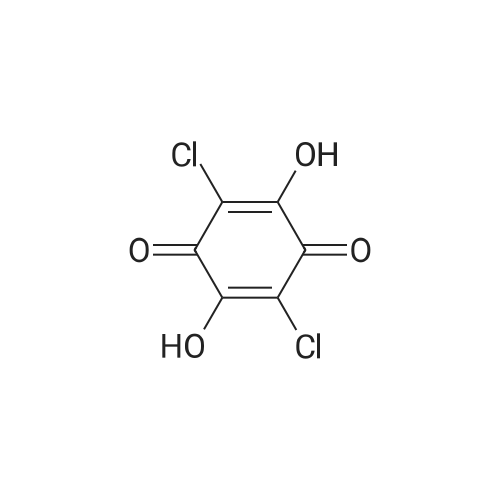
[ 186826-86-8 ]
Yield
Reaction Conditions
Operation in experiment
77%
With hydrogenchloride In methanol at 0 - 5℃; for 1 h;
Moxifloxacin base 50.0 g was stirred in 200.0 ml methanol for 10-15 minutes at 25-3-0C. The pH was adjusted to 1.0 -2.0 using methanolic hydrochloric acid. The reaction mass was chilled to 0-50C and maintained for 1 hour. The solids were filtered and dried under vacuum at 85-900C to yield 52.5 g (105percent) of moxifloxacin hydrochloride; 70.0 g of moxifloxacin base was stirred with 350.0 ml of methanol at 25- 30°. The pH was adjusted to 1.0 - 2.0 using methanolic hydrochloric acid and contents were chilled to 0-5°C and stirred for 1 hour at the same temperature. The solid was filtered and dried under vacuum at 85-9O°C to yield 78.Og (77percent) of moxifloxacin hydrochloride
54.84%
With hydrogenchloride; edetate disodium In methanol; water at 0 - 38℃; for 2 h;
The compound obtained from the example 6(85g) was taken in methanol (250 ml) and purified water (150 ml) followed by the addition of hydrochloric acid (17.14 g) and EDTA disodium salt (57 mg) the contents was heated to 35-38°C and maintained for 1 hour ,cooled to 0-5°C for 1 hour. The product was filtered washed with methanol treated with activated carbon (5.0 g) to get the desired product. Dry wt: 53.0g, Yield: 54.84percent
44.3 g
With hydrogenchloride In ethanol; water at 20 - 60℃; for 6 h;
68.2 g of purified Moxifloxacin Tosylate of example 2 (1.0 mol. Equiv.) (HPLC purity equivalent to 99.30percent and impurity of formula (VII) equivalent to 0.27percent), 409.2 mL of saturated NaHCO3 solution, 136.4 mL of purified water, 409.2 mL of dichloromethane and 68.2 mL of isopropanol were introduced into a 4-neck flask provided with a mechanical stirrer, thermometer, cooler and thermostat. Heating is carried out at 30-35°C and stirring is carried out for one hour up to complete dissolution. Then the phases are separated and the aqueous phase is extracted using 136.4 mL of Dichloromethane. The organic phases are collected and dried. The obtained solid is recovered with 954.8 mL of Ethanol and 136.4 mL of purified water. Heating is carried out at reflux up to complete dissolution, then it is brought to 60°C and a solution of 12.3 mL of concentrated hydrochloric acid (32percent) (1.05 mol. equiv.) and 136.4 mL of Ethanol is dripped in 2 hours. It is brought to r.t in 4 hours, it is left at room temperature for at least 4 hours, then it is cooled at 0°C and it is left at 0°C for another 4 hours. The obtained crystal is filtered washing it with 68.2 mL of Ethanol. The obtained salt is dried at 50°C under vacuum for 24 hours. 44.3 g of Moxifloxacin hydrochloride with a molar yield equivalent to 85.1percent with HPLC purity equivalent to 99.79percent and a formula (VII) impurity content equivalent to 0.08percent are obtained. The obtained product does not contain, even at ppm level, any ester of the sulfonic acids as an impurity, which is an essential aspect it being widely known that such impurities are genotoxic.#10;
44.3 g
With hydrogenchloride In ethanol; water at 20 - 60℃; for 10 h;
68.2 g of purified moxifloxacin tosylate of example 2 (1.0 mol. Equiv.) (HPLC purity equivalent to 99.30percent and impurity of formula (VII) equivalent to 0.27percent), 409.2 mL of saturated NaHCO3 solution, 136.4 mL of purified water, 409.2 mL of dichloromethane and 68.2 mL of isopropanol were introduced into a 4-neck flask provided with a mechanical stirrer, thermometer, cooler and thermostat. Heating was carried out at 30-35°C and stirring was carried out for one hour up to complete dissolution. Then the phases were separated and the aqueous phase was extracted using 136.4 mL of Dichloromethane. The organic phases were collected and dried. The obtained solid was recovered with 954.8 mL of Ethanol and 136.4 mL of purified water. Heating was carried out at reflux up to complete dissolution, then it was brought to 60°C and a solution of 12.3 mL of concentrated hydrochloric acid (32percent) (1.05 mol. equiv.) and 136.4 mL of Ethanol was dripped in 2 hours. It was brought to r.t in 4 hours, it was left at room temperature for at least 4 hours, then it was cooled at 0°C and it was left at 0°C for another 4 hours. The obtained crystal was filtered washing it with 68.2 mL of Ethanol. The obtained salt was dried at 50° C. under vacuum for 24 hours. 44.3 g of moxifloxacin hydrochloride with a molar yield equivalent to 85.1percent with HPLC purity equivalent to 99.79percent and a formula (VII) impurity content equivalent to 0.08percent was obtained. The obtained product does not contain, even at ppm level, any ester of sulfonic acids as an impurity. This is important because it is well-known that such impurities are genotoxic.
With tetrabutyl-ammonium chloride In <i>N</i>-methyl-acetamide; hydrogenchloride; ethanol; water
Example 1 Synthesis of Moxifloxacin HCl (I-HCl)-Exemplifying the Invention In a 250 mL three-neck flask provided with a mechanical stirrer, thermometer and thermostat, 20.0 g of 1-cyclopropyl-6,7-difluoro-1,4-dihydro-8-methoxy-4-oxo-3-quinolinic acid (67.74 mmol), 7.37 g of magnesium methoxide (85.3 mmol, 1.26 mol. eq.), 4.33 g of magnesium hydroxide (74.2 mmol, 1.10 mol. eq.) and 120 mL of dimethylformamide (DMF) (6 vol.) were loaded under inert atmosphere. The mixture is heated at 30° C. for one hour and the methanol which is formed is removed by means of low pressure distillation. To the solution is then added 0.5 g of tetrabutylammonium chloride (TBAC) (2.5percent by weight on the substrate. 10.25 g of (4aS, 7aS)-Octahydro-1H-pyrrole[3,4-b]pyridine (81.29 mmol, 1.20 mole equiv.) is then added. The reaction mixture is heated at 45° C. and maintained at T=45-50° C. until the HPLC monitored reaction is completed, i.e. about 16 hours. The solvent is vacuum distilled and about 110 mL DMF are recovered. To the residual suspension 200 mL water and 25 mL HCl 12 N (300 mmol) is added, thereby bringing the solution pH to 3-4. The suspension is heated at 30° C. and stirred at that temperature for 30 minutes, then it is cooled at T<10° C. and, after the product has precipitated, it is stirred at this temperature for 30 minutes. The suspension is filtered and the solid is washed with water. The crude is dissolved in 150 mL hydrochloric acid 5.5 mol/L. The whole is stirred for at least 20 minutes until it is dissolved. The small amount of insolute material, being an impurity, is eliminated through filtration. The filtrate is concentrated to a small volume under low pressure. To the obtained solution is added 70 mL anhydrous ethanol. The mixture is stirred and cooled below 5° C. for at least one hour after solid precipitation. The suspension is filtrated and the solid is washed with mL ethyl acetate. The solid is vacuum dried for 3 hours at 25° C. then at Tmax.=70° C. for 4 hours. 26.4 g of Moxifloxacin hydrochloride is obtained (molar yield=89percent) with HPLC (A percent) purity>99.0percent.
Reference:
[1] Patent: US2011/230661, 2011, A1,
8
[ 139693-52-0 ]
[ 151213-40-0 ]
[ 186826-86-8 ]
Yield
Reaction Conditions
Operation in experiment
45 g
Stage #1: With triethylamine In ethanol at 70℃; for 4 h; Stage #2: With hydrogenchloride In water at 30℃; for 1 h; Heating
The main ring chelate compound 50g, anhydrous ethanol 200g, (3,3)-2,8-diazabicyclo[4,3,0]nonane 15.7g and triethylamine 12.0g were added to the reaction flask. Temperature 70 °C reaction 4. Oh; the end of the reaction, temperature control 30 ° C to the solution was added dropwise 12mol / L of hydrochloric acid, stirring while stirring, adjust the pH to 1, stirring for 1h, temperature control to 10 °C, Crystal growth 2h; After crystal growth, filtration, washing with 95 percent ethanol twice, each time 50g; Wet powder 60 °C vacuum drying to 2.8percent moisture, to obtain moxifloxacin hydrochloride dry powder 45.0g, a single impurity 0.1percent, The total impurity is 0.2percent; after refining with ethanol and water, the content is 99percent, which meets the quality standards of the European Pharmacopoeia EP-7.0 and the total impurity is less than 0.1percent
Stage #1: With 1,8-diazabicyclo[5.4.0]undec-7-ene In acetonitrile at 20 - 85℃; for 36 h; Inert atmosphere Stage #2: With hydrogenchloride In water at 15℃; for 1 h; Inert atmosphere
Under nitrogen protection, 1-cyclopropyl-6, 7-difluoro-1, 4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid 10.0 g (0·0339 mοl) and (S,S) -2,8-diazabicyclo[4,3,0] nonane 5 .13g(0.0406mol)were added to 70 mL acetonitrile, it was stirred at 20 ° C~30 ° C and 1,8-diazabicyclo[5·4·0]undec-7-ene 7.74 g (0.0508 mol) was added, it was stirred at 75 ° C~85 ° C for 36 h. Concentrated in vacuo (temperature 45 ° C ~ 65 ° C, pressure -0 · 08MPa ~ -0.1MPa) to remove most of the solvent, it was then dissolved in chloroform, the mass concentration is 2percent aqueous acetic acid solution (the mass concentration refers to the mass of acetic acid as a percentage of the total mass of the aqueous acetic acid solution), the mass concentration was 10percent saline (the mass concentration refers to the mass of sodium chloride as a percentage of the total mass of brine were sequentially added, respectively, then the mixture was stirred, allowed to stand, and the aqueous layer was separated. The organic layer was concentrated in vacuo (temperature 35 ° C ~ 55 ° C, pressure -0.08MPa ~ -0.1MPa) to remove most of the solvent, ethyl acetate was added, and the mixture was cooled to 10 ° C to 20 ° C and stirred for 2 hours to 3 hours. Centrifugation, rinsed three times with ethyl acetate, dried in vacuo (vacuum degree -0.01MPa~-0.1MPa, temperature 45~55°C) dried for 14 to 18 hours, to obtain 4.01g of moxifloxacin , yield 29.5percent.HPLC purity 93.54percent. Under nitrogen protection, add 4.0 g of moxifloxacin to 2.5 mL of water and 10 mL of methanol.Stir at 170 ° C for 1 hour, filter, and cool to 35 ° C.The pH was adjusted to 1.4 to 1.8 by the addition of 6N hydrochloric acid. Cool to 15 ° C and stir for 1 hour.It was filtered, washed with water, and stirred at 15 ° C for 1 hour in a solution of water 5 mL and concentrated hydrochloric acid.Filtered, washed with water, vacuum dried (vacuum degree - 0.01 MPa ~ -0.1 MPa, temperature 45 ~ 55 ° C) for 16 hours, to obtain 3.3 g of moxifloxacin hydrochloride,Yield 75.6percent (total yield 22.3percent, based on 1-cyclopropyl-6,7-difluoro-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid ). The HPLC purity was 99.23percent, and the maximum single impurity was 0.47percent.
Reference:
[1] Spectrochimica Acta - Part A: Molecular and Biomolecular Spectroscopy, 2014, vol. 121, p. 254 - 258
[2] Patent: CN109096276, 2018, A, . Location in patent: Paragraph 0090-0093
10
[ 1403836-23-6 ]
[ 186826-86-8 ]
Reference:
[1] Patent: CN105524060, 2016, A, . Location in patent: Paragraph 0020; 0024; 0025; 0026
11
[ 1028205-69-7 ]
[ 186826-86-8 ]
Yield
Reaction Conditions
Operation in experiment
6.4 g
Stage #1: With water; sodium hydroxide In ethylene glycol at 115℃; Inert atmosphere Stage #2: With hydrogenchloride In water at 15℃; for 1 h; Inert atmosphere
Under nitrogen protection, add 35g (0.875mol) of sodium hydroxide to 165mL of water and 150mL of ethylene glycol.And 7-((S,S)-2,8-diazabicyclo[4,3,0]decane-8-yl)-1-cyclopropyl-6-fluoro-1,4-dihydro- 8-methoxy-4-oxo-3-quinolinecarboxylic acid dimethylamide 12.0 g (0.0263 mol),Stir for 15 hours while heating to 115 °C.Cool to 5 ° C and adjust the pH to 5-6 with concentrated hydrochloric acid. Stir at 0.5 ° C for 0.5 hours.Filtration, water washing, vacuum drying (vacuum degree -0.01MPa~-0.1MPa, temperature 45°C55 ° C) for 16 hours, 8.0 g of moxifloxacin was obtained, and the yield was 75.8percent. The HPLC purity was 93.25percent.Under nitrogen protection, 8.0 g of moxifloxacin was added to 5 mL of water and 20 mL of methanol, and the mixture was stirred at 70 ° C for 1 hour, filtered, cooled to 35 ° C, and adjusted to pH 1.4 to 1.8 by adding 6 N hydrochloric acid.Cool to 15 ° C and stir for 1 hour. It was filtered, washed with water, and stirred at 15 ° C for 1 hour in 10 mL of water and 0.2 mL of concentrated hydrochloric acid.Filtration, washing with water, vacuum drying (vacuum degree - 0.01 MPa ~ -0.1 MPa, temperature 45 ~ 55 ° C) for 16 hours, 6.4 g of moxifloxacin hydrochloride was obtained, the yield was 73.3percent.(Total yield 29.4percent based on 1-cyclopropyl-6,7-difluoro-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid). The HPLC purity was 99.16percent and the maximum single impurity was 0.54percent.
Reference:
[1] Patent: CN109096276, 2018, A, . Location in patent: Paragraph 0084; 0085; 0088; 0089
12
[ 496919-99-4 ]
[ 151213-40-0 ]
[ 186826-86-8 ]
Yield
Reaction Conditions
Operation in experiment
75%
Stage #1: at 10 - 100℃; for 3 h; Stage #2: With hydrogenchloride In methanol; butan-1-ol at 25 - 30℃; for 2 h;
1 -cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylic acid- O3,O4 bis(propyloxy-O)borate (100.Og) was suspended in n-butanol (500.0 ml) to which (S,S)-2,8-diazabicyclo (4,3,0) nonane (29.0 g) diluted with 100.0 ml n-butanol was added slowly at 10-15°C. The contents were heated to 100°C and maintained for 3 hours. After the completion of reaction it was cooled to 25-3O0C. 200.0 ml methanol was added and pH was adjusted 1.0-2.0 using methanolic hydrochloric acid. The contents were stirred at 25-3O0C for 2 hours. After completion of reaction the reaction mass was distilled to residue. Purified water 500 ml was added and pH was adjusted to 7.5-9.0 using liquor ammonia. The reaction mass was then extracted with dichloromethane. The organic layer was dried using sodium <n="15"/>sulphate and concentrated to residue. The residue was stripped with 100 ml methanol. 300 ml methanol was charged and pH was adjusted to 1.0-2.0 using methanolic hydrochloric acid, the contents were further cooled to 0-50C and maintained for 1 hour. The solid was filtered and washed with chilled methanol (50.0 ml) and dried under vacuum at 85-9O0C to yield 75.0 g (75percent) of moxifloxacin hydrochloride.
10 g of crude moxifloxacin hydrochloride are suspended in a mixture of 72.5 ml of methyl alcohol/water (1:1; vol/vol) . The mixture is heated at reflux until the dissolution of the product is finished and then 72.5 ml of acetone are added slowly. The resulting dissolution is heated at T = 40-450C and it is kept at that conditions until the precipitation of the product, and then it is cooled at T 15-25C during one hour. Then, the suspension is filtered, the solid is washed with 10 ml of a mixture of acetone/methyl alcohol/water (2:1:1; vol/vol/vol) and it is dried at 400C with vacuum until a constant weight is obtained. 6.28 g of crystalline form of moxifloxacin hydrochloride are obtained.
2
(1-cyclopropyl-6,7-difluoro-1,4-dihydro-8-methoxy-4-(oxo-κO)-3-quinoline-carboxylato-κO3)difluoro-Boron[ No CAS ]
[ 151213-40-0 ]
[ 186826-86-8 ]
Yield
Reaction Conditions
Operation in experiment
Example 4 5Preparation of l-cycIopropyl-6-fluoro-l,4-dihydro-8-methoxy-7-[(4a5>,7a5')- octahydro-6H-pyrroIo[3,4-£]pyridin-6-yl]-4-oxo-3-quinolinecarboxylic acid hydrochloride (Moxifloxacin hydrochloride (Form A))0 l-CyclopiOpyl-6,7-difluoiO-8-methoxy-4-oxo-l,4-dihydroquinoline-3-carboxylic acid boron difluoride chelate (III) (150g, 0.44 moles) was suspended in methanol EPO <DP n="18"/>(750 ml) and (4£uS',7ai9)octahydiO-l//-pynOlo[3,4-6]pyridino (66 g, 0.52 moles) followed by triethylamine (60.9g, 0.60 moles) were added. The reaction mixture was stirred at 3O0C for 10 h. Thereafter triethylamine (220.8 g, 2.19 moles) was added and the suspension was heated to reflux for 22 h. After completion of> reaction the solvent and excess triethylamine were distilled off under reduced pressure at 40-50C. The crude residue was suspended in a mixture of acetonitrile (600 ml) and methanol (600 ml) and concentrated hydrochloric acid was added till a clear solution was formed at pH ~7. Carbon (7.5 g) was added, the reaction mixture was stirred at 20-250C and filtered. The clear filtrate was slowly) acidified with the remaining amount of concentrated hydrochloric acid ( 136 g, 1.3 moles) at 2O0C and the precipitated solid was stirred at 20-250C for 2h followed by another hour at 0-50C. The product was filtered under nitrogen atmosphere, washed with a mixture of acetonitrile-methanol (1 :1, 150 ml) and dried under reduced pressure at 60-700C till moisture content was less than 2 %. The dry i weight of the product was 97 g, with chromatographic purity >99.7 %.
With hydrogenchloride; In methanol; at 0 - 5℃; for 1h;pH 1.0 - 2.0;Product distribution / selectivity;
<strong>[151096-09-2]Moxifloxacin</strong> base 50.0 g was stirred in 200.0 ml methanol for 10-15 minutes at 25-3-0C. The pH was adjusted to 1.0 -2.0 using methanolic hydrochloric acid. The reaction mass was chilled to 0-50C and maintained for 1 hour. The solids were filtered and dried under vacuum at 85-900C to yield 52.5 g (105%) of <strong>[151096-09-2]moxifloxacin</strong> hydrochloride; 70.0 g of <strong>[151096-09-2]moxifloxacin</strong> base was stirred with 350.0 ml of methanol at 25- 30. The pH was adjusted to 1.0 - 2.0 using methanolic hydrochloric acid and contents were chilled to 0-5C and stirred for 1 hour at the same temperature. The solid was filtered and dried under vacuum at 85-9OC to yield 78.Og (77%) of <strong>[151096-09-2]moxifloxacin</strong> hydrochloride
54.84%
With hydrogenchloride; edetate disodium; In methanol; water; at 0 - 38℃; for 2h;
The compound obtained from the example 6(85g) was taken in methanol (250 ml) and purified water (150 ml) followed by the addition of hydrochloric acid (17.14 g) and EDTA disodium salt (57 mg) the contents was heated to 35-38C and maintained for 1 hour ,cooled to 0-5C for 1 hour. The product was filtered washed with methanol treated with activated carbon (5.0 g) to get the desired product. Dry wt: 53.0g, Yield: 54.84%
With hydrogenchloride; In ethanol; at 0 - 10℃; for 2h;Product distribution / selectivity;
EXAMPLE 3; I UPreparation of 1 -cyclopropyl-6-fluoro-l,4-dihydro-8-methoxy-7-f (4aS,7aS)- octahydro-H-pyrroloP^-^lpyridin--ylJ^-oxo-S-quinoIinecarboxylic acid hydrochloride (<strong>[151096-09-2]Moxifloxacin</strong> hydrochloride Form A)1 5 <strong>[151096-09-2]Moxifloxacin</strong> ( 1 g) was suspended in absolute alcohol (20 ml) and cooled to0-50C with stirring. Dry hydrogen chloride gas was bubbled into the reaction mixture till saturation upon which a clear solution resulted. After stirring for 1 hour at 0-10 0C the reaction mixture turned hazy and crystallization of product was observed. The reaction was maintained for another hour at 0-10 0C, the 0 solution concentrated under reduced pressure and further dried for 12 hours at 75- 80 0C under reduced pressure (-10 mm Hg) to yield a new crystalline form of <strong>[151096-09-2]Moxifloxacin</strong> hydrochloride (Form A) (1.1 g).
With hydrogenchloride; In ethanol; toluene; benzyl alcohol; at 20℃; for 1h;
Example 4 Obtaining <strong>[151096-09-2]Moxifloxacin</strong> hydrochloride In a four-mouth 500 ml flask with refrigerant, thermometer, mechanical stirring and nitrogen bubbler with the outlet connected to a washing flask with a solution of H2SO4 is placed 50 g (0.1694 moles, 1 eq) of the compound (II), 150 ml of acetonitryl (3 volumes), and this is heated to T = 76-80 C (reflux). Once reflux has been reached, and maintaining T = 76-80 C, a compensated addition funnel is used to add 19.14 g (0.1186 moles, 0.7 eq) of hexamethyldisilazane. Once addition has been completed, the reaction is maintained with stirring at T = 76-80 C for 1 hour. Once that period has elapsed the reaction mixture is cooled to T = 0-15 C and 28.85 g (0.2033 moles, 1.2 eq) of Boron trifluoride etherate is added, maintaining T<15 C. Once the addition has been completed, it is maintained at T = 0-15 C until the starting product has disappeared (approximately 2 hours) and it is adjusted pH = 8-9 with triethylamine (approximately 30 ml). To the resulting suspension is added a solution of 32.07 g (0.2541 moles, 1.5 eq) of (S,S)-2,8-diazabicyclo[4.3.0]nonane and 25.71 g of triethylamine (0.2541 moles, 1.5 eq) in 150 ml of acetonitryl, maintaining T = 15-25 C. The resulting amber solution is maintained with stirring under those conditions until the starting product has disappeared (approximately 5-6 hours). Once the reaction is completed it is distilled until a stirrable paste is obtained, 250 ml of methanol is added, the resulting solution is brought to T = 63-67 C (reflux) and is maintained under those conditions until the boron compound (VIII) has disappeared, (approximately 5-6 hours). The reaction completed, the methanol is distilled at low pressure and to the resulting concentrate is added 340 ml of water. The resulting suspension is taken to pH = 12 with NaOH 30% and then to pH = 8.0-8.2 with HCl 35%. The dark solution obtained is extracted with benzyl alcohol (3 x 100 ml). The combined organic extracts are washed with 200 ml of water, and to the resulting organic phase is added 72.54 g of solution of hydrogen chloride in ethyl alcohol 8.52% (6.18 g, 0.1694 moles, 1 eq). 1320 ml of toluene is then added to the solution obtained, producing precipitation of <strong>[151096-09-2]Moxifloxacin</strong>·HCl as a yellow solid. The resulting suspension is maintained with stirring at ambient temperature for one hour, following which it is filtered and the solid is washed with toluene (2 x 70 ml). The moist product is dried at 40 C to provide 59.82 g of <strong>[151096-09-2]Moxifloxacin</strong>·HCl. Yield: 80.6%.
With hydrogenchloride; In methanol; water; at 15 - 35℃; for 0.75h;pH 1.6;
A mixture of 8 grams of <strong>[151096-09-2]moxifloxacin</strong>, 16 ml of water and 64 ml of methanol was heated to reflux temperature of 700C. The reaction mixture was filtered to remove undissolved material. Filtrate was cooled to 35C. The pH of the filtrate was adjusted to 1.6 using hydrochloric acid. The reaction mixture was cooled to 150C. The reaction mixture was stirred for 45 minutes at 150C. The solid obtained was filtered and washed with methanol and dried at 40-450C to get crystalline Form-Y of <strong>[151096-09-2]moxifloxacin</strong> hydrochloride monohydrate. Yield: 5.5 grams
With hydrogenchloride; In methanol; water; at 5 - 35℃; for 1.25h;pH 1.6;
A mixture of 25 grams of <strong>[151096-09-2]moxifloxacin</strong>, 16 ml of water and 64 ml of methanol was heated to reflux temperature of 700C. The reaction mixture was filtered to remove undissolved material. Filtrate was cooled to 35C. The pH of the filtrate was adjusted to 1.6 using hydrochloric acid. The reaction mixture was cooled to 150C. The reaction mixture was stirred for 45 minutes at 150C. The obtained solid was taken into a mixture of 22 ml of water and 0.5ml of hydrochloric acid and stirred 30 min at 5C. The compound was filtered and washed with water, dried at 40-450C to get <strong>[151096-09-2]moxifloxacin</strong> hydrochloride monohydrate particles having oval shape. Yield: 20 grams
With hydrogenchloride; In methanol;pH 1 - 2;Product distribution / selectivity;
<strong>[151096-09-2]Moxifloxacin</strong> base 50.0 g was stirred in 200.0 ml methanol for 10-15 minutes at 253 C. The pH was adjusted to 1.0-2.0 using methanolic hydrochloric acid. The reaction mass was chilled to 0-5 C. and maintained for 1 hour. The solids were filtered and dried under vacuum at 85-90 C. to yield 52.5 g (105%) of <strong>[151096-09-2]moxifloxacin</strong> hydrochloride.
Example 1: Preparation of novel crystalline Form -Y of <strong>[151096-09-2]moxifloxacin</strong> hydrochloride.Suspended 95grams of <strong>[151096-09-2]Moxifloxacin</strong> obtained as per the above mentioned process in a mixture of 600ml of Methanol and 66 ml of water. Heated the reaction mixture to 50- 60C.Dissolved the compound by adjusted the pH to 8.5 with aqueous sodium hydroxide solution then filtered through a hyflow bed. Adjusted the filtrate pH to 0.6 using aqueous hydrochloric acid at below 150C and the Isolate the solid by filtration and dried the compound at 60-700C for 4-6 hours to get the title compound crystalline Form-Y of <strong>[151096-09-2]Moxifloxacin</strong> hydrochloride. EPO <DP n="14"/>
Example 5: Preparation of novel crystalline form X of anhydrous <strong>[151096-09-2]Moxifloxacin</strong> hydrochloride5Og of 1-cyclopropyl -6,7-difluoro -1,4-difluoro-l, 4 - dihydro - 4 - oxo-8-methoxy quinolone-3-carboxilic acid , (S, S) diazabicyclo nonane and 5gr of 1,8-diazabicyclo [5,4,0] undec-7-ene (5g) in 300 ml acetonitrile heated to 65-75C and stirred. Evaporated the solvent completely and added 5 % aqueous isopropyl alcohol. Adjusted the pH to 8.5 with caustic solution, then the reaction mass filtered. Adjusted the pH filtrate to 5 with aqueous hydrochloric acid. Cooled the reaction mixture to 10- 150C. Isolated the separated solid to afford <strong>[151096-09-2]moxifloxacin</strong>. This wet <strong>[151096-09-2]Moxifloxacin</strong>, suspended in 400 ml Toluene and heated 110-1120C to reflux azeotropically and then reaction mass is cooled to 25-35C. Quenched the reaction mass with 200 ml of isopropyl alcohol and adjusted pH to .98 with hydrochloric acid in Isopropyl alcohol. Isolated the solid by filtration and washed the material with isopropyl alcohol. Dried at 80-90C under vacuum to afford the novel crystalline form -X of anhydrous <strong>[151096-09-2]Moxifloxacin</strong> hydrochloride. (Weight: 33.0 grams, M.C by KF method is 0.09%w/w, Purity by HPLC: 99.94%)
44.3 g
With hydrogenchloride; In ethanol; water; at 20 - 60℃; for 6h;
68.2 g of purified <strong>[151096-09-2]Moxifloxacin</strong> Tosylate of example 2 (1.0 mol. Equiv.) (HPLC purity equivalent to 99.30% and impurity of formula (VII) equivalent to 0.27%), 409.2 mL of saturated NaHCO3 solution, 136.4 mL of purified water, 409.2 mL of dichloromethane and 68.2 mL of isopropanol were introduced into a 4-neck flask provided with a mechanical stirrer, thermometer, cooler and thermostat. Heating is carried out at 30-35C and stirring is carried out for one hour up to complete dissolution. Then the phases are separated and the aqueous phase is extracted using 136.4 mL of Dichloromethane. The organic phases are collected and dried. The obtained solid is recovered with 954.8 mL of Ethanol and 136.4 mL of purified water. Heating is carried out at reflux up to complete dissolution, then it is brought to 60C and a solution of 12.3 mL of concentrated hydrochloric acid (32%) (1.05 mol. equiv.) and 136.4 mL of Ethanol is dripped in 2 hours. It is brought to r.t in 4 hours, it is left at room temperature for at least 4 hours, then it is cooled at 0C and it is left at 0C for another 4 hours. The obtained crystal is filtered washing it with 68.2 mL of Ethanol. The obtained salt is dried at 50C under vacuum for 24 hours. 44.3 g of <strong>[151096-09-2]Moxifloxacin</strong> hydrochloride with a molar yield equivalent to 85.1% with HPLC purity equivalent to 99.79% and a formula (VII) impurity content equivalent to 0.08% are obtained. The obtained product does not contain, even at ppm level, any ester of the sulfonic acids as an impurity, which is an essential aspect it being widely known that such impurities are genotoxic.
44.3 g
With hydrogenchloride; In ethanol; water; at 20 - 60℃; for 10h;
68.2 g of purified <strong>[151096-09-2]moxifloxacin</strong> tosylate of example 2 (1.0 mol. Equiv.) (HPLC purity equivalent to 99.30% and impurity of formula (VII) equivalent to 0.27%), 409.2 mL of saturated NaHCO3 solution, 136.4 mL of purified water, 409.2 mL of dichloromethane and 68.2 mL of isopropanol were introduced into a 4-neck flask provided with a mechanical stirrer, thermometer, cooler and thermostat. Heating was carried out at 30-35C and stirring was carried out for one hour up to complete dissolution. Then the phases were separated and the aqueous phase was extracted using 136.4 mL of Dichloromethane. The organic phases were collected and dried. The obtained solid was recovered with 954.8 mL of Ethanol and 136.4 mL of purified water. Heating was carried out at reflux up to complete dissolution, then it was brought to 60C and a solution of 12.3 mL of concentrated hydrochloric acid (32%) (1.05 mol. equiv.) and 136.4 mL of Ethanol was dripped in 2 hours. It was brought to r.t in 4 hours, it was left at room temperature for at least 4 hours, then it was cooled at 0C and it was left at 0C for another 4 hours. The obtained crystal was filtered washing it with 68.2 mL of Ethanol. The obtained salt was dried at 50 C. under vacuum for 24 hours. 44.3 g of <strong>[151096-09-2]moxifloxacin</strong> hydrochloride with a molar yield equivalent to 85.1% with HPLC purity equivalent to 99.79% and a formula (VII) impurity content equivalent to 0.08% was obtained. The obtained product does not contain, even at ppm level, any ester of sulfonic acids as an impurity. This is important because it is well-known that such impurities are genotoxic.
(1-cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-(oxo-κO3)-3-quinolinecarboxylato-κO3)difluoroboron[ No CAS ]
[ 186826-86-8 ]
Yield
Reaction Conditions
Operation in experiment
55 - 74%
Stage II; Preparation of Moxifloxacin hydrochlorideThe residue obtained from step Il was dissolved in a mixture of methanol (50 ml) and lrielhylamine (7.4 g, 0.0733 moles) and was heated to reflux at 60-650C for 6 h. Alter the completion of reaction, solvent and excess of triethylamine were distilled off at 40-500C under reduced pressure to obtain a residue, which was suspended in a mixture of acetonitrile (20 ml) and methanol (20 ml) and concentrated hydrochloric acid (4.0 ml) was added at 20-250C. The reaction mass was stirred for 30 niin at 20-250C and thereafter cooled further to 0-50C. After stirring for 2 h at 0-50C, the resulting product Moxifloxacin hydrochloride was filtered and washed with acetonitrile-methanol mixture (1 : 1 , 5 ml). The product was dried at 45-500C till moisture content was less than < 2% w/w. The dry weight of the product 3.5 g (55%).; Stage IIPreparation of l-cycIopropyI-6-fluoiO-l,4-dihydro-8-methoxy-7-|(4aS',7a.S)- octahydro-6H-pyrrolo[3,4-6]pyridin-6-yl]-4-oxo-3-quinoline carboxylic acid hydrochloride (Moxiiloxacin hydrochloride)The Moxifloxacin borondifluoride chelate (V) obtained as above (310 g, 0.69 moles) was suspended in methanol (1240 ml), triethylamine (69.8 g, 0.69 moles) was added and the mixture was heated to reflux for twenty-four hours. After cooling, the methanol was distilled off under reduced pressure and the solid residue was suspended in water (2480 ml) and dichloromethane (2480 ml). This reaction mixture was neutralized with 10 % aqueous sodium hydroxide solution and organic layer was separated and the aqueous layer was further extracted with dichloromethane (930 ml). The combined organic layer was distilled under reduced pressure at 30-350C and the crude solid residue was suspended in a mixture of acetonitrile-methanol [(1 :1), 1240 ml]. Concentrated hydrochloric acid (216 g, 2.01 moles) was added dropwise at 0-50C. The precipitated yellow crystals were stirred for one hour at O0C to 5C, filtered and dried under vacuum at 60-650C (224 g, 74%)
Example 7: Preparation of Anhydrous Moxifloxacin hydrochloride.A suspension of 10 gr of Moxifloxacin hydrochloride monohydrate and toluene was heated to reflux in azeotropic mode till to get complete water traces. Cooled the mass to 25 -350C and filtered the solid, washed with isopropyl alcohol to get anhydrous Moxifloxacin hydrochloride.Yield: 8 gr
1 -cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylic acid- O3,O4 bis(propyloxy-O)borate (100.Og) was suspended in n-butanol (500.0 ml) to which (S,S)-2,8-diazabicyclo (4,3,0) nonane (29.0 g) diluted with 100.0 ml n-butanol was added slowly at 10-15C. The contents were heated to 100C and maintained for 3 hours. After the completion of reaction it was cooled to 25-3O0C. 200.0 ml methanol was added and pH was adjusted 1.0-2.0 using methanolic hydrochloric acid. The contents were stirred at 25-3O0C for 2 hours. After completion of reaction the reaction mass was distilled to residue. Purified water 500 ml was added and pH was adjusted to 7.5-9.0 using liquor ammonia. The reaction mass was then extracted with dichloromethane. The organic layer was dried using sodium <n="15"/>sulphate and concentrated to residue. The residue was stripped with 100 ml methanol. 300 ml methanol was charged and pH was adjusted to 1.0-2.0 using methanolic hydrochloric acid, the contents were further cooled to 0-50C and maintained for 1 hour. The solid was filtered and washed with chilled methanol (50.0 ml) and dried under vacuum at 85-9O0C to yield 75.0 g (75%) of moxifloxacin hydrochloride.
With sodium hydroxide; In water; acetone; at 25 - 60℃; for 0.666667h;Product distribution / selectivity;
Moxifloxacin hydrochloride (120 grams) was suspended in 600 ml of water. The pH of the reaction mixture was adjusted to 7.9 with aqueous sodium hydroxide solution.The reaction mixture was stirred for 10 minutes at 25-300C. The reaction mixture was extracted with methylene chloride. The solvent completely was distilled off under reduced pressure at below 6O0C. 100 ml of acetone was added and the reaction mixture was stirred for 30 minutes at 25-30C. The solid obtained was filtered and washed with acetone and dried at 60C.Yield: 86 grams.Water content: 2.30% w/v Chloride content: Not Detected
With sodium hydroxide; In cyclohexane; water; acetone; at 25 - 60℃; for 13.1667 - 15.1667h;Heating / reflux;Product distribution / selectivity;
Moxifloxacin hydrochloride (120 grams) was suspended in 600 ml of water. The pH of the reaction mixture was adjusted to 7.9 with aqueous sodium hydroxide solution.The reaction mixture was stirred for 10 minutes at 25-300C. The reaction mixture was extracted with methylene chloride. The solvent completely was distilled off under reduced pressure at below 60C. 100 ml of acetone was added and the reaction mixture was stirred for 30 minutes at 25-30C. The solid obtained was filtered and washed with acetone. The obtained wet solid taken in 1200 ml of cyclohexane. The reaction mixture was heated to reflux and partially distilled off the solvent to remove the traces of acetone.The reaction mixture stirred at azeotropic reflux for 12-14 hours. The traces of water were removed at reflux temperature. The reaction mixture was cooled to 25-300C under nitrogen atmosphere. The reaction mixture was stirred at 25-300C under nitrogen atmosphere for 30 minutes. The precipitated solid was filtered, washed with isopropyl alcohol and dried at 90-1000C. <n="29"/>Yield: 78 gramsWater content: 0.25% w/vChloride content: 110 ppm
The mixture of (S,S)2,8-diazabicyclo[4,3,0]nonane (32.8 gm), (1- cyclopropyl-6,7-difluoro-1 ,4-dihydro-8-methoxy-4-oxoquinoline-3-carboxylic acid- O3,O4) bis (acyloxy-O)borate (100 gm), 1 ,8-diazabicyclo[5,4,0]undec-7-ene (12.6 gm) and toluene (500 ml) is heated to 75 - 800C for 1 hour 30 minutes. Distilled off the solvent completely under vacuum and methanol (400 ml) and water (100 ml) are added to reaction mass. The pH is adjusted to 1.0 - 2.0 with concentrated hydrochloric acid at 250C and cooled to 50C. The solid obtained is collected by filtration and the solid is dried at 60-650C for 5 hours to obtain 72.5 gm moxifloxacin hydrochloride monohydrate polymorph IV. The product obtained above may further be processed, if required, to obtain moxifloxacin hydrochloride monohydrate polymorph IV in higher chromatographic purity as follows:The product obtained above (50 gm) is suspended in water (600 ml). The pH is adjusted to 7.5 - 8.0 with 50 % aqueous sodium hydroxide solution and extracted moxifloxacin free base with methylene dichloride (500 ml). Distilled off the methylene dichloride layer to get moxifloxacin free base as oily residue. To the oily residue, methanol (360 ml) and water (100 ml) are added and the contents are heated to 600C. The clear solution obtained is subjected to carbon treatment and the reaction mass is filtered through cellite bed and the bed is washed with hot methanol and water mixture (50ml, 4:1 by volume). The filtrate is cooled to 250C. The pH is adjusted to 1.0 - 2.0 with concentrated hydrochloric acid at 250C and cooled to O0C. The solid obtained is collected by filtration and the solid is dried at 60-650C for 5 hours to obtain 46 gm of moxifloxacin hydrochloride monohydrate polymorph IV.
To a 5 L reactor was charged 215 g of (S, S) -2,8-diazabicyclo [4.3.0] nonane,400 ml of methanol and 300 g of triethylamine,A solution of 1-cyclopropyl-6,7-difluoro-8-methoxy-1,4-dihydro-4-oxoquinoline-3-carboxylic acid-O3, O4- diacetate boron Ester of methanol solution,Drop finished,Reflux 6 ~ 10h.Reaction is completed,The solvent was distilled off under reduced pressure,The residue was dissolved by the addition of 500 ml of ethanol,With concentrated hydrochloric acid to adjust the pH to acidic,Cooling crystallization,filter,To give 610 g of a yellow solid,That is, crude moxifloxacin hydrochloride.
45 g
The main ring chelate compound 50g, anhydrous ethanol 200g, (3,3)-2,8-diazabicyclo[4,3,0]nonane 15.7g and triethylamine 12.0g were added to the reaction flask. Temperature 70 C reaction 4. Oh; the end of the reaction, temperature control 30 C to the solution was added dropwise 12mol / L of hydrochloric acid, stirring while stirring, adjust the pH to 1, stirring for 1h, temperature control to 10 C, Crystal growth 2h; After crystal growth, filtration, washing with 95 % ethanol twice, each time 50g; Wet powder 60 C vacuum drying to 2.8% moisture, to obtain moxifloxacin hydrochloride dry powder 45.0g, a single impurity 0.1%, The total impurity is 0.2%; after refining with ethanol and water, the content is 99%, which meets the quality standards of the European Pharmacopoeia EP-7.0 and the total impurity is less than 0.1%
<strong>[186826-86-8]Moxifloxacin hydrochloride</strong> monohydrate (10 gm) as disclosed in the US Patent No. 5,849,752 is suspended in methanol (70 ml) and water (20 ml). The pH is adjusted to 7.5 - 8.5 with 50 % aqueous sodium hydroxide solution. The clear solution obtained is subjected to carbon treatment and the reaction mass is filtered through cellite bed and the bed is washed with hot methanol and water mixture (10ml, 4:1 by volume). The filtrate is cooled to 250C. The pH is adjusted to 1.0 - 2.0 with concentrated hydrochloric acid at 250C and cooled to O0C. The solid obtained is collected by filtration and the solid is dried at 60-650C for 6 hours to obtain 9.4 gm of <strong>[186826-86-8]moxifloxacin hydrochloride</strong> monohydrate polymorph IV.
<strong>[186826-86-8]Moxifloxacin hydrochloride</strong> 100.0 gm was stirred with methanol 800.0 ml and triethyl amine 25-30 C. The reaction mass was concentrated partially. Further 300.0 ml of methanol was added and the pH was adjusted to 1.0-2.0 using hydrochloric acid gas dissolved in methanol at 20-25 C. The contents were cooled to 0-5 C. and maintained at 0-5 C. for 2 hours. The resulting solids were filtered and washed with 50.0 ml chilled methanol. The product was dried under vacuum at 80-90 C. to obtain 90-95.0 gm of <strong>[186826-86-8]moxifloxacin hydrochloride</strong> Form C.It will be appreciated that the invention described above may be modified within the scope of the claims.
12
(4aS-cis)-1-cyclopropyl-7-(2,8 diazabicyclo[4.3.0]non-8-yl)-6-fluoro-8-methoxy-4-oxo-1,4-dihydro-3-quinoline carboxylic aid-O3,O4-bis(propyloxy-O)borate.[ No CAS ]
[ 186826-86-8 ]
Yield
Reaction Conditions
Operation in experiment
With hydrogenchloride; In methanol; at 25 - 30℃; for 2h;pH 1 - 2;
1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydro-3-quinoline carboxylic acid-O3,O4 bis(propyloxy-O)borate (100.0 g) was suspended in n-butanol (500.0 ml) to which (S,S)-2,8-diazabicyclo(4,3,0)nonane (29.0 g) diluted with 100.0 ml n-butanol was added slowly at 10-15 C. The contents were heated to 100 C. and maintained for 3 hours. After the completion of reaction it was cooled to 25-30 C. 200.0 ml methanol was added and pH was adjusted 1.0-2.0 using methanolic hydrochloric acid. The contents were stirred at 25-30 C. for 2 hours. After completion of reaction the reaction mass was distilled to residue. Purified water 500 ml was added and pH was adjusted to 7.5-9.0 using liquor ammonia. The reaction mass was then extracted with dichloromethane. The organic layer was dried using sodium sulphate and concentrated to residue. The residue was stripped with 100 ml methanol. 300 ml methanol was charged and pH was adjusted to 1.0-2.0 using methanolic hydrochloric acid, the contents were further cooled to 0-5 C. and maintained for 1 hour. The solid was filtered and washed with chilled methanol (50.0 ml) and dried under vacuum at 85-90 C. to yield 75.0 g (75%) of moxifloxacin hydrochloride.
1-cyclopropyl-6,7-difluoro-1,4-dihydro-8-methoxy-4-oxo-3-quinolinic acid[ No CAS ]
octahydro-1H-pyrrole[3,4-b]pyridine[ No CAS ]
[ 109-88-6 ]
Moxifloxacin HCl (I-HCl)-Exemplifying[ No CAS ]
[ 186826-86-8 ]
Yield
Reaction Conditions
Operation in experiment
89%
With tetrabutyl-ammonium chloride; In N-methyl-acetamide; hydrogenchloride; ethanol; water;
Example 1 Synthesis of Moxifloxacin HCl (I-HCl)-Exemplifying the Invention In a 250 mL three-neck flask provided with a mechanical stirrer, thermometer and thermostat, 20.0 g of 1-cyclopropyl-6,7-difluoro-1,4-dihydro-8-methoxy-4-oxo-3-quinolinic acid (67.74 mmol), 7.37 g of magnesium methoxide (85.3 mmol, 1.26 mol. eq.), 4.33 g of magnesium hydroxide (74.2 mmol, 1.10 mol. eq.) and 120 mL of dimethylformamide (DMF) (6 vol.) were loaded under inert atmosphere. The mixture is heated at 30 C. for one hour and the methanol which is formed is removed by means of low pressure distillation. To the solution is then added 0.5 g of tetrabutylammonium chloride (TBAC) (2.5% by weight on the substrate. 10.25 g of (4aS, 7aS)-Octahydro-1H-pyrrole[3,4-b]pyridine (81.29 mmol, 1.20 mole equiv.) is then added. The reaction mixture is heated at 45 C. and maintained at T=45-50 C. until the HPLC monitored reaction is completed, i.e. about 16 hours. The solvent is vacuum distilled and about 110 mL DMF are recovered. To the residual suspension 200 mL water and 25 mL HCl 12 N (300 mmol) is added, thereby bringing the solution pH to 3-4. The suspension is heated at 30 C. and stirred at that temperature for 30 minutes, then it is cooled at T<10 C. and, after the product has precipitated, it is stirred at this temperature for 30 minutes. The suspension is filtered and the solid is washed with water. The crude is dissolved in 150 mL hydrochloric acid 5.5 mol/L. The whole is stirred for at least 20 minutes until it is dissolved. The small amount of insolute material, being an impurity, is eliminated through filtration. The filtrate is concentrated to a small volume under low pressure. To the obtained solution is added 70 mL anhydrous ethanol. The mixture is stirred and cooled below 5 C. for at least one hour after solid precipitation. The suspension is filtrated and the solid is washed with mL ethyl acetate. The solid is vacuum dried for 3 hours at 25 C. then at Tmax.=70 C. for 4 hours. 26.4 g of Moxifloxacin hydrochloride is obtained (molar yield=89%) with HPLC (A %) purity>99.0%.
With sodium hydroxide; In methanol; at 60℃; for 2h;
General procedure: All the complexes were prepared in a similar manner. The complexes were prepared by the addition of a 2 mmol of methanolic solution of MXnH2O (M = Co, Cu, Ni, Zn, Mn, Ag and Cr, X = Cl or NO3; n = 0, 2, 4 or 6) to 0.875 g (2 mmol) of moxifloxacin (MOX) dissolved in (2 mmol) of NaOH solution, after which, 0.136 g(2 mmol) of imidazole (Him) in 20 mL of methanol in 1:1 ratio was added. The reaction mixture was stirred at 60 C for 2 h. The solution was left for slow evaporation, the precipitates formed were filtered off, washed with distilled water several times and dried over calcium chloride in a descicator. Unfortunately we were not able to prepare single crystals of the complexes. It is well known that low solubility of quinolones and their complexes presents a great difficulty in preparing single crystals.The complexes were characterized by elemental analyses,Table 1, infrared, electronic absorption spectra, ESR for the Cu(II)complex, 1H NMR and 13C NMR spectra for the Ni(II) complex and thermal analyses. All the complexes under investigation arenon-electrolytes.
With sodium hydroxide; In methanol; at 60℃; for 2h;
General procedure: All the complexes were prepared in a similar manner. The complexes were prepared by the addition of a 2 mmol of methanolic solution of MXnH2O (M = Co, Cu, Ni, Zn, Mn, Ag and Cr, X = Cl or NO3; n = 0, 2, 4 or 6) to 0.875 g (2 mmol) of moxifloxacin (MOX) dissolved in (2 mmol) of NaOH solution, after which, 0.136 g(2 mmol) of imidazole (Him) in 20 mL of methanol in 1:1 ratio was added. The reaction mixture was stirred at 60 C for 2 h. The solution was left for slow evaporation, the precipitates formed were filtered off, washed with distilled water several times and dried over calcium chloride in a descicator. Unfortunately we were not able to prepare single crystals of the complexes. It is well known that low solubility of quinolones and their complexes presents a great difficulty in preparing single crystals.The complexes were characterized by elemental analyses,Table 1, infrared, electronic absorption spectra, ESR for the Cu(II)complex, 1H NMR and 13C NMR spectra for the Ni(II) complex and thermal analyses. All the complexes under investigation arenon-electrolytes.
With sodium hydroxide; In methanol; at 60℃; for 2h;
General procedure: All the complexes were prepared in a similar manner. The complexes were prepared by the addition of a 2 mmol of methanolic solution of MXnH2O (M = Co, Cu, Ni, Zn, Mn, Ag and Cr, X = Cl or NO3; n = 0, 2, 4 or 6) to 0.875 g (2 mmol) of moxifloxacin (MOX) dissolved in (2 mmol) of NaOH solution, after which, 0.136 g(2 mmol) of imidazole (Him) in 20 mL of methanol in 1:1 ratio was added. The reaction mixture was stirred at 60 C for 2 h. The solution was left for slow evaporation, the precipitates formed were filtered off, washed with distilled water several times and dried over calcium chloride in a descicator. Unfortunately we were not able to prepare single crystals of the complexes. It is well known that low solubility of quinolones and their complexes presents a great difficulty in preparing single crystals.The complexes were characterized by elemental analyses,Table 1, infrared, electronic absorption spectra, ESR for the Cu(II)complex, 1H NMR and 13C NMR spectra for the Ni(II) complex and thermal analyses. All the complexes under investigation arenon-electrolytes.
With sodium hydroxide; In methanol; at 60℃; for 2h;
General procedure: All the complexes were prepared in a similar manner. The complexes were prepared by the addition of a 2 mmol of methanolic solution of MXnH2O (M = Co, Cu, Ni, Zn, Mn, Ag and Cr, X = Cl or NO3; n = 0, 2, 4 or 6) to 0.875 g (2 mmol) of moxifloxacin (MOX) dissolved in (2 mmol) of NaOH solution, after which, 0.136 g(2 mmol) of imidazole (Him) in 20 mL of methanol in 1:1 ratio was added. The reaction mixture was stirred at 60 C for 2 h. The solution was left for slow evaporation, the precipitates formed were filtered off, washed with distilled water several times and dried over calcium chloride in a descicator. Unfortunately we were not able to prepare single crystals of the complexes. It is well known that low solubility of quinolones and their complexes presents a great difficulty in preparing single crystals.The complexes were characterized by elemental analyses,Table 1, infrared, electronic absorption spectra, ESR for the Cu(II)complex, 1H NMR and 13C NMR spectra for the Ni(II) complex and thermal analyses. All the complexes under investigation arenon-electrolytes.
[Cu(II)(moxifloxacin)(imidazole)Cl*H2O]2*2H2O[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sodium hydroxide; In methanol; at 60℃; for 2h;
General procedure: All the complexes were prepared in a similar manner. The complexes were prepared by the addition of a 2 mmol of methanolic solution of MXnH2O (M = Co, Cu, Ni, Zn, Mn, Ag and Cr, X = Cl or NO3; n = 0, 2, 4 or 6) to 0.875 g (2 mmol) of moxifloxacin (MOX) dissolved in (2 mmol) of NaOH solution, after which, 0.136 g(2 mmol) of imidazole (Him) in 20 mL of methanol in 1:1 ratio was added. The reaction mixture was stirred at 60 C for 2 h. The solution was left for slow evaporation, the precipitates formed were filtered off, washed with distilled water several times and dried over calcium chloride in a descicator. Unfortunately we were not able to prepare single crystals of the complexes. It is well known that low solubility of quinolones and their complexes presents a great difficulty in preparing single crystals.The complexes were characterized by elemental analyses,Table 1, infrared, electronic absorption spectra, ESR for the Cu(II)complex, 1H NMR and 13C NMR spectra for the Ni(II) complex and thermal analyses. All the complexes under investigation arenon-electrolytes.
With sodium hydroxide; In methanol; at 60℃; for 2h;
General procedure: All the complexes were prepared in a similar manner. The complexes were prepared by the addition of a 2 mmol of methanolic solution of MXnH2O (M = Co, Cu, Ni, Zn, Mn, Ag and Cr, X = Cl or NO3; n = 0, 2, 4 or 6) to 0.875 g (2 mmol) of moxifloxacin (MOX) dissolved in (2 mmol) of NaOH solution, after which, 0.136 g(2 mmol) of imidazole (Him) in 20 mL of methanol in 1:1 ratio was added. The reaction mixture was stirred at 60 C for 2 h. The solution was left for slow evaporation, the precipitates formed were filtered off, washed with distilled water several times and dried over calcium chloride in a descicator. Unfortunately we were not able to prepare single crystals of the complexes. It is well known that low solubility of quinolones and their complexes presents a great difficulty in preparing single crystals.The complexes were characterized by elemental analyses,Table 1, infrared, electronic absorption spectra, ESR for the Cu(II)complex, 1H NMR and 13C NMR spectra for the Ni(II) complex and thermal analyses. All the complexes under investigation arenon-electrolytes.
With sodium hydroxide; In methanol; at 60℃; for 2h;
General procedure: All the complexes were prepared in a similar manner. The complexes were prepared by the addition of a 2 mmol of methanolic solution of MXnH2O (M = Co, Cu, Ni, Zn, Mn, Ag and Cr, X = Cl or NO3; n = 0, 2, 4 or 6) to 0.875 g (2 mmol) of moxifloxacin (MOX) dissolved in (2 mmol) of NaOH solution, after which, 0.136 g(2 mmol) of imidazole (Him) in 20 mL of methanol in 1:1 ratio was added. The reaction mixture was stirred at 60 C for 2 h. The solution was left for slow evaporation, the precipitates formed were filtered off, washed with distilled water several times and dried over calcium chloride in a descicator. Unfortunately we were not able to prepare single crystals of the complexes. It is well known that low solubility of quinolones and their complexes presents a great difficulty in preparing single crystals.The complexes were characterized by elemental analyses,Table 1, infrared, electronic absorption spectra, ESR for the Cu(II)complex, 1H NMR and 13C NMR spectra for the Ni(II) complex and thermal analyses. All the complexes under investigation arenon-electrolytes.
Under nitrogen protection, 1-cyclopropyl-6, 7-difluoro-1, 4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid 10.0 g (0·0339 momicronl) and (S,S) -2,8-diazabicyclo[4,3,0] nonane 5 .13g(0.0406mol)were added to 70 mL acetonitrile, it was stirred at 20 C~30 C and 1,8-diazabicyclo[5·4·0]undec-7-ene 7.74 g (0.0508 mol) was added, it was stirred at 75 C~85 C for 36 h. Concentrated in vacuo (temperature 45 C ~ 65 C, pressure -0 · 08MPa ~ -0.1MPa) to remove most of the solvent, it was then dissolved in chloroform, the mass concentration is 2% aqueous acetic acid solution (the mass concentration refers to the mass of acetic acid as a percentage of the total mass of the aqueous acetic acid solution), the mass concentration was 10% saline (the mass concentration refers to the mass of sodium chloride as a percentage of the total mass of brine were sequentially added, respectively, then the mixture was stirred, allowed to stand, and the aqueous layer was separated. The organic layer was concentrated in vacuo (temperature 35 C ~ 55 C, pressure -0.08MPa ~ -0.1MPa) to remove most of the solvent, ethyl acetate was added, and the mixture was cooled to 10 C to 20 C and stirred for 2 hours to 3 hours. Centrifugation, rinsed three times with ethyl acetate, dried in vacuo (vacuum degree -0.01MPa~-0.1MPa, temperature 45~55C) dried for 14 to 18 hours, to obtain 4.01g of moxifloxacin , yield 29.5%.HPLC purity 93.54%. Under nitrogen protection, add 4.0 g of moxifloxacin to 2.5 mL of water and 10 mL of methanol.Stir at 170 C for 1 hour, filter, and cool to 35 C.The pH was adjusted to 1.4 to 1.8 by the addition of 6N hydrochloric acid. Cool to 15 C and stir for 1 hour.It was filtered, washed with water, and stirred at 15 C for 1 hour in a solution of water 5 mL and concentrated hydrochloric acid.Filtered, washed with water, vacuum dried (vacuum degree - 0.01 MPa ~ -0.1 MPa, temperature 45 ~ 55 C) for 16 hours, to obtain 3.3 g of moxifloxacin hydrochloride,Yield 75.6% (total yield 22.3%, based on <strong>[112811-72-0]1-cyclopropyl-6,7-difluoro-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid</strong> ). The HPLC purity was 99.23%, and the maximum single impurity was 0.47%.
(4aS-cis)-1-cyclopropyl-7-(2,8-diazabicyclo-[4.3.0]non-8-yl)-6-fluoro-1,4-dihydro-8-methoxy-4-oxo-3-quinoline carboxylic acid-O3,O4(bis(acyloxy-O)) borate[ No CAS ]
[ 186826-86-8 ]
Yield
Reaction Conditions
Operation in experiment
96.2%
With hydrogenchloride; In methanol; water; at 0 - 8℃; for 0.333333h;
At room temperature, add 30.00g (0.071mol) MX-1 to a 500ml four-necked flask, add 74.5ml acetonitrile, and stir until dissolved; slowly add 9.84g (0.075mol) (S, S) -2,8-diazabicyclo [4,3,0] Nonane, triethylamine 10g (0.099mol), temperature controlled 45 C, and stirred for 1.5 hours; after the reaction was completed, the temperature was lowered to 0 C and a cooled methanolic hydrochloric acid solution (74g (2.31mol) of methanol, mass concentration) was added. 36 g (0.36 mol) of 37% concentrated hydrochloric acid) was stirred at 8 C for 20 minutes, 20 g (0.217 mol) of glycerol was added and stirred for 10 minutes, and crystal form 2 moxifloxacin having a purity of 99.5% or more was added as a seed crystal And stirring for 1 hour, adding isopropyl alcohol 116g (1.86mol) and stirring for 3 hours; filtering and washing the filter cake with isopropyl alcohol; 29.9g of yellow-green solid dried at 40 C for 15 hours, yield 96.2%, The purity is 99.87%, and the boron content is 0.05%.
93.1%
With hydrogenchloride; In methanol; at -5 - 20℃; for 2h;pH 1;
Under the protection of nitrogen, the boric acid 6.2g (100mmol) and second grade acid anhydride 35.7g (350mmol) added to the three-port flask is heated to 85 C contact reaction 1 hour, cooling to 75 C; adding glycine (2.6g) then adding 1-cyclopropyl -6, 7-difluoro -1, 4-dihydro-8-methoxy-4-oxo-3-quinoline carboxylic acid ethyl ester 21.7g (67mmol), in the 75 C to continue stirring for 2 hours, after TLC monitoring reaction, cooling to room temperature, by adding acetonitrile 80 ml (nonane weight of 2 times) and N-methyl morpholine 23.8g (235mmol), with (S, S)-2, 8-diazabicyclo [4.3.0] nonane 8.1g (64mmol) in 60 C reaction under 1 hour, to room temperature, filtering the insoluble matter, by adding methanol (160 ml), dropping concentrated hydrochloric acid at room temperature, adjusting the pH value to 1, stirring 2 hours after cooling to -5 C crystallization, filtration, cold ethanol washing (50 ml × 3 times), vacuum drying, to obtain white solid moxifloxacin hydrochloride 26.1g, yield: 93.1%, purity 99.67% (HPLC area unitary method).
84.75%
With sodium hydroxide; at 80℃; for 2.5h;
After the solid was added to 630mL of 20% sodium hydroxide solution was heated to 80 , stirring 2.5h HPLC showed the starting material substantially complete the reaction, cooled to room temperature (25 ), was treated with glacial acetic acid PH = 7.0 (spent about 70mL of glacial acetic acid).500ml of absolute ethanol was added, stirring was continued for 1h, then concentrated hydrochloric PH = 1.0 (with a 215ml), stirred a lot of the precipitated solid.At room temperature and then stirred for 1h, and filtered.40ml with a mixed solvent (ethanol: water = 1: 1) washing the filter cake, 40 vacuum drying 9h,40 vacuum drying 9h, was 101.7g moxifloxacin hydrochloride, quality Yield: 84.75%, 99.65% purity obtained moxifloxacin hydrochloride,
118 g
With hydrogenchloride; In water; at 10 - 15℃; for 6h;pH 1.0 - 2.0;
Experiment 2: Preparation of Moxifloxacin Hydrochloride in a solvent system comprising of 8.2%wt of water and Acetonitrile. To the solution of 650ml of Acetonitrile and 203g (0.33 lmol) of wet cake of 1 -cyclopropyl-6.7- difluoro- 1 ,4-dihydro-8-methoxy-4-oxo-3-quinoline carbox lic acid-0J,04) bis(acetato- 0)-borate (having 31% moisture content) was added 40.8g (0.323moi) of [S,S]-2,8- diazabicyclo-[4,3,0]nonane dissolved in 50ml of acetonitrile at 10-15C, To the above reaction mass added 33.4g (0.330mol) of triethyl amine and stirred at 15-20C for 6-8 h, After completion of reaction, distilled the reaction mixture under vacuum at 30C followed by addition of 300 ml of isopropyl alcohol at 10~15C. Adjusted the pH of the reaction mass to 1.0-2.0 with dilute hydrochloric acid and stirred for 6h. Cooled the reaction mass to 0-5C and filtered the solid mass. The crude mass so obtained was crystallized in methanol and water (3: 1) to get wet Moxifloxacin hydrochloride, which in turn, was dried under vacuum to get 118g of Moxifloxacin hydrochloride having purity 99.97% by HPLC.
48 g
With hydrogenchloride; In ethanol; water; at 25℃; for 1h;pH 1;
A solution of the main chelate (1-cyclopropyl-6,7-difluoro-8-methoxy-1,4-dihydro-4-oxoquinoline-3-carboxylic acid-0.3, 04-diacetate boron ester), 50 g, Acetonitrile 198 g, (S, S) -2,8-diazabicyclo [4,3,0] nonane 15.78 and triethylamine (12.0 g) at a temperature of 50 C for 4 h. The reaction was completed at 50 C, After the concentration was complete, 200 g of absolute ethanol was added to the concentrated residue, and the mixture was stirred and dissolved. Then, 12 ml of 1 / L hydrochloric acid was added dropwise to the solution at 25 C, and the mixture was stirred, PH to 1, insulation stirring 1h; Cooling to 5 C, stirring crystal 2h; After the end of the crystal, filter, washed with 95% ethanol twice, each 50g; wet powder 60 C vacuum dry to 2.6% moisture, moxifloxacin hydrochloride powder 48.0g, a single impurity ? 0.1 , total impurities ? 0.2 ; refined with ethanol, the content of ? 99%, in line with the European Pharmacopoeia EP-7.0 of the product quality standards, total impurities
With cerium(IV) sulphate; In water; at 25℃; for 0.666667h;Kinetics;
Aliquots of standard MOXF solution (0.038-0.8 mL, 0.5 mg mL-1) were transferred into a series of 10 mL calibrated volumetric flasks. Then 0.75 mL of MBTH solution was added. After that, 1.0 mL of Ce(SO4)2 solution was added. The volume was made up to the mark with distilled water. After mixing, the contents of each flask were immediately transferred to the spectrophotometric cell and the increase in absorbance was recorded at 623 nm as a function of time between 0 and 45 min against reagent blank treated similarly. The initial rate of the reaction (gamma) at different concentrations was obtained from the slope of the tangent to the absorbance-time curve. The calibration curve was constructed by plotting the logarithm of the initial rate (log gamma) versus the logarithmof the molar concentration of the MOXF (logC). The amount of the drug was obtained either from the calibration graphs or the regression equation.
General procedure: A simple synthetic protocol was used for the preparationof nanostructured complexes of MOX. A typicalprocedure for the preparation is briefly described asfollows. First, 2 mmol of MOX in a methanol solvent(20 mL) was added to 20 mL of a solution containing2 mmol of the acceptor (either PA, CLA or CHL) in the same solvent. The resulting mixture was stirred at roomtemperature for approximately half an hour. A change incolor occurred, and the solution was allowed to evaporateslowly at room temperature, resulting in the precipitationof the solid complexes. The precipitates were isolated,filtered and further purified using the same system and arecrystallization process to obtain the pure product. Theproducts were then collected and dried in vacuo for 48 h.The excellent agreement between the experimental andcalculated values of C, H and N indicated that thesynthesized nanoparticles were free of impurities. Thestoichiometry of the interaction between the drug MOXand the acceptors was found to have a 1:1 ratio.
General procedure: A simple synthetic protocol was used for the preparationof nanostructured complexes of MOX. A typicalprocedure for the preparation is briefly described asfollows. First, 2 mmol of MOX in a methanol solvent(20 mL) was added to 20 mL of a solution containing2 mmol of the acceptor (either PA, CLA or CHL) in the same solvent. The resulting mixture was stirred at roomtemperature for approximately half an hour. A change incolor occurred, and the solution was allowed to evaporateslowly at room temperature, resulting in the precipitationof the solid complexes. The precipitates were isolated,filtered and further purified using the same system and arecrystallization process to obtain the pure product. Theproducts were then collected and dried in vacuo for 48 h.The excellent agreement between the experimental andcalculated values of C, H and N indicated that thesynthesized nanoparticles were free of impurities. Thestoichiometry of the interaction between the drug MOXand the acceptors was found to have a 1:1 ratio.
General procedure: A simple synthetic protocol was used for the preparationof nanostructured complexes of MOX. A typicalprocedure for the preparation is briefly described asfollows. First, 2 mmol of MOX in a methanol solvent(20 mL) was added to 20 mL of a solution containing2 mmol of the acceptor (either PA, CLA or CHL) in the same solvent. The resulting mixture was stirred at roomtemperature for approximately half an hour. A change incolor occurred, and the solution was allowed to evaporateslowly at room temperature, resulting in the precipitationof the solid complexes. The precipitates were isolated,filtered and further purified using the same system and arecrystallization process to obtain the pure product. Theproducts were then collected and dried in vacuo for 48 h.The excellent agreement between the experimental andcalculated values of C, H and N indicated that thesynthesized nanoparticles were free of impurities. Thestoichiometry of the interaction between the drug MOXand the acceptors was found to have a 1:1 ratio.
With water; magnesium chloride; In methanol; for 65h;
General procedure: CaII, MgII, and ZnII <strong>[186826-86-8]moxifloxacin hydrochloride</strong> complexes were prepared by refluxing a mixture of a metal salt and mox-HCl(molar ratio 1 : 2) dissolved in methanol on a hot plate for 3-4 h at ca 65C. The precipitate formed immediately upon cooling. The corresponding complex ofFeIII complex was synthesized according to the sameprocedure with molar ratio FeCl3·6H2O to mox-HCl(1 : 3). The colored precipitates were filtered off andwashed with methanol and diethyl ether and dried in avacuum desiccator over CaCl2. Yields of the productswere 80-85%
With water; calcium chloride; In methanol; for 65h;
General procedure: CaII, MgII, and ZnII <strong>[186826-86-8]moxifloxacin hydrochloride</strong> complexes were prepared by refluxing a mixture of a metal salt and mox-HCl(molar ratio 1 : 2) dissolved in methanol on a hot plate for 3-4 h at ca 65C. The precipitate formed immediately upon cooling. The corresponding complex ofFeIII complex was synthesized according to the sameprocedure with molar ratio FeCl3·6H2O to mox-HCl(1 : 3). The colored precipitates were filtered off andwashed with methanol and diethyl ether and dried in avacuum desiccator over CaCl2. Yields of the productswere 80-85%
General procedure: CaII, MgII, and ZnII <strong>[186826-86-8]moxifloxacin hydrochloride</strong> complexes were prepared by refluxing a mixture of a metal salt and mox-HCl(molar ratio 1 : 2) dissolved in methanol on a hot plate for 3-4 h at ca 65C. The precipitate formed immediately upon cooling. The corresponding complex ofFeIII complex was synthesized according to the sameprocedure with molar ratio FeCl3·6H2O to mox-HCl(1 : 3). The colored precipitates were filtered off andwashed with methanol and diethyl ether and dried in avacuum desiccator over CaCl2. Yields of the productswere 80-85%
General procedure: CaII, MgII, and ZnII <strong>[186826-86-8]moxifloxacin hydrochloride</strong> complexes were prepared by refluxing a mixture of a metal salt and mox-HCl(molar ratio 1 : 2) dissolved in methanol on a hot plate for 3-4 h at ca 65C. The precipitate formed immediately upon cooling. The corresponding complex ofFeIII complex was synthesized according to the sameprocedure with molar ratio FeCl3·6H2O to mox-HCl(1 : 3). The colored precipitates were filtered off andwashed with methanol and diethyl ether and dried in avacuum desiccator over CaCl2. Yields of the productswere 80-85%
2g of <strong>[186826-86-8]moxifloxacin hydrochloride</strong> was added to 50ml of methanol, stirred to dissolve, was added 18g peroxy acid, 50 deg.] C the reaction was refluxed for 8h, point to the raw material disappeared, the reaction end, by filtration, the filtrate was collected, concentrated under reduced pressure 30 deg.]
With hydrogenchloride; In ethanol; water; at 25 - 30℃; for 1h;
The 50g tartaric acid moxifloxacin is added to250ml water and 250ml of ethanol and stirred, was added 16. 6ml of concentratedhydrochloric acid at 25~30 C, the reaction was stirred at this temperaturefor 1 hour, suction filtered, the filter cake was washed with 100ml of ethanol,50~55 C for 3 hours in vacuo give moxifloxacin hydrochloride 35. 10g (88.3%). Yield: 86 61% isomer content: 0?05%
With hydrogenchloride; In water; at -5℃; for 2h;pH 2;
Comprising the steps of:Under nitrogen protection,A solution of 1.1 g (40 mmol) of CuCl,(110 mmol) of ethylene glycol,Isobutylamine (12.4 g, 170 mmol)And 1-cyclopropyl-6,7-difluoro-1,4-dihydro-8-methoxy-4-oxo-3-quinoline-carboxylate 32.3g (100mmol)Was charged into a reaction vessel equipped with 320 mL of methanol,The reaction was carried out at 50 C for 3 hours,The temperature was then raised to 68 C,(S, S) -2,8-diazabicyclo [4.3.0] nonane 13.9 g (110 mmol)The reaction was continued for 4 hours,The insoluble matter was filtered off by hot filtration (temperature: 58 C)Concentrated hydrochloric acid was added dropwise at room temperature,Adjust the pH to 2,Stirring for 2 hours after cooling to -5 crystallization,Filtration,Cold ethanol washing,Vacuum drying,Moxifloxacin hydrochloride as a white solid (40.7 g)The yield was 93.0%Purity 99.82% (HPLC area normalization method).
With hydrogenchloride; In methanol; at 20℃; for 2h;pH 3;
Under an argon atmosphere, boric acid 6.2g (100mmol) and propionic anhydride 39g (300mmol) was added to the three-necked flask was added Thermal contact with the reaction to 98 C for 1 hour and cooled to 80 C; glycine (2g) was added and then 1-cyclopropyl-6,7-difluoro-1,4 Hydrogen-8-methoxy-4-oxo-3-quinoline-carboxylate 20.4g (63mmol), stirring was continued at 80 C in 1.5 hours, TLC Monitor completion of the reaction, cooled to room temperature, acetonitrile (75ml) and N- methyl morpholine 16.2g (160mmol), and then (S, S) 2,8-diazabicyclo [4.3.0] nonane 7.3g (58mmol) for 2 hours at 75 C, the cooled to room temperature, insolubles were filtered off, Methanol was added (150ml), concentrated hydrochloric acid was added dropwise at room temperature, adjusted to pH 3 and stirred for 2 hours after cooling to -10 C crystallization, filtration, Washed with cold ethanol (50ml × 3 times), and dried in vacuo to give a white solid moxifloxacin hydrochloride 23.6g, yield: 92.9%, purity 99.45% (HPLC area normalization).
In a 2000 mL three-necked round-bottomed flask, 150.00 g of acetic anhydride was added, stirred, heated to 80 C,boric acid 28. 00g, stirring evenly, slowly warming to 110 C, stirring reaction 2 hours. Cooled to 60-70 C, ethyl 1-cyclopropyl-6,7-difluoro-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylate 100 was added. And the reaction was continued at 90 to 100 C for 3 hours. After completion of the reaction, the reaction mixture was cooled to room temperature. 650 mL of acetonitrile and 592 mL of triethylamine were added to the reaction solution, and the mixture was stirred for 30 minutes. To the reaction solution was added 39.39 g of (S, S) -2,8-diazabicyclo [4.3.0] nonane (1.01eq), heating reflux reaction 3 hours, TLC detection reaction is completed, down to room temperature, stirring 30 minutes, ice bath temperature 5 ~ 10 C 90mL concentrated hydrochloric acid, adjust PH = 1. 2, continue ice bath Stirring and crystallization for 8 hours, filtration, ice-ethanol 50mLX2 washing filter cake, filter cake 50 ~ 60 C / _0.095MPa vacuum drying 12 hours to obtain crude moxifloxacin hydrochloride 121. 48g light yellow powder, yield: 89 78%, HPLC: 99.7%.
To the other reactor was added 60 g of (S, S) -2,8-diazabicyclo [4.3.0] nonane,40 (^ acetonitrile,4 (^ triethylamine,120 g of the above-prepared moxifloxacin hydrochloride intermediate was added with stirring,The reaction temperature was 15 C,The tracking reaction was monitored by TLC,To (S, S) -2,8-diazabicyclo [4.3.0] nonane;After the reaction is complete,The filtrate was added with 50 g of purified water, Keeping the temperature of the reaction solution at about 20 C,Concentrated hydrochloric acid,To a pH of 0.5, Stirring crystallization centrifugal filtration, And the resulting solid was dried at 70 C for about 7 hours,Moxifloxacin hydrochloride was crude 114g,The crude yield of moxifloxacin hydrochloride was 91.8% and the purity of HPCL was 99.45%.
[La(moxifloxacin hydrochloride)(2,2'-bipyridine)(H2O)2]Cl3.8H2O[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80.81%
With sodium hydroxide; In ethanol; for 3h;Reflux;
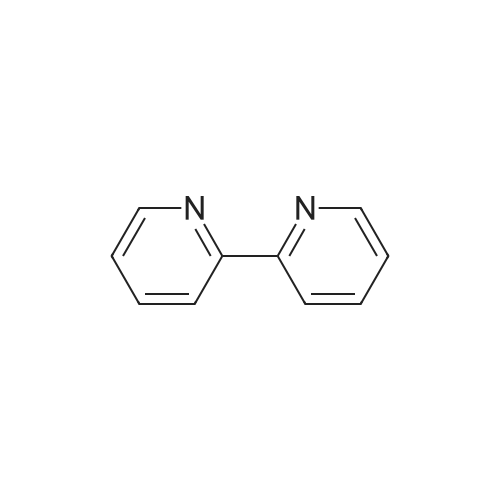
General procedure: The yellow solid complex [Y(MOX)(Bipy)(H2O)2]Cl38H2O wasprepared by mixing 1 mmol (0.437 g) of MOX (L1) with 1 mmol(0.04 g) NaOH and 1 mmol (0.156 g) of Bipy (L2) in 50 ml ethanolwith the same ratio 1 mmol (0.303 g) of yttrium(III) chloridehexahydrate. The mixture was refluxed for 3 h. The yellow precipitatewas filtered off and dried under vacuum over anhydrousCaCl2. The white brown, pale yellow, gray and dark yellow solidcomplexes [ZrO(MOX)(Bipy)(H2O)](NO3)2H2O, [La(MOX)(Bipy)(H2O)2]Cl38H2O, [Ce(MOX)(Bipy)(H2O)2](SO4)26H2O and[UO2(MOX)(Bipy)](CH3COO)2H2O were prepared in a similarmanner described above by using ethanol as a solvent andZrO(NO3)2, LaCl37H2O, Ce(SO4)2 and UO2(CH3COO)22H2O,respectively, in 1:1:1 (Mn:L1:L2) molar ratio. Single crystal suitablefor X-ray crystallographic measurements was not obtained.
[UO2(moxifloxacin hydrochloride)(2,2'-bipyridine)](CH3COO)2.H2O[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85.57%
With sodium hydroxide; In ethanol; for 3h;Reflux;
General procedure: The yellow solid complex [Y(MOX)(Bipy)(H2O)2]Cl38H2O wasprepared by mixing 1 mmol (0.437 g) of MOX (L1) with 1 mmol(0.04 g) NaOH and 1 mmol (0.156 g) of Bipy (L2) in 50 ml ethanolwith the same ratio 1 mmol (0.303 g) of yttrium(III) chloridehexahydrate. The mixture was refluxed for 3 h. The yellow precipitatewas filtered off and dried under vacuum over anhydrousCaCl2. The white brown, pale yellow, gray and dark yellow solidcomplexes [ZrO(MOX)(Bipy)(H2O)](NO3)2H2O, [La(MOX)(Bipy)(H2O)2]Cl38H2O, [Ce(MOX)(Bipy)(H2O)2](SO4)26H2O and[UO2(MOX)(Bipy)](CH3COO)2H2O were prepared in a similarmanner described above by using ethanol as a solvent andZrO(NO3)2, LaCl37H2O, Ce(SO4)2 and UO2(CH3COO)22H2O,respectively, in 1:1:1 (Mn:L1:L2) molar ratio. Single crystal suitablefor X-ray crystallographic measurements was not obtained.
[ZrO(moxifloxacin hydrochloride)(2,2'-bipyridine)(H2O)](NO3)2.H2O[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
75.6%
With sodium hydroxide; In ethanol; for 3h;Reflux;
General procedure: The yellow solid complex [Y(MOX)(Bipy)(H2O)2]Cl38H2O wasprepared by mixing 1 mmol (0.437 g) of MOX (L1) with 1 mmol(0.04 g) NaOH and 1 mmol (0.156 g) of Bipy (L2) in 50 ml ethanolwith the same ratio 1 mmol (0.303 g) of yttrium(III) chloridehexahydrate. The mixture was refluxed for 3 h. The yellow precipitatewas filtered off and dried under vacuum over anhydrousCaCl2. The white brown, pale yellow, gray and dark yellow solidcomplexes [ZrO(MOX)(Bipy)(H2O)](NO3)2H2O, [La(MOX)(Bipy)(H2O)2]Cl38H2O, [Ce(MOX)(Bipy)(H2O)2](SO4)26H2O and[UO2(MOX)(Bipy)](CH3COO)2H2O were prepared in a similarmanner described above by using ethanol as a solvent andZrO(NO3)2, LaCl37H2O, Ce(SO4)2 and UO2(CH3COO)22H2O,respectively, in 1:1:1 (Mn:L1:L2) molar ratio. Single crystal suitablefor X-ray crystallographic measurements was not obtained.
[Ce(moxifloxacin hydrochloride)(2,2'-bipyridine)(H2O)2](SO4)2.6H2O[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
77.37%
With sodium hydroxide; In ethanol; for 3h;Reflux;
General procedure: The yellow solid complex [Y(MOX)(Bipy)(H2O)2]Cl38H2O wasprepared by mixing 1 mmol (0.437 g) of MOX (L1) with 1 mmol(0.04 g) NaOH and 1 mmol (0.156 g) of Bipy (L2) in 50 ml ethanolwith the same ratio 1 mmol (0.303 g) of yttrium(III) chloridehexahydrate. The mixture was refluxed for 3 h. The yellow precipitatewas filtered off and dried under vacuum over anhydrousCaCl2. The white brown, pale yellow, gray and dark yellow solidcomplexes [ZrO(MOX)(Bipy)(H2O)](NO3)2H2O, [La(MOX)(Bipy)(H2O)2]Cl38H2O, [Ce(MOX)(Bipy)(H2O)2](SO4)26H2O and[UO2(MOX)(Bipy)](CH3COO)2H2O were prepared in a similarmanner described above by using ethanol as a solvent andZrO(NO3)2, LaCl37H2O, Ce(SO4)2 and UO2(CH3COO)22H2O,respectively, in 1:1:1 (Mn:L1:L2) molar ratio. Single crystal suitablefor X-ray crystallographic measurements was not obtained.
[Y(moxifloxacin hydrochloride)(2,2'-bipyridine)(H2O)2]Cl3.8H2O[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
88.71%
With sodium hydroxide; In ethanol; for 3h;Reflux;
General procedure: The yellow solid complex [Y(MOX)(Bipy)(H2O)2]Cl38H2O wasprepared by mixing 1 mmol (0.437 g) of MOX (L1) with 1 mmol(0.04 g) NaOH and 1 mmol (0.156 g) of Bipy (L2) in 50 ml ethanolwith the same ratio 1 mmol (0.303 g) of yttrium(III) chloridehexahydrate. The mixture was refluxed for 3 h. The yellow precipitatewas filtered off and dried under vacuum over anhydrousCaCl2. The white brown, pale yellow, gray and dark yellow solidcomplexes [ZrO(MOX)(Bipy)(H2O)](NO3)2H2O, [La(MOX)(Bipy)(H2O)2]Cl38H2O, [Ce(MOX)(Bipy)(H2O)2](SO4)26H2O and[UO2(MOX)(Bipy)](CH3COO)2H2O were prepared in a similarmanner described above by using ethanol as a solvent andZrO(NO3)2, LaCl37H2O, Ce(SO4)2 and UO2(CH3COO)22H2O,respectively, in 1:1:1 (Mn:L1:L2) molar ratio. Single crystal suitablefor X-ray crystallographic measurements was not obtained.The chemical Structures of synthesized metal complexes wereconfirmed as follows:1-Cyclopropyl-6-fluro-8-methoxy-4-oxo-7-(tetrahydro-1Hpyrrolo[3,4-b]pyridin) 6(2H,7H,7aH)-yl)-1,4-dihydroquionoline-3-carboxylato 2,20-bipyridine diaquo yttrium chloride octahydrate(1, YC31H52FN5O14Cl3)Yellow; Yield 88.71%; m.p.: 310 C; Elemental analysis: found, C39.88, H 5.60, N 7.48, M 9.51. Calc. for YC31H52FN5O14Cl3 (933.0) C39.91%, H 5.62%, N 7.51%, M 9.53%; Lm 280.2 S cm2 mol1; meff:diam.; IR (KBr): n 3366mbr (OeH, H2O, COOH), 1619s (asymmetricCOO), 1570m (C]O, pyridone group), 1523m (C]N),1357m (symmetric COO), 654w, 506w, 517vw, 465vw (M-O) and(M-N). UVe Vis. (DMSO-d6): l (253 nm) (39.525 cm1) pi-pi*transitions, l (296 nm) (33.783 cm1) n-pi* transitions, l(538 nm) (18.587 cm1) (epsilon 118 M1 cm1) Ligand-metal chargetransfer. 1H NMR (DMSO-d6): delta 0.89 (1,2) (m, 4H, -CH2 cyclopropane),1.08 (3) (m, 1H, -CH cyclopropane), 1.15-1.86 (10,11) (m,2H, -CH2), 2.11-2.13 (12) (m, 1H, -CH), 2.51-2.66 (13) (d, 2H,-CH2), 3.21-3.30 (9) (t, 2H, -CH2), 3.30 (6) (d, 2H, -CH2), 3.61 (7)(m, 1H, -CH), 3.86-4.51 (s, 2H, H2O), 3.74 (14) (s, 3H, -CH3methoxy), 7.11 (8) (s, 2H, -NH2), 7.44-7.98 (4,5) (s, 2H, HAr),8.38-8.81 (1//-8//) (m, 8H, Hpy)
With triethylamine; In acetonitrile; at 100℃; for 24h;
10 ml of acetonitrile was added to the flask, 2 mmol of moxifloxacin hydrochloride was added, 2 mmol of N-phenyl 2-iodoacetamide and 4.4 mmol of triethylamine were added, and the mixture was refluxed at 100 C. for 24 h.The crude product was obtained by rotary evaporation under reduced pressure and recrystallized from acetonitrile to obtain a pale yellow solid with a yield of 90%.
90%
With triethylamine; In acetonitrile; at 100℃; for 24h;
10 ml of acetonitrile was added to the flask, 2 mmol of moxifloxacin hydrochloride was added, 2 mmol of N-phenyl 2-iodoacetamide and 4.4 mmol of triethylamine were added, and the mixture was refluxed at 100 C. for 24 h.The crude product was obtained by rotary evaporation under reduced pressure and recrystallized from acetonitrile to obtain a pale yellow solid with a yield of 90%.
(1) The 100L reactor was sequentially charged with 24.49 kg of Im-1 and 60 L of acetonitrile.Further, 8.77 kg of SM2 and 11.71 kg of triethylamine were added, and the mixture was heated to reflux at 80 to 90 C with stirring.(2) Reflow reaction for 5 h, TLC monitoring (developing solvent: ethyl acetate).(3) After the reaction is completed, the temperature is lowered to room temperature, and the reaction liquid is transferred to a 500 L reaction vessel.60 L of methanol was added with stirring.(4) Add hydrochloric acid dropwise under stirring to adjust pH ?0.5, about 18.44L concentrated hydrochloric acid, dark green solid precipitated.(5) After the completion of the dropwise addition, stirring at room temperature for 2 h; cooling to 0 to 5 C, stirring for 1 h;(6) Centrifugation, the filter cake is washed with 24 L of methanol cooled to 0-5 C.Drying at 50-55 C for 20 to 24 hours, to obtain crude product Im-2 19.35kg,Yellow powder solid, yield 76.3%.
Adding 160 grams of purified water to a 500 ml four-necked flask, stirring and heating to raise the purified water to 25 C, adding 21 grams of <strong>[186826-86-8]moxifloxacin hydrochloride</strong> (0.046 mol), raising the temperature to 45 C; adding under stirring Sodium hydroxide solution (sodium hydroxide 3.68 g + purified water 20 g), after dissolution and clarification, 0.5 g of activated carbon was added for decolorization for 20 minutes.
Adding 160 grams of purified water to a 500 ml four-necked flask, stirring and heating to raise the purified water to 20 C, adding 21 grams of <strong>[186826-86-8]moxifloxacin hydrochloride</strong> (0.046 mol), raising the temperature to 70 C; adding under stirring Sodium hydroxide solution (sodium hydroxide 3.68 g + purified water 20 g), after dissolution and clarification, 0.5 g of activated carbon was added for decolorization for 20 minutes.
Adding 160 grams of purified water to a 500 ml four-necked flask, stirring and heating to raise the purified water to 30 C, adding 21 grams of <strong>[186826-86-8]moxifloxacin hydrochloride</strong> (0.046 mol), raising the temperature to 80 C; adding under stirring Sodium hydroxide solution (sodium hydroxide 3.68 g + purified water 20 g), after dissolution and clarification, 0.5 g of activated carbon was added for decolorization for 20 minutes.
In a 500ml four-necked flask, 160g of purified water was added, the stirring was started and heated to raise the purified water to 27 C, 21 g (0.046 mol) of <strong>[186826-86-8]moxifloxacin hydrochloride</strong> was added, and the temperature was raised to 40 C; Sodium hydroxide solution (sodium hydroxide 3.68 g + purified water 20 g), after dissolution and clarification, 0.5 g of activated carbon was added for decolorization for 20 minutes.
Adding 160 grams of purified water to a 500 ml four-necked flask, stirring and heating to raise the purified water to 23 C, adding 21 grams of <strong>[186826-86-8]moxifloxacin hydrochloride</strong> (0.046 mol), raising the temperature to 60 C; adding under stirring Sodium hydroxide solution (sodium hydroxide 3.68 g + purified water 20 g), after dissolution and clarification, 0.5 g of activated carbon was added for decolorization for 20 minutes.
7-((4aS,7aS)-1-benzyloctahydro-6H-pyrrolo[3,4-b]pyridin-6-yl)-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
<strong>[186826-86-8]Moxifloxacin hydrochloride</strong> (6.1; 80 mg, 0.199 mmol, 1 eq) was added to a 1:1 mix ofacetonitrile and distilled water ( mL total). After stirring for minutes, potassiumcarbonate (82.63 mg, o.8 mmol, 3 eq) was added and the mixture stirred for a furtherminutes. Once fully dissolved, bromomethylbenzene (30.68 mg, 0.18 mmol, 0.90 eq)was added slowly over the course of 1 hour and the mixture subsequently stirred for 18hours. Upon completion, the product was extracted with dichloromethane (2 x 20 mL)using a 1M solution of citric acid to neutralise the aqueous phase. Combined organicfractions were washed with distilled water (20 mL) and dried over MgSO4, filtered and concentrated in vacuo to give the pure product 6.4.
1-cyclopropyl-6-fluoro-7-((4aS,7aS)-1-(4-(hydroxymethyl)benzyl)octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl)-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Moxifloxacin hydrochloride (6.1; 80 mg, 0.199 mmol, 1 eq) was added to a 1:1 mix of acetonitrile and distilled water ( mL total). After stirring for minutes, potassium carbonate (82.63 mg, o.8 mmol, 3 eq) was added and the mixture stirred for a further minutes. Once fully dissolved, (<strong>[71831-21-5](4-(bromomethyl)phenyl)methanol</strong> (36.06 mg, 0.18 mmol, 0.90 eq) was added slowly over the course of 1 hour and the mixturesubsequently stirred for 18 hours. Upon completion, the product was extracted with dichloromethane (2 x 20 mL) using a 1M solution of citric acid to neutralise the aqueous phase. Combined organic fractions were washed with distilled water (20 mL) and dried over MgSO4, filtered and concentrated in vacuo to give the pure product 6.6.
1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-7-((4aS,7aS)-1-(4-(pyrrolidin-1-yl)benzyl)octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl)-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
<strong>[186826-86-8]Moxifloxacin hydrochloride</strong> (6.1; 80 mg, 0.199 mmol, 1 eq) was added to a 1:1 mix ofacetonitrile and distilled water ( mL total). After stirring for s minutes, potassiumcarbonate (82.63 mg, o.8 mmol, 3 eq) was added and the mixture stirred for a further minutes. Once fully dissolved, 1-(4-(bromomethyl)phenyl)pyrrolidine (3 mg, 0.18 mmol, 0.90 eq) was added slowly over the course of 1 hour and the mixture subsequently stirred for 18 hours. Upon completion, the product was extracted with dichloromethane (2 x 20 mL) using a 1M solution of citric acid to neutralise the aqueous phase. Combined organic fractions were washed with distilled water (20 mL)and dried over MgSO4, filtered and concentrated in vacuo to give the pure product 6.8.
1-cyclopropyl-6-fluoro-8-methoxy-7-((4aS,7aS)-1-(naphthalen-1-ylmethyl)octahydro-6H-pyrrolo[3,4-b]pyridine-6-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
84.6%
<strong>[186826-86-8]Moxifloxacin hydrochloride</strong> (6.1; 100 mg, 0.23 mmol, 1 eq) was added to a 1:1 mix of acetonitrile and distilled water (10 mL total). After stirring for minutes, potassiumcarbonate (?s mg, 0.69 mmol, 3 eq) was added and the mixture stirred for a further s minutes. Once fully dissolved, i-(bromomethyl)naphthalene (48 mg, 0.22 mmol, 0.95 eq) was added slowly over the course of 1 hour and the mixture subsequently stirred for 24 hours. Upon completion, the product was extracted with dichloromethane (2 x 20 mL) using a 1M solution of citric acid to neutralise the aqueous phase. Combinedorganic fractions were washed with distilled water (20 mL) and dried over MgSO4, filtered and concentrated in vacuo to give the crude product. Purification was achieved using an SCX-2 catch and release cartridge (see Solid Phase Extraction method) to afford 6 .2.
Under nitrogen protection, add 35g (0.875mol) of sodium hydroxide to 165mL of water and 150mL of ethylene glycol.And 7-((S,S)-2,8-diazabicyclo[4,3,0]decane-8-yl)-1-cyclopropyl-6-fluoro-1,4-dihydro- 8-methoxy-4-oxo-3-quinolinecarboxylic acid dimethylamide 12.0 g (0.0263 mol),Stir for 15 hours while heating to 115 C.Cool to 5 C and adjust the pH to 5-6 with concentrated hydrochloric acid. Stir at 0.5 C for 0.5 hours.Filtration, water washing, vacuum drying (vacuum degree -0.01MPa~-0.1MPa, temperature 45C55 C) for 16 hours, 8.0 g of moxifloxacin was obtained, and the yield was 75.8%. The HPLC purity was 93.25%.Under nitrogen protection, 8.0 g of moxifloxacin was added to 5 mL of water and 20 mL of methanol, and the mixture was stirred at 70 C for 1 hour, filtered, cooled to 35 C, and adjusted to pH 1.4 to 1.8 by adding 6 N hydrochloric acid.Cool to 15 C and stir for 1 hour. It was filtered, washed with water, and stirred at 15 C for 1 hour in 10 mL of water and 0.2 mL of concentrated hydrochloric acid.Filtration, washing with water, vacuum drying (vacuum degree - 0.01 MPa ~ -0.1 MPa, temperature 45 ~ 55 C) for 16 hours, 6.4 g of moxifloxacin hydrochloride was obtained, the yield was 73.3%.(Total yield 29.4% based on 1-cyclopropyl-6,7-difluoro-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid). The HPLC purity was 99.16% and the maximum single impurity was 0.54%.
With hydrogenchloride; In ethanol; at 30 - 40℃;Inert atmosphere; Industrial scale;
Under nitrogen protection, stirring to ethanol30L and 2.0mol/L hydrogen chloride ethanol solution 30L (60mol)Moxifloxacin hydrochloride intermediate III 8.00Kg (17.6 mol) was added to the mixture.After the addition, the mixture is stirred at 30 C to 40 C for 4 hours to 6 hours.Most of the solvent is removed by vacuum concentration (temperature 35 C ~ 55 C, pressure -0.08 MPa ~ -0.1 MPa), and cooled to 10 C ~ 15 C,40 L of dichloromethane was added, and the mixture was stirred at 0 to 5 C for 1 hour to 2 hours. Filtration, washing with 10 L of dichloromethane, wet product under vacuum (pressure -0.01 MPa ~ -0.1 MPa)The drying oven was dried at 45 C to 55 C for 8 hours to 12 hours to obtain a crude solid of 7.71 K g of moxifloxacin hydrochloride I in a yield of 100%, and the HPLC purity was 99.32%.Under a nitrogen atmosphere, a whole batch of crude moxifloxacin hydrochloride I was charged into 38 L of methanol, heated to 40 C to 45 C and stirred for 1 hour to 2 hours. 15 L of dichloromethane was added, and the mixture was cooled to 0 to 5 C and stirred for 3 to 4 hours.Filtration, washing with 10 L of dichloromethane, and drying the wet product in a vacuum (pressure -0.01 MPa to -0.1 MPa) drying oven at 45 C to 55 C for 8 hours to 12 hours.5.78 Kg of pale yellow solid moxifloxacin hydrochloride I was obtained in a yield of 75.0% (total yield 56.7%, 1-cyclopropyl-6,7-difluoro-1,4-dihydro-8-methoxy- 4-oxo-3-quinolinecarboxylic acid count).The HPLC purity was 99.87% and the maximum single impurity was 0.04%.
7-[3-(3-aminopropyl)-1H-pyrrol-1-yl]-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
pH 7;Irradiation;
Taking <strong>[186826-86-8]moxifloxacin hydrochloride</strong> injection, Adjust the pH of the solution to 7, placed under cool white fluorescent light for 30 days, The light intensity is 4500 ± 500 lux, take out after the end of the irradiation. The light-damaged sample is then extracted with dichloromethane. Freeze-dried into a powder, Recrystallization from purified water as a solvent, The recrystallization method is 300 mg of lyophilized powder plus 4 ml of purified water. Mix and heat in a 50 C water bath. And stir at the right time. After 1 hour, take it out and cool it at 4 C. The supernatant was taken after centrifugation. The supernatant was then purified on a reverse phase silica gel column. The stationary phase is octadecylsilane bonded silica gel, The mobile phase was methanol-0.1% formic acid in water (45:55). Isocratic elution and collection of the eluent. Concentrated by rotary evaporation under reduced pressure, And placed in a vacuum oven at 40 C overnight to completely remove the moisture, A pale yellow powder photodegradation product was obtained. The normalized purity is greater than 97%.
7-((4aS,7aS)-1-benzyloctahydro-6H-pyrrolo[3,4-b]pyridin-6-yl)-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sodium hydroxide; In tetrahydrofuran; water; at 50℃; for 24h;
General procedure: Moxifloxacin.HCl (1.50 mmol) was stirred in tetrahydrofuran(30 mL) in the presence of aq. 5 N NaOH (0.6 mL). Appropriate benzylchloride (2.25 mmol) was added to the medium gradually afterwards.Reaction was maintained at 50 C for 24 h. When moxifloxacin spotdisappeared from TLC sheet, reaction medium was treated with ice-coldwater. Solution was neutralized with 10% acetic acid. The precipitatewas then filtered and washed with water. Crude product was recrystallizedfrom EtOH or acetone.4.1.1.1. 6-Fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-benzyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylic acid1. (CAS Number: 2254156-04-0) M.p. 150 C. Elemental analysiscalculated for C28H30FN3O4; C 68.42, H 6.15, N 8.55; found C 68.12,H 5.67, N 8.51. IR (cm-1): 1732, 1622 (C]O). 1H NMR delta ppm(300 MHz, CDCl3): 15.11 (s, 1H, COOH), 8.80 (s, 1H, C2-H), 7.93 (m,1H, Ar-H'), 7.83 (d, 1H, C5-H, J=13.8 Hz), 7.62-7.29 (m, 3H, Ar-H'),7.32-3-7.28 (m, 1H), 3.59 (s, 3H, OCH3), 4.08-0.82 (aliphatic CeH).HRMS (ESI+) m/z calculated for C28H30FN3O4 [M+Na]+, 514.2118,found 514.2124
6-fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-(4-methylphenyl)methyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sodium hydroxide; In tetrahydrofuran; water; at 50℃; for 24h;
General procedure: Moxifloxacin.HCl (1.50 mmol) was stirred in tetrahydrofuran(30 mL) in the presence of aq. 5 N NaOH (0.6 mL). Appropriate benzylchloride (2.25 mmol) was added to the medium gradually afterwards.Reaction was maintained at 50 C for 24 h. When moxifloxacin spotdisappeared from TLC sheet, reaction medium was treated with ice-coldwater. Solution was neutralized with 10% acetic acid. The precipitatewas then filtered and washed with water. Crude product was recrystallizedfrom EtOH or acetone
6-fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-(4-fluorophenyl)methyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sodium hydroxide; In tetrahydrofuran; water; at 50℃; for 24h;
General procedure: Moxifloxacin.HCl (1.50 mmol) was stirred in tetrahydrofuran(30 mL) in the presence of aq. 5 N NaOH (0.6 mL). Appropriate benzylchloride (2.25 mmol) was added to the medium gradually afterwards.Reaction was maintained at 50 C for 24 h. When moxifloxacin spotdisappeared from TLC sheet, reaction medium was treated with ice-coldwater. Solution was neutralized with 10% acetic acid. The precipitatewas then filtered and washed with water. Crude product was recrystallizedfrom EtOH or acetone
6-fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-(4-methoxyphenyl)methyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sodium hydroxide; In tetrahydrofuran; water; at 50℃; for 24h;
General procedure: Moxifloxacin.HCl (1.50 mmol) was stirred in tetrahydrofuran(30 mL) in the presence of aq. 5 N NaOH (0.6 mL). Appropriate benzylchloride (2.25 mmol) was added to the medium gradually afterwards.Reaction was maintained at 50 C for 24 h. When moxifloxacin spotdisappeared from TLC sheet, reaction medium was treated with ice-coldwater. Solution was neutralized with 10% acetic acid. The precipitatewas then filtered and washed with water. Crude product was recrystallizedfrom EtOH or acetone
6-fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-(4-chlorophenyl)methyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sodium hydroxide; In tetrahydrofuran; water; at 50℃; for 24h;
General procedure: Moxifloxacin.HCl (1.50 mmol) was stirred in tetrahydrofuran(30 mL) in the presence of aq. 5 N NaOH (0.6 mL). Appropriate benzylchloride (2.25 mmol) was added to the medium gradually afterwards.Reaction was maintained at 50 C for 24 h. When moxifloxacin spotdisappeared from TLC sheet, reaction medium was treated with ice-coldwater. Solution was neutralized with 10% acetic acid. The precipitatewas then filtered and washed with water. Crude product was recrystallizedfrom EtOH or acetone
6-fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-(4-nitrophenyl)methyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sodium hydroxide; In tetrahydrofuran; water; at 50℃; for 24h;
General procedure: Moxifloxacin.HCl (1.50 mmol) was stirred in tetrahydrofuran(30 mL) in the presence of aq. 5 N NaOH (0.6 mL). Appropriate benzylchloride (2.25 mmol) was added to the medium gradually afterwards.Reaction was maintained at 50 C for 24 h. When moxifloxacin spotdisappeared from TLC sheet, reaction medium was treated with ice-coldwater. Solution was neutralized with 10% acetic acid. The precipitatewas then filtered and washed with water. Crude product was recrystallizedfrom EtOH or acetone
6-fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-(4-bromophenyl)methyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sodium hydroxide; In tetrahydrofuran; water; at 50℃; for 24h;
General procedure: Moxifloxacin.HCl (1.50 mmol) was stirred in tetrahydrofuran(30 mL) in the presence of aq. 5 N NaOH (0.6 mL). Appropriate benzylchloride (2.25 mmol) was added to the medium gradually afterwards.Reaction was maintained at 50 C for 24 h. When moxifloxacin spotdisappeared from TLC sheet, reaction medium was treated with ice-coldwater. Solution was neutralized with 10% acetic acid. The precipitatewas then filtered and washed with water. Crude product was recrystallizedfrom EtOH or acetone
6-fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-(phenyl)carbonyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: To a solution of moxifloxacin.HCl (1.50 mmol) in DMF (30 mL),K2CO3 (3.00 mmol) was added. The mixture was allowed to stir at 60 Cfor 2 h. Related benzoyl chloride (2.25 mmol) was then added to themedium dropwise. When the reaction was complete, the mixture waspoured into ice-water. The precipitate was filtered and the resultantraw material was purified by column chromatography using DCM:MeOH (90:10, v/v) as mobile phase.4.1.2.1. 6-Fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-(phenyl)carbonyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylicacid 8. M.p. 162 C. Elemental analysis calculated for C28H28FN3O5.3/2H2O; C 63.15, H 5.87, N 7.89; found C 63.63, H 5.55, N 7.85. IR(cm-1): 3536 (OeH); 1732, 1618 (C]O). 1H NMR delta ppm (300 MHz,DMSO-d6): 15.14 (s, 1H, COOH), 8.66 (s, 1H, C2-H), 7.66 (d, 1H, C5-H,J=13.8 Hz), 7.73-7.45 (m, 5H, Ar-H'), 3.58 (s, 3H, OCH3), 4.16-0.87(aliphatic CeH). HRMS (ESI+) m/z calculated for C28H28FN3O5[M+Na]+, 528.1911, found 528.1920.
6-fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-(4-methylphenyl)carbonyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: To a solution of moxifloxacin.HCl (1.50 mmol) in DMF (30 mL),K2CO3 (3.00 mmol) was added. The mixture was allowed to stir at 60 Cfor 2 h. Related benzoyl chloride (2.25 mmol) was then added to themedium dropwise. When the reaction was complete, the mixture waspoured into ice-water. The precipitate was filtered and the resultantraw material was purified by column chromatography using DCM:MeOH (90:10, v/v) as mobile phase.
6-fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-(4-fluorophenyl)carbonyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: To a solution of moxifloxacin.HCl (1.50 mmol) in DMF (30 mL),K2CO3 (3.00 mmol) was added. The mixture was allowed to stir at 60 Cfor 2 h. Related benzoyl chloride (2.25 mmol) was then added to themedium dropwise. When the reaction was complete, the mixture waspoured into ice-water. The precipitate was filtered and the resultantraw material was purified by column chromatography using DCM:MeOH (90:10, v/v) as mobile phase.
6-fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-(4-methoxyphenyl)carbonyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: To a solution of moxifloxacin.HCl (1.50 mmol) in DMF (30 mL),K2CO3 (3.00 mmol) was added. The mixture was allowed to stir at 60 Cfor 2 h. Related benzoyl chloride (2.25 mmol) was then added to themedium dropwise. When the reaction was complete, the mixture waspoured into ice-water. The precipitate was filtered and the resultantraw material was purified by column chromatography using DCM:MeOH (90:10, v/v) as mobile phase.
6-fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-(4-chlorophenyl)carbonyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: To a solution of moxifloxacin.HCl (1.50 mmol) in DMF (30 mL),K2CO3 (3.00 mmol) was added. The mixture was allowed to stir at 60 Cfor 2 h. Related benzoyl chloride (2.25 mmol) was then added to themedium dropwise. When the reaction was complete, the mixture waspoured into ice-water. The precipitate was filtered and the resultantraw material was purified by column chromatography using DCM:MeOH (90:10, v/v) as mobile phase.
6-fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-(4-nitrophenyl)carbonyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: To a solution of moxifloxacin.HCl (1.50 mmol) in DMF (30 mL),K2CO3 (3.00 mmol) was added. The mixture was allowed to stir at 60 Cfor 2 h. Related benzoyl chloride (2.25 mmol) was then added to themedium dropwise. When the reaction was complete, the mixture waspoured into ice-water. The precipitate was filtered and the resultantraw material was purified by column chromatography using DCM:MeOH (90:10, v/v) as mobile phase.
6-fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-(4-bromophenyl)carbonyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: To a solution of moxifloxacin.HCl (1.50 mmol) in DMF (30 mL),K2CO3 (3.00 mmol) was added. The mixture was allowed to stir at 60 Cfor 2 h. Related benzoyl chloride (2.25 mmol) was then added to themedium dropwise. When the reaction was complete, the mixture waspoured into ice-water. The precipitate was filtered and the resultantraw material was purified by column chromatography using DCM:MeOH (90:10, v/v) as mobile phase.
6-fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-phenylsulfonyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With triethylamine; In acetonitrile; at 20℃; for 4h;
General procedure: To moxifloxacin. HCl (1.50 mmol) and triethlyamine (2.25 mmol)solution in acetonitrile (50 mL), related arylsulfonyl chloride(2.25 mmol) was added. The reaction medium was maintained at roomtemperature for 4 h. At the end of reaction, ACN was evaporated andthe resultant precipitate was washed with water. Final crude productwas recrystallized from EtOH. 4.1.3.1. 6-Fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-phenylsulfonyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylic acid15. M.p. 165-168 C. Elemental analysis calculated forC27H28FN3O6S·H2O; C 57.95, H 5.40, N 7.51, S 5.73; found C 57.70,H 5.55, N 7.45, S 5.76. IR (cm-1): 1722, 1616 (C]O). 1H NMR delta ppm(300 MHz, CDCl3): 8.80 (s, 1H, C2-H), 7.87-7.33 (m, 6H, Ar-H', C5-H),3.52 (s, 3H, OCH3), 4.67-0.78 (aliphatic CeH). HRMS (ESI+) m/zcalculated for C27H28FN3O6S [M+Na]+, 564.1575, found 564.1581.
With triethylamine; In acetonitrile; at 20℃; for 4h;
General procedure: To moxifloxacin. HCl (1.50 mmol) and triethlyamine (2.25 mmol)solution in acetonitrile (50 mL), related arylsulfonyl chloride(2.25 mmol) was added. The reaction medium was maintained at roomtemperature for 4 h. At the end of reaction, ACN was evaporated andthe resultant precipitate was washed with water. Final crude productwas recrystallized from EtOH.
6-fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-(4-fluorophenyl)sulfonyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With triethylamine; In acetonitrile; at 20℃; for 4h;
General procedure: To moxifloxacin. HCl (1.50 mmol) and triethlyamine (2.25 mmol)solution in acetonitrile (50 mL), related arylsulfonyl chloride(2.25 mmol) was added. The reaction medium was maintained at roomtemperature for 4 h. At the end of reaction, ACN was evaporated andthe resultant precipitate was washed with water. Final crude productwas recrystallized from EtOH.
6-fluoro-8-methoxy-4-oxo-1-cyclopropyl-7-(2-(4-methoxyphenyl)sulfonyl-2,8-diazabicyclo[4.3.0]nonan-8-yl)-1,4-dihydroquinoline-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With triethylamine; In acetonitrile; at 20℃; for 4h;
General procedure: To moxifloxacin. HCl (1.50 mmol) and triethlyamine (2.25 mmol)solution in acetonitrile (50 mL), related arylsulfonyl chloride(2.25 mmol) was added. The reaction medium was maintained at roomtemperature for 4 h. At the end of reaction, ACN was evaporated andthe resultant precipitate was washed with water. Final crude productwas recrystallized from EtOH.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






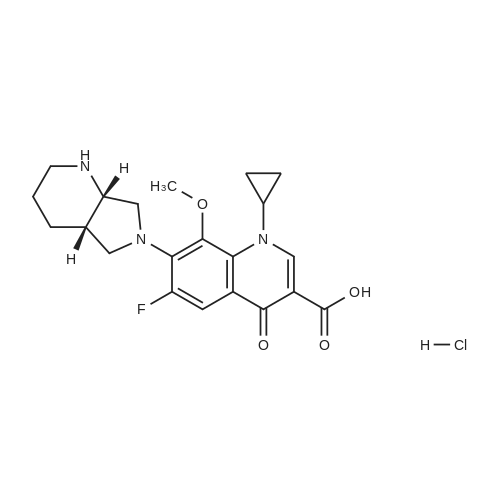

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping