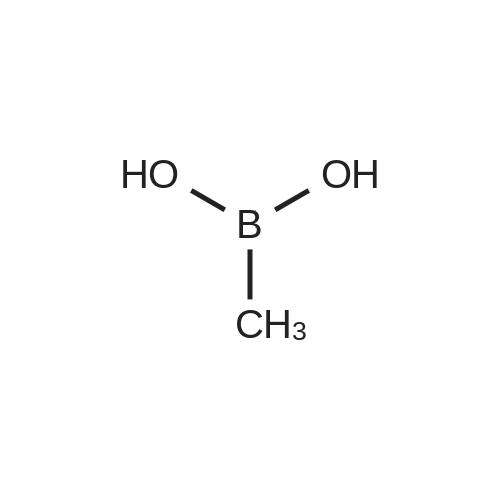
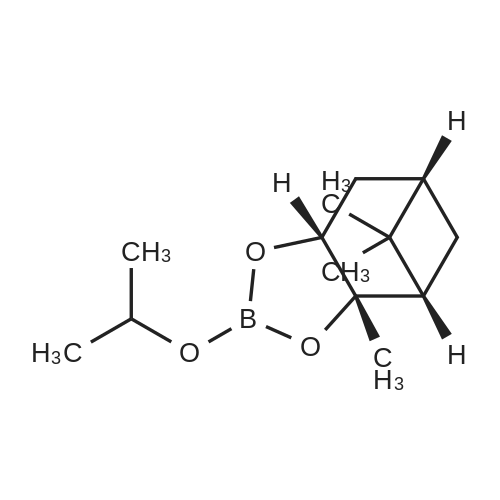
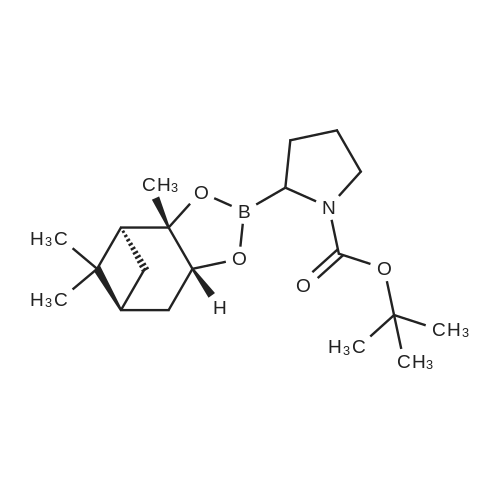
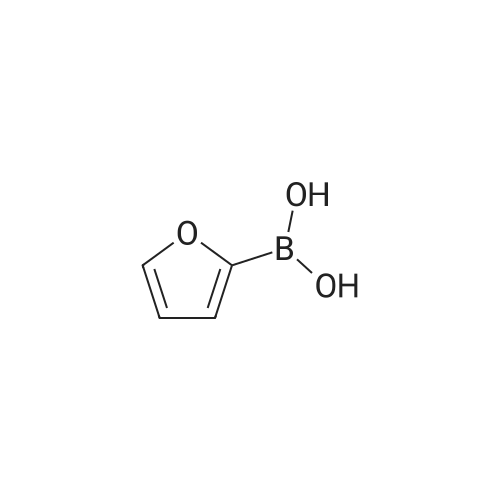
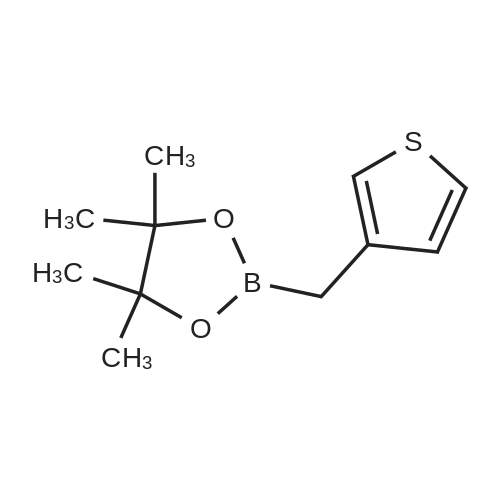
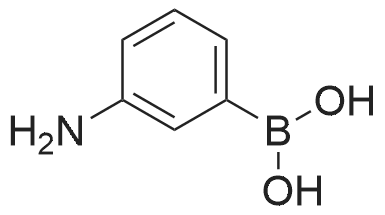
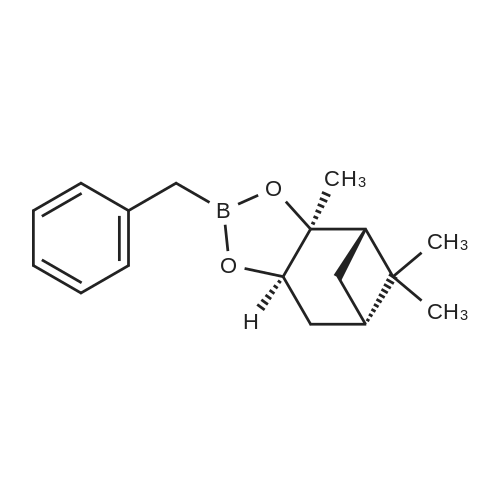
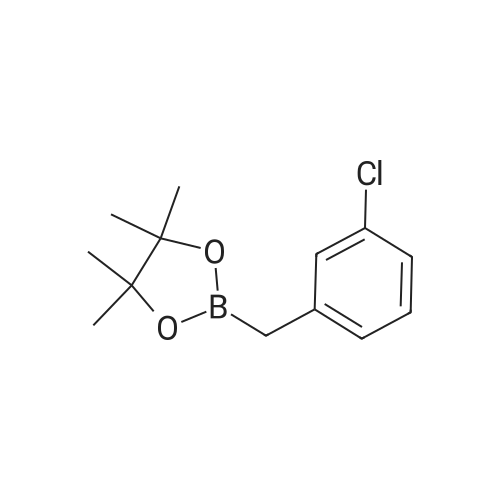
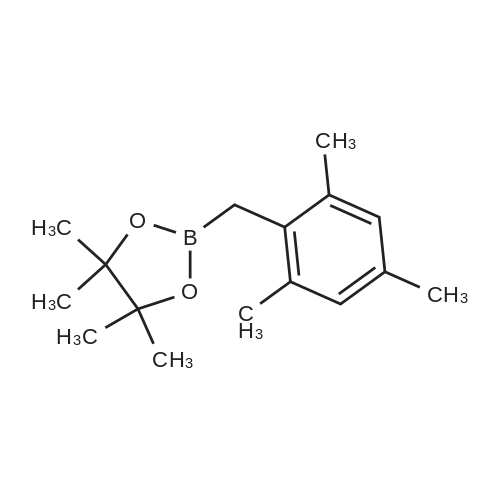
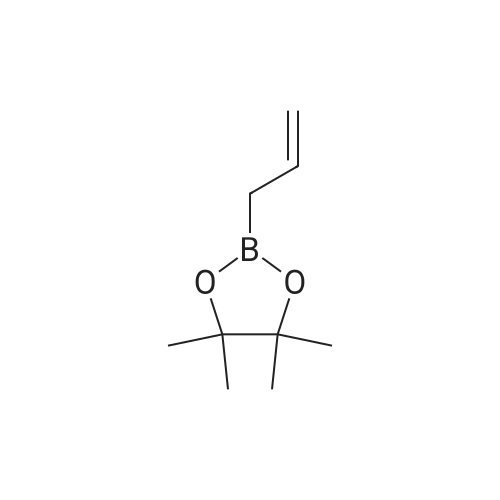
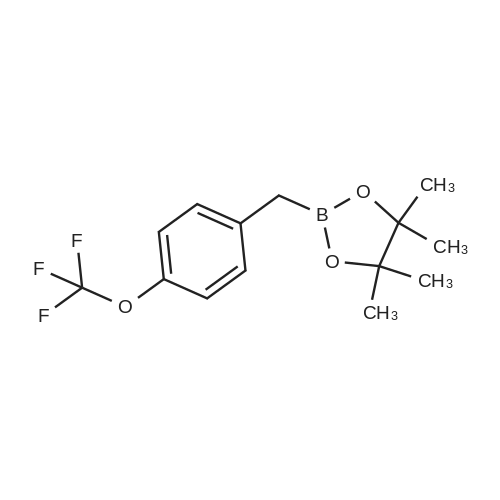
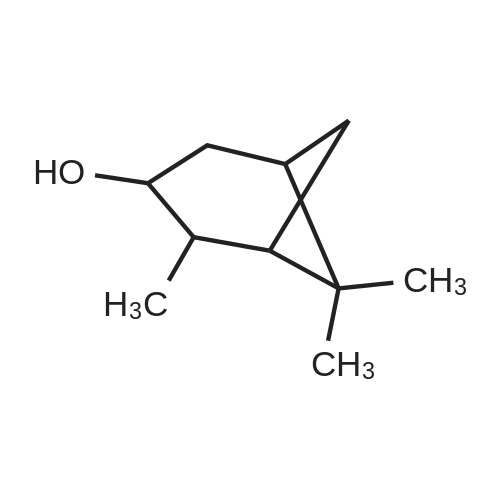
| 82% |
In diethyl ether at 20℃; for 12h; |
1a.4 Step 4: 2-(benzofuran-3-ylmethyl)boronic acid (+)-pinanediol ester
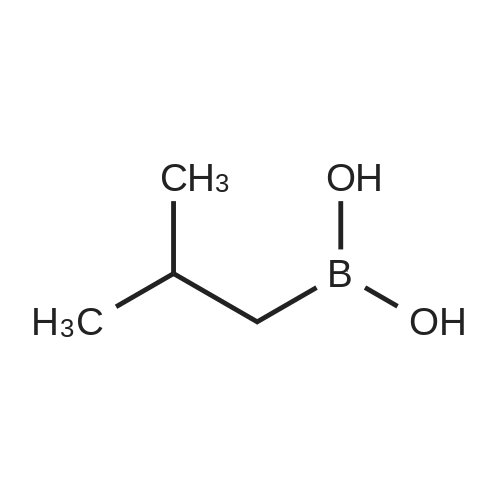
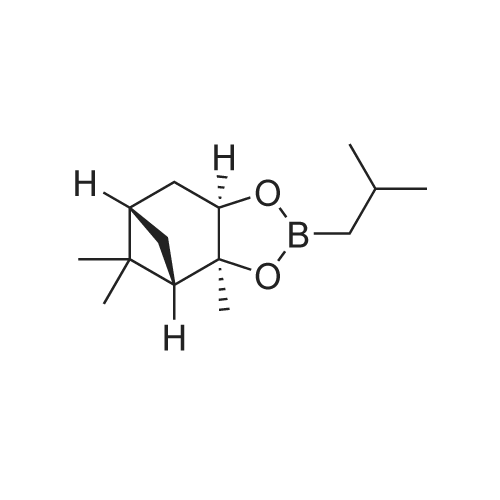
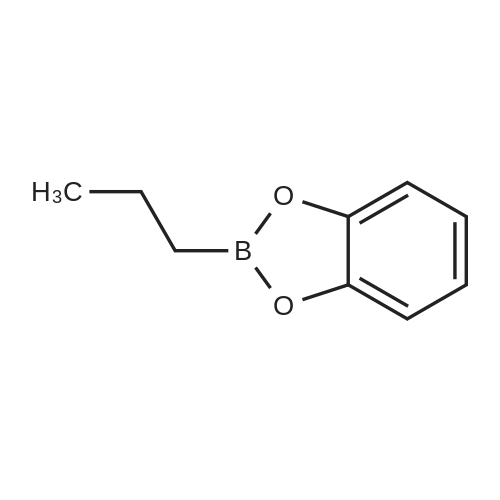
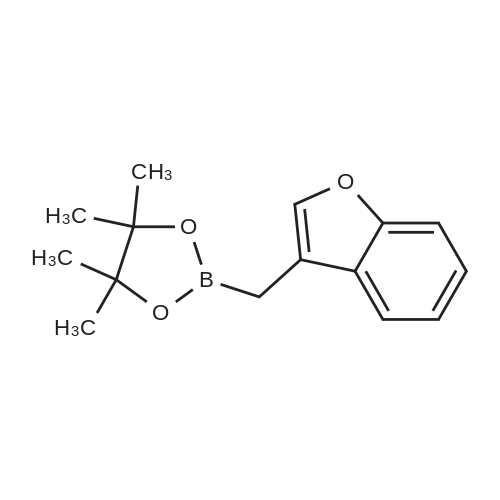
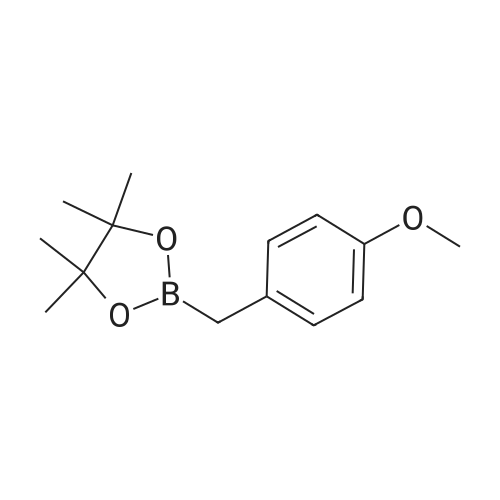
Step 4: 2-(benzofuran-3-ylmethyl)boronic acid (+)-pinanediol ester A solution of 2-(benzofuran-3-ylmethyl)-4,4l5,5-tetramethyl-1,3>2-dioxaborolane (6.1 g, 23.6 mmol) in diethyl ether (60 ml) was treated with (1S, 2S, 3R, 5S)-(+)-pinanediol (6.0 g, 35.4 mmol). The reaction mixture was stirred at room temperature for 12 h then the mixture was washed with water twice, then with brine and dried over anhydrous sodium sulphate, then concentrated. The crude product was purified by flash column chromatography on silica gel, eluting with 5% of ethyl acetate in petroleum ether, to afford the title compound (6.3 g, 82%). 1H NMR (400 MHz, CDC ): δ 7.58-7.56 (m, 1 H), 7.55-7.53 (m, 1 H), 7.46-7.44 (m, 1H), 7.28-7.23 (m, 2H), 4.33 (dd, J = 1.88, 8.76 Hz, 1 H), 2.34-2.32 (m, 1 H), 2.28 (s, 2H), 2.22- 2.21 (m, 1H), 2.08 (t, J = 5.88 Hz, 1 H), 1.42 (s, 3H), 1.29 (s, 3H), 1.13 (d, J = 10.92 Hz, 1H), 0.85 (s, 3H). GCMS: m/z: 310. |
| 82% |
In diethyl ether at 20℃; for 12h; |
2.4 Step 4: 2-(benzofuran-3-ylmethyl)boronic acid (+)-pinanediol ester
A solution of 2-(benzofuran-3-ylmethyl)-4,4,5,5-tetramethyl-1 ,3,2-dioxaborolane (6.1 g, 23.6 mmol) in diethyl ether (60 ml) was treated with (1S, 2S, 3R, 5S)-(+)-pinanediol (6.0 g, 35.4 mmol). The reaction mixture was stirred at room temperature for 12 h then the mixture was washed with water twice, then with brine and dried over anhydrous sodium sulphate, then concentrated. The crude product was purified by flash column chromatography on silica gel, eluting with 5% of ethyl acetate in petroleum ether, to afford the title compound (6.3 g, 82%). 1H NMR (400 MHz, CDCI3): δ 7.58-7.56 (m, 1 H), 7.55-7.53 (m, 1 H), 7.46-7.44 (m, 1 H), 7.28-7.23 (m, 2H), 4.33 (dd, J = 1.88, 8.76 Hz, 1 H), 2.34-2.32 (m, 1H), 2.28 (s, 2H), 2.22- 2.21 (m, 1H), 2.08 (t, J = 5.88 Hz, 1 H), 1.42 (s, 3H), 1.29 (s, 3H), 1.13 (d, J = 10.92 Hz, 1 H), 0.85 (s, 3H). GCMS: m/z: 310. |
| 82% |
In diethyl ether at 20℃; for 12h; |
4 Step 4: 2-(benzofuran-3-ylmethyl)boronic acid (+)-pinanediol ester
A solution of 2-(benzofuran-3-ylmethyl)-4,4,5,5-tetramethyl-1 ,3,2- dioxaborolane (6.1 g, 23.6 mmol) in diethyl ether (60 ml) is treated with (1 S, 2S, 3R, 5S)-(+)-pinanediol (6.0 g, 35.4 mmol). The reaction mixture is stirred at room temperature for 12 h then the mixture is washed with water twice, then with brine and dried over anhydrous sodium sulphate, then concentrated. The crude product is purified by flash column chromatography on silica gel, eluting with 5% of ethyl acetate in petroleum ether, to afford the title compound (6.3 g, 82%). 1H NMR (400 MHz, CDCI3): δ 7.58-7.56 (m, 1 H), 7.55-7.53 (m, 1 H), 7.46- 7.44 (m, 1 H), 7.28-7.23 (m, 2H), 4.33 (dd, J = 1 .88, 8.76 Hz, 1 H), 2.34-2.32 (m, 1 H), 2.28 (s, 2H), 2.22-2.21 (m, 1 H), 2.08 (t, J = 5.88 Hz, 1 H), 1 .42 (s, 3H), 1 .29 (s, 3H), 1 .13 (d, J = 10.92 Hz, 1 H), 0.85 (s, 3H). GCMS: m/z: 310. |
| 82% |
In diethyl ether at 20℃; for 12h; |
1.4 Step 4: 2-(benzofuran-3-ylmethyl)boronic acid (+)-pinanediol ester.
A solution of 2-(benzofuran-3-ylmethyl)-4,4,5,5-tetramethyl-1,3,2- dioxaborolane (6.1 g, 23.6 mmol) in diethyl ether (60 ml_) is treated with (1S,2S,3R,5S)-(+)-pinanediol (6.0 g, 35.4 mmol). The reaction mixture is stirred at room temperature for 12 h then the mixture is washed with water (twice), then with brine and resulting solution is dried over anhydrous sodium sulphate and concentrated. The crude product is purified by flash column chromatography on silica gel, eluting with 5% of ethyl acetate in petroleum ether to obtain 2-(benzofuran-3-ylmethyl)boronic acid (+)-pinanediol ester (6.3 g, 82%). 1H NMR (400 MHz, CDCIs): d 7.58-7.56 (m, 1H), 7.55-7.53 (m, 1H), 7.46-7.44 (m, 1H), 7.28-7.23 (m, 2H), 4.33 (dd, J = 1.88, 8.76 Hz, 1H), 2.34-2.32 (m, 1H), 2.28 (s, 2H), 2.22-2.21 (m, 1H), 2.08 (t, J = 5.88 Hz, 1H), 1.42 (s, 3H), 1.29 (s, 3H), 1.13 (d, J = 10.92 Hz, 1H), 0.85 (s, 3H). GCMS: m/z: 310. |
| 82% |
In diethyl ether at 20℃; for 12h; Inert atmosphere; |
|
| 80% |
In diethyl ether at 20℃; |
31; 41; 47
A solution of 2-(benzofuran-3-ylmethyl)-4,4,5,5-tetramethyl-l,3,2-dioxaborolane (4.58 g, 17.7 mmol) in diethyl ether (30 mL) was treated with (lS,2S,3R,5S)-2,6,6- trimethylbicyclo[3.l.l]heptane-2,3-diol (3.9 g, 23.1 mmol), the mixture was stirred at rt for 12 h. Then the mixture was concentrated and the crude was purified by column chromatography on silica gel, eluting with 5% of ethylacetate in petroleum ether to afford (3aS,4S,6S,7aR)-2-(benzofuran-3-ylmethyl)-3a,5,5- trimethylhexahydro-4,6-methanobenzo[d][l,3,2]dioxaborole (4.5 g, 80%) as a yellow oil. |
| 34% |
In diethyl ether at 20℃; |
16.4
Into a 50-mL round-bottom flask, was placed a solution of 2-(I -benzofuran-3- ylmethyl)-4,4,5,5-tetramethyl-l,3,2-dioxaborolane (490 mg, 1.90 mmol, 1.00 eq.) in ether (5 mL), and ( lS,2S,3R,5S)-2,6,6-tiimethylbicyclo[3.1.1]heptane-2,3~diol (420 mg, 2.47 mmol, 1.30 eq.). The resulting solution was stirred overnight at rt. The resulting mixture was concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (3:97). This resulted in 200 mg (34%) of (lS,2S,6R,8S)-4-( l-benzofuran-3-ylmethyl)-2,9,9-trimethyl-3,5-dioxa-4- boratricyclo[6.1.1.0A[2,6]]decane as a yellow oil. |
|
In diethyl ether at 20℃; Inert atmosphere; |
|
| 10.5 g |
In diethyl ether at 20℃; |
1.D Step D: Benzofuran-3-ylmethylboronic acid (1S,2S,3R,5S)-(+)-pinane-2,3-diol ester
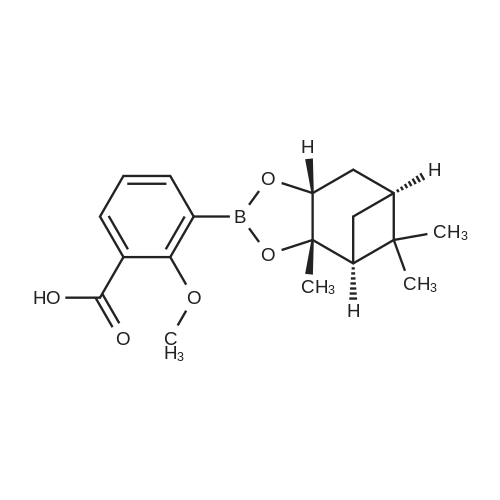
Pinacol benzofuran-3-ylmethylboronic acid (14.0g)And (1S, 2S, 3R, 5S)-(+)-Pinane-2,3-diol (18.4g) were added to anhydrous ether (200mL),The suspension was stirred overnight at room temperature,After the detection reaction is completed, wash with water three times, dry the organic phase with anhydrous sodium sulfate, evaporate the solvent after filtration, and purify the residue by silica gel column chromatography (50% dichloromethane/petroleum ether) to obtain the product (10.5 g). |
| 10.5 g |
In diethyl ether at 20℃; |
1.D Step D: Benzofuran-3-ylmethylboronic acid-(1S,2S,3R,5S)-(+)-pinene-2,3-diol ester
Pinacol benzofuran-3-ylmethylboronic acid (14.0g)And (1S, 2S, 3R, 5S)-(+)-Pinane-2,3-diol (18.4g) were added to anhydrous ether (200mL),The suspension was stirred overnight at room temperature. After the LC/MS detection of the reaction was completed,Wash three times with water, dry the organic phase with anhydrous sodium sulfate,After filtration, the solvent was evaporated to dryness, and the residue was purified by silica gel column chromatography (50% dichloromethane/petroleum ether) to obtain the product (10.5 g). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping