There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
Structure of 1820-80-0 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
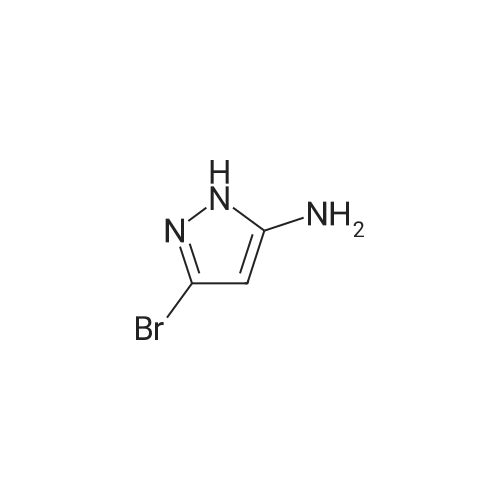
With caesium carbonate; copper(ll) bromide In N,N-dimethyl-formamide at 190℃; for 0.333333 h; Microwave irradiation; Sealed tube
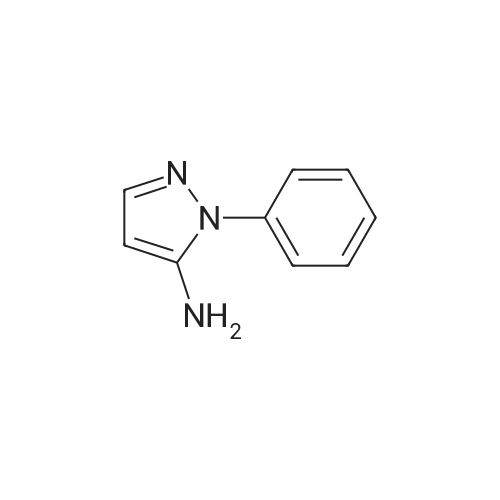
1 -phenyl- 1 H -pyrazol-3-amine 3-Amino-pyrazole (1 .0 g, 12.03 mmol), Cs2CO3 (3.92 g, 12.03 mmol), iodobenzene (3.68 g, 18.05 mmol), CuBr2 (0.268 g, 0.1 mmol), and DMF (4 ml_) were added to a 10-mL microwave vial. The vial was sealed and heated to 190 °C for 20 min (monitored by TLC). After cooling, the reaction mixture was diluted with saturated aqueous ammonium chloride and extracted with ethyl acetate (50 imL x 3). The organic layers were dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash column chromatography, eluting with 0-40percent ethyl acetate and petroleum benzine, afforded the desired product, 1 -phenyl-1 -/-pyrazol-3-amine, as a brown solid (1 .53 g, 80percent).
Reference:
[1] Bulletin of the Korean Chemical Society, 2012, vol. 33, # 6, p. 2067 - 2070
[2] Patent: WO2015/172196, 2015, A1, . Location in patent: Paragraph 00225
With sodium ethanolate In ethanol at 80℃; for 16 h; Inert atmosphere
Step A - Synthesis of pyrazolo[ l,5-a]pyrimidine-5,7-diol (lnt-9a)To lH-pyrazol-3-amine (12.3 g, 148.0 mmol) in EtOH (50 mL) was added diethyl malonate (25.0 mL, 164.7 mmol), 21 wtpercent NaOEt in EtOH (1 10 mL, 294.6 mmol) and additional EtOH (50 mL). The resulting reaction mixture was then heated at 800C under an atmosphere of argon for 16 hours, at which time the reaction was allowed to cool to room temperature. The reaction mixture was then concentrated in vacuo until almost dry, before H2O (500 mL) was added. Vigorous stirring aided the dissolution of solids, at which time cone. HCl was added until pH~2 was attained (precipitate formed). The precipitate was collected and dried by vacuum filtration giving pyrazolo[l,5-a]pyrimidine-5,7-diol (Int-9a) as a tan solid (17.13 g, 1 13.4 mmol, 77percent).
77%
With sodium ethanolate In ethanol at 80℃; for 16 h; Inert atmosphere
To lH-pyrazol-3-amine (12.3 g, 148.0 mmol) in EtOH (50 mL) was added diethyl malonate (25.0 mL5 164.7 mmol), 21wtpercent NaOEt in EtOH (1 10 mL, 294.6 mmol) and additional EtOH (50 mL). The resulting mixture was then heated at 80 C under an atmosphere of argon for 16 hours, at which time the reaction was allowed to cool to room temperature. The reaction mixture was then concentrated in vacuo until almost dry, before 3/40 (500 mL) was added. Vigorous stirring aided the dissolving of solids, at which time cone. HC1 was added until pH~2 was attained (solid precipitate formed). The precipitate was collected and dried by vacuum filtration giving pyrazolo[l,5-a]pyrimidine-5,7-diol as a tan solid (17.13 g, 113.4 mmol, 77percent).
Stage #1: With hydrogenchloride; zinc(II) chloride In ethanolReflux Stage #2: for 1 h;
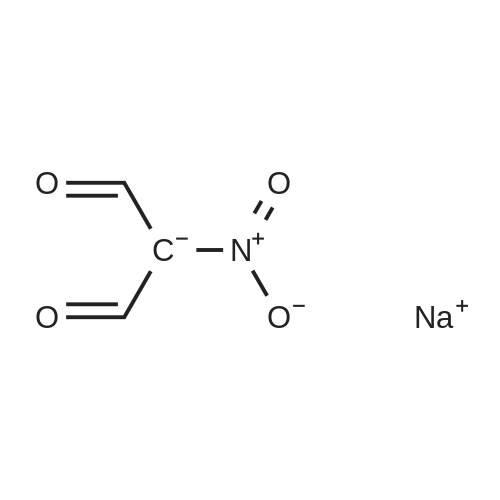
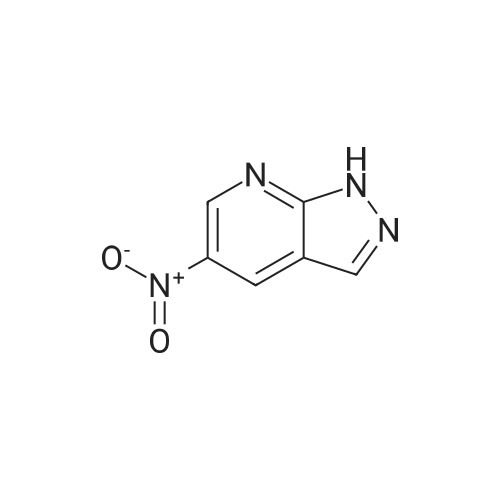
1H-pyrazol-3-amine (427 mg, 5.139 mmol) was dissolved in ethanol (10 mL) and 12M HCl (0.5 mL) was added to the stirred solution, followed by granulated zinc chloride (300 mg). The mixture was heated to reflux, and to the mixture was added a solution of 5.139 mmol of the source of the appropriate 1,3-dicarbonyl compound (sodium 2-nitro-1,3-dioxopropan-2-ide) in ethanol (5 mL). After 1 h the reaction mixture was poured into ice-cold water (15 mL), the resultant solution was made alkaline with concentrated aqueous ammonia, and the product was isolated by trichloromethane extraction. The product 5-nitro-1H-pyrazolo[3,4-b]pyridine (279 mg, 33percent) ( A) was isolated and purified by column chromatography.
27%
at 90℃; for 16 h;
To a solution of sodium nitromalonaldehyde monohydrate(1.23 g, 7.81 mmol) in water was added 1H-pyrazol-3-amine(0.84 g, 7.43 mmol). The mixture was stirred at 90 °C for 16 h andthen cooled to room temperature. The reaction solution wasadjusted to pH 5 and extracted by ethyl acetate (200 mL). Theorganic layer was dried over Na2SO4 and evaporated. The residuewas purified by silica gel chromatography (1percent MeOH in dichloromethane)to give 5-nitro-1H-pyrazolo [4,3-b]pyridine as a whitesolid (118 mg, 27percent). Rf: 0.23 (DCM/MeOH, 100/1, v/v). Mp:206-208 °C. 1H NMR (DMSO-d6, 400 MHz) d 14.40 (s, 1H), 9.35 (d,J 2.5 Hz, 1H), 9.21 (d, J 2.5 Hz, 1H), 8.46 (s, 1H). MS (ESI) m/z:162.8 [MH]-.
Reference:
[1] Patent: US2013/29995, 2013, A1, . Location in patent: Paragraph 0372-0373
[2] European Journal of Medicinal Chemistry, 2017, vol. 131, p. 1 - 13
13
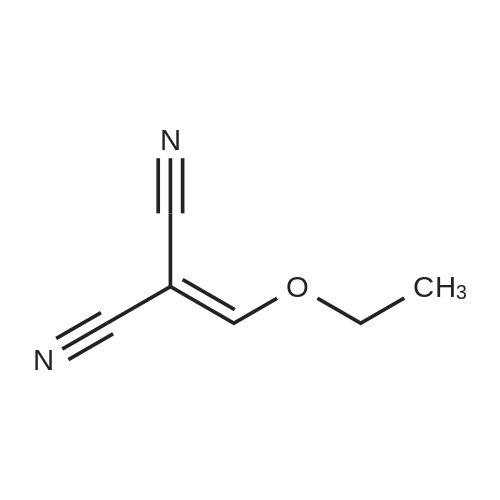
[ 1820-80-0 ]
[ 34461-00-2 ]
[ 63572-73-6 ]
Yield
Reaction Conditions
Operation in experiment
33%
With hydrogenchloride; zinc(II) chloride In ethanol for 1 h; Reflux
1H-pyrazol-3-amine (427 mg, 5.139 mmol) was dissolved in ethanol (10 mL) and 12M HCl (0.5 mL) was added to the stirred solution, followed by granulated zinc chloride (300 mg). The mixture was heated to reflux, and to the mixture was added a solution of 5.139 mmol of the source of the appropriate 1,3-dicarbonyl compound (sodium 2-nitro-1,3-dioxopropan-2-ide) in ethanol (5 mL). After 1 h the reaction mixture was poured into ice-cold water (15 mL), the resultant solution was made alkaline with concentrated aqueous ammonia, and the product was isolated by trichloromethane extraction. The product 5-nitro-1H-pyrazolo[3,4-b]pyridine (279 mg, 33 percent) (A) was isolated and purified by column chromatography.
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1980, p. 1347 - 1351
[2] Patent: US4081545, 1978, A,
16
[ 1820-80-0 ]
[ 14339-33-4 ]
Yield
Reaction Conditions
Operation in experiment
42%
With hydrogenchloride; copper dichloride In water; acetonitrile at 0℃; for 0.5 h;
3-Chloro-1H-pyrazole (1): Copper (II) chloride (65.0 g, 481 mmol) and cone. HCl (20 mL) were added to a solution of lH-pyrazol-3-amine (20.0 g, 241 mmol) in acetonitrile (600 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 30 min, then isopentyl nitrite (56.4 g, 481 mmol) was added dropwise. The mixture was stirred at room temperature for 2 days, and then quenched with aq. ammonia (10percent, 1 L). The aqueous phase was extracted with EtOAc (5 x 500 mL) and the combined organic phases were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated. The residue was purified by silica chromatography (hexane: EtOAc= 20: 1) to give the title compound (10.3 g, 42percent) as a green oil. 1H NMR (400 MHz, CDC13): δ 12.84 (bs, 1H), 7.62 (s, 1H), 6.29 (s, 1H).
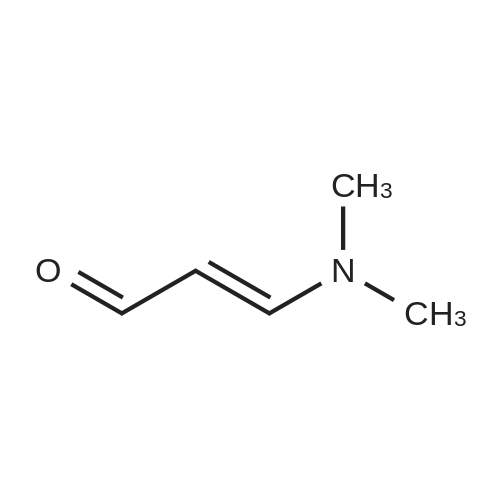
Preparation of compound 23a: pyrazolo[1,5-a]pyrimidineA mixture of 1 H-pyrazol-3-ylamine (32 g, 0.386 mol) and (E)-3-dimethylamino- propenal (38.2 g, 0.386 mol) in EtOH (500 ml_) was refluxed for 6 h. The solvent was removed in vacuo and the residue was purified via column chromatography (petroleum ether/EtOAc = 10:1 -2:1 ) which gave the title compound 23a as a white solid (30 g, 65percent).
Reference:
[1] Organic Process Research and Development, 2014, vol. 18, # 7, p. 886 - 890
19
[ 1820-80-0 ]
[ 623-47-2 ]
[ 29274-22-4 ]
Yield
Reaction Conditions
Operation in experiment
36%
at 110℃; Inert atmosphere
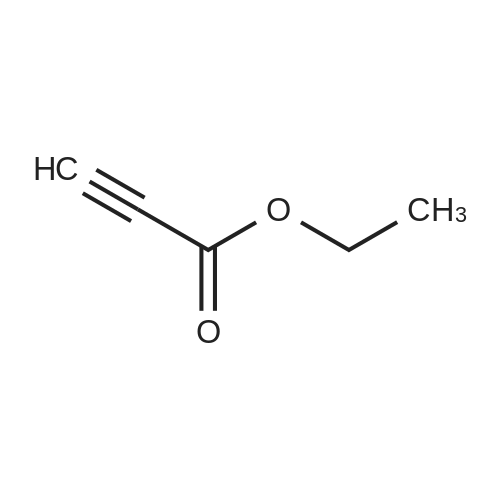
A solution of 1H-pyrazol-3-ylamine (7 g, 84.24 mmol, 1 .00 equiv) and ethyl prop-2-ynoate (50 mL) in dioxane ( 1 0 g, 1 21 equiv) was stirred under nitrogen overnight at 110 "C. The reaction mixture was cooled to rt and the precipitated product was collected by filtration to give 4 g (36percent) of the title compound as a light brown solid 1H NMR (300 MHz, DMSO-d6) δ 12.04 (s, 1H), 8.41 -8.44 (m, 1H), 7.71 (d. J = 18 Hz, 1H), 5.88 (d, J = 8. 1Hz, 1H), 5.77 (m, 1H)
36%
at 110℃; Inert atmosphere
A solution of 1H-pyrazol-3-ylamine (7 g, 84.24 mmol, 1 .00 equiv) and ethyl prop-2-ynoate (50 mL) in dioxane ( 1 0 g, 1 21 equiv) was sti rred under nitrogen overnight at 1 1 0 "C. The reaction mixture was cooled to rt and the precipitated product was collected by filtration to give 4 g (36percent) of the title compound as a light brown sol id1HNMR ( 300 MHz, DMSO-d6) δ 1 2.04 (s, 1H), 8.41 -8.44 (m, 1H), 7.71 (d. J = 18 Hz, 1H ), 5.88 (d, J = 8. 1Hz, 1H), 5.77 (m, 1H)
36%
at 110℃; Inert atmosphere
A solution of 1H-pyrazol-3-ylamine (7 g, 84.24 mmol, 1 .00 equiv) and ethyl prop-2-ynoate (50 mL) in dioxane ( 1 0 g, 1 21 equiv) was stirred under nitrogen overnight at 110 "C. The reaction mixture was cooled to rt and the precipitated product was collected by filtration to give 4 g (36percent) of the title compound as a light brown solid 1H NMR (300 MHz, DMSO-d6) δ 12.04 (s, 1H), 8.41 -8.44 (m, 1H), 7.71 (d. J = 18 Hz, 1H), 5.88 (d, J = 8. 1Hz, 1H), 5.77 (m, 1H)
36%
at 110℃; Inert atmosphere
0217] Step 1. 4H-Pyrazolor 5-alpyrimidin-5-one. A solution of lH-pyrazol-3-ylamine (7 g, 84.24 mmol, 1.00 equiv) and ethyl prop-2-ynoate (50 mL) in dioxane (10 g, 1.21 equiv) was stirred under nitrogen overnight at 110 °C. The reaction mixture was cooled to rt and the precipitated product was collected by filtration to give 4 g (36percent) of the title compound as a light brown solid. 1H NMR (300 MHz, DMSO-J6) δ 12.04 (s, 1H), 8.41-8.44 (m, 1H), 7.71 (d, J = 1.8 Hz, 1H), 5.88 (d, J = 8.1 Hz, 1H), 5.77 (m, 1H).
Reference:
[1] European Journal of Organic Chemistry, 2017, vol. 2017, # 41, p. 6168 - 6178
[2] Patent: WO2013/127266, 2013, A1, . Location in patent: Page/Page column 142
[3] Patent: WO2013/127267, 2013, A1, . Location in patent: Page/Page column 93
[4] Patent: WO2013/127268, 2013, A1, . Location in patent: Page/Page column 68
[5] Patent: WO2013/130943, 2013, A1, . Location in patent: Paragraph 0217
20
[ 1820-80-0 ]
[ 623-47-2 ]
[ 29274-22-4 ]
Reference:
[1] European Journal of Organic Chemistry, 2017, vol. 2017, # 41, p. 6168 - 6178
21
[ 874-14-6 ]
[ 1820-80-0 ]
[ 29274-22-4 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2011, vol. 21, # 18, p. 5521 - 5527
22
[ 1820-80-0 ]
[ 105-53-3 ]
[ 57489-77-7 ]
Reference:
[1] Journal of Medicinal Chemistry, 2012, vol. 55, # 15, p. 6700 - 6715
23
[ 1820-80-0 ]
[ 16461-94-2 ]
Yield
Reaction Conditions
Operation in experiment
88%
Stage #1: With bromine In acetic acid at 0 - 5℃; for 2 h; Stage #2: With sodium hydrogencarbonate In water
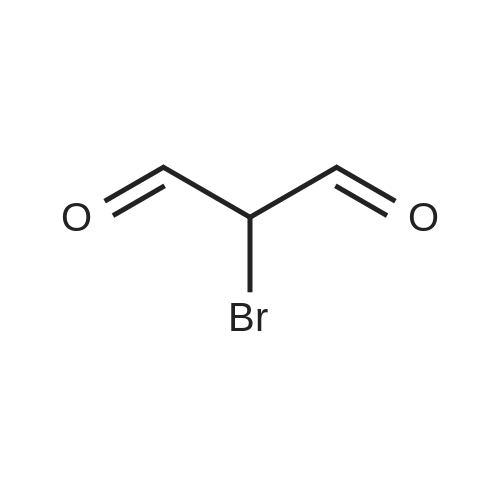
At 0-50C, a solution of 3-amino pyrazole (120mmol) in AcOH (22mL) was slowly added a solution of Br2 in AcOH (22mL) over a period of 2h. The reaction was complete immediately after the addition of Br2 solution (120mmol). To the reaction mixture was added CCl4 (8mL), stirred for 30min at rt. The precipitated solid was filtered and washed with additional CCl4 (8mL). The solid so obtained was dissolved in water (40 mL), adjusted to pH ~7.5 (using aq. NaHCO3 solution) and the precipitated solid was filtered and washed with water (8mL). The combined filtrates were also adjusted to pH ~8 (aq. Na2CO3 solution), extracted with EtOAc (80OmL), washed with brine solution <n="70"/>(20OmL), dried (Na2SO4), filtered and evaporated to obtain the desired compound as a yellow solid. The crude compound was stirred with CCl4 (2OmL), filtered and washed with acetone (5mL) and CCl4 (8mL), and dried under vacuum. The product was obtained as a pale yellow solid (17.2 g, 88 percent yield). TLC system: DCM/MeOH (9: 1). Rf value: 0.5. (M + H): 162.3.
At room temperature,3-amino-1H-pyrazole (120 mmol, 10 g)A solution in 50 ml of anhydrous acetic acid was added dropwise to bromomalonaldehyde (120 mmol, 18.9 g)In a suspension of 50 mL of anhydrous acetic acid,The resulting brown solution was stirred at room temperature for 3 hours.Evaporating acetic acid under reduced pressure,Adding chloroform to the residue;The organic phase is saturated with NaHCO3 solution,Washed with brine and dried over sodium sulfate.After the column, the product was obtained (14g, 58percent).
58%
With acetic acid In ethanol at 80℃; for 1.5 h;
Synthesis of 4-[6-(4-Piperazin-l-ylphenyl)pyrazolo[l ,5-a]pyrimidin-3-yllquinoline hydrochloride salt (13 HCH; A mixture of 2-bromomalondialdehyde (1.5 g, 10 mmol) and lH-pyrazol-3- ylamine (4b, 0.83 g, 10 mmol) in a mixture of EtOH (15 mL) and acetic acid (5 mL) was heated at 80 °C for 1.5 h. The reaction mixture was concentrated and the resulting residue purified by column chromatography using hexane/EtOAc (5:1 ) to give 15a (1.15 g, 58percent yield) as light yellow crystals.
42%
at 20℃; for 3 h;
Preparation 16-Bromo-pyrazolo[1 ,5-a]pyrimidine[00117] A solution of 3-amino-1 H-pyrazole (120 mmol, 10g) in 50ml of anhydrous acetic acid is added dropwise to a suspension of bromomalonaldehyde (120 mmol, 18.9 g) in 50 mL anhydrous acetic acid at room temperature. The resulting brown solution is stirred at room temperature for 3 hours, acetic acid is evaporated at reduced pressure and chloroform is added to the residue. The organic phase is washed with saturated solution of NaHCO3, brine, and dried over sodium sulfate. The product is purified by flash column chromatography (CH2CI2 : Et3N 100 : 0.5 as eluent, Rf=0.3) to yield the title compound (5 g, 42percent).
11%
for 2 h; Heating / reflux
Dissolve bromomalonaldehyde (Aldrich; 2.5 g, 16.5 mmol) and 3-aminopyrazole (Aldrich; 1.38 g, 16.5 mmol) in glacial acetic acid (25 ML) and reflux the resulting mixture under nitrogen for 2 h. Concentrate under reduced pressure. Dissolve the residue in absolute methanol (150 mL), vacuum filter through a pad diatomaceous earth and concentrate under reduced pressure. Chromatograph on flash silica using a gradient from neat hexane to 50percent ethyl acetate/50percent hexane to obtain 365 mgs (11percent) of the subtitled compound as a light yellow solid. High Resolution Mass Spectrum: 197.9674 (M+1).
Reference:
[1] Patent: CN108586464, 2018, A, . Location in patent: Paragraph 0031; 0038; 0039
[2] Patent: WO2009/114180, 2009, A1, . Location in patent: Page/Page column 71
[3] Patent: WO2011/15343, 2011, A1, . Location in patent: Page/Page column 38-39
[4] Patent: WO2004/50659, 2004, A1, . Location in patent: Page 72
[5] Patent: WO2008/78100, 2008, A2, . Location in patent: Page/Page column 111-112
[6] Patent: WO2004/74290, 2004, A1, . Location in patent: Page 83
[7] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 11, p. 3248 - 3252
Preparation 6; 5 -Bromo- 1 H-pyrazolo [3 ,4-b] pyridine; Dissolve 2-bromomalonaldehyde (Aldrich; 2.5 g, 16.5 mmol) and3-aminopyrazole (Aldrich; 1.38 g, 16.5 mmol) in glacial acetic acid (25 mL) and reflux under nitrogen for 2 h. Concentrate under reduced pressure and dissolve the residue in absolute methanol (150 mL), vacuum filter through a pad of diatomaceous earth and concentrate under reduced pressure. Purify via chromatography (silica gel, hexane to 50percent ethyl acetate/50percent hexane) to obtain 365 mg (11percent) of the title compound as a light EPO <DP n="25"/>yellow solid. TOF MS ES+ exact mass calculated for C6H5N3Br (p+H): m/z =197.9667, Found: 197.9674.
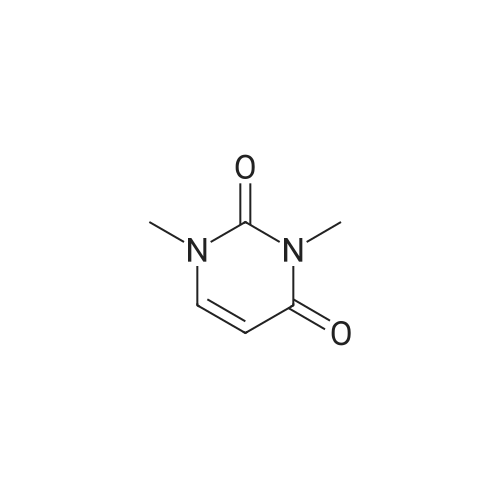
A mixture of <strong>[874-14-6]1,3-dimethyluracil</strong> (4.7 g, 1.4 eq.) and 3-aminopyrazole (2.0 g, 24.1 mmol) was heated to reflux in 1M NaOEt in EtOH (31 mL) for one hour. The resulting solution was cooled to 0° C. The resulting precipitate was dissolved in water and neutralized with acetic acid and cooled to 0° C. The precipitate was collected and dried under reduced pressure to yield a cream-colored solid (2.4 g, 72percent yield): M+H=136.
(a) Ethyl 7-hydroxypyrazolo[1,5-a]pyrimidine-6-carboxylate3 A mixture of diethyl ethoxymethylenemalonate (5.25g, 24mM) and 3-aminopyrazole (2.0g, 24mM) in glacial acetic acid (20ml) was heated under reflux for two hours the resulting precipitated product was filtered and dried in vacuo. Yield 2.70g (54.0%). m.p. 292-4 C (decomposition). nu max (nujol) 1718 (ester C=O) and 1665 (lactam C=O)cm-1.
Preparation of compound 23a: pyrazolo[1,5-a]pyrimidineA mixture of 1 H-pyrazol-3-ylamine (32 g, 0.386 mol) and (E)-3-dimethylamino- propenal (38.2 g, 0.386 mol) in EtOH (500 ml_) was refluxed for 6 h. The solvent was removed in vacuo and the residue was purified via column chromatography (petroleum ether/EtOAc = 10:1 -2:1 ) which gave the title compound 23a as a white solid (30 g, 65%).
At room temperature,3-amino-1H-pyrazole (120 mmol, 10 g)A solution in 50 ml of anhydrous acetic acid was added dropwise to bromomalonaldehyde (120 mmol, 18.9 g)In a suspension of 50 mL of anhydrous acetic acid,The resulting brown solution was stirred at room temperature for 3 hours.Evaporating acetic acid under reduced pressure,Adding chloroform to the residue;The organic phase is saturated with NaHCO3 solution,Washed with brine and dried over sodium sulfate.After the column, the product was obtained (14g, 58%).
58%
With acetic acid; In ethanol; at 80℃; for 1.5h;
Synthesis of 4-[6-(4-Piperazin-l-ylphenyl)pyrazolo[l ,5-a]pyrimidin-3-yllquinoline hydrochloride salt (13 HCH; A mixture of 2-bromomalondialdehyde (1.5 g, 10 mmol) and lH-pyrazol-3- ylamine (4b, 0.83 g, 10 mmol) in a mixture of EtOH (15 mL) and acetic acid (5 mL) was heated at 80 C for 1.5 h. The reaction mixture was concentrated and the resulting residue purified by column chromatography using hexane/EtOAc (5:1 ) to give 15a (1.15 g, 58% yield) as light yellow crystals.
42%
With acetic acid; at 20℃; for 3.0h;
Preparation 16-Bromo-pyrazolo[1 ,5-a]pyrimidine[00117] A solution of 3-amino-1 H-pyrazole (120 mmol, 10g) in 50ml of anhydrous acetic acid is added dropwise to a suspension of bromomalonaldehyde (120 mmol, 18.9 g) in 50 mL anhydrous acetic acid at room temperature. The resulting brown solution is stirred at room temperature for 3 hours, acetic acid is evaporated at reduced pressure and chloroform is added to the residue. The organic phase is washed with saturated solution of NaHCO3, brine, and dried over sodium sulfate. The product is purified by flash column chromatography (CH2CI2 : Et3N 100 : 0.5 as eluent, Rf=0.3) to yield the title compound (5 g, 42%).
11%
In acetic acid; for 2.0h;Heating / reflux;
Dissolve bromomalonaldehyde (Aldrich; 2.5 g, 16.5 mmol) and 3-aminopyrazole (Aldrich; 1.38 g, 16.5 mmol) in glacial acetic acid (25 ML) and reflux the resulting mixture under nitrogen for 2 h. Concentrate under reduced pressure. Dissolve the residue in absolute methanol (150 mL), vacuum filter through a pad diatomaceous earth and concentrate under reduced pressure. Chromatograph on flash silica using a gradient from neat hexane to 50% ethyl acetate/50% hexane to obtain 365 mgs (11%) of the subtitled compound as a light yellow solid. High Resolution Mass Spectrum: 197.9674 (M+1).
General Scheme to svnthesise pyrazoloH ,5-aipyrimidines; Procedure K - General ring formation; To a solution of 2-Bromo-malonaldehyde (12.8 g, 80 mmol) in EtOH (150 ml) was added 3- aminopyrazole (6 g, 37 mmol) followed by glacial acetic acid (10 ml). The mixture was refluxed for 4 h then allowed to cool, solid was filtered off and the filtrate was evaporated under reduced pressure. The residue was partitioned between 1M NaOH (50ml) and EtOAc (200ml) [some insoluble material was filtered off]. The organic layer was washed with brine, dried (MgSO4), filtered and concentrated under reduced pressure. The solid was recrystallised from MeOH, filtered warm and washed with further MeOH and dried to afford 4.5g of product. MS: [M+H]+ 198
In ethanol; acetic acid; for 4.0h;Heating / reflux;
Bromomalonaldehyde (12 g, 0.08 mol) and 3-aminopyrazole (6 g, 0. 087 mol) in acetic acid (10 ml) and EtOH (150 ml) was heated at reflux for 4 h. After cooling to room temperature the insolubles were removed by filtration and the filtrate concentrated. The residue was partitioned between 1N NAOH solution (50 ml) and EtAc (3X100 ml). The organic phase was dried (MgSO4) and concentrated to give a yellow gum which was purified by column chromatography (SILICA ; EtAc: Hexane 1: 4) to give 6 g of product as pale yellow crystals, MS : (ES (M+1)) 198,200.
With acetic acid; In ethanol; for 12.0h;Reflux;
To a solution of 2-bromomalonaldehyde, 8, (5.0g, 33.1 mmol, 1.2 eq) in EtOH (65 mL) was added 1H-pyrazol-3-amine, 5 (2.3g, 28.2 mmol, 1.0 eq), followed by acetic acid (4.0 mL). The mixture was refluxed for 12h and cooled to rt. The reaction was filtered and the filtrate was evaporated under reduced pressure. The residue was partitioned between 1M NaOH (25 mL) and EtOAc (150 mL). The organic layer was separated and washed with Brine, dried (MgSO4), filtered and concentrated. Material was taken through without purification. LCMS: RT = 0.415min, >98% (at) 215 and 254 nM, m/z = 198.0 [M + H]+.
4-(2,1,3-benzoxadiazol-4-yl)-5-cyano-4,7-dihydro-6-(1,4-dioxa-spiro[4,5]decan-6-yl)-2H-pyrazolo[3,4-b]-pyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In acetonitrile;Heating / reflux;
To a solution of ethyl 2-cyclohexanonecarboxylate (25 g) in toluene (200 mL) was added ethyleneglycol (10.1 g) and p-toluenesulfonic acid (2.8 g) at room temperature and the mixture was heated under reflux with Dean-Stark apparatus for five hours. The reaction mixture was cooled to room temperature, the solvent was evaporated under reduced pressure and the residue was purified by silica gel column chromatography (eluent: hexane-ethyl acetate (10: 1) ) to give ethyl 1, 4-dioxa-spiro [4,5] decane-6-carboxylate (31 g) as a colorless oil. To a solution of acetonitrile (7.2 g) in THF (700 mL) was added n-BuLi (160 mmol) [AT-78C.] Further, ethyl 1, 4-dioxa-spiro [4,5] decane-6-carboxylate (31 g) was added and the mixture was stirred for an hour. After acidification with hydrochloric acid, the mixture was extracted with ethyl acetate. The solvent was evaporated under reduced pressure and the residue was purified by silica gel column chromatography (eluent: hexane-ethyl acetate (10: 1) ) to give [1-CYANO-2- (1,] 4-dioxa-spiro [4,5]- decan-6-yl) ethan-2-one (14.5 g) as a colorless oil. A solution of 2, 1, 3-benzoxadiazole-4-aldehyde (0.8 g), 3- aminopyrazole (0.5 g) and [1-CYANO-2- (1,] 4-dioxa-spiro [4,5]- decan-6-yl) ethan-2-one (1.2 g) in acetonitrile (10 mL) was heated under reflux overnight. The reaction mixture was cooled to room temperature, and the precipitated crystals were collected by filtration to give [4- (2, 1,] 3-benzoxa- [ DIAZOL-4-YL) -5-CYANO-4, 7-DIHYDRO-6- (1, 4-DIOXA-SPIRO [4,5]-] decan-6-yl)-2H-pyrazolo [3, 4-b]pyridine (1.3 g) as colorless crystals. To a solution of [4- (2, 1,] 3-Benzoxadiazol-4-yl) -5-cyano- 4, [7-DIHYDRO-6- (1,] 4-dioxa-spiro [4,5] [DECAN-6-YL)-2H-PYRAZOLO-] [3, [4-B] PYRIDINE] (1.0 g) in methanol (30 [ML)] was added 4N HC1 dioxane solution (6.0 mL) at room temperature and the mixture was heated at [60C] for two hours. After alkalification with sodium bicarbonate, the mixture was extracted with chloroform. The solvent was evaporated under reduced pressure and the residue was purified by silica gel column chromatography (eluent: hexane-ethyl acetate (1: 1) ) to give the title compound (20 mg) as colorless crystals. MP: >270C. MS [(EI)] : 360 [(M+).] [H-NMR] [(400MHZ,] DMSO-d6) [6] [(PPM)] : 1. [74-1.,] 80 (5H, m), 2.60-2. 65 (3H, m), 3.31-3. 35 (lH, [M),] 5.98 (1H, s), 6.92 (1H, d, J=6. 6Hz), 7.39 [(LH,] s), 7.47 [(LH,] dd, J=9. OHz and 6. [6HZ),] 7.84 [(LH,] d, J=9. [OHZ),] 9.33 [(LH,] brs), 12.15 [(LH,] brs).
4-(2,1,3-benzoxadiazol-4-yl)-6-(1-t-butoxycarbonyl-piperidin-4-yl)-5-cyano-4,7-dihydro-2H-pyrazolo[3,4-b]-pyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In acetonitrile;Heating / reflux;
To a solution of ethyl isonipecotate (10.0 g) in THF (200 mL) was added triethylamine (7.8 g), 4-dimethylamino- pyridine (0.8 g) and [DI-TERT-BUTYLDICARBONATE] (15.3 g) at [0C] and the mixture was stirred for an hour. The mixture was extracted with ethyl acetate and the solvent was evaporated under reduced pressure to give ethyl N-Boc- [PIPERIDINE-4-CARBOXYLATE] (16.3 g) as a colorless oil. To a solution of acetonitrile (3.2 g) in THF (300 [ML)] was added [N-BULI] (44 mmol) at-78C and stirred for three hours. Further, ethyl N-Boc-piperidine-4-carboxylate (16.3 g) was added and the mixture was stirred for an hour. After acidification with hydrochloric acid, the mixture was extracted with ethyl acetate. The solvent was evaporated under reduced pressure and the residue was purified by silica gel column chromatography (eluent: hexane-ethyl acetate (5: 1) ) to give [1-] (N-Boc-piperidin-4-yl)-2- [CYANOETHAN-1-ONE] (11.6 g) as a colorless oil. A solution of 2, 1, 3-benzoxadiazole-4-aldehyde (1.0 g), 3-aminopyrazole (0.6 g) and 2- (N-Boc-piperidin-4-yl)-l-cyanoethan-2-one (1.7 g) in acetonitrile (10 mL) was heated under reflux overnight. The reaction mixture was cooled to room temperature, and the precipitated crystals were collected by filtration to give the title compound (2.0 g) as colorless crystals. MP: [226C.] Anal. Calcd. For: [C23H25N703] : C, 61.73 ; H, 5.63 ; N, 21.97. Found: C, 61. 45 ; H, 5. 82 ; N, 21.61. MS [(EI)] : 447 (M+). H-NMR [(400MHZ,] [DMSO-D6) B (PPM)] : 1.42 (9H, m), 1.59-1. 62 (2H, m), 1.89-1. 92 (2H, [M),] 2.62-2. 86 (3H, m), 4.05-4. 08 (2H, m), 5.40 [(LH,] [S),] 7.26 [(LH,] s), 7.41 [(LH,] d, J=6.6Hz), 7.58 [(1H,] dd, J=9. OHz and 6.6Hz), 7.92 (1H, d, J=9. [OHZ),] 9.81 (lH, brs), 12.24 (lH, brs).
4-(2,1,3-benzoxadiazol-4-yl)-5-cyano-4,7-dihydro-6-(1-phenylpiperidin-4-yl)-2H-pyrazolo[3,4-b]-pyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In acetonitrile;Heating / reflux;
To a solution of ethyl isonipecotate (8.9 g) in [CH2C12] (500 mL) was added triphenyl bismus (25 g) and Copper (II) acetate (10.3 g) at room temperature, the mixture was stirred overnight. After filteration, the mixture was extracted with [CH2CL2.] The solvent was evaporated under reduced pressure and the residue was purified by silica gel column chromatography (eluent: hexane-ethyl acetate (10: 1) ) to give ethyl 1-phenylpiperidine-4-carboxylate (8.6 g) as colorless crystals. To a solution of acetonitrile (1.9 g) in THF (200 mL) was added n-BuLi (41 mmol) [AT-78C.] Further, ethyl 1-phenylpiperidine-4-carboxylate (8. [6] g) was added and the mixture was stirred for an hour. After acidification with hydrochloric acid, the mixture was extracted with ethyl acetate. The solvent was evaporated under reduced pressure and the residue was purified by silica gel column chromatography (eluent: hexane-ethyl acetate (10: 1) ) to give [1- (L-PHENYLPIPERIDIN-4-YL)-2-] [CYANOETHAN-1-ONE] (2.0 g) as colorless crystals. A solution of 2, 1, 3-benzoxadiazole-4-aldehyde (0.3 g), 3-aminopyrazole (0.2 g) and [1-(1-PHENYLPIPERIDIN-4-YL)-2-CYANOETHAN-1-ONE] (0.5 g) in acetonitrile (10 mL) was heated under reflux overnight. The reaction mixture was cooled to room temperature, and the precipitated crystals were collected by filtration to give the title compound (0.6 g) as colorless crystals. MS (FAB): [424 (M++1).] [H-NMR] [(400MHZ, DMSO-D6) 6 (PPM)] : 1. [73-1. 76 (2H,] m), 2.14- 2.18 (2H, m), 2.62-2. 66 (2H, m), 2.81-2. 84 (lH, m), 3.80- 3. 84 (2H, m), 5.41 (lH, s), 6. [75 (LH,] dd, J=7.3Hz and 7. [2HZ),] 6.94-6. 96 (2H, m), 7.18-7. 27 (3H, m), 7.42 (lH, d, J=6.6Hz), 7.59 (lH, dd, J=9. OHz and 6. 6Hz), 7.92 [(1H,] [D,] J=9. [OHZ),] 9. 81 (lH, brs), 12.17 (lH, brs).
6-(1-acetylpiperidine-4-yl)-4-(2,1,3-benzoxadiazol-4-yl)-5-cyano-4,7-dihydro-2H-pyrazolo[3,4-b]-pyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In acetonitrile; at 20℃;
To a solution of ethyl isonipecotate (8.0 g) in THF (100 [ML)] was added triethylamine (5.7 g), dimethylaminopyridine (0.6 g) and acetyl chloride (4.4 g) at [0C] and the mixture was stirred for an hour. The mixture was extracted with ethyl acetate and the solvent was evaporated under reduced pressure to give ethyl 1-acetylpiperidine-4-carboxylate (10 g) as a colorless oil. To a solution of acetonitrile (2.5 g) in THF (300 mL) was added n-BuLi (57 mmol) [AT-78C.] Further, ethyl 1-acetylpiperidine-4-carboxylate (10 g) was added and the mixture was stirred for an hour. After acidification with hydrochloric acid, the mixture was extracted with ethyl acetate. The solvent was evaporated under reduced pressure and the residue was purified by silica gel column chromatography (eluent: hexane-ethyl acetate (10: 1) ) to give [1- (L-ACETYLPIPERIDIN-4-YL)-2-] [CYANOETHAN-1-ONE] (7.5 g) as a colorless oil. A solution of 2, 1, 3-benzoxadiazole-4-aldehyde (0.3 g), 3-aminopyrazole (0.17 g) and [1-(1-ACETYLPIPERIDIN-4-YL)-2-CYANOETHAN-1-ONE] (0.4 g) in acetonitrile (10 mL) was heated under reflux overnight. The reaction mixture was cooled to room temperature, and the precipitated crystals were collected by filtration to give the title compound (0.49 g) as yellow crystals. MP: [248C.] MS (FAB): [340 (M++1).] [H-NMR] [(400MHZ,] [DMSO-D6)] [6] [(PPM)] : 1.62-1. 64 (2H, m), 1.82-1. 84 [(LH,] m), 2.00-2. 02 (4H, m), 2.49-2. 50 (lH, m), 2.94-3. 07 (2H, [M),] 3.89-3. 92 [(LH,] m), 4.48-4. 51 (lH, m), 5.40 [(LH,] s), 7.27 (lH, s), 7.42 [(LH,] d, J=6.6Hz), 7.59 [(LH,] dd, J=9. OHz and 6.6Hz), 7.92 (1H, d, J=9. [OHZ),] 9.81 (lH, brs), 12.18 (lH, brs).
Preparation 6; 5 -Bromo- 1 H-pyrazolo [3 ,4-b] pyridine; Dissolve 2-bromomalonaldehyde (Aldrich; 2.5 g, 16.5 mmol) and3-aminopyrazole (Aldrich; 1.38 g, 16.5 mmol) in glacial acetic acid (25 mL) and reflux under nitrogen for 2 h. Concentrate under reduced pressure and dissolve the residue in absolute methanol (150 mL), vacuum filter through a pad of diatomaceous earth and concentrate under reduced pressure. Purify via chromatography (silica gel, hexane to 50% ethyl acetate/50% hexane) to obtain 365 mg (11%) of the title compound as a light EPO <DP n="25"/>yellow solid. TOF MS ES+ exact mass calculated for C6H5N3Br (p+H): m/z =197.9667, Found: 197.9674.
5-n-butyl-7-hydroxypyrazolo[1,5-a]pyrimidine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In diethyl ether; toluene;
Step (1) A suspension (120 ml) of 100 g of 3-aminopyrazole and 190 g of <strong>[39815-78-6]methyl 3-oxoheptanoate</strong> in toluene was prepared and refluxed with heating at 100 C. for 3 hours. After cooling, toluene was distilled off under reduced pressure and diethyl ether was added to the residue. The crystals precipitated were collected and washed with diethyl ether and then with acetonitrile to provide 184 g of 5-n-butyl-7-hydroxypyrazolo[1,5-a]pyrimidine as colorless crystals.
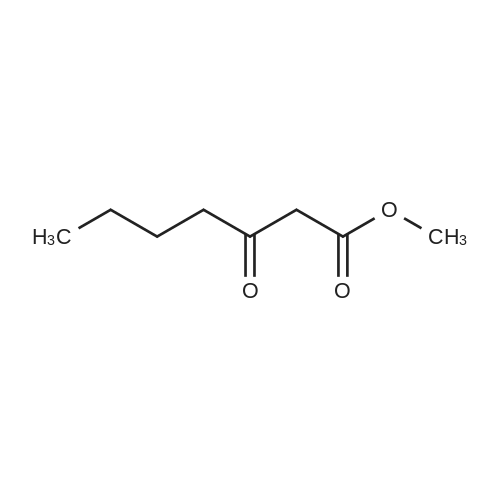
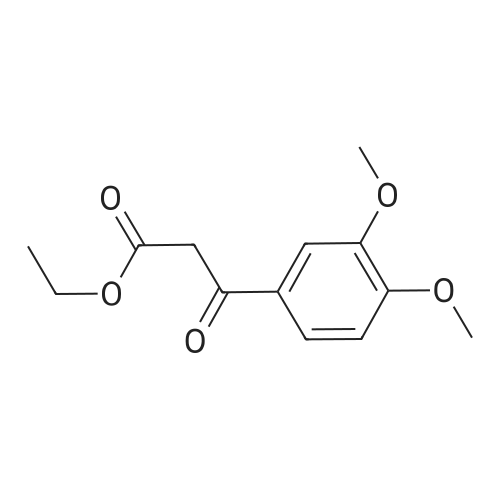
5-(3,4-dimethoxyphenyl)pyrazolo[1,5-a]pyrimidin-7-ol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In acetic acid;
The (6R,7R)-7-(2-amino-4-thiazoleglyoxylamido)-3-[[[5-(3,4-dihydroxyphenyl)pyrazolo[1,5-a]pyrimidin-7-yl]thio]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid used as the starting material can be prepared as follows: 3-Aminopyrazole (33.2 g) and 110 g of <strong>[4687-37-0]ethyl 3,4-dimethoxybenzoylacetate</strong> are stirred in 500 ml of glacial acetic acid for 6 hours at 80 C. After cooling the crystallized-out product is filtered off under suction, washed with diethyl ether and dried. There are obtained 45 g of 5-(3,4-dimethoxyphenyl)pyrazolo[1,5-a]pyrimidin-7-ol of m.p. 268-270 C.
5-n-butyl-7-hydroxypyrazolo[1,5-a]pyrimidine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In diethyl ether; toluene;
Production Example 1 Preparation of 5-n-butyl-7-hydroxypyrazolo[1,5-a]pyrimidine A suspension of 100 g of 3-aminopyrazole and 190 g of <strong>[39815-78-6]methyl 3-oxoheptanoate</strong> in 120 ml of toluene was heated under reflux at 100 C. for 3 hours and cooled. Toluene was distilled off under reduced pressure, and diethyl ether was added to the residue. The crystals precipitated were collected and sequentially washed with diethyl ether and acetonitrile to provide 184 g of 5-n-butyl-7-hydroxypyrazolo[1,5-a]pyrimidine as colorless crystals. The structure and melting point of the compound obtained are shown in Table 1 below.
9-(3,4-dibromophenyl)-5,6,7,9-tetrahydropyrazolo[5,1-b]quinazolin-8(4H)-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
0.23 g (56%)
In ethanol;
EXAMPLE 5 9-(3 4-dibromophenyl)-5,6,7,9-tetrahydropyrazolo[5,1-b]quinazolin-8(4H)-one A solution of 1,3-cyclohexanedione(0.11 g, 1 mmol), <strong>[74003-55-7]3,4-dibromobenzaldehyde</strong>(0.26 g, 1 mmol), and 3-aminopyrazole(0.11 g, 1.27 mmol) in ethanol (10 mL) was heated at 80 C. in a sealed 20 mL vial for 3 days. After the reaction mixture was allowed to cool to ambient temperature, the resulting solid was isolated by filtration and recrystallization from acetone to provide 0.23 g (56%) of the title compound. 1H-NMR(DMSO-d6) 1.91(m, 2H), 2.25(m, 2H), 2.64(m,2H), 5.78(d, 1H), 6.19(s, 1H), 6.95-7.65(m, 4H), 10.59(s, 1H); MS(APCI+) m/z: 423(M+H)+; Analysis Calculated for C16H13Br2N3O: C, 45.42; H, 3.10; N, 9.93; Br, 37.77. Found: C, 45.17; H, 3.22; N, 9.88; Br, 37.59.
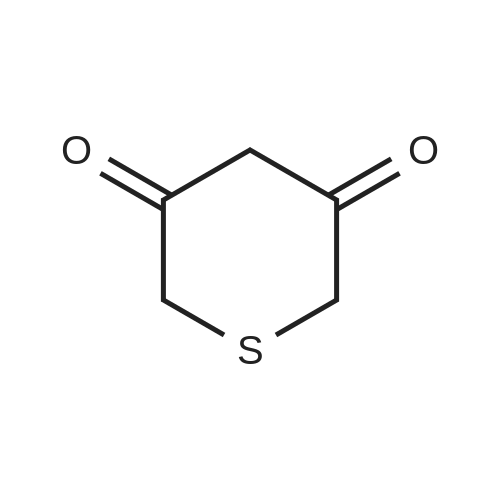
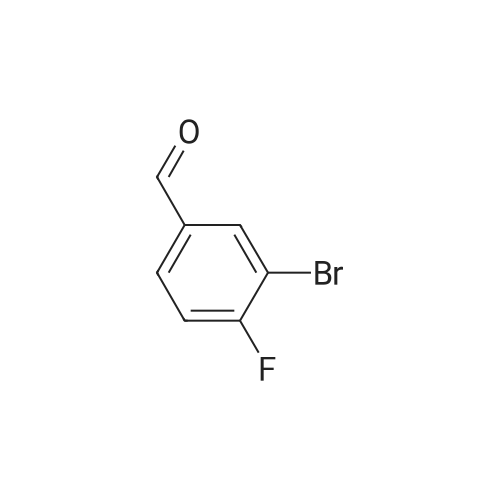
9-(3-bromo-4-fluorophenyl)-5,9-dihydro-4H-pyrazolo[1,5-a]thiopyrano[3,4-d]pyrimidin-8(7H)-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
0.045 g (12%)
In ethanol;
EXAMPLE 2 9-(3-bromo-4-fluorophenyl)-5,9-dihydro-4H-pyrazolo[1,5-a]thiopyrano[3,4-d]pyrimidin-8(7H)-one A solution of 3,5-thiopyrandione (0.13 g, 1 mmol), 3-bromo-4-fluorobenzaldehyde(0.203 g, 1 mmol), and 3-aminopyrazole (0.082 g, 1 mmol) in ethanol (2 mL) was heated at reflux for 24 hours. After the reaction mixture was allowed to cool to ambient temperature, the volatiles were evaporated at reduced pressure and the resulting residue was chromatographed on silica gel, eluding with 5% ethanol/methylene chloride to provide 0.045 g (12%) of the title compound. mp 160-163 C.; 1H NMR (DMSO-d6) delta 3.15 (d, 1H), 3.5 (d, 1H), 3.6 (d, 1H),3.9 (d, 1H), 5.8 (d, 1H), 6.26 (s, 1H), 7.13 (m, 1H), 7.29 (t, 1H), 7.38 (d, 1H), 7.42 (dd, 1H), 10.86 (s, 1H); MS (ESI-) m/z: 380 (M-H)-; Anal. Calcd for C15H11BrFN3OS.0.25C2H6O: C, 47.52; H, 3.22; N, 10.73. Found: C, 47.57; H, 2.89; N, 10.29.
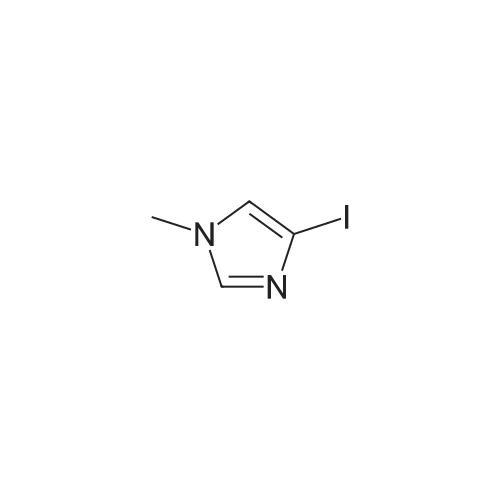
Example 4N-(4-[l-(l-methyl-lH-pyrazol-3-yl)-lH-indol-5-yl]oxy}rhoyrintauidin-2-yl)benzene-l,3-diamine (Compound 75)Scheme 4Step 1: 3-iodo-lH-pyrazole.To a stirred suspension of lH-pyrazol-3-amine (1.0 g, 12 mMol) in concentrated HCl (16 mL) was added a solution of sodium nitrite (1.7 g, 24 mMol) in water (3.0 mL) over 5 minutes at 00C. To the resulting orange reaction mixture was added a solution of KI (5.0 g, 30 mMol) in water (7.0 mL) over 10 minutes, resulting in nitrogen evolution. The reaction mixture was stirred for 5 minutes, upon which nitrogen evolution ceased. TetaF (25 mL) was added, followed by water (25 mL). The aqueous mixture was extracted with Et2O (3 x 30 mL) and the combined organic extracts were washed with Na2S2O3 (2 x 30 mL), dried over magnesium sulfate, filtered and concentrated to afford the title compound. LRMS (ESI) calculated for C3H3IN2 [M+HJ+, 194.9; found 194.9.
With hydrogenchloride; potassium iodide; sodium nitrite; In water; at 0 - 28℃; for 2.75h;
To a stirred suspension of 118 (2.00g, 24.O7mmol) in concentrated HC1 (32mL) was added a solution ofNaNO2 (3.32g. 48.i4mmoi) in water (5mL) over 5 minutes at 0C. To the resulting orange reaction mixture was added a solution of KI (9.99g, 60 iSmmol) in water (l0mL) over 10 minutes. The reaction mixture was stirred at 0C for 30 minutes and then kept at 28C for another 2 hours, TLC showed the reaction was complete, then, solvent THF (3OmL) was added,followed by water (3OmL). The aqueous mixture was extracted with EtOAc (3 x8OmL) and the combined organic extracts were washed with Na2S2O3 (2 x4OmL), dried over Na2SO4, filtered and concentrated in vacuum to afford product 180 (200 g, crude), the crude product was used directly for the next step without purification.LCMS: m/z, i94.9M+H)??.
8
Example 8 (E)-2-(3-Chloro-4-methanesulfonyl-phenyl)-2-isopropoxyimino-N-(1H-pyrazol-3-yl)-acetamide 3-Amino-pyrazole (2 g, 24.1 mmol) was dissolved in 1,4-dioxane (60.25 mL) and triethylamine (6.77 mL, 48.2 mmol) was added followed by the dropwise addition of di-tert-butyl dicarbonate (5.78 g, 26.5 mmol). The solution stirred at 25° C. for 4 h. The mixture was concentrated in vacuo, diluted with ethyl acetate (100 mL), washed with water (2*50 mL), saturated aqueous brine solution (2*50 mL), dried over magnesium sulfate, filtered and concentrated in vacuo. Flash column chromatography (Merck silica gel 60, 40-63 μm; 20% ethyl acetate/hexanes to 50% ethyl acetate/hexanes) afforded 5-amino-pyrazole-1-carboxylic acid tert-butyl ester (less polar product, 2.53 g, 57.3%) as a white solid: H1-NMR (400 MHz, CDCl3) δ 1.656 (9H, s, 5.1-5.45 (2H, bs), 5.39 (1H, d, J=2 Hz), 7.37 (1H, d, J=2 Hz).
At 0-50C, a solution of 3-amino pyrazole (120mmol) in AcOH (22mL) was slowly added a solution of Br2 in AcOH (22mL) over a period of 2h. The reaction was complete immediately after the addition of Br2 solution (120mmol). To the reaction mixture was added CCl4 (8mL), stirred for 30min at rt. The precipitated solid was filtered and washed with additional CCl4 (8mL). The solid so obtained was dissolved in water (40 mL), adjusted to pH ~7.5 (using aq. NaHCO3 solution) and the precipitated solid was filtered and washed with water (8mL). The combined filtrates were also adjusted to pH ~8 (aq. Na2CO3 solution), extracted with EtOAc (80OmL), washed with brine solution <n="70"/>(20OmL), dried (Na2SO4), filtered and evaporated to obtain the desired compound as a yellow solid. The crude compound was stirred with CCl4 (2OmL), filtered and washed with acetone (5mL) and CCl4 (8mL), and dried under vacuum. The product was obtained as a pale yellow solid (17.2 g, 88 % yield). TLC system: DCM/MeOH (9: 1). Rf value: 0.5. (M + H): 162.3.
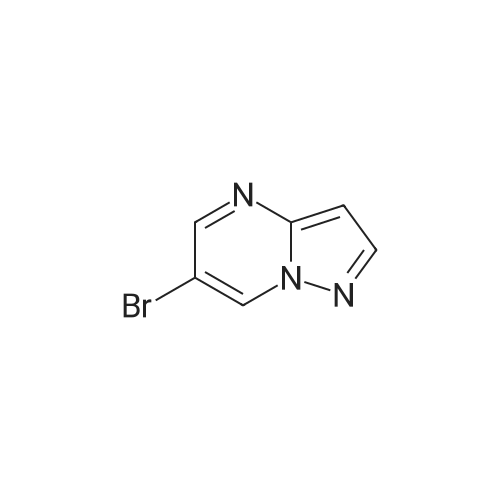
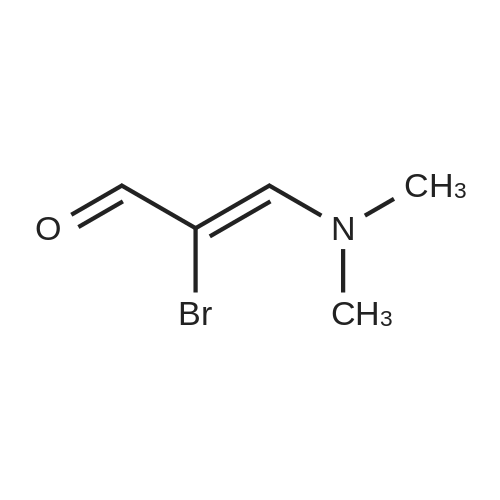
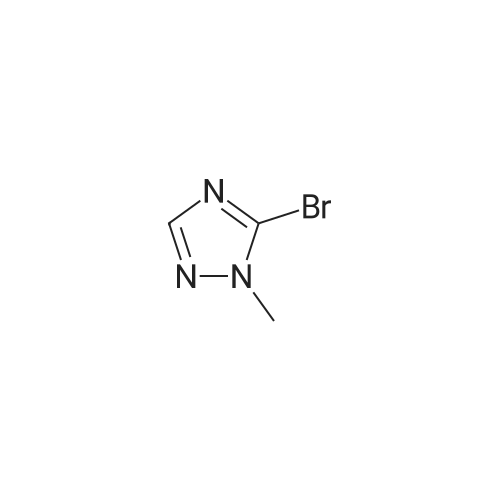
3-Aminopyrazole (0.75 g, 9.04 mmol) was mixed with (2Z)-2-bromo-3- (dimethylamino)acrylaldehyde obtained at the previous step (1.85 g, 10.4 mmol). Absolute ethanol (20 mL) and glacial acetic acid (2 mL) were added. The reaction mixture was stirred under reflux for 16 h and evaporated to dryness. The residue was washed on a filter with cold ethanol/hexane mixture (3:1) and dried in a vacuum oven at 30 C to give 6-bromopyrazolo[l,5- tf]pyrimidine.
With N-ethyl-N,N-diisopropylamine; In butan-1-ol; at 90℃; for 3h;
To a suspension of 2,4-dichloro-7-tosyl-7H-pyrrolo[2,3-<i]pyrirnidine (100 mg, 0.29 mmol) in nBuOeta (1 mL) was added 3-aminopyrazole (26 mg, 0.32 mmol) and <n="155"/>DIPEA (0.057 mL, 0.32 mmol). After heating at 90 C for 3 h, the mixture was diluted with EtOAc, washed with Sat. NaHCO3, brine, dried and concentrated to give a mixture of 2- chloro-N-(lH-pyrazol-5-yl)-7-tosyl-7H-pyrrolo[2,3-T|pyrimidine-4-amine (100 mg).
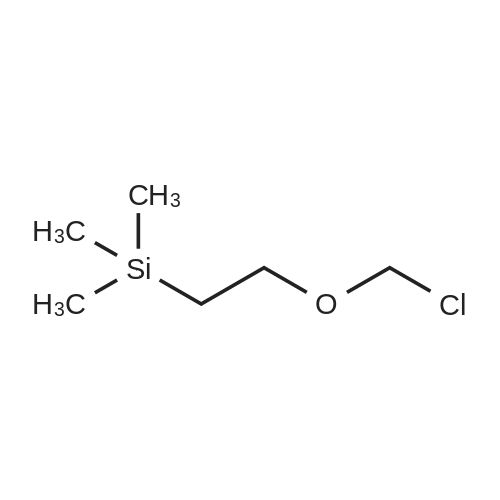
Reference Example 3 Synthesis of 1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazol-3-amine To a solution of 10 g of 1H-pyrazol-3-amine in 100 ml of N,N-dimethylformamide was added 9.6 g of sodium hydride (60%, in oil) under cooling with ice. The reaction mixture was stirred for 30 minutes, and then 21.3 ml of 2-(trimethylsilyl)ethoxymethyl chloride was added thereto. After stirring the resulting mixture at room temperature for 1 hour, aqueous ammonium chloride was added thereto, and the mixture was extracted with chloroform. The resulting organic layer was washed with water and brine, and then dried over magnesium sulfate. The organic layer was filtered and concentrated in vacuo. The resulting residue was purified by a silica gel column chromatography (eluent: hexane/ethyl acetate = 4/1 to 1/2) to give the title compound.
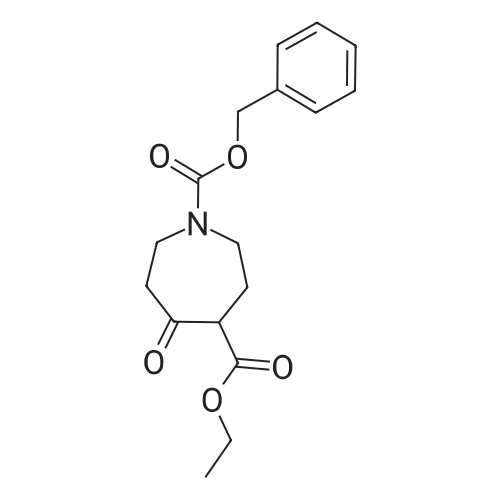
To 5.87 g (18.38 mmol) 5-oxo-azepane-1 ,4-dicarboxylic acid 1 -benzyl ester 4-ethyl ester in 3 mL acetic acid was added 1.53 g (18.38 mmol) 3-aminopyrazole and the reaction mixture was heated at 80 C for 15 minutes. The resulting solid was collected by filtration and washed with TBME (3 x 10 mL) to give the desired product. Yield: 5.9 g (95% of theory) Ci8H18N4O3 (M= 338.37) predicted: Molecular ion (M+H)+: 339 observed: Molecular ion (M+H)+: 339 HPLC-MS: 1.5 minutes (Method A)
With sodium ethanolate; In ethanol; for 1h;Reflux; Inert atmosphere;
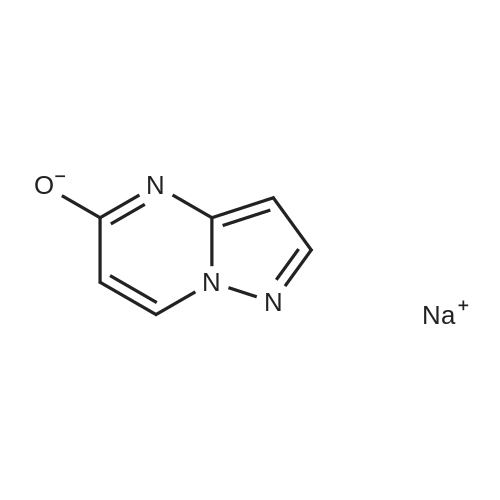
A mechanically stirred mixture of 3-aminopyrazole (9.38 g, 0.11 mM, 1.0 equiv), <strong>[874-14-6]1,3-dimethyluracil</strong> (14.7 g, 0.11 mM, 1.0 equiv) and 21percent sodium ethoxide in ethanol (170 mL, 5.0 equiv) was heated to reflux. Within minutes, a heavy precipitate formed. After refluxing for 1 hour, <strong>[874-14-6]1,3-dimethyluracil</strong> could no longer be detected by thin layer chromatography (tic) (92:8 dichloromethane (dichloromethane):MeOH). The reaction mixture was cooled, filtered, washed with cold ethanol and vacuum dried to give 13.47 g (95percent) of sodium pyrazolo[l,5-alpha]pyrimidin-5-olate. LCMS (ESI) m+H = 136.0; 1H NMR (400 MHz, OMSO-d6) delta: 8.0 (d, 1 H), 7.43 (d, 1 H), 5.65 (d, IH), 5.37 (d, 1 H).
95%
With sodium ethanolate; In ethanol; for 1h;Reflux; Inert atmosphere;
A mechanically stirred mixture of 3-aminopyrazole (9.38 g, 0.11 mM, 1.0 equiv), <strong>[874-14-6]1,3-dimethyluracil</strong> (14.7 g, 0.11 mM, 1.0 equiv) and 21percent sodium ethoxide in ethanol (170 mL, 5.0 equiv) was heated to reflux. Within minutes, a heavy precipitate formed. After refluxing for 1 hour, <strong>[874-14-6]1,3-dimethyluracil</strong> could no longer be detected by thin layer chromatography (tlc) (92:8 dichloromethane (dichloromethane):MeOH). The reaction mixture was cooled, filtered, washed with cold ethanol and vacuum dried to give 13.47 g (95percent) of sodium pyrazolo[1,5-a]pyrimidin-5-olate. LCMS (ESI) m+H=136.0; 1H NMR (400 MHz, DMSO-d6) delta: 8.0 (d, 1H), 7.43 (d, 1H), 5.65 (d, 1H), 5.37 (d, 1H).
With N-ethyl-N,N-diisopropylamine; potassium iodide; In N,N-dimethyl-formamide; at 20 - 50℃;Product distribution / selectivity;
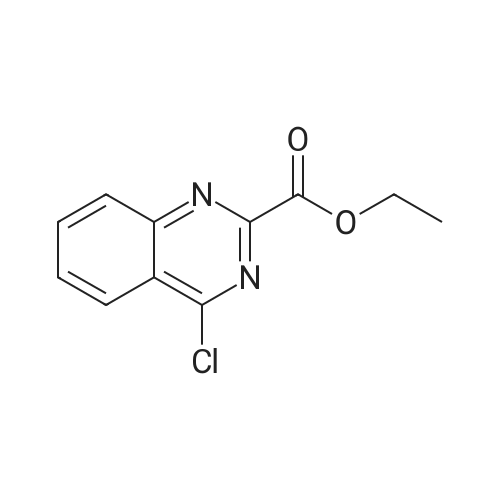
[00384] Step A: A stirred mixture of <strong>[34632-69-4]ethyl 4-chloroquinazoline-2-carboxylate</strong> (709 mg, 3 mmol), 3-aminopyrazole (274 mg, 3.3 mmol), potassium iodide (498 mg, 3 mmol), and DIEA (574 muL, 3.3 mmol) in N, N-dimethylformamide (5 mL) was heated at 50 0C for 2 h and then stirred at rt overnight. Water was added to the mixture and the precipitated solid was filtered, washed with water and dried under high vacuum at 50 0C for 3 h to afford ethyl 4-(lH-pyrazol-3-ylamino)quinazoline-2- carboxylate (595 mg, 70%). LC-MS (ESI) m/z 284 (M+H)+.
70%
With potassium iodide; DIEA; In water; N,N-dimethyl-formamide;
Step A: A stirred mixture of <strong>[34632-69-4]ethyl 4-chloroquinazoline-2-carboxylate</strong> (709 mg, 3 mmol), 3-aminopyrazole (274 mg, 3.3 mmol), potassium iodide (498 mg, 3 mmol), and DIEA (574 muL, 3.3 mmol) in N,N-dimethylformamide (5 mL) was heated at 50 C. for 2 hrs and then stirred at room temperature overnight. Water was added to the mixture and the precipitated solid was filtered, washed with water and dried under high vacuum at 50 C. for 3 hrs to afford ethyl 4-(1H-pyrazol-3-ylamino)quinazoline-2-carboxylate (595 mg, 70%). LC-MS (ESI) m/z 284 (M+H)+.
With potassium iodide; DIEA; In N,N-dimethyl-formamide;
Step A: To a solution of <strong>[34632-69-4]ethyl 4-chloroquinazoline-2-carboxylate</strong> (0.250 g, 1.05 mmol) in DMF (2.5 mL) at room temperature under argon were added potassium iodide (0.192 g, 1.16 mmol), DIEA (0.238 mL, 1.37 mmol), and 1H-pyrazol-3-amine (0.113 g, 1.37 mmol). The mixture was stirred at room temperature for 5 hrs and diluted with H2O (5 mL). The precipitate was collected by filtration, washed with H2O, and dried under high vacuum for several hours to afford ethyl 4-(1H-pyrazol-3-ylamino)quinazoline-2-carboxylate as a tan solid (0.190 g, 64%). 1H NMR (300 MHz, DMSO-d6) delta 12.52 (s, 1H), 10.58 (s, 1H), 8.72 (d, 1H), 7.90 (d, 2H), 7.78 (s, 1H), 7.68 (m, 1H), 7.18 (s, 1H), 4.48 (q, 2H), 1.48 (t, 3H); LC-MS (ESI) m/z 284 (M+H)+.
With sodium ethanolate; In ethanol; at 80℃; for 16.0h;Inert atmosphere;
Step A - Synthesis of pyrazolo[ l,5-a]pyrimidine-5,7-diol (lnt-9a)To lH-pyrazol-3-amine (12.3 g, 148.0 mmol) in EtOH (50 mL) was added diethyl malonate (25.0 mL, 164.7 mmol), 21 wt% NaOEt in EtOH (1 10 mL, 294.6 mmol) and additional EtOH (50 mL). The resulting reaction mixture was then heated at 800C under an atmosphere of argon for 16 hours, at which time the reaction was allowed to cool to room temperature. The reaction mixture was then concentrated in vacuo until almost dry, before H2O (500 mL) was added. Vigorous stirring aided the dissolution of solids, at which time cone. HCl was added until pH~2 was attained (precipitate formed). The precipitate was collected and dried by vacuum filtration giving pyrazolo[l,5-a]pyrimidine-5,7-diol (Int-9a) as a tan solid (17.13 g, 1 13.4 mmol, 77%).
77%
With sodium ethanolate; In ethanol; at 80℃; for 16.0h;Inert atmosphere;
To lH-pyrazol-3-amine (12.3 g, 148.0 mmol) in EtOH (50 mL) was added diethyl malonate (25.0 mL5 164.7 mmol), 21wt% NaOEt in EtOH (1 10 mL, 294.6 mmol) and additional EtOH (50 mL). The resulting mixture was then heated at 80 C under an atmosphere of argon for 16 hours, at which time the reaction was allowed to cool to room temperature. The reaction mixture was then concentrated in vacuo until almost dry, before ¾0 (500 mL) was added. Vigorous stirring aided the dissolving of solids, at which time cone. HC1 was added until pH~2 was attained (solid precipitate formed). The precipitate was collected and dried by vacuum filtration giving pyrazolo[l,5-a]pyrimidine-5,7-diol as a tan solid (17.13 g, 113.4 mmol, 77%).
41.24%
With sodium; In ethanol; at 80℃; for 5.0h;
To a solution of 58 (20.0 g, 240.96 mmol, 1.0 eq)58.1 (38.5 g, 240.96 mmol, 1.0 eq) in ethanol (200 mE) was added sodium metal (6.6 g, 289.15 mmol, 1.2 eq) and reflux at 80 C. for 5 h. After completion of reaction, reaction mixture was cooled to room temperature, transferred into water and product was extracted with ethyl acetate. Organic layer was combined, washed with brine solution, dried over sodium sulphate and concentrated under reduced pressure to obtain crude material. This was further purified by column chromatography and compound was eluted in 22% ethyl acetate in hexane to obtain pure 58.2. (15.0 g, Yield:41.24%). MS(ES): mlz 152.04 [M+H].
3-Amino-pyrazole (534 mg, 6.43 mM) was dissolved in sodium ethoxide (1M ethanol solution, 12.9 mL, 12.9 mM) and then diethyl malonate (1.07 mL, 7.07M) was added to the solution. This reaction liquid was stirred at 150 C. for 40 minutes under the irradiation with microwaves. This reaction mixture was diluted with water, acidified (pH 2) by the addition of hydrochloric acid and then extracted with ethyl acetate. The extracts were combined together, dried over anhydrous sodium sulfate and then the solvent was distilled off. Phosphoryl chloride (10 mL) was added to the resulting solid with ice-cooling and then the resulting suspension was stirred for 2 hours and a half, while refluxing the same with heating. The phosphoryl chloride was distilled off from this reaction liquid, ethanol was added to the resulting residue with ice-cooling and the mixture was subsequently stirred for 15 minutes. After the concentration of this reaction liquid, the latter was diluted with a saturated aqueous sodium bicarbonate solution and then extracted with methylene chloride. The extracts obtained were combined, dried over anhydrous sodium sulfate and then purified by the silica gel column chromatography (methylene chloride/hexane=3:1) to thus obtain the title compound (403 mg, overall yield of these two steps: 34%).1H-NMR (300 MHz, CDCl3): delta 6.74 (d, 1H, J=2.3 Hz), 6.99 (s, 1H), 8.22 (d, 1H, J=2.3 Hz); MS (ESI) m/z 188 (M+H)+.
With sodium ethanolate; In ethanol; at 80℃; for 16.0h;Inert atmosphere;
To lH-pyrazol-3 -amine (123 g, 148.0 mmol) in EtOH (50 mL) was added diethyl malonate (25.0 mL, 164.7 mmol), 21 wt% NaOEt in EtOH (110 mL, 294.6 mmol) and additional EtOH (50 mL). The resulting mixture was then heated at 80C under an atmosphere of argon for 16 hours, at which time the reaction was allowed to cool to room temperature. The reaction mixture was then concentrated in vacuo until almost dry, before H20 (500 mL) was added. Vigorous stirring aided the dissolving of solids, at which time cone. HCI was added until pH~2 was attained (solid precipitate formed). The precipitate was collected and dried by vacuum filtration giving pyrazolo[l,5-a]pyrimidine~5,7-diol as a tan solid.
With sodium ethanolate; In ethanol; at 80℃; for 16.0h;Inert atmosphere;
To lH-pyrazol-3 -amine (12,3 g, 148.0 mmol) in EtOH (50 mL) was added diethyl malonate (25.0 mL, 164.7 mmol), 21wt% NaOEt in EtOH (110 mL, 294.6 mmol) and additional EtOH (50 mL). The resulting mixture was then heated at 80C under an atmosphere of argon for 16 hours, at which time the reaction was allowed to cool to room temperature. The reaction mixture was then concentrated in vacuo until almost dry, before H20 (500 mL) was added. Vigorous stirring aided the dissolving of solids, at which time cone. HCl was added until pH~2 was attained (solid precipitate formed). The precipitate was collected and dried by vacuum filtration giving pyrazolo[l,5-a]pyrimidine-5,7-diol as a tan solid.
With sodium ethanolate; In ethanol; at 80℃; for 16.0h;Inert atmosphere;
To lH-pyrazol-3 -amine (12.3 g, 148.0 mmol) in EtOH (50 mL) was added diethyl malonate (25.0 mL, 164.7 mmol), 21wt% NaOEt in EtOH (110 mL, 294.6 mmol) and additional EtOH (50 mL). The resulting mixture was then heated at 80C under an atmosphere of argon for 16 hours, at which time the reaction was allowed to cool to room temperature. The reaction mixture was then concentrated in vacuo until almost dry, before ¾0 (500 mL) was added. Vigorous stirring aided the dissolving of solids, at which time cone. HC1 was added until pH~2 was attained (solid precipitate formed). The precipitate was collected and dried by vacuum filtration giving pyrazolo[l,5-a]pyrimidine-5,7-diol as a tan solid.
To lH-pyrazol-3 -amine (12.3 g, 148.0 mmol) in EtOH (50 mL) was added diethyl malonate (25.0 mL, 164.7 mmol), 21wt% NaOEt in EtOH (110 mL, 294.6 mmol) and additional EtOH (50 mL). The resulting mixture was then heated at 80C under an atmosphere of argon for 16 hours, at which time the reaction was allowed to cool to room temperature. The reaction mixture was then concentrated in vacuo until almost dry, before H20 (500 mL) was added. Vigorous stirring aided the dissolving of solids, at which time cone. HC1 was added until pH~2 was attained (solid precipitate formed). The precipitate was collected and dried by vacuum filtration giving pyrazolo[l,5-a]pyrimidine-5,7-diol as a tan solid.
With sodium ethanolate; In ethanol; for 20.0h;Reflux;
Metal sodium (7.62 g, 331.0 mmol) was reacted with 150 mL of ethanol at 0C. 3-Aminopyrazole (11.00 g, 132.38 mmol) and diethyl malonate (21.20 g, 132.38 mmol) were added to the above solution of sodium ethoxide in ethanol. The reaction mixture was refluxed for 20 h, then cooled down to room temperature. The precipitate was filtered off and washed with EtOH. The dried solid was dissolved in water (400 mL), and the solution was acidified to pH 2 with 10 N HCl. The solid precipitate was collected by filtration and washed with water to give pyrazolo[1,5-a]pyrimidine-5,7-diol as a light yellow solid, yield 63.5%. Thus obtained crude product was used in the next step without further purification.
A solution of sodium metal (11.1 g, 481 mmol) in EtOH (344 mL) was added 1H-pyrazol-3-amine (20.0 g, 241 mmol) and <strong>[874-14-6]1,3-dimethylpyrimidine-2,4(1H,3H)-dione</strong> (35.4 g, 253 mmol) at room temperature. The reaction mixture was refluxed overnight and cooled to room temperature. A precipitated solid was collected by filtration, washed with cold EtOH and dried under vacuum to afford pyrazolo[1,5-a]pyrimidin-5-ol (36.0 g, >99percent) as a white solid. 1H-NMR (DMSO-d6, Varian, 400 MHz): delta 5.35 (1H, d, J=1.6 Hz), 5.63 (1H, d, J=7.2 Hz), 7.43 (1H, d, J=1.6 Hz), 7.97 (1H, d, J=7.2 Hz). * A proton from OH was not observed.
86%
With sodium methylate; In methanol; at 65 - 70℃; for 4h;Large scale;
Step 1, under anhydrous, oxygen-free and nitrogen-free protection, add 1.9kg to a mixture of 1kg of compound A and 2.7L of sodium methoxide in methanol (in which methanolic sodium methoxide solution, sodium methoxide mass fraction is 30percent) Compound B, heating, the temperature was controlled at 65-70 ° C, and the reaction was refluxed for 4 hours. After the reaction was completed, the reaction liquid was cooled and filtered, and the solvent was evaporated under reduced pressure to give a white solid, and then white solid was dissolved in 1.5 L at 10 ° C. In water, the pH was adjusted to 2, and 1.4 kg of white solid compound C was obtained by filtration, yield 86percent
53%
To a solution of dry EtOH (500 mL) was added sodium (22.1 g, 481 mmol) under a cold water bath. After complete dissolution, lH-pyrazol-3 -amine (40.0 g, 481 mmol) and l ,3-dimethylpyrimidine-2,4(lH,3H)-dione (101 g, 722 mmol) were added. The reaction mixture was heated at reflux for 4 hours. After cooling, the white solid was collected by filtration, washed with cold EtOH and dried under vacuum. The crude solid was suspended in minimum amount of water and the pH of the solution was adjusted to 5 by dropwise addition of acetic acid. The precipitated solid was collected by filtration, re- dissolved in MeOH and concentrated by rotary evaporation to azeotrope any remaining water to afford pyrazolo[l ,5-a]pyrimidin-5-ol (34.3 g, 53percent>).
With potassium carbonate; In N,N-dimethyl-formamide; at 100℃; for 24h;
-(8-Huoro-benzothiazoi-2-yS}-1 H-pyrazoS-3-ySamine 2A mixture of 2-Chloro-6-fluoro benzothiazoie (200 mg, 1 .07 mmol), 3-Amino pyrazole (178 mg, 2, 14 mmol) and K2C03 (885 mg, 6,40 mmol) in DMF (5 mL) was stirred at100 for 24 h. The reaction mixture was poured inf o water and extracted with ether.The combined organic phases were washed with brine, dried over sodium sulphate, filtrated and concentrated. The residue was purified by chromatography on silica gel to give 130 mg (52%) of compound 2,1H-N R (300 MHz, DMSO-d6): delta = 8.26 (d, 1 H), 7.92 (dd, 1 H), 7.77 (dd, 1 H), 7.32 (ddd,1 H), 5.97 (d, 1 H), 5.73 (br s, 2H) ppm. ESi-fWfS m/z 235 (M+1 ).
With potassium carbonate; In N,N-dimethyl-formamide; at 100℃; for 24h;
A mixture of 2-Chloro-6-methoxy benzothiazoie (500 mg, 2.5 mmol), 3-Amino pyrazole (416 mg, 5.0 mmol) and K2C03 (1.38 g, 10 mmol) in DMF (10 mL) was stirred at 100 for 24 h. The reaction mixture was poured into water and extracted with dichloromethane. The combined organic phases were washed with brine, dried over sodium sulphate, filtrated and concentrated. The residue was purified by chromatography on silica gel to give 270 mg (44%) of 1-(6-methoxy-benzothiazoi-2-yl)- 1 H-pyrazol-3-ylamine .1H-N R (300 MHz, d6-DMSG): delta = 8.18 (d, 1 H), 7.62 (d, 1 H), 7.55 (d, 1 H), 7.00 (dd, 1 H), 5.88 (d, 1 H), 5.60 (br s, 2H), 3.77 (s, 3H) ppm. ESI-MS m/z 247 (M+1 ).
With potassium carbonate; In N,N-dimethyl-formamide; at 120℃; for 3h;Microwave irradiation;
1 -(8-Bromo-benzothiazoi-2-yi)-1 H-pyrazo.-3-ylamine 1A mixture of 2-Ch.oro~6-bromo benzothiazoie (1 .0 g, 4.02 mmo.), 3-Amino pyrazole (670 mg, 8.05 mmol) and K2C03 (1.38 g, 10 mmol) in DMF (16 mL) was stirred at 120 for 3 h under microwave heating. The reaction mixture was poured into water, the precipitate was filtered off, washed with water and purified by chromatography on silica gel to give 470 mg (40%) of compound 1.1H-N R (400 MHz, CDCi3): delta = 8.19 (d, 1 H), 7.92 (d, 1 H), 7.65 (d, 1 H), 7.52 (dd, 1 H), 5.97 (d, 1 H), 4.04 (br s, 2H) ppm. LC/ S ES? m/z 295.27 (M+1 ).
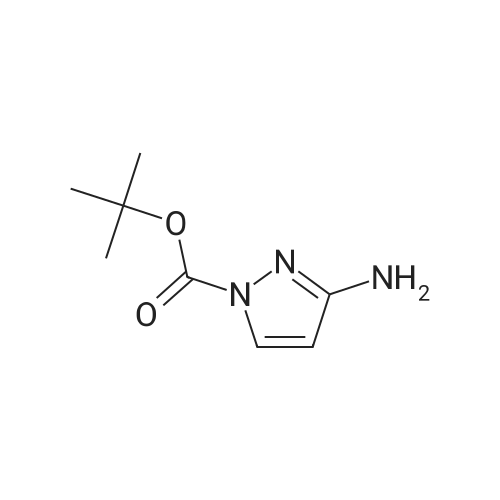
With caesium carbonate; copper(ll) bromide; In N,N-dimethyl-formamide; at 190℃; for 0.333333h;Microwave irradiation; Sealed tube;
1 -phenyl- 1 H -pyrazol-3-amine 3-Amino-pyrazole (1 .0 g, 12.03 mmol), Cs2CO3 (3.92 g, 12.03 mmol), iodobenzene (3.68 g, 18.05 mmol), CuBr2 (0.268 g, 0.1 mmol), and DMF (4 ml_) were added to a 10-mL microwave vial. The vial was sealed and heated to 190 C for 20 min (monitored by TLC). After cooling, the reaction mixture was diluted with saturated aqueous ammonium chloride and extracted with ethyl acetate (50 imL x 3). The organic layers were dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash column chromatography, eluting with 0-40% ethyl acetate and petroleum benzine, afforded the desired product, 1 -phenyl-1 -/-pyrazol-3-amine, as a brown solid (1 .53 g, 80%).
6-hydroxy-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid ethyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
28%
With acetic acid; In water; at 85℃; for 15h;
1.1: 6-Hydroxy-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid ethyl ester To a solution of 3-aminopyrazole (224 g, 2.7 mol) in water (1 L) were added a solution of diethyl oxalacetate sodium salt (567 g, 2.7 mol) in water (1.3 L) and then acetic acid in 4 portions (0.8 L). The reaction mixture was heated at 85C for 15 hours then cooled to room temperature and filtered. The precipitate was washed with water and then dried to afford the title compound as an orange solid (159 g, 28%). 1 H NMR (250 MHz, DMSO): delta 1.35 (t, 3H), 4.36 (q, 2H,), 6.69 (s, 1H), 8.16 (s, 1H).
28%
With acetic acid; In water; at 85℃; for 15h;
1.1: 6-Hydroxy-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid ethyl ester To a solution of 3-aminopyrazole (224 g, 2.7 mol) in water (1 L) were added a solution of diethyl oxalacetate sodium salt (567 g, 2.7 mol) in water (1.3 L) and then acetic acid in 4 portions (0.8 L). The reaction mixture was heated at 85 C. for 15 hours then cooled to room temperature and filtered. The precipitate was washed with water and then dried to afford the title compound as an orange solid (159 g, 28%). 1H NMR (250 MHz, DMSO): delta 1.35 (t, 3H), 4.36 (q, 2H), 6.69 (s, 1H), 8.16 (s, 1H).
28%
With acetic acid; In water; at 85℃; for 15h;
1 .1 : 6-Hydroxy-1 H-pyrazolo[3,4-b]pyridine-4-carboxylic acid ethyl esterTo a solution of 3-aminopyrazole (224 g, 2.7 mol) in water (1 L) were added a solution of diethyl oxalacetate sodium salt (567 g, 2.7 mol) in water (1 .3 L) and then acetic acid in 4 portions (0.8 L). The reaction mixture was heated at 85C for 15 hours then cooled to room temperature and filtered. The precipitate was washed with water and then dried to afford the title compound as an orange solid (159 g, 28%).1 H NMR (250 MHz, DMSO): ? 1 .35 (t, 3H), 4.36 (q, 2H,), 6.69 (s, 1 H), 8.16 (s, 1 H).
22%
With acetic acid; In water; at 0 - 100℃;
[0713] Step 2: Synthesis of ethyl 6-hydroxy-lH-pyrazolo[3,4-b]pyridine-4-carboxylate [0714] A stirred solution of lH-pyrazol-3 -amine (45 g, 542 mmol) in acetic acid (297 mL) and water (900 mL) was cooled to 0 C and <strong>[40876-98-0]diethyl oxaloacetate sodium salt</strong> (1 13.85 g:542.16 mmol) was added to it. Resulting solution was heated at 100 C for overnight. After completion of reaction, solid was filtered and dried to obtain the desired intermediate (25 g, 22%).
22%
In water; acetic acid; at 100℃; for 16h;
A stirred solution of lH-pyrazol-3-amine (45 g, 542.16 mmol) in acetic acid (297 mL) and water (900 mL) was cooled to 0 C and <strong>[40876-98-0]diethyl oxaloacetate sodium salt</strong> (113.85 g, 542.16 mmol) was added. The resulting solution was heated at 100 C for 16 h. After which time the solids were filtered and dried to obtain the title compound (22% yield).
General Procedure A:A mixture of a 3-aminopyrazole (1 equivalent) and a dialkyl acetylsuccinate (1.1 equivalent) in toluene (1 mL/mmol of default reagent) was heated to reflux under with Dean Stark system until the theoric volume of water distilled in the trap. The precipitate was filtered-off, washed with toluene and diethylether to afford the expected alkyl 2-(7-hydroxy-5-methylpyrazolo[1,5-a]pyrimidin-6-yl)acetate, which was used for the next step without any further purification.; Intermediate 1Preparation of Methyl 2-(7-hydroxy-5-methylpyrazolo[1,5-a]pyrimidin-6-yl)acetateThis intermediate was prepared according to the procedure A from <strong>[10420-33-4]dimethyl acetylsuccinate</strong> (4.1 g; 22 mmol) and 3-aminopyrazole (1.66 g; 20 mmol) in toluene (20 mL) for 18 h. The 4.2 g of the title compound (95%) was obtained as a white solid. ESI/APCI(+): 222 (M+H).
With hydrogenchloride; zinc(II) chloride; In ethanol; for 1h;Reflux;
1H-pyrazol-3-amine (427 mg, 5.139 mmol) was dissolved in ethanol (10 mL) and 12M HCl (0.5 mL) was added to the stirred solution, followed by granulated zinc chloride (300 mg). The mixture was heated to reflux, and to the mixture was added a solution of 5.139 mmol of the source of the appropriate 1,3-dicarbonyl compound (sodium 2-nitro-1,3-dioxopropan-2-ide) in ethanol (5 mL). After 1 h the reaction mixture was poured into ice-cold water (15 mL), the resultant solution was made alkaline with concentrated aqueous ammonia, and the product was isolated by trichloromethane extraction. The product 5-nitro-1H-pyrazolo[3,4-b]pyridine (279 mg, 33 %) (A) was isolated and purified by column chromatography.
1H-pyrazol-3-amine (427 mg, 5.139 mmol) was dissolved in ethanol (10 mL) and 12M HCl (0.5 mL) was added to the stirred solution, followed by granulated zinc chloride (300 mg). The mixture was heated to reflux, and to the mixture was added a solution of 5.139 mmol of the source of the appropriate 1,3-dicarbonyl compound (sodium 2-nitro-1,3-dioxopropan-2-ide) in ethanol (5 mL). After 1 h the reaction mixture was poured into ice-cold water (15 mL), the resultant solution was made alkaline with concentrated aqueous ammonia, and the product was isolated by trichloromethane extraction. The product 5-nitro-1H-pyrazolo[3,4-b]pyridine (279 mg, 33%) ( A) was isolated and purified by column chromatography.
27%
In water; at 90℃; for 16h;
To a solution of sodium nitromalonaldehyde monohydrate(1.23 g, 7.81 mmol) in water was added 1H-pyrazol-3-amine(0.84 g, 7.43 mmol). The mixture was stirred at 90 C for 16 h andthen cooled to room temperature. The reaction solution wasadjusted to pH 5 and extracted by ethyl acetate (200 mL). Theorganic layer was dried over Na2SO4 and evaporated. The residuewas purified by silica gel chromatography (1% MeOH in dichloromethane)to give 5-nitro-1H-pyrazolo [4,3-b]pyridine as a whitesolid (118 mg, 27%). Rf: 0.23 (DCM/MeOH, 100/1, v/v). Mp:206-208 C. 1H NMR (DMSO-d6, 400 MHz) d 14.40 (s, 1H), 9.35 (d,J 2.5 Hz, 1H), 9.21 (d, J 2.5 Hz, 1H), 8.46 (s, 1H). MS (ESI) m/z:162.8 [MH]-.
A solution of 1H-pyrazol-3-ylamine (7 g, 84.24 mmol, 1 .00 equiv) and ethyl prop-2-ynoate (50 mL) in dioxane ( 1 0 g, 1 21 equiv) was stirred under nitrogen overnight at 110 "C. The reaction mixture was cooled to rt and the precipitated product was collected by filtration to give 4 g (36%) of the title compound as a light brown solid 1H NMR (300 MHz, DMSO-d6) delta 12.04 (s, 1H), 8.41 -8.44 (m, 1H), 7.71 (d. J = 18 Hz, 1H), 5.88 (d, J = 8. 1Hz, 1H), 5.77 (m, 1H)
36%
In 1,4-dioxane; at 110℃;Inert atmosphere;
A solution of 1H-pyrazol-3-ylamine (7 g, 84.24 mmol, 1 .00 equiv) and ethyl prop-2-ynoate (50 mL) in dioxane ( 1 0 g, 1 21 equiv) was sti rred under nitrogen overnight at 1 1 0 "C. The reaction mixture was cooled to rt and the precipitated product was collected by filtration to give 4 g (36%) of the title compound as a light brown sol id1HNMR ( 300 MHz, DMSO-d6) delta 1 2.04 (s, 1H), 8.41 -8.44 (m, 1H), 7.71 (d. J = 18 Hz, 1H ), 5.88 (d, J = 8. 1Hz, 1H), 5.77 (m, 1H)
36%
In 1,4-dioxane; at 110℃;Inert atmosphere;
A solution of 1H-pyrazol-3-ylamine (7 g, 84.24 mmol, 1 .00 equiv) and ethyl prop-2-ynoate (50 mL) in dioxane ( 1 0 g, 1 21 equiv) was stirred under nitrogen overnight at 110 "C. The reaction mixture was cooled to rt and the precipitated product was collected by filtration to give 4 g (36%) of the title compound as a light brown solid 1H NMR (300 MHz, DMSO-d6) delta 12.04 (s, 1H), 8.41 -8.44 (m, 1H), 7.71 (d. J = 18 Hz, 1H), 5.88 (d, J = 8. 1Hz, 1H), 5.77 (m, 1H)
36%
In 1,4-dioxane; at 110℃;Inert atmosphere;
0217] Step 1. 4H-Pyrazolor 5-alpyrimidin-5-one. A solution of lH-pyrazol-3-ylamine (7 g, 84.24 mmol, 1.00 equiv) and ethyl prop-2-ynoate (50 mL) in dioxane (10 g, 1.21 equiv) was stirred under nitrogen overnight at 110 C. The reaction mixture was cooled to rt and the precipitated product was collected by filtration to give 4 g (36%) of the title compound as a light brown solid. 1H NMR (300 MHz, DMSO-J6) delta 12.04 (s, 1H), 8.41-8.44 (m, 1H), 7.71 (d, J = 1.8 Hz, 1H), 5.88 (d, J = 8.1 Hz, 1H), 5.77 (m, 1H).
General procedure: A stirring solution of ketone (1, 1.0 mmol), aldehyde (2, 1.0 mmol) and KOtBu (2.1 mmol) in anhydrous ethanol (5 mL) was refluxed for an hour. Then aminopyrazole (3, 1.0 mmol) was added to the reaction mixture and the reaction mixture was refluxed for another two hours. The residue obtained after removal of the solvent was extracted with ethyl acetate, washed with water, brine and dried over anhydrous sodium sulphate. The crude product obtained after removal of the solvent was purified by silica gel column chromatography using ethyl acetate/hexane as the eluent to obtain pure product 4.
With copper(I) oxide; caesium carbonate; hydroxybenzaldoxime; In N,N-dimethyl-formamide; at 95℃;Inert atmosphere;
Step 1: 1-(4-Bromo-3-methoxyphenyl)-1H-pyrazol-3-amine A mixture of <strong>[755027-18-0]1-bromo-4-iodo-2-methoxybenzene</strong> (2.5 g, 7.99 mmol), 3-aminopyrazole (0.797 g, 9.59 mmol), salicylaldoxime (0.219 g, 1.598 mmol), Cu2O (91 mg, 0.479 mmol), and Cs2CO3 (3.9 g, 11.98 mmol) in DMF (8 mL) was degassed with N2 and heated at 95 C. overnight. After cooling to RT, the mixture was filtered through celite and rinsed with EtOAc. The filtrate was washed with water and brine. The organic solution was dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel chromatography (10%-60% EtOAc in Heptane) to give 1-(4-bromo-3-methoxyphenyl)-1H-pyrazol-3-amine (800 mg, MS: 270.3 [M+H+]).
1-(5,6-Difluoropyrimidin-4-yl)-1H-pyrazole-3-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With potassium carbonate; In cyclohexane; ethyl acetate; acetonitrile;
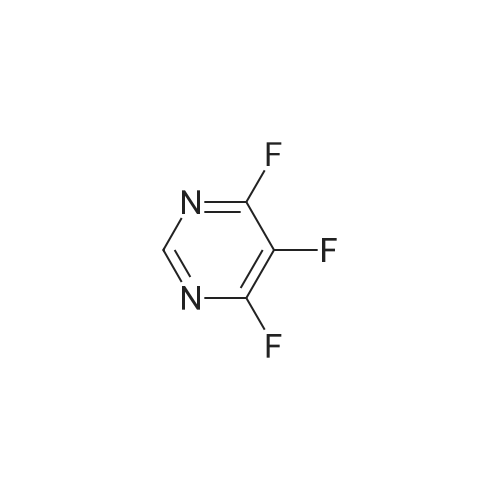
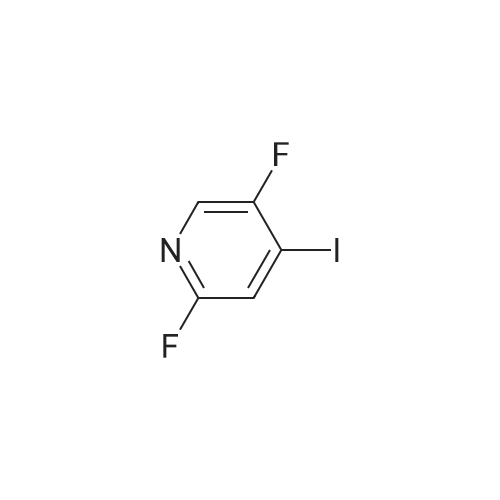
Step 1: 1-(5,6-Difluoropyrimidin-4-yl)-1H-pyrazole-3-amine (via route A-1) 1H-Pyrazole-3-amine (1.25 g) was initially charged in acetonitrile (75 ml), <strong>[17573-78-3]4,5,6-trifluoropyrimidine</strong> (2.02 g) and potassium carbonate (4.16 g) were added and the mixture was heated under reflux overnight. The reaction mixture was then diluted with dichloromethane, washed with water, dried over sodium sulphate and concentrated to dryness under reduced pressure. The residue was purified by column chromatography on silica gel using the mobile phase cyclohexane/ethyl acetate (gradient=2 h from 100% cyclohexane to 100% ethyl acetate). This gave 379 mg of the title compound. HPLC-MS: log P=0.86; mass (m/z): 198.1 (M+H)+; 1H-NMR (DMSO-D6) 5.73 (br. s, 2H), 6.01 (d, 1H), 8.34 (d, 1H), 8.46 (s, 1H).
1-(3-fluoro-4-iodopyridin-2-yl)-1H-pyrazole-3-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With potassium carbonate; In methanol; water; ethyl acetate;
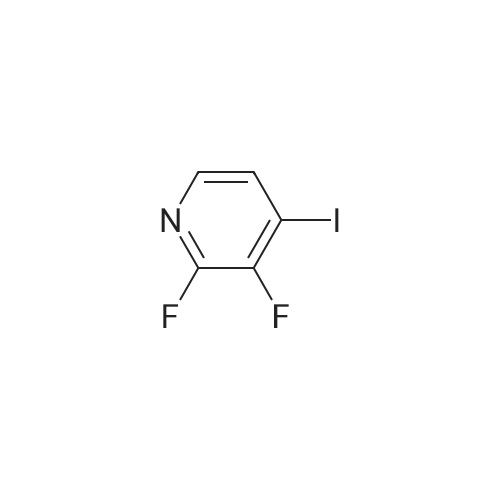
Step 1: 1-(3-Fluoro-4-iodopyridin-2-yl)-1H-pyrazole-3-amine (via A-1) A mixture of 3-aminopyrazole (0.50 g), <strong>[851386-34-0]2,3-difluoro-4-iodopyridine</strong> (1.45 g), potassium carbonate (3.3 g) and 10 ml anhydrous dimethyl sulphoxide was stirred at 85° C. for 48 hours. For work-up, 100 ml of water were added at room temperature and the mixture was extracted with ethyl acetate. The organic phase was dried with magnesium sulphate and then concentrated completely and chromatographed on silica gel using an ethyl acetate/methanol gradient. This gave 705 mg of the title compound. HPLC-MS: log P=1.38; mass (m/z): 304.9 (M+H)+; 1H-NMR (DMSO-D6) 5.91 (m, 1H), 7.62 (m, 1H), 7.85 (m, 1H), 8.08 (m, 1H).
1-(3,5-Difluoropyridin-2-yl)-1H-pyrazole-3-amine[ No CAS ]
1-(2,5-difluoropyridin-3-yl)-1H-pyrazole-3-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With potassium carbonate; In cyclohexane; ethyl acetate; acetonitrile;
Step 1: 1-(3,5-Difluoropyridin-2-yl)-1H-pyrazole-3-amine (via route A-1) 1H-Pyrazole-3-amine (1.80 g) was initially charged in acetonitrile (50 ml), <strong>[76469-41-5]2,3,5-trifluoropyridine</strong> (2.88 g) and potassium carbonate (5.99 g) were added and the mixture was heated under reflux overnight. The reaction mixture was then concentrated under reduced pressure, taken up in dichloromethane, washed with water, dried over sodium sulphate and concentrated to dryness under reduced pressure. The residue was purified by column chromatography on silica gel using the mobile phase cyclohexane/ethyl acetate (gradient=2 h from 100% cyclohexane to 100% ethyl acetate). This gave 610 mg of the title compound. HPLC-MS: log P=0.81; mass (m/z): 197.1 (M+H)+; 1H-NMR (CD3CN) 4.27 (br. s, 2H), 5.88 (d, 1H), 7.56-7.62 (m, 1H), 7.99-8.00 (m, 1H), 8.17-8.18 (m, 1H). 410 mg of the isomeric 1-(2,5-difluoropyridin-3-yl)-1H-pyrazole-3-amine were isolated as a further product. HPLC-MS: log P=1.15; mass (m/z): 197.0 (M+H)+; 1H-NMR (CD3CN) 4.39 (br. s, 2H), 5.92 (d, 1H), 7.80-7.82 (m, 1H), 7.94-7.95 (m, 1H), 8.02-8.07 (m, 1H).
1-(3,5-Difluoropyridin-2-yl)-1H-pyrazole-3-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
610 mg
With potassium carbonate; In acetonitrile;Reflux;
Step 1 : l-(3,5-Difluoropyridin-2-yl)-lH-pyrazole-3-amine (intermediate (Ib-1) lH-Pyrazole-3 -amine (1.80 g) was initially charged in acetonitrile (50 ml), <strong>[76469-41-5]2,3,5-trifluoropyridine</strong> (2.88 g) and potassium carbonate (5.99 g) were added and the mixture was heated under reflux overnight. The reaction mixture was then concentrated under reduced pressure, taken up in dichloromethane, washed with water, dried over sodium sulphate and concentrated to dryness under reduced pressure. The residue was purified by column chromatography on silica gel using the mobile phase cyclohexane/ethyl acetate (gradient = 2 h from 100% cyclohexane to 100% ethyl acetate). This gave 610 mg of the title compound. HPLC-MS: logP = 0.81 ; mass (m/z): 197.1 (M+H)+; 1H-NMR (CD3CN) 4.27 (br. s, 2H), 5.88 (d, 1H), 7.56 - 7.62 (m, 1H), 7.99 - 8.00 (m, 1H), 8.17 - 8.18 (m, 1H).
610 mg
With potassium carbonate; In acetonitrile;Reflux;
1H-Pyrazole-3-amine (1.80 g) was initially charged in acetonitrile (50 ml), <strong>[76469-41-5]2,3,5-trifluoropyridine</strong> (2.88g) and potassium carbonate (5.99 g) were added and the mixture was heated under reflux overnight. The reaction mixture was then concentrated under reduced pressure, taken up in dichloromethane, washed with water, dried over sodium sulphate and concentrated to dryness under reduced pressure. The residue was purified by column chromatography on silica gel using the mobile phase cyclohexane/ethyl acetate (gradient = 2 h from 100% cyclohexane to 100% ethyl acetate). This gave 610 mg of the title compound.HPLC-MS: logP = 0.81; mass (m/z): 197.1 (M+H)+; 1H-NMR (CD3CN) 4.27 (br. s, 2H), 5.88 (d, 1H),7.56 -7.62 (m, 1H), 7.99 - 8.00 (m, 1H), 8.17 - 8.18 (m, 1H).
With caesium carbonate; copper(l) chloride; In N,N-dimethyl-formamide; at 130℃; for 0.5h;Microwave irradiation;
To a solution of uSc g, 24O7mmol), CuCI (0238g. 24O7mmoi) and Cs2C03 (8.63g. 26.Smmoi) in 5mL of DMF was added 224(6.93g, 24.O7mmoi) and the resulting mixture was heated at130 C via MW irradiation for 30 minutes. The mixture was cooled to room temperature, and diluted with EA, washed with [120 (20 mLx3). The combined organic layer was washed with saturated NaCI and dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel to afford 230(lg, yield: 17.08%).
Next, ethyl acrylate (24.12 ml, 159 mmol) and 3-aminopyrazole (12.00 g, 144 mmol) were added, and the reaction mixture was heated to reflux for 3 h. The reaction mixture was cooled to room temperature, filtered, and the collected solids rinsed with excess EtOH. The isolated solids were dissolved in water (? 300 mL), which was cooled with and ice water bath. The aqueous mixture was acidified to pH= 1-2 with concentrated HCl. The resulting precipitate was filtered and rinsed with excess water. The isolated solid was dried in a vacuum oven at 30 C over the weekend to afford the title compound (J1) as an off-white solid (13.5 g, 62%). LRMS m/z (M+H+H2O) 170.1 found, 152.0 required.
7-(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-5(4H)-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
63%
General procedure: In a microwave vial, the 3-aminopyrazole 1 (1.0 mmol) was dissolved in 1,4-dioxane (4 mL). Ethyl 4,4,4-trifluoro-2-butynoate (2) (1.2mmol) was then added, and the mixture was degassed by bubbling with argon for 10 min. The sealed tube was heated at 110 C for 2 husing a microwave reactor. After cooling, MeONa (2.0 mmol) was added and the mixture was stirred for 12 h at r.t. The reaction mixture was neutralized with 10 mL of a solution of ethereal HCl (2 M) andthe solvent was removed under reduced pressure. The crude residue was purified using a silica gel column eluting with the mobile phases given below.
With caesium carbonate; In N,N-dimethyl-formamide; at 110℃;
To a solution of 1H-pyrazol-3-amine (500.6 mg) in N,N-dimethylformamide (10 mL) were added ethyl (2E)-3-ethoxyacrylate (1.3 mL) and cesium carbonate (2.93 g), and the mixture was stirred overnight at 110 C. The reaction mixture was neutralized with 2M aqueous hydrogen chloride solution, and extracted three times with ethyl acetate. Then, to the aqueous layer was added sodium chloride, and the mixture was extracted three times with tetrahydrofuran (15 mL)/ethyl acetate (15 mL). The obtained organic layer was washed with saturated brine (20 mL), and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane/ethyl acetate/methanol) to give the title compound (226.3 mg). MS: [M+H]+ 136.0.
With hydrogenchloride; copper dichloride; In water; acetonitrile; at 0℃; for 0.5h;
3-Chloro-1H-pyrazole (1): Copper (II) chloride (65.0 g, 481 mmol) and cone. HCl (20 mL) were added to a solution of lH-pyrazol-3-amine (20.0 g, 241 mmol) in acetonitrile (600 mL) at 0 C. The reaction mixture was stirred at 0 C for 30 min, then isopentyl nitrite (56.4 g, 481 mmol) was added dropwise. The mixture was stirred at room temperature for 2 days, and then quenched with aq. ammonia (10%, 1 L). The aqueous phase was extracted with EtOAc (5 x 500 mL) and the combined organic phases were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated. The residue was purified by silica chromatography (hexane: EtOAc= 20: 1) to give the title compound (10.3 g, 42%) as a green oil. 1H NMR (400 MHz, CDC13): delta 12.84 (bs, 1H), 7.62 (s, 1H), 6.29 (s, 1H).
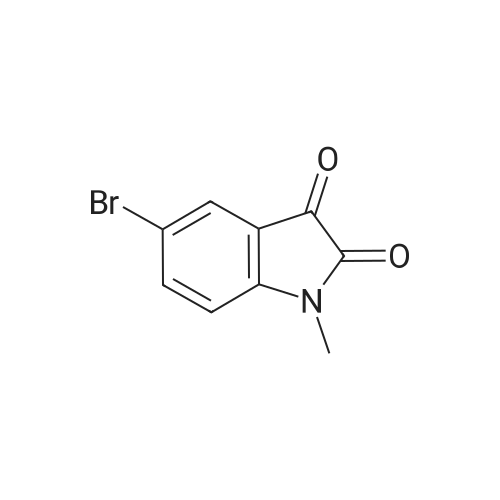
3-[(1H-pyrazol-3-yl)imino]-5-bromo-1-methylindolin-2-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
64%
With acetic acid; In ethanol;
General procedure: The general synthetic approach involved condensation of an equimolar mixture of corresponding substituted indole-2,3-dione (0.01 mol) and substituted pyrazole (0.01 mol) in absolute ethanol in the presence of 2,3-drops of glacial acetic acid for 3-4 h. On cooling, flakes separated out which were filtered and recrystallized from hot ethanol to give shining bright needles of Schiff base.
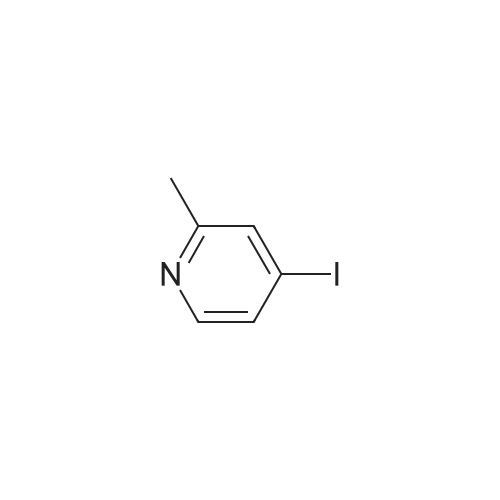
1-(2-methylpyridin-4-yl)-1H-pyrazol-3-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
63%
With caesium carbonate; copper(I) bromide; In N,N-dimethyl-formamide; at 120℃; for 14h;Sealed tube;
[253] 1H-pyrazol-3-amine (300 mg, 3.61 mmol), <strong>[22282-65-1]4-iodo-2-methyl-pyridine</strong> (817 mg, 3.73 mmol), copper (I) bromide (60 mg, 0.42 mmol), cesium carbonate (1.3 g, 3.99 mmol) were combined in DMF (4.0 mL) and heated in a sealed vessel at 120C for 14 h. The reaction mixture was partitioned into 1:1 ethyl acetate/water and filtered through a plug of silica gel. The layers were separated, and the aqueous further extracted with ethyl acetate (2 x 10 mL). The combined organics were washed with brine, dried (Na2SO4), filtered, and concentrated. The crude residue was purified by silica gel chromatography (40 g silica gel column; linear gradient of 10 - 100% ethyl acetate/heptane) to provide 1-(2-methylpyridin-4-yl)-1H-pyrazol-3-amine (420 mg; 63% yield) as a colorless solid. 1H NMR (400 MHz, DMSO-d6) delta 8.33 (d, J = 5.6 Hz, 1H), 8.27 (d, J = 2.7 Hz, 1H), 7.47 (d, J = 2.1Hz, 1H), 7.44 - 7.36 (m, 1H), 5.85 (d, J = 2.7 Hz, 1H), 5.32 (s, 2H), 2.45 (s, 3H) ppm. ESI-MS m/z calc. 174.09, found 175.58 (M+1).
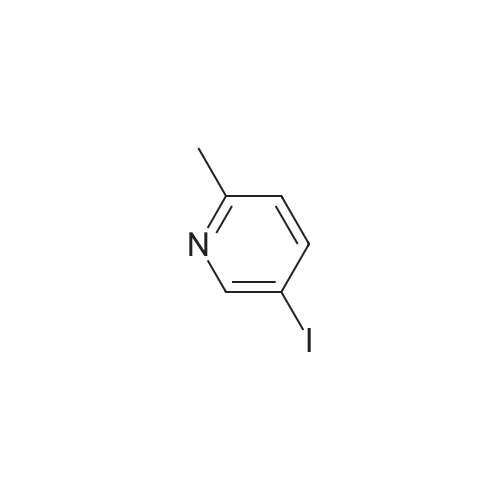
1-(2,5-difluoropyridin-4-yl)-1H-pyrazol-3-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
20%
With caesium carbonate; copper(I) bromide; In N,N-dimethyl-formamide; at 100℃; for 42h;Sealed tube;
[254] 1H-pyrazol-3-amine (500 mg, 6.02 mmol), <strong>[1017793-20-2]2,5-difluoro-4-iodo-pyridine</strong> (1.450 g, 6.02 mmol), copper (I) bromide (300 mg, 2.09 mmol), and cesium carbonate (3.03 g, 9.30 mmol) were combined and suspended in DMF (5.1 mL). The resultant reaction mixture was heated in a sealed vessel at 100C for 42 h. The reaction mixture was partitioned into 1:1 ethyl acetate/water (150 mL) and filtered through a plug of Celite. The layers were separated, and the aqueous further extracted with ethyl acetate (100 mL). The combined organics were dried (Na2SO4), filtered, and concentrated. The crude residue was purified by silica gel chromatography (80 g silica gel column; linear gradient of 10 - 50% ethyl acetate/heptane) to provide 1-(2,5-difluoropyridin-4-yl)-1H-pyrazol-3-amine (263 mg, 20% yield). 1H NMR (400 MHz, DMSO-d6) delta 8.27 (d, J = 4.1Hz, 1H), 8.06 (s, 1H), 7.34 (d, J = 5.4 Hz, 1H), 5.99 (d, J = 2.6 Hz, 1H), 5.61 (s, 2H) ppm. ESI-MS m/z calc. 196.06, found 197.10 (M+1).
1-(6-methylpyridin-3-yl)-1H-pyrazol-3-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
43%
With caesium carbonate; copper(I) bromide; In N,N-dimethyl-formamide; at 120℃; for 24h;Sealed tube;
[256] 1H-pyrazol-3-amine (500 mg, 6.02 mmol), <strong>[695-17-0]5-iodo-2-methylpyridine</strong> (1.32 g, 6.12 mmol), copper (I) bromide (300 mg, 2.09 mmol) and cesium carbonate (3.03 g, 9.30 mmol were combined and suspended in DMF (5.0 mL). The resultant reaction mixture was heated in a sealed vessel at 120C for 24 h. The reaction mixture was partitioned into 1:1 ethyl acetate/water (150 mL) and filtered through a plug of Celite. The layers were separated, and the aqueous further extracted with ethyl acetate (3 x 50 mL). The combined organics were dried (Na2SO4), filtered, and concentrated to provide a regioisomeric mixture of products. The residue was purified twice by reverse phase chromatography: the first time using an ISCO 150g C18 column and a linear gradient of 10-50% acetonitrile/water with TFA modifier, and the second time using an ISCO 150g C18Aq column and a linear gradient of 0-70% acetonitrile/water with TFA modifier. The resultant TFA salt was dissolved in dichloromethane and washed with saturated aqueous NaHCO3. The layers were separated, and the aqueous layer was further extracted with dichloromethane. The combined organics were dried (Na2SO4), filtered, and concentrated to provide 1-(6-methylpyridin-3-yl)-1H-pyrazol-3-amine (220 mg, 43% yield) as a colorless glass. 1H NMR (400 MHz, CDCl3) delta 7.91 (d, J = 2.6 Hz, 1H), 7.30 (d, J = 2.4 Hz, 1H), 7.03 (dd, J = 8.5, 2.7 Hz, 1H), 6.41 (d, J = 8.3 Hz, 1H), 4.90 (d, J = 2.4 Hz, 1H), 4.28 (s, 2H), 1.59 (s, 3H) ppm. ESI- MS m/z calc. 174.09, found 175.12 (M+1).
1-(2-(difluoromethoxy)pyridin-4-yl)-1H-pyrazol-3-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With caesium carbonate; copper(I) bromide; In N,N-dimethyl-formamide; at 110℃; for 16h;Inert atmosphere; Sealed tube;
[265] 1H-pyrazol-3-amine (200 mg, 2.41 mmol), <strong>[832735-56-5]4-bromo-2-(difluoromethoxy)pyridine</strong> (539 mg, 2.41 mmol), cesium carbonate (784 mg, 2.41 mmol), copper(I) bromide (69 mg, 0.48 mmol) and DMF (2.0 mL) were combined under nitrogen. The vessel was sealed and heated to 110 C for 16 h. The crude reaction mixture was filtered through Celite, washing filter pad with methanol. The filtrate was concentrated, and the residue was dissolve in dichloromethane and washed with 1N NaOH. The organics were collected and evaporated to provide 1-(2-(difluoromethoxy)pyridin-4-yl)-1H-pyrazol-3- amine, which was used without further manipulation. 1H NMR (400 MHz, DMSO-d6) delta 8.36 (d, J = 2.8 Hz, 1H), 8.16 (d, J = 5.8 Hz, 1H), 7.52 - 7.48 (m, 1H), 7.21 (d, J = 1.9 Hz, 1H), 5.89 (d, J = 2.7 Hz, 1H), 5.47 (s, 2H) ppm.
1-(2-methyl-1,2,4-triazol-3-yl)pyrazol-3-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
19%
With caesium carbonate; copper(I) bromide; In N,N-dimethyl-formamide; at 120℃;Sealed tube;
[280] 1H-pyrazol-3-amine (305 mg, 3.671 mmol, 1.0 eq), <strong>[16681-72-4]5-bromo-1-methyl-1,2,4-triazole</strong> (600 mg, 3.704 mmol, 1.01 eq), copper(I) bromide (106 mg, 0.739 mmol, 0.2 eq), cesium carbonate (1.26 g, 3.852 mmol, 1.05 eq), and N,N-dimethylformamide (2.2 mL) were combined. The reaction vessel was sealed and stirred overnight at 120 C. The mixture was diluted with dichloromethane and methanol, and the mixture was filtered though a layer of Celite. The filtrate was concentrated. The crude residue was purified by silica gel chromatography (linear gradient of 0-15% [2229] methanol/dichloromethane) to provide 1-(2-methyl-1,2,4-triazol-3-yl)pyrazol-3-amine (118 mg, 19% yield). 1H NMR (400 MHz, CDCl3) delta 7.99 (d, J = 2.7 Hz, 1H), 7.68 (s, 1H), 5.89 (d, J = 2.7 Hz, 1H), 4.18 (s, 3H), 3.91 (s, 2H) ppm. ESI-MS m/z calc. 164.08, found 165.23 (M+1).
1-(3-fluoropyridin-4-yl)-1H-pyrazol-3-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
48%
With caesium carbonate; copper(I) bromide; In 1-methyl-pyrrolidin-2-one; at 120℃; for 18h;Inert atmosphere; Sealed tube;
[289] 1H-pyrazol-3-amine (500 mg, 6.02 mmol), <strong>[22282-75-3]3-fluoro-4-iodo-pyridine</strong> (1.5 g, 6.73 mmol), copper (I) bromide (100 mg, 0.70 mmol), and cesium carbonate (3.0 g, 9.21 mmol) were combined and suspended in NMP (7.0 mL). The resultant mixture was heated in a sealed vessel at 120°C under an atmosphere of nitrogen for 18 h. The reaction mixture was partitioned into 1:1 ethyl acetate/water. The layers were separated, and the aqueous further extracted with ethyl acetate (2 x 20 mL). The combined organics were dried (Na2SO4), filtered, and concentrated. The crude residue was purified by reverse phase chromatography (ISCO C18 Aq 150g column; linear gradient of 10- 50percent acetonitrile in water with TFA modifier). Pure fractions were washed with saturated sodium bicarbonate and extracted with dichloromethane. The combined organic extracts were dried (Na2SO4), filtered, and concentrated to provide a yellow solid. The solid was further purified by trituration with warm ethyl acetate/heptane to provide 1-(3-fluoropyridin-4-yl)-1H-pyrazol-3-amine (431 mg; 48percent yield) as a yellow powder. 1H NMR (300 MHz, DMSO-d6) delta 8.70 (d, J = 5.1Hz, 1H), 8.42 (d, J = 5.6 Hz, 1H), 8.07 (t, J = 2.5 Hz, 1H), 7.82 (dd, J = 7.5, 5.6 Hz, 1H), 6.00 (d, J = 2.8 Hz, 1H), 5.44 (s, 2H) ppm. ESI-MS m/z calc. 178.07, found 179.00 (M+1).
With caesium carbonate; copper(I) bromide; In 1-methyl-pyrrolidin-2-one; at 120℃; for 60h;Inert atmosphere; Sealed tube;
[291] 1H-pyrazol-3-amine (600 mg, 7.22 mmol), <strong>[3034-55-7]<strong>[3034-55-7]5-bromothiazol</strong>e</strong> (1.30 g, 7.93 mmol), copper (I) bromide (240 mg, 1.67 mmol), and cesium carbonate (4.0 g, 12.28 mmol) were combined and suspended in NMP (6.0 mL). The resultant mixture was heated in a sealed vessel at 120C under an atmosphere of nitrogen for 60 h. The reaction mixture was partitioned into 1:1 ethyl acetate/brine. The layers were separated, and the aqueous further extracted with ethyl acetate. The combined organics were dried (Na2SO4), filtered, and concentrated. The crude residue was purified by silica gel chromatography (40g column, linear gradient of 0-50% ethyl acetate/heptane) to provide 1-(thiazol- 5-yl)-1H-pyrazol-3-amine (55 mg, 4% yield). ESI-MS m/z calc. 166.03, found 166.93 (M+1).
With caesium carbonate; copper(I) bromide; In 1-methyl-pyrrolidin-2-one; at 120℃; for 8h;Inert atmosphere; Sealed tube;
[292] To a solution of <strong>[92525-10-5]3-iodo-1-methyl-1H-pyrazole</strong> (4.0 g, 19.23 mmol) in NMP (60 mL) was added 1H- pyrazol-3-amine (1.6 g, 19.23 mmol), copper (I) bromide (3.0 g, 21 mmol) and cesium carbonate (15.6 g, 48.07 mmol). The resultant mixture was heated in a sealed vessel at 120C under an atmosphere of nitrogen for 8 h. The reaction mixture was partitioned into 1:1 ethyl acetate/brine. The layers were separated, and the aqueous further extracted with ethyl acetate. The combined organics were dried (Na2SO4), filtered, and concentrated to provide 1'-methyl-1'H-[1,3'-bipyrazol]-3-amine (2.0 g, 64% yield) as a brown oil which was used without further purification.
1-(2-methylpyrimidin-5-yl)-1H-pyrazol-3-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
31%
With caesium carbonate; copper(I) bromide; In 1-methyl-pyrrolidin-2-one; at 120℃; for 16h;Inert atmosphere; Sealed tube;
[294] 1H-pyrazol-3-amine (440 mg, 5.30 mmol), <strong>[7752-78-5]5-bromo-2-methyl-pyrimidine</strong> (1.0 g, 5.78 mmol), copper (I) bromide (80 mg, 0.56 mmol), and cesium carbonate (2.4 g, 7.37 mmol) were combined and suspended in NMP (6.0 mL). The resultant mixture was heated in a sealed vessel under nitrogen at 120 °C for 16 h. The reaction mixture was partitioned into 1:1 ethyl acetate/water. The layers were separated, and the aqueous further extracted with ethyl acetate (2 x 25 mL). The combined organics were washed with brine (20 mL), dried (Na2SO4), filtered, and concentrated to yield an orange crystalline solid of 90percent purity. The solid was triturated with ethyl acetate/heptane to provide 1-(2- methylpyrimidin-5-yl)-1H-pyrazol-3-amine (303.9 mg, 31percent yield). 1H NMR (300 MHz, DMSO-d6) delta 8.98 (d, J = 2.0 Hz, 2H), 8.25 (d, J = 2.6 Hz, 1H), 5.82 (d, J = 2.6 Hz, 1H), 5.30 (s, 2H), 2.60 (d, J = 1.8 Hz, 3H) ppm. ESI-MS m/z calc. 175.09, found 176.07 (M+1).
With copper(I) oxide; potassium hydroxide; In dimethyl sulfoxide; at 120℃; for 12h;Inert atmosphere;
[297] 1H-pyrazol-3-amine (1.7 g, 20.5 mmol, 1.0 eq), <strong>[73583-39-8]3-bromo-5-chloropyridine</strong> (5.9 g, 30.8 mmol, 1.5 eq), cuprous oxide (300 mg, 2.1 mmol, 0.1 eq), potassium hydroxide (2.3 g, 41.0 mmol, 2.0 eq), and anhydrous DMSO (80 mL) were combined and heated at 120 °C for 12 h under an atmosphere of argon. The mixture was poured into 200 mL of water and extracted with ethyl acetate (3 100 mL). The organic layer was dried (Na2SO4), filtered, and concentrated. The residue was purified by silica gel chromatography (isocratic 1:1 ethyl acetate/heptane) to provide an impure product. The material was further purified by reverse phase HPLC (acetonitrile/water with NH4HCO3 modifier) to provide 1- (5-chloro-3-pyridyl)pyrazol-3-amine (1.0 g, 25.1 percent).
N-(2-methyl-4-nitrophenyl)-1H-pyrazole-3-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
3-Aminopyrazole (1.25 g, 15.00 mmol), potassium carbonate (4.14 g, 30.00 mmol) and N,N-dimethylformylThe amine (50 mL) was added to a 100 mL single-necked flask and heated to 65 ° C for 2 hours. Slowly add <strong>[455-88-9]2-fluoro-5-nitrotoluene</strong>(2.79g, 18.00mmol)A solution of N,N-dimethylformamide (20 mL) was stirred for 10 h. Cool to room temperature and mixThe mixture was poured into ice water (100 mL) and stirred for 30 minutes, a solid precipitated, filtered, and the filter cake was washed with water (100 mL x 3) to giveThe title compound (brown solid, 2.71 g, yield: 83percent).
6-fluoropyrazolo[1,5-a]pyrimidine-5,7-diol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
55.7%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In 1,4-dioxane; at 110℃; for 18h;Inert atmosphere;
Under nitrogen protection,Add 2.0 g (24.1 mmol) of raw material 1a,4.3 g (24.1 mmol) of the starting material 1b and 7.3 g (48.2 mmol) of DBU were dissolved in 40 mL of 1,4-dioxane and reacted at 110 C. for 18 hours.TLC detection. After the reaction, the solution was concentrated by rotary evaporation, 20 mL of ice water was added, and under the protection of nitrogen, about 14 mL of 5M HCl was added to adjust the pH of the solution to about 2, and a solid precipitated.Stir at low temperature, suction filter, and vacuum dry to obtain 2.27 g of the product as a gray solid with a yield of 55.7%.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping