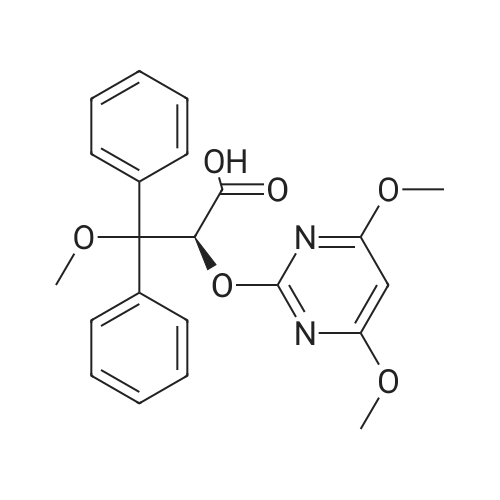
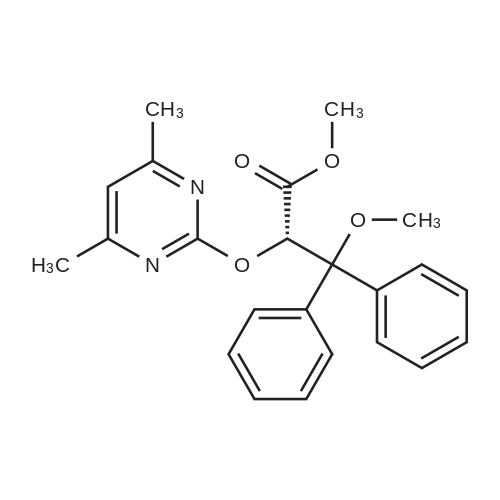
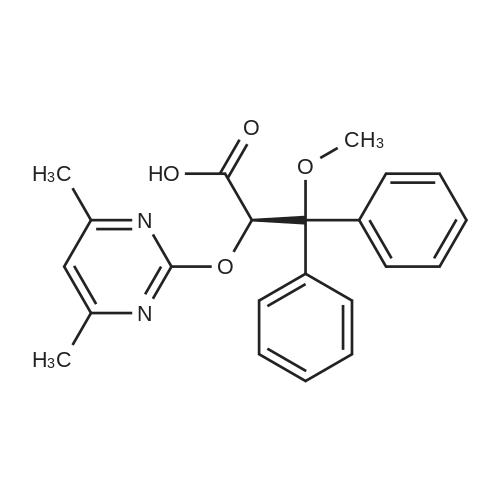
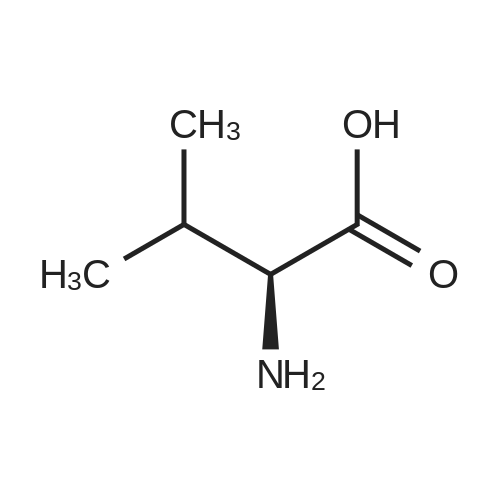
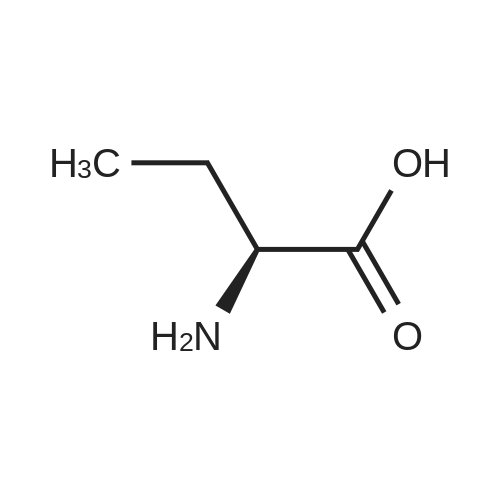
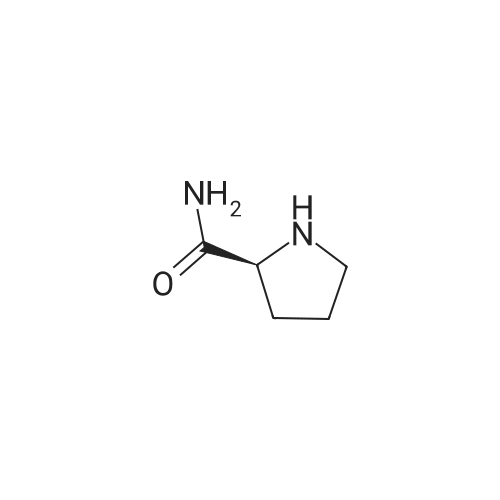
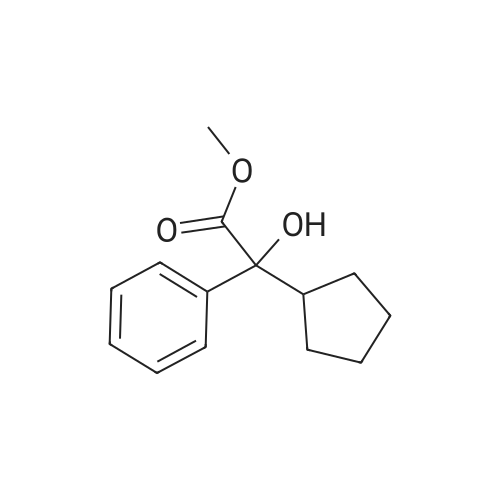
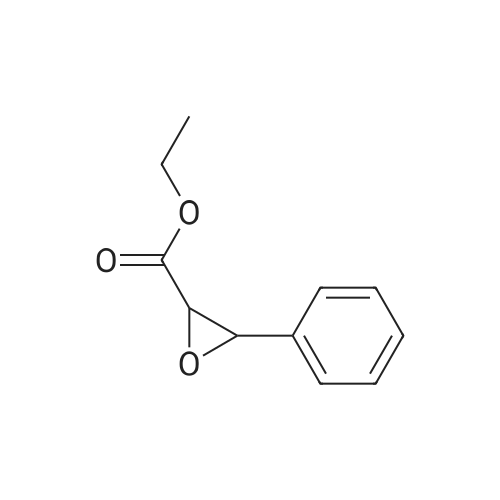
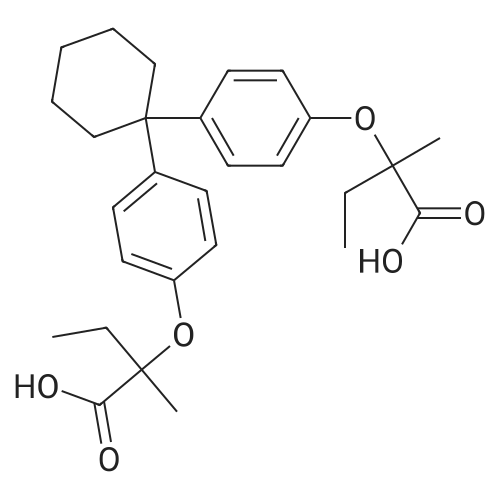
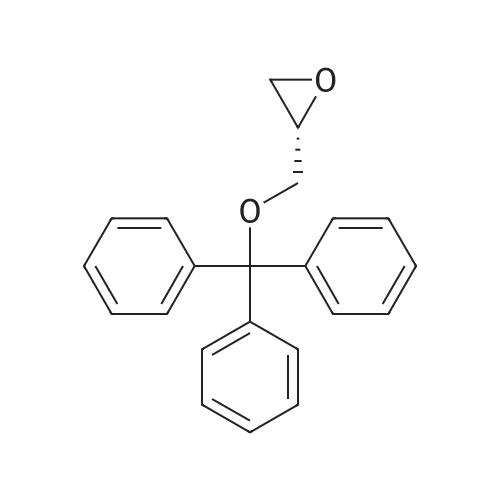
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
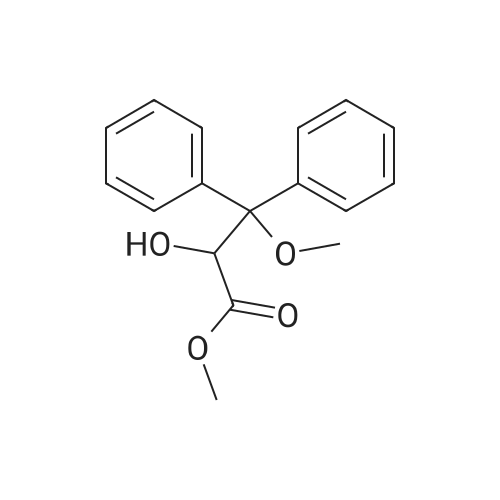
500 g (1.75 mol) of (RS) 2-hydroxy-3-methoxy-3,3-diphenyl methyl propanoate Suspended and agitated in 1,000 mL of water, 950 mL (1.90 mol) of 2 mol / L sodium hydroxide was added, and the mixture was stirred at 100 ° C. for 1 hour. The reaction solution was cooled to room temperature, 1500 mL of tert-butyl methyl ether was added, 700 mL of 10percent hydrochloric acid was added, and the mixture was stirred for 20 minutes to dissolve the precipitated solid. After liquid-liquid separation, the organic layer was washed with 1000 mL of water. 1200 mL of heptane was added, and the solvent was distilled off under reduced pressure (200 mmHg) at 40 ° C. to precipitate crystals. 1000 mL of heptane was added, and the mixture was stirred at 0 ° C. for 1 hour. The crystals were collected by filtration and dried under reduced pressure at 40 ° C. for 7 hours,(RS) -2-hydroxy-3-methoxy-3,3-diphenylpropanoic acid 431 g (yield 91percent) was obtained.
90%
at 90 - 95℃; for 1.5 h;
Step-HI : Preparation of 2-hydroxy-3-methoxy-3,3-diphenyl propionic acid of the formula -(IV) :Into a 5L round bottomed flask a mixture water(l.OL) and compound of formula -III (200g) from step-II were charged and stirred for 15 minutes. IN aqueous sodium hydroxide solution was charged and the reaction mass was stirred for 15 minutes.Reaction mass was heated to 90-95°C and maintained at the same temperature for one hour. The reaction mass was brought to room temperature adjustment of pH was carried out with IN hydrochloric acid solution(1.6L) to 2-3. The product slurry was cooled to 5- 10°C and maintained at the same temperature for 2hours. The product was filtered and dried at 60-65°CDry weight : 172g(90percent)Purity by HPLC : 99.88percentMelting range : 100- 102 °C
123 g
With water; sodium hydroxide In methanol at 45 - 50℃; for 1 h;
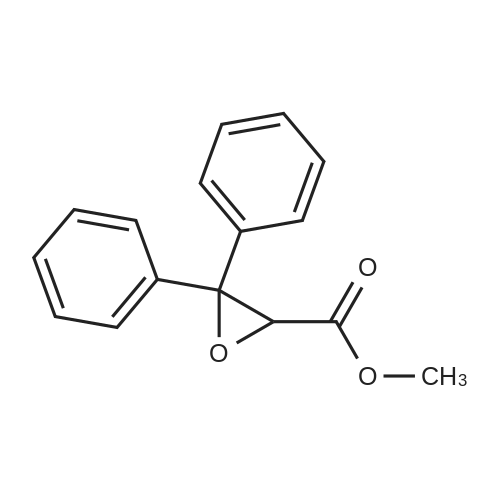
140 g of methyl 3,3-diphenyloxirane-2-carboxylate was dissolved in 280 ml of methanol and then cooled to 0°C .6.9 ml of BF3OEt2 was added slowly and stirred at 0°C for 2 hours. When the methyl 3,3-diphenyloxirane-2-carboxylate disappears completely and methyl 2-hydroxy-3-methoxy-3,3-diphenylpropionate is produced,65.8 g of NaOH was dissolved in 980 ml of water and slowly added dropwise. Methanol (140 ml) was added, the temperature was raised to 45 to 50°C, and the mixture was stirred for 1 hour. When the reaction was completed, the methanol was distilled off under reduced pressure, and diluted with 700 ml of dichloromethane. 300 ml of 6N HCl aqueous solution was added to adjust the pH to 2, and the organic layer was separated. The organic layer was washed with 300 ml of brine, dried, filtered and distilled under reduced pressure, and recrystallized from 500 ml of hexane to obtain 123 g of 2-hydroxy-3-methoxy-3,3-diphenylpropionic acid (yield: 82percent).
Reference:
[1] Patent: JP2017/128528, 2017, A, . Location in patent: Paragraph 0005; 0040
[2] Patent: WO2012/17441, 2012, A1, . Location in patent: Page/Page column 11
[3] Patent: WO2010/70658, 2010, A2, . Location in patent: Page/Page column 18-19
[4] Patent: US2011/263854, 2011, A1, . Location in patent: Page/Page column 8
[5] Organic Process Research and Development, 2001, vol. 5, # 1, p. 16 - 22
[6] Patent: KR2016/39907, 2016, A, . Location in patent: Paragraph 0094; 0095
3
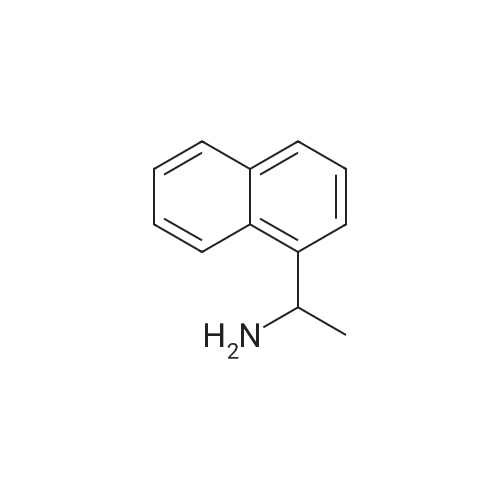
[ 119-61-9 ]
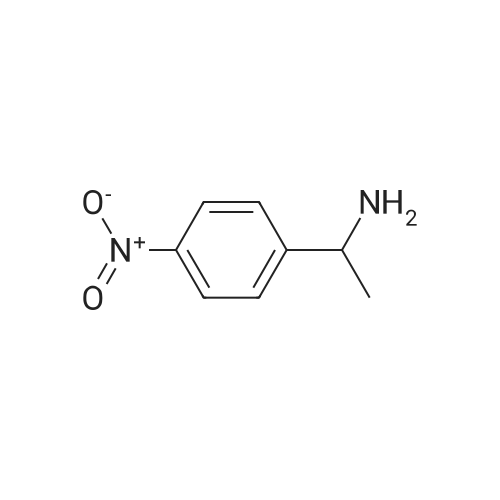
[ 124-41-4 ]
[ 96-34-4 ]
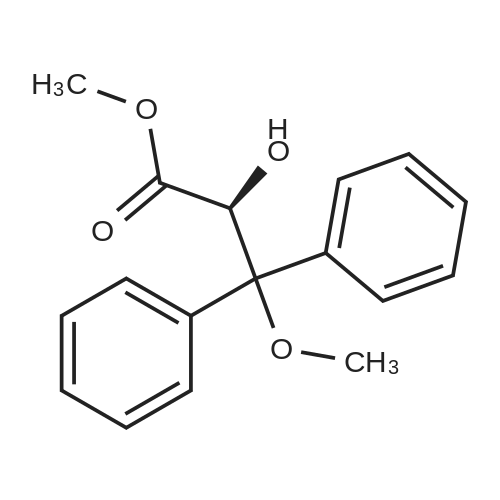
[ 178306-51-9 ]
Yield
Reaction Conditions
Operation in experiment
80%
Stage #1: at -10 - 40℃; for 1 h; Stage #2: With toluene-4-sulfonic acid In methanol at 20℃; for 1 h; Stage #3: at 97℃;
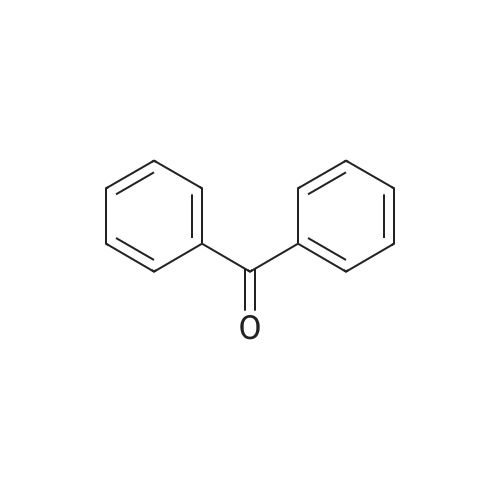
91.11 g (0.5 mol) of benzophenone and 45.92 g (0.85 mol) of sodium methoxide were added,Was suspended in 150 ml of methyl t-butyl ether (MTB) at room temperature. After cooling to -10 ° C.,92.24 g (0.85 mol) of methyl chloroacetate,In such a way that the internal temperature rose to 40 ° C. while the outside was cooled in a bath at -10 ° C.next,The mixture was stirred for 1 hour at naturally occurring temperature without cooling.250 ml of water was added,After stirring for a while,The aqueous phase was separated.The MTB phase was washed with 250 ml of an aqueous dilute sodium chloride solution.After changing the solvent to methanol (250 ml)A solution of 1 g of p-toluenesulfonic acid in 10 ml of methanol was added at room temperature.The mixture was stirred at naturally occurring temperature for 1 hour,Followed by heating under reflux.While methanol was distilled off,400 g of 10percent strength sodium hydroxide solution was added dropwise,Finally 60 ml of water was added.This methanol was added drop-Until the bottom temperature reached 97 ° C,And distilled.After cooling to 55 ° C., 190 ml of MTB was added and the mixture was acidified to pH 2 with about 77 ml of concentrated hydrochloric acid.After cooling to room temperature, the aqueous phase was distilled off, and the organic phase was concentrated by distilling 60 ml of MTB.The product was crystallized by adding 500 ml of heptane and gradually cooled to room temperature.The crude crystalline solid was filtered off with suction, washed with heptane and dried in a vacuum oven at 40 ° C. to constant weight.Yield: 108.9 g (80percent), HPLC> 99.5percent area.
Reference:
[1] Patent: US2011/263854, 2011, A1,
[2] Patent: WO2012/17441, 2012, A1,
[3] Organic Process Research and Development, 2001, vol. 5, # 1, p. 16 - 22
[4] Patent: KR2016/39907, 2016, A,
[5] Patent: JP2017/128528, 2017, A,
5
[ 76527-25-8 ]
[ 178306-51-9 ]
Reference:
[1] Patent: WO2012/17441, 2012, A1,
[2] Patent: KR2016/39907, 2016, A,
[3] Patent: JP2017/128528, 2017, A,
6
[ 178306-52-0 ]
[ 178306-51-9 ]
Reference:
[1] European Journal of Organic Chemistry, 2006, # 20, p. 4573 - 4577
7
[ 178306-51-9 ]
[ 178306-52-0 ]
Yield
Reaction Conditions
Operation in experiment
89%
With (S)-1,2,3,4-tetrahydronapht-1-yl-amine In tert-butyl methyl ether for 1 h; Reflux
500 mg (1.84 mmol) of (RS) -2-hydroxy-3-methoxy-3,3-diphenylpropanoic acid was dissolved in 8 mL of 2-propanol to prepare , 135 mg (0.92 mmol) of (S) - (+) -1,2,3,4-tetrahydro-1-naphthylamine was added. After stirring for 1 hour under heating reflux, it was cooled to room temperature and stirred for 2 hours. Precipitated crystals were collected by filtration, washed with 2-propanol, and dried under reduced pressure.The same operation as in Example 1 was carried out using the solvents in Table 1.
55%
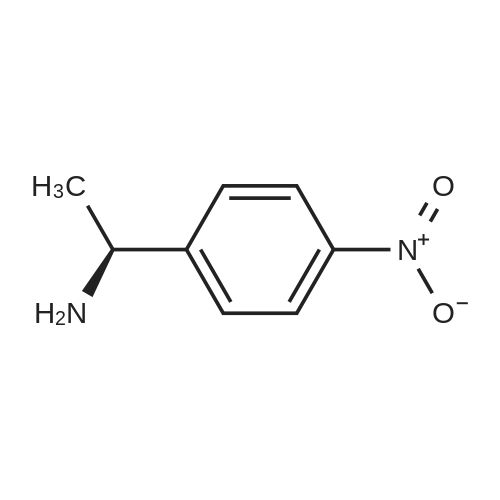
With (S)-1-(p-nitrophenyl)ethylamine In tert-butyl methyl ether; acetone at 10 - 55℃; for 13 h; Resolution of racemate
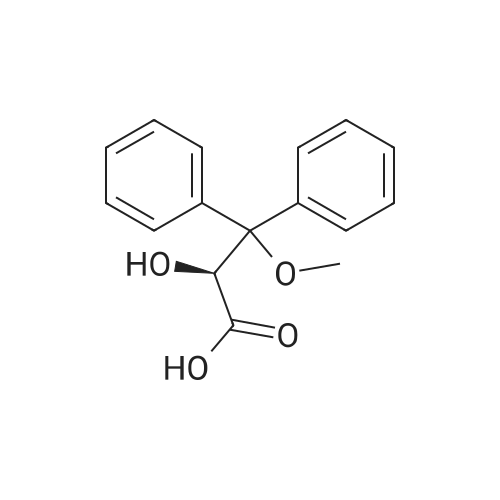
Step-IV : preparation of S-2-hydroxy-3-methoxy-3,3-diphenyl propionic acid of the formula -(I) :Into a 3L round bottomed flask compound of formula(IV)from step-III(200g) was added to a mixture methyl tert-butyl ether(1.25L) and acetone(1.5L). Reaction mass was heated to reflux temperature (50-55°C) and S-(-)p-nitro phenyl ethyl amine(61g dissolved in 250ml of MTB) was added slowly during 30minutes at reflux temperature. Reaction mass was maintained at the same temperature for one hour and cooled to 10- 15°C and maintained at the same temperature for 12hours. It was filtered and washed with MTB(500ml) . The wet diastereomeric salt was suspended in a mixture of MTB(500ml) and water(1.5L) and acidified with concentrated hydrochloric acid (30ml). the resulting mixture was stirred for 30minutes and the organic layer was separated. Aqueous layer was extracted with MTB(500ml) and the combined MTB layer was washed with water and distilled completely under vacuum. Reaction mass was brought to 40°C and mixture of MTB (138ml)and n-heptane(322ml) were charged and the cooled to 25-30°CThe crystalline compound of formula -I was filtered and dried at 50-60°C Dry weight : 55g(55percent)Purity by HPLC : 99.97percent(chemical purity)99.98percent(chiral purity)Melting range : 123-125°C
35.6%
With sodium methylate; L-proline methyl ester monohydrochloride In methanol; tert-butyl methyl ether at 15 - 30℃; for 20 h; Resolution of racemate; Autoclave; Industrial scale
L-Proline methyl ester hydrochloride (6.5 kg, 39.25 mol) and anhydrous methanol (3.5 L) were added to a 10 L reaction flask. After stirring well, sodium methoxide (2.12 kg, 39.25 mol) Of anhydrous methanol (5.0L) mixture, after completion of the addition, the reaction was stirred at (15 ~ 30 ° C) for 20 ~ 30min,The mixture was charged to a 300-L reaction vessel, and then a solution of a raw material A (10.68 kg, 39.25 mol) in t-butyl methyl ether (90 L) was also charged to the above reaction vessel to control the temperature at 15-30 ° C ) Was stirred for 20h,Methyl tert-butyl ether (180L) was added to the reaction kettle, and the internal temperature was controlled by stirring at -5 ~ 0 ° C for 30min by circulating cooling. The mixture was filtered and the filtrate was transferred to a 500L reactor.Add hydrochloric acid 3.5L with stirring, purified water 130L, stirred for 3 ~ 5min, allowed to stand for 10 ~ 15min, discard the water layer,The organic layer was dried over anhydrous magnesium sulfate (10 kg), filtered, and the filtrate was concentrated and n-heptane wasadded(37L) crystallization 1.5h.Filtration and drying gave 3.81 kg, yield 35.6percent.HPLC Purity: 99.1percent, Chiral purity: 96.3percent.
35%
With sodium methylate; L-proline methyl ester monohydrochloride In methanol; tert-butyl methyl ether at 20℃;
148.8 g (0.826 mol) of a methanol solution of 30percent concentration sodium methanolate,Was added dropwise to 240 g (0.826 mol) of methanol solution of L-proline methyl ester hydrochloric acid at 57percent concentration at room temperature,And 2.4 l MTB and 225 g (0.826 mol)Of 2-hydroxy-3-methoxy-3,3-diphenylpropionic acid were added.While dropping 2.4 L of MTB at room temperature,At the same time, 2680 ml of the MTB / methanol mixture was distilled off,The mixture was slowly cooled at room temperature,The crystals (R-2-hydroxy-3-methoxy-3,3-diphenylpropionic acid × L-proline methyl ester)This solid was washed with 150 ml of MTB.From this, 1.5 l of MTB was distilled off, the filtrate was concentrated,Then 1.0 liter of water was added.Concentrated hydrochloric acid was added at room temperature to adjust the pH to 1.2,After stirring and phase separation, the aqueous phase was separated and extracted with 0.4 l MTB.The combined organic phases were extracted with 0.4 l of water.After MTB was stripped, the residue was dissolved in toluene (65.0 ml) under reflux,And the seed was given to crystallize the product and allowed to cool slowly.The filtrate was suction filtered, washed with toluene,When dried in a vacuum oven,78.7 g of S-2-hydroxy-3-methoxy-3,3-diphenylpropionic acid were obtained (yield 35percent based on racemate).
32.91%
Stage #1: With (S)-1-(1-Naphthyl)ethylamine In methanol; tert-butyl methyl ether at 25 - 30℃; for 1 h; Stage #2: With hydrogenchloride In tert-butyl methyl ether; water for 0.5 h;
To a stirred solution of 2-hydroxy-3-methoxy-3,3-diphenylpropionic acid (compound VII; 240 gms/ 0.88 moles) in a mixture of tert-butyl methyl ether (1.2 lit) and methanol (1.2 lit) was added (S)-(-)- l-(l-Naphthyl) ethyl amine (82.8 gms/ 0.484 moles). The reaction mass was farther stirred for 1 hour at 25-30°C. The solid was isolated by filtration, washed with tert-butyl methyl ether (500 ml) and dried.The solid was stirred in a mixture of distilled water (1.2 lit) and tert-butyl methyl ether (1.2 lit) and cooled to 10-15°C. The reaction mass was acidified with cone. HC1 and stirred for 30 minutes. The organic phase was separated; aqueous phase was extracted with tert-butyl methyl , ether (1.0 lit). The organic phases were combined together, washed with brine, and concentrated under vacuum at 25-30°C. The residue was stirred in n-Hepatne (720 ml). The solid was isolated by filtration and dried to give 79 g of the title compound (VIII).Efficiency: 32.91 percentPurity by HPLC: 99.5percentChiral purity: 98.1percent
With (S)-1,2,3,4-tetrahydronapht-1-yl-amine; In tert-butyl methyl ether; for 1h;Reflux;
500 mg (1.84 mmol) of (RS) -2-hydroxy-3-methoxy-3,3-diphenylpropanoic acid was dissolved in 8 mL of 2-propanol to prepare , 135 mg (0.92 mmol) of (S) - (+) -1,2,3,4-tetrahydro-1-naphthylamine was added. After stirring for 1 hour under heating reflux, it was cooled to room temperature and stirred for 2 hours. Precipitated crystals were collected by filtration, washed with 2-propanol, and dried under reduced pressure.The same operation as in Example 1 was carried out using the solvents in Table 1.
55%
With (S)-1-(p-nitrophenyl)ethylamine; In tert-butyl methyl ether; acetone; at 10 - 55℃; for 13h;Resolution of racemate;
Step-IV : preparation of S-2-hydroxy-3-methoxy-3,3-diphenyl propionic acid of the formula -(I) :Into a 3L round bottomed flask compound of formula(IV)from step-III(200g) was added to a mixture methyl tert-butyl ether(1.25L) and acetone(1.5L). Reaction mass was heated to reflux temperature (50-55C) and S-(-)p-nitro phenyl ethyl amine(61g dissolved in 250ml of MTB) was added slowly during 30minutes at reflux temperature. Reaction mass was maintained at the same temperature for one hour and cooled to 10- 15C and maintained at the same temperature for 12hours. It was filtered and washed with MTB(500ml) . The wet diastereomeric salt was suspended in a mixture of MTB(500ml) and water(1.5L) and acidified with concentrated hydrochloric acid (30ml). the resulting mixture was stirred for 30minutes and the organic layer was separated. Aqueous layer was extracted with MTB(500ml) and the combined MTB layer was washed with water and distilled completely under vacuum. Reaction mass was brought to 40C and mixture of MTB (138ml)and n-heptane(322ml) were charged and the cooled to 25-30CThe crystalline compound of formula -I was filtered and dried at 50-60C Dry weight : 55g(55%)Purity by HPLC : 99.97%(chemical purity)99.98%(chiral purity)Melting range : 123-125C
35.6%
With sodium methylate; L-proline methyl ester monohydrochloride; In methanol; tert-butyl methyl ether; at 15 - 30℃; for 20h;Resolution of racemate; Autoclave; Industrial scale;
L-Proline methyl ester hydrochloride (6.5 kg, 39.25 mol) and anhydrous methanol (3.5 L) were added to a 10 L reaction flask. After stirring well, sodium methoxide (2.12 kg, 39.25 mol) Of anhydrous methanol (5.0L) mixture, after completion of the addition, the reaction was stirred at (15 ~ 30 C) for 20 ~ 30min,The mixture was charged to a 300-L reaction vessel, and then a solution of a raw material A (10.68 kg, 39.25 mol) in t-butyl methyl ether (90 L) was also charged to the above reaction vessel to control the temperature at 15-30 C ) Was stirred for 20h,Methyl tert-butyl ether (180L) was added to the reaction kettle, and the internal temperature was controlled by stirring at -5 ~ 0 C for 30min by circulating cooling. The mixture was filtered and the filtrate was transferred to a 500L reactor.Add hydrochloric acid 3.5L with stirring, purified water 130L, stirred for 3 ~ 5min, allowed to stand for 10 ~ 15min, discard the water layer,The organic layer was dried over anhydrous magnesium sulfate (10 kg), filtered, and the filtrate was concentrated and n-heptane wasadded(37L) crystallization 1.5h.Filtration and drying gave 3.81 kg, yield 35.6%.HPLC Purity: 99.1%, Chiral purity: 96.3%.
35%
With sodium methylate; L-proline methyl ester monohydrochloride; In methanol; tert-butyl methyl ether; at 20℃;
148.8 g (0.826 mol) of a methanol solution of 30% concentration sodium methanolate,Was added dropwise to 240 g (0.826 mol) of methanol solution of L-proline methyl ester hydrochloric acid at 57% concentration at room temperature,And 2.4 l MTB and 225 g (0.826 mol)Of 2-hydroxy-3-methoxy-3,3-diphenylpropionic acid were added.While dropping 2.4 L of MTB at room temperature,At the same time, 2680 ml of the MTB / methanol mixture was distilled off,The mixture was slowly cooled at room temperature,The crystals (R-2-hydroxy-3-methoxy-3,3-diphenylpropionic acid × L-proline methyl ester)This solid was washed with 150 ml of MTB.From this, 1.5 l of MTB was distilled off, the filtrate was concentrated,Then 1.0 liter of water was added.Concentrated hydrochloric acid was added at room temperature to adjust the pH to 1.2,After stirring and phase separation, the aqueous phase was separated and extracted with 0.4 l MTB.The combined organic phases were extracted with 0.4 l of water.After MTB was stripped, the residue was dissolved in toluene (65.0 ml) under reflux,And the seed was given to crystallize the product and allowed to cool slowly.The filtrate was suction filtered, washed with toluene,When dried in a vacuum oven,78.7 g of S-2-hydroxy-3-methoxy-3,3-diphenylpropionic acid were obtained (yield 35% based on racemate).
32.91%
To a stirred solution of 2-hydroxy-3-methoxy-3,3-diphenylpropionic acid (compound VII; 240 gms/ 0.88 moles) in a mixture of tert-butyl methyl ether (1.2 lit) and methanol (1.2 lit) was added (S)-(-)- l-(l-Naphthyl) ethyl amine (82.8 gms/ 0.484 moles). The reaction mass was farther stirred for 1 hour at 25-30C. The solid was isolated by filtration, washed with tert-butyl methyl ether (500 ml) and dried.The solid was stirred in a mixture of distilled water (1.2 lit) and tert-butyl methyl ether (1.2 lit) and cooled to 10-15C. The reaction mass was acidified with cone. HC1 and stirred for 30 minutes. The organic phase was separated; aqueous phase was extracted with tert-butyl methyl , ether (1.0 lit). The organic phases were combined together, washed with brine, and concentrated under vacuum at 25-30C. The residue was stirred in n-Hepatne (720 ml). The solid was isolated by filtration and dried to give 79 g of the title compound (VIII).Efficiency: 32.91 %Purity by HPLC: 99.5%Chiral purity: 98.1%
ExampIe-3: Preparation of (S)-2-hydroxy-3-methoxy-3,3-diphenylpropionic acid compound of formula-7:Methanolic sodium methoxide solution (33 grams) was added to the solution of L-proline methyl ester hydrochloride (30.38 grams) in methanol (28 ml), stirred for 15 minutes at 25-35C then filtered to remove the unwanted solid. The solvent from the reaction mixture was distilled off under reduced pressure at 6O0C. The solution of 2-hydroxy-3-methoxy-3,3-diphenylpropionic acid compound of formula-6 (50 grams) in methyltertiarybutylether (530 ml) was added to the above residue. The reaction mixture was heated to reflux temperature (55-60C), stirred for 45 minutes and then cooled to 25-35C and further stirred for 30 minutes. The solid separated was filtered off and washed with MTBE. Water (220 ml) was added to the filtrate and pH was adjusted to 1.2 with hydrochloric acid. The aqueous and organic layers were separated and then aqueous layer extracted with MTBE. The total organic layer washed with water and then distilled off the solvent completely under reduced pressure at 60C. Toluene (100 ml) was added to the residue, heated to reflux temperature for 15 minutes then cooled to 25-350C and stirred for 45 minutes. The obtained solid was filtered off and washed with toluene then dried at 50-600C to get the title compound. Yield: 17 grams M.R: 118-1220C. S.O.R: + 26 (C= 0.5; MeOH)
With (R)-1-phenyl-ethyl-amine; In chloroform; at 40℃; for 0.5h;Product distribution / selectivity;
Preparation of (S)-2-hydroxy-3-methoxy-3,3-diphenylpropionic acid R(+)-phenyl ethyl amine (11.12 grams) was added to a solution of 2-hydroxy-3-methoxy-3,3-diphenylpropionic acid (25 grams) and chloroform (25 ml) at 40 C. and stirred for 30 minutes. The reaction mixture was cooled, and the solid obtained was filtered and washed with chloroform. Ethyl acetate (50 ml) and water (50 ml) was added to the obtained solid and then the reaction mixture was acidified with concentrated hydrochloric acid. The layers were separated and the aqueous layer extracted with ethyl acetate. The solvent from combined organic layer was distilled off completely under reduced pressure at below 60 C. The obtained residue was dissolved in toluene (30 ml) at 60-70 C. and then cooled to 25-30 C. The solid was filtered, washed with toluene and then dried to get the title compound.Yield: 5.5 grams; S.O.R: +28.32
With sodium methylate; L-proline methyl ester monohydrochloride; In methanol; tert-butyl methyl ether; at 25 - 60℃; for 1.25h;Reflux;Product distribution / selectivity;
Preparation of (S)-2-hydroxy-3-methoxy-3,3-diphenylpropionic acid compound of formula-7 Methanolic sodium methoxide solution (33 grams) was added to the solution of L-proline methyl ester hydrochloride (30.38 grams) in methanol (28 ml), stirred for 15 minutes at 25-35 C. then filtered to remove the unwanted solid. The solvent from the reaction mixture was distilled off under reduced pressure at 60 C. The solution of 2-hydroxy-3-methoxy-3,3-diphenylpropionic acid compound of formula-6 (50 grams) in methyltertiarybutylether (530 ml) was added to the above residue. The reaction mixture was heated to reflux temperature (55-60 C.), stirred for 45 minutes and then cooled to 25-35 C. and further stirred for 30 minutes. The solid separated was filtered off and washed with MTBE. Water (220 ml) was added to the filtrate and pH was adjusted to 1.2 with hydrochloric acid. The aqueous and organic layers were separated and then aqueous layer extracted with MTBE. The total organic layer washed with water and then distilled off the solvent completely under reduced pressure at 60 C. Toluene (100 ml) was added to the residue, heated to reflux temperature for 15 minutes then cooled to 25-35 C. and stirred for 45 minutes. The obtained solid was filtered off and washed with toluene then dried at 50-60 C. to get the title compound.Yield: 17 gramsM.R: 118-122 C.S.O.R: +26 (C=0.5; MeOH)
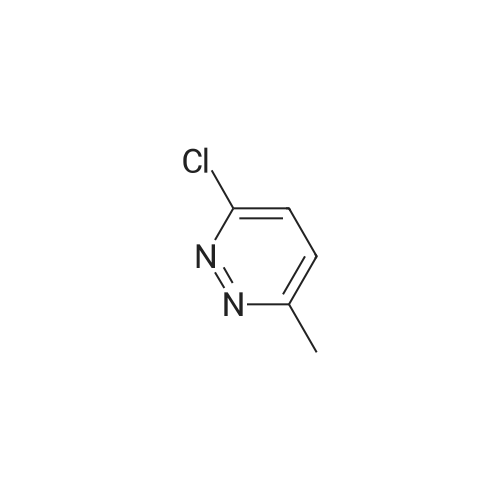
Example 1 2-(6-Methylpyridazin-3-yloxy)-3-methoxy-3,3-diphenylpropionic acid (I-517) 1.3 g (4.8 mmol) of <strong>[178306-51-9]2-hydroxy-3-methoxy-3,3-diphenylpropionic acid</strong> dissolved in DMF were added dropwise to a suspension of 0.43 g of NaH (14.3 mmol, 80% in white oil) in 10 ml of DMF. After stirring to room temperature for 30 minutes, the mixture was treated with 0.6 g (4.8 mmol) of 3-chloro-6-methylpyridazine in 10 ml of DMF and stirred overnight at room temperature. To complete the reaction, 0.6 g (4.8 mmol) of 3-chloro-6-methylpyridazine were then added again and the mixture was kept at 60 C. for 5 hours. It was poured onto ice water, extracted three times with ethyl acetate, the aqueous phase was brought to pH 2 with half-concentrated hydrochloric acid and the precipitate which was deposited was extracted with ethyl acetate. These ethyl acetate phases were dried with magnesium sulfate and then filtered and the solvent was stripped off under reduced pressure. 800 mg of the brown residue (1.19 g) were purified by means of MPLC, it being possible to isolate 199 mg of the desired product as a white solid. 1H-NMR (200 MHz, DMSO): 7.5 ppm (1 H, d), 7.2-7.3 (10 H, m), 7.1 (1 H, d), 6.3 (1 H, s), 3.3 (3 H, s), 2.5 (3 H, S). FAB-MS: 365 (M+H+)
Example 9 2-Hydroxy-3-methoxy-3,3-diphenylpropionic acid 91.11 g (0.5 mol) of benzophenone and 45.92 g (0.85 mol) of sodium methoxide were suspended in 150 ml of methyl tert-butyl ether (MTB) at room temperature. After cooling to -100 C., 92.24 g (0.85 mol) of methyl chloroacetate were added in such a way that the internal temperature rose to 40 C. while continuing to cool in a bath at -10 C. The mixture was then stirred without cooling at the autogenous temperature for one hour. After addition of 250 ml of water and brief stirring, the aqueous phase was separated off. The MTB phase was washed with 250 ml of dilute sodium chloride solution. After the solvent had been changed to methanol (250 ml), a solution of 1 g of p-toluenesulfonic acid in 10 ml of methanol was added at room temperature. The mixture was stirred at autogenous temperature for one hour and then heated to reflux. While distilling out the methanol, 400 g of a 10% strength sodium hydroxide solution was added dropwise, and finally 60 ml of water were added. The methanol was distilled out until the bottom temperature reached 97 C. After cooling to 55 C., 190 ml of MTB were added and the mixture was acidified to pH 2 with about 77 ml of concentrated HCl. After cooling to room temperature, the aqueous phase was separated off and the organic phase was concentrated by distilling out 60 ml of MtB [sic]. The product was crystallized by adding 500 ml of heptane and slowly cooling to room temperature. The coarsely crystalline solid was filtered off with suction, washed with heptane and dried to constant weight in a vacuum oven at 40 C. Yield: 108.9 g (80%), HPLC >99.5% area.
EXAMPLE 9 2-Hydroxy-3-methoxy-3,3-diphenylpropionic acid 91.11 g (0.5 mol) of benzophenone and 45.92 g (0.85 mol) of sodium methoxide were suspended in 150 ml of methyl tert-butyl ether (MTB) at room temperature. After cooling to -10 C., 92.24 g (0.85 mol) of methyl chloroacetate were added in such a way that the internal temperature rose to 40 C. while continuing to cool in a bath at -10 C. The mixture was then stirred without cooling at the autogenous temperature for one hour. After addition of 250 ml of water and brief stirring, the aqueous phase was separated off. The MTB phase was washed with 250 ml of dilute sodium chloride solution. After the solvent had been changed to methanol (250 ml), a solution of 1 g of p-toluenesulfonic acid in 10 ml of methanol was added at room temperature. The mixture was stirred at autogenous temperature for one hour and then heated to reflux. While distilling out the methanol, 400 g of a 10% strength sodium hydroxide solution was added dropwise, and finally 60 ml of water were added. The methanol was distilled out until the bottom temperature reached 97 C. After cooling to 55 C., 190 ml of MTB were added and the mixture was acidified to pH 2 with about 77 ml of concentrated HCl. After cooling to room temperature, the aqueous phase was separated off and the organic phase was concentrated by distilling out 60 ml of MTB. The product was crystallized by adding 500 ml of heptane and slowly cooling to room temperature. The coarsely crystalline solid was filtered off with suction, washed with heptane and dried to constant weight in a vacuum oven at 40 C. Yield: 108.9 g (80%), HPLC >99.5% area.
9
[ 100-39-0 ]
[ 178306-51-9 ]
[ 178306-59-7 ]
Yield
Reaction Conditions
Operation in experiment
With potassium carbonate; In N,N-dimethyl-formamide;
Example 12 Benzyl 3-methoxy-2-(4-methoxy-6,7-dihydro-5H-cyclopentapyrimidin-2-yloxy)-3,3-diphenylpropionate 24.48 g (90 mmol) of 3-methoxy-3,3-diphenyl-2-hydroxypropionic acid were dissolved in 150 ml of DMF, and 13.7 g (99 mmol) of potassium carbonate were added. The suspension was stirred at room temperature for 30 min. Then 10.7 ml (90 mmol) of benzyl bromide were added dropwise over the course of 5 min, and the mixture was stirred for 1 h, during which the temperature rose to 32 C. To this mixture were successively added 24.84 g (180 mmol) of K2 CO3 and 20.52 g (90 mmol) of 2-methanesulfonyl-4-methoxy-6,7-dihydro-5H-cyclopentapyridine [sic], and the mixture was stirred at 80 C. for 3 h. For workup, the contents of the flask were diluted with about 600 ml of H2 0 and cautiously acidified with concentrated HCl, and 250 ml of ethyl acetate were added. 31.4 g of pure product precipitated and were filtered off. The ethyl acetate phase was separated from the mother liquor, the aqueous phase was extracted again with ethyl acetate, and the combined organic phases were concentrated. The oily residue (19 g) was purified by chromatography (cyclohexane/ethyl acetate=9/1) to result in a further 10.5 g of pure product. Total yield: 41.9 g (82.2 mmol)=91%; Melting point 143-147 C.; MS: MH+ =511
2-methanesulfonyl-4-methoxy-6,7-dihydro-5H-Ocyclopentapyrimidine[ No CAS ]
[ 7732-18-5 ]
[ 100-39-0 ]
[ 178306-51-9 ]
[ 178306-59-7 ]
Yield
Reaction Conditions
Operation in experiment
With potassium carbonate; In N,N-dimethyl-formamide;
EXAMPLE 12 Benzyl 3-methoxy-2-(4-methoxy-6,7-dihydro-5H-cyclopentapyrimidin-2-yloxy)-3,3-diphenylpropionate 24.48 g (90 mmol) of 3-methoxy-3,3-diphenyl-2-hydroxypropionic acid were dissolved in 150 ml of DMF, and 13.7 g (99 mmol) of potassium carbonate were added. The suspension was stirred at room temperature for 30 min. Then 10.7 ml (90 mmol) of benzyl bromide were added dropwise over the course of 5 min., and the mixture was stirred for 1 h., during which the temperature rose to 32 C. To this mixture were successively added 24.84 g (180 mmol) of K2 CO3 and 20.52 g (90 mmol) of 2-methanesulfonyl-4-methoxy-6,7-dihydro-5H-Ocyclopentapyrimidine, and the mixture was stirred at 80 C. for 3 h. For workup, the contents of the flask were diluted with about 600 ml of H2 O and cautiously acidified with concentrated HCl, and 250 ml of ethyl acetate were added. 31.4 g of pure product precipitated and were filtered off. The ethyl acetate phase was separated from the mother liquor, the aqueous phase was extracted again with ethyl acetate, and the combined organic phases were concentrated. The oily residue (19 g) was purified by chromatography (cyclohexane/ethyl acetate=9/1) to result in a further 10.5 g of pure product. Total yield: 41.9 g (82.2 mmol)=91% Melting point 143-147 C. MS: MH+=511
500 g (1.75 mol) of (RS) 2-hydroxy-3-methoxy-3,3-diphenyl methyl propanoate Suspended and agitated in 1,000 mL of water, 950 mL (1.90 mol) of 2 mol / L sodium hydroxide was added, and the mixture was stirred at 100 C. for 1 hour. The reaction solution was cooled to room temperature, 1500 mL of tert-butyl methyl ether was added, 700 mL of 10% hydrochloric acid was added, and the mixture was stirred for 20 minutes to dissolve the precipitated solid. After liquid-liquid separation, the organic layer was washed with 1000 mL of water. 1200 mL of heptane was added, and the solvent was distilled off under reduced pressure (200 mmHg) at 40 C. to precipitate crystals. 1000 mL of heptane was added, and the mixture was stirred at 0 C. for 1 hour. The crystals were collected by filtration and dried under reduced pressure at 40 C. for 7 hours,(RS) -2-hydroxy-3-methoxy-3,3-diphenylpropanoic acid 431 g (yield 91%) was obtained.
90%
With water; sodium hydroxide; at 90 - 95℃; for 1.5h;
Step-HI : Preparation of 2-hydroxy-3-methoxy-3,3-diphenyl propionic acid of the formula -(IV) :Into a 5L round bottomed flask a mixture water(l.OL) and compound of formula -III (200g) from step-II were charged and stirred for 15 minutes. IN aqueous sodium hydroxide solution was charged and the reaction mass was stirred for 15 minutes.Reaction mass was heated to 90-95C and maintained at the same temperature for one hour. The reaction mass was brought to room temperature adjustment of pH was carried out with IN hydrochloric acid solution(1.6L) to 2-3. The product slurry was cooled to 5- 10C and maintained at the same temperature for 2hours. The product was filtered and dried at 60-65CDry weight : 172g(90%)Purity by HPLC : 99.88%Melting range : 100- 102 C
Example-2: Preparation of 2-hydroxy-3-methoxy-3,3-diphenylpropionic acid compound of formula-6:Mixture of <strong>[178306-47-3]2-hydroxy-3-methoxy-3,3-diphenylpropionic acid methyl ester</strong> (100 grams) and aqueous sodium hydroxide solution (22 grams in 200 ml of water) was heated to 95-1000C and stirred for 120 minutes. The reaction mixture was cooled to 40-450C and quenched with water. The pH of the reaction mixture was adjusted to 1.3 with concentrated hydrochloric acid and extracted with ethyl acetate. The solvent from the ethyl acetate layer was distilled off completely under reduced pressure at 600C. The reaction mixture was cooled to 4O0C and cyclohexane (230 ml) was added and stirred at reflux for 30 minutes. The reaction mixture was cooled to.25-35C and stirred for 40 minutes at 25-35C. The solid formed was filtered off and washed with cyclohexane then dried at 60-70C to get the title compound. Yield: 92 grams; M.R: 108-112C
Preparation of 2-hydroxy-3-methoxy-3,3-diphenylpropionic acid compound of formula-6 Mixture of <strong>[178306-47-3]2-hydroxy-3-methoxy-3,3-diphenylpropionic acid methyl ester</strong> (100 grams) and aqueous sodium hydroxide solution (22 grams in 200 ml of water) was heated to 95-100 C. and stirred for 120 minutes. The reaction mixture was cooled to 40-45 C. and quenched with water. The pH of the reaction mixture was adjusted to 1.3 with concentrated hydrochloric acid and extracted with ethyl acetate. The solvent from the ethyl acetate layer was distilled off completely under reduced pressure at 60 C. The reaction mixture was cooled to 40 C. and cyclohexane (230 ml) was added and stirred at reflux for 30 minutes. The reaction mixture was cooled to 25-35 C. and stirred for 40 minutes at 25-35 C. The solid formed was filtered off and washed with cyclohexane then dried at 60-70 C. to get the title compound.Yield: 92 grams; M.R: 108-112 C.
123 g
With water; sodium hydroxide; In methanol; at 45 - 50℃; for 1h;
140 g of methyl 3,3-diphenyloxirane-2-carboxylate was dissolved in 280 ml of methanol and then cooled to 0C .6.9 ml of BF3OEt2 was added slowly and stirred at 0C for 2 hours. When the methyl 3,3-diphenyloxirane-2-carboxylate disappears completely and <strong>[178306-47-3]methyl 2-hydroxy-3-methoxy-3,3-diphenylpropionate</strong> is produced,65.8 g of NaOH was dissolved in 980 ml of water and slowly added dropwise. Methanol (140 ml) was added, the temperature was raised to 45 to 50C, and the mixture was stirred for 1 hour. When the reaction was completed, the methanol was distilled off under reduced pressure, and diluted with 700 ml of dichloromethane. 300 ml of 6N HCl aqueous solution was added to adjust the pH to 2, and the organic layer was separated. The organic layer was washed with 300 ml of brine, dried, filtered and distilled under reduced pressure, and recrystallized from 500 ml of hexane to obtain 123 g of 2-hydroxy-3-methoxy-3,3-diphenylpropionic acid (yield: 82%).
Similarly, different diastereomeric salt of (S)-2-hydroxy-3 -methoxy 3, 3-diphenyl propionate was prepared using different chiral amine in different batches and the results are summarized in Table 2 given below.Table 2;* Input refers to 3, 3-diphenyl-2-hydroxy-3-methoxy propionic acid
In acetonitrile; for 1.75h;Reflux;Product distribution / selectivity;
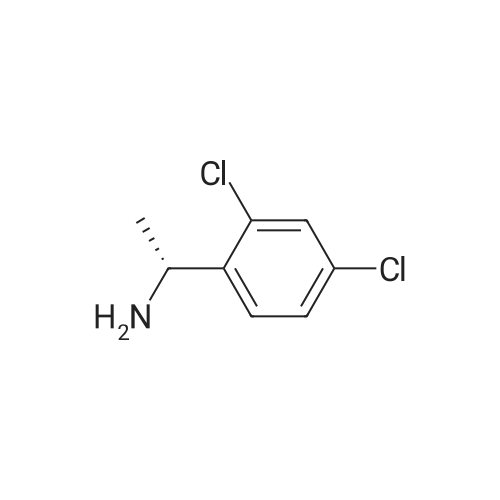
In a 5 liter three necked flask, (302 g) (1.10 mole) 3,3-diphenyl-2-hydroxy-3-methoxy propanoic acid and 3.02 liter acetonitrile was taken. Subsequently, the reaction mixture was stirred and heated at reflux temperature. (105.4 g) (0.55 mole) (R)-2,4-dichloro PEA, in 50 mL acetonitrile was added drop wise in 45 min. time interval Solid material was precipitated during this addition. The reaction mixture was stirred under reflux for 1 hr and cooled to room temperature. The reaction mixture was further stirred at room temperature for 1 hour. Solid material was filtered, washed with acetonitrile and dried. Yield: 219 g, (85.41%), HPLC purity: 99.94%, Chiral purity: 95.01%. Similarly, different diastereomeric salt of (S)-2-hydroxy-3-methoxy 3,3-diphenyl propionate was prepared using different chiral amine in different batches and the results are summarized in Table 2 given below
60%
In acetonitrile;Resolution of racemate;Product distribution / selectivity;
Similarly, different diastereomeric salt of (S)-2-hydroxy-3 -methoxy 3, 3-diphenyl propionate was prepared using different chiral amine in different batches and the results are summarized in Table 2 given below.Table 2;* Input refers to 3, 3-diphenyl-2-hydroxy-3-methoxy propionic acid
In methanol; tert-butyl methyl ether; at 20℃; for 4h;Reflux;Product distribution / selectivity;
(R)-2-hydroxy-3-methoxy 3,3 diphenyl propionate In a 50 mL three neck flask, attached with stirrer, thermometer, condenser with guard tube, over water bath (1 g) racemic 2-hydroxy-3-methoxy 3,3 diphenyl propanoic acid and 10 mL methyl t-butyl ether at room temperature was added and stirred for 5 min. To the clear yellow solution, 10 mL methanol was added and then heated. Solution was refluxed gently. To this reaction, mixture of (0.454 g) (S)-2,4-dichloro PEA and 6 mL methyl t-butyl ether was added. The solution was refluxed for 1 hour and hazy solution was allowed to cool naturally. The hazy solution was stirred at room temperature for 3 hrs. Solid material was filtered, washed with 5 mL methyl t-butyl ether and dried. Yield: 298 mg. (35%). HPLC purity: 99.9%. Chiral purity: 99.3%. Similarly, (S)-2,4-dichlorophenylethylammonium (R)-2-hydroxy-3-methoxy 3,3 diphenyl propionate was prepared in different batches and the results are summarized in table 1 given below
35%
In methanol; tert-butyl methyl ether; for 1h;Reflux; Resolution of racemate;Product distribution / selectivity;
Example 2 : Preparation of (S)-2, 4-dichlorophenylethylammonium (R)-2-hydroxy-3- methoxy 3. 3 diphenyl propionateIn a 50 mL three neck flask, attached with stirrer, thermometer, condenser with guard tube, over water bath (1 g) racemic 2-hydroxy-3-methoxy 3, 3 diphenyl propanoic acid and 10 mL methyl t-butyl ether at room temperature was added and stirred for 5 min. To the clear yellow solution, 10 mL methanol was added and then heated. Solution was refluxed gently. To this reaction, mixture of (0.454 g) (S)-2, 4-dichloro PEA and 6 mL methyl t- butyl ether was added. The solution was refluxed for 1 hour and hazy solution was allowed to cool naturally. The hazy solution was stirred at room temperature for 3 hrs. Solid material was filtered, washed with 5 mL methyl t-butyl ether and dried. Yield: 298 mg. (35 %). HPLC purity: 99.9 %. Chiral purity: 99.3 %.Similarly, (S)-2, 4-dichlorophenylethylammonium (R)-2-hydroxy-3-methoxy 3, 3 diphenyl propionate was prepared in different batches and the results are summarized in table 1 given below.Table-1* Input refers to propionic acid deriv.
In acetonitrile; at 20℃; for 2h;Reflux;Product distribution / selectivity;
In 2 liter three necked flask, (90.0 g) (0.33 mole) 3,3-diphenyl-2-hydroxy-3-methoxy propionic acid and 0.9 liter acetonitrile was taken. Subsequently, the reaction mixture was stirred and heated at reflux temperature. (30 g) (0.198 mole) (S)-3-methoxy PEA, in 90 mL acetonitrile was added drop wise in 10 minute time interval. Further, the reaction mixture was stirred under reflux for 0.5 hr. The reaction mixture was cooled at room temperature and was stirred for 1.5 hour. Solid material was filtered, washed with acetonitrile and dried. Yield: 65.0 g, (92.9%), HPLC purity: 99.82%, Chiral purity: 96.57%
85%
In ethanol; at 20℃; for 2.16667h;Reflux; Resolution of racemate;Product distribution / selectivity;
Example-12: Preparation of (S)-3-methoxyphenylethylammonium (S)-2-hvdroxy-3- methoxy 3, 3-diphenyl propionateIn 100 mL three necked flask, (5g) (0.018 mole) 3, 3-diphenyl-2-hydroxy-3- methoxy propionic acid and 50 mL rectified spirit was taken. Subsequently, the reaction mixture was stirred and heated at reflux temperature. (1.8g) (0.01 1 mole) (S)- 3- methoxy PEA, in 5 mL rectified spirit was added drop wise in 10 min. time intervalThe reaction mixture was stirred under reflux for 0.5 hr and cooled to room temperature and again stirred at room temperature for 1.5 hour. Solid material was filtered, washed with rectified spirit and dried.Yield: 3.3 g, (85.0 %), HPLC purity: 99.91 %, Chiral purity: 99.38 %.
In water; isopropyl alcohol; at 100℃;Resolution of racemate;
To a round-bottom flask equipped with magnetic stirrer containing 2-hydroxy- 3-methoxy-3,3-diphenylpropanoic acid (1 .65 g, 6.06 mmol) and L-valine (0.35 g, 2.99 mmol, 0.5 eq.) was added a mixture of water/IPA (3:2) (16 mL). The resulting suspension was heated at 100 C (bath temperature) until complete dissolution and then cooled down to room temperature. The solution was seeded with Form I, obtained in Example 2, at 60-70 C. A white precipitate was formed and the slurry was stirred at room temperature for 2 hours. The solid was filtered with a sintered funnel (porosity 3), washed with water/IPA (3:2) (2 x 4 mL) and dried under vacuum at room temperature. (S)-2-hydroxy- 3-methoxy-3,3-diphenylpropanoic acid - L-valine (2:1 ) (Form I) was obtained as a white powder (0.79 g, 40% yield, 99% ee). According to 1H NMR (MeOD- d4, 400 MHz) and XRPD it corresponds to the compound of the title.
(R)-2-hydroxy-3-methoxy-3,3-diphenylpropanoic acid[ No CAS ]
2C16H16O4*C5H11NO2[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In water; for 0.75h;Resolution of racemate;
To a 2 ml_ eppendorf containing <strong>[178306-51-9]2-hydroxy-3-methoxy-3,3-diphenylpropanoic acid</strong> (35 mg, 0.13 mmol), L-valine (15 mg, 0.13 mmol) and three 4 mm stainless steel grinding balls, one drop of water was added. The reactor was stirred 45 minutes at a rate of 30 Hz (3 x 15 minutes). The product was dried under vacuum at room temperature. A mixture of (S)-2-hydroxy-3-methoxy- 3,3-diphenylpropanoic acid - L-valine (2:1 ) (Form I), (R)-2-hydroxy-3- methoxy-3,3-diphenylpropanoic acid and L-valine was obtained as a white powder. According to XRPD, the compound of the title is present in the mixture. The same experiment was carried out with MeOH instead of water with the same result.
(R)-2-hydroxy-3-methoxy-3,3-diphenylpropanoic acid[ No CAS ]
2C16H16O4*C4H9NO2[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In water; for 0.75h;Resolution of racemate;
To a 2 mL eppendorf containing <strong>[178306-51-9]2-hydroxy-3-methoxy-3,3-diphenylpropanoic acid</strong> (25 mg, 0.09 mmol), L-2-aminobutyric acid (10 mg, 0.10 mmol) and three 4 mm stainless steel grinding balls, one drop of water was added. The reactor was stirred 45 minutes at a rate of 30 Hz (3 x 15 minutes). The product was dried under vacuum at room temperature. A mixture of (S)-2-hydroxy-3- methoxy-3,3-diphenylpropanoic acid - L-2-aminobutyric acid (2:1 ) (Form II), (R)-<strong>[178306-51-9]2-hydroxy-3-methoxy-3,3-diphenylpropanoic acid</strong> and L-2-aminobutyric acid was obtained as a white powder. According to XRPD the compound of the title is present in the mixture. The same experiment was carried out with IPA, AcOEt or methyl isobutyl ketone with the same result.
In water; isopropyl alcohol; at 60℃;Resolution of racemate;
To a round-bottom flask equipped with magnetic stirrer containing 2-hydroxy- 3-methoxy-3,3-diphenylpropanoic acid (420 mg, 1 .54 mmol) and L-2- aminobutyric acid (80 mg, 0.78 mmol) was added a mixture of water/IPA (3:2) (4 mL). The resulting suspension was heated at 60 C (bath temperature) until complete dissolution and then cooled down to room temperature. The solution was seeded with Form II at 40 C. A white precipitate was formed and the slurry was stirred at room temperature for 2 hours. The solid was filtered with a sintered funnel (porosity 3), washed with water/IPA (3:2) (2 x 1 mL) and dried under vacuum at room temperature. (S)-2-hydroxy-3-methoxy-3,3- diphenylpropanoic acid - L-2-aminobutyric acid (2:1 ) (Form II) was obtained as a white powder (138 mg, 28% yield, 97% ee).
(a) Resolution using (S)-(-)-1-(1-Naphthyl)ethyl amine To a stirred solution of 2-hydroxy-3-methoxy-3,3-diphenylpropionic acid (compound VII; 240 gms/0.88 moles) in a mixture of tert-butyl methyl ether (1.2 lit) and methanol (1.2 lit) was added (S)-(-)-1-(1-Naphthyl) ethyl amine (82.8 gms/0.484 moles). The reaction mass was further stirred for 1 hour at 25-30 C. The solid was isolated by filtration, washed with tert-butyl methyl ether (500 ml) and dried. The solid was stirred in a mixture of distilled water (1.2 lit) and tert-butyl methyl ether (1.2 lit) and cooled to 10-15 C. The reaction mass was acidified with conc. HCl and stirred for 30 minutes. The organic phase was separated; aqueous phase was extracted with tert-butyl methyl ether (1.0 lit). The organic phases were combined together, washed with brine, and concentrated under vacuum at 25-30 C. The residue was stirred in n-Hepatne (720 ml). The solid was isolated by filtration and dried to give 79 g of the title compound (VIII). Efficiency: 32.91% Purity by HPLC: 99.5% Chiral purity: 98.1%
(b) Resolution using (S)-1-(4-nitrophenyl)ethyl amine To a stirred solution of 2-hydroxy-3-methoxy-3,3-diphenylpropionic acid (compound VII; 240 gms/0.88 moles) in tert-butyl methyl ether (1.2 lit) was added S-1-(4-nitro phenyl)ethyl amine (73.5 gms/0.44 moles) at 45-50 C. The reaction mass was further stirred for 1 hour at 45-50 C., cooled to 25-30 C. and stirred for 1 hour. The solid was isolated by filtration, washed with tert-butyl methyl ether (500 ml) and dried. The solid was stirred in a mixture of distilled water (1.2 lit) and tert-butyl methyl ether (1.2 lit) and cooled to 10-15 C. The reaction mass was acidified with conc. HCl and stirred for 30 minutes. The organic phase was separated; aqueous phase was extracted with tert-butyl methyl ether (1.0 lit). The organic phases were combined together, washed with brine, dried on sodium sulphate and concentrated under vacuum at 25-30 C. The residue was stirred in n-Heptane (720 ml). The solid was isolated by filtration, and dried to give 66 g of the title compound (VIII). Efficiency: 27.5% Purity by HPLC: 99.78% Chiral purity: 99.9%
(S)-2-hydroxy-3-methoxy-3,3-diphenylpropionic acid L-prolinamide salt[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
73%
In tetrahydrofuran; for 1h;Reflux; Resolution of racemate;
100 g of <strong>[178306-51-9]2-hydroxy-3-methoxy-3,3-diphenylpropionic acid</strong> was dissolved in 1000 ml of THF, and 41.9 g of L-proline amide was added. The reaction was refluxed with stirring for 1 hour, cooled to room temperature and stirred for 30 minutes. The resulting crystals were filtered and washed with 100 ml of THF. The resulting solid was vacuum-dried to obtain 64 g (yield: 45%, optical purity: 93% ee) of (S) -<strong>[178306-51-9]2-hydroxy-3-methoxy-3,3-diphenylpropionic acid</strong> L-prolinamide salt. S) -<strong>[178306-51-9]2-hydroxy-3-methoxy-3,3-diphenylpropionic acid</strong> L-prolineamide salt was suspended in 600 ml of THF and stirred at reflux for 1 hour. The reaction was cooled to ambient temperature and stirred for 30 min. The resulting crystals were filtered. 55 g of the obtained compound was suspended in 550 ml of a mixed solvent of THF: H2O = 30: 1, and the mixture was refluxed with stirring for 1 hour. The reaction was cooled to ambient temperature and stirred for 30 minutes. The resulting crystals were filtered and dried in vacuo to give 44g of (S) -<strong>[178306-51-9]2-hydroxy-3-methoxy-3,3-diphenylpropionic acid</strong> L-prolinamide salt (yield: 73%).
In methanol; tert-butyl methyl ether; at 10 - 30℃;Resolution of racemate; Industrial scale;
Raw material A (11kg, 40.4mol), anhydrous methanol (51L), methyl tert-butyl ether (51L) was added to a 200L reactor,(R) - (+) - alpha-phenylethylamine (4.4kg, 36.3mol) was added dropwise at 10-30 C. After the addition was completed, the mixture was stirred at 10-30 C for 3-4h and filtered. The filter cake was washed with methyl tert-butyl Ether (12 L × 2), dried to give 3.55 kg, a yield of 64.5%. HPLC Purity: 99.5%, Chiral purity: 68.3%.
(1) 17.2 g of (R/S)-<strong>[178306-51-9]2-hydroxy-3-methoxy-3,3-diphenylpropionic acid</strong> was placed in a 250 mL reaction flask.Add 136 mL of isopropanol and stir.Add 7.8 g of (S)-(-)-1-(2-chlorophenyl)ethylamine,Stir at 20 C for 2 h.Chiral product(S)-<strong>[178306-51-9]2-hydroxy-3-methoxy-3,3-diphenylpropionic acid</strong>Precipitation of (S)-(-)-2-chlorophenylethylamine salt; suction filtration,The filter cake was washed with isopropyl alcohol, drained and set aside. (R)-<strong>[178306-51-9]2-hydroxy-3-methoxy-3,3-diphenylpropionic acid</strong> in an isopropanol solution can be recycled.The filter cake was dissolved in 65 mL of ethyl acetate.Stir,Slowly add 4.05g sodium methoxide, slowly cool to 0 C, keep warm for 1 h,product(2s) 2-hydroxy-3-methoxy-3,3-diphenylpropanate precipitated; suction filtration, wash the filter cake with a small amount of isopropyl alcohol,Drying at 40-50 C to obtain 13.9 g of product, the yield was 45.1%.HPLC purity: 99.22%, chiral purity: 99.80%. The resolving agent (S)-(-)-2-chlorophenylethylamine in the ethyl acetate solution was recycled.
2-(methylsulfonyl)-4-methoxy-6,7-dihydro-5H-cyclopentapyrimidine[ No CAS ]
[ 178306-51-9 ]
[ 178306-59-7 ]
Yield
Reaction Conditions
Operation in experiment
91%
24.48 g (90 mmol) of 3-methoxy-3,3-diphenyl-2-hydroxypropionic acid were dissolved in 150 ml of DMF,Then, 13.7 g (99 mmol) of potassium carbonate was added.The suspension was stirred at room temperature for 30 minutes.next,10.7 ml (90 mmol) of benzyl bromide was added dropwise over 5 minutes,When this mixture was stirred for 1 hour,During this time the temperature rose to 32 C.Following this mixture,24.84 g (180 mmol) of K 2 CO 3And 20.52 g (90 mmol) of 2-methanesulfonyl-4-methoxy-6,7-dihydro-5H-cyclopentapyridine were added,The mixture was stirred at 80 C. for 3 hours.For purification,The contents of the flask were diluted with about 600 ml of water,And carefully acidified with concentrated hydrochloric acid,250 ml of ethyl acetate was then added.As 31.4 g of pure product precipitated,This was collected by filtration.The ethyl acetate phase was separated from the mother liquor,The aqueous phase was again extracted with ethyl acetate,The collected organic phase was concentrated.The oily residue was purified by chromatography (cyclohexane / ethyl acetate = 9/1) to give an additional 10.5 g of pure product.Total yield: 41.9 g (82.2 mmol) 91%
91.11 g (0.5 mol) of benzophenone and 45.92 g (0.85 mol) of sodium methoxide were added,Was suspended in 150 ml of methyl t-butyl ether (MTB) at room temperature. After cooling to -10 C.,92.24 g (0.85 mol) of methyl chloroacetate,In such a way that the internal temperature rose to 40 C. while the outside was cooled in a bath at -10 C.next,The mixture was stirred for 1 hour at naturally occurring temperature without cooling.250 ml of water was added,After stirring for a while,The aqueous phase was separated.The MTB phase was washed with 250 ml of an aqueous dilute sodium chloride solution.After changing the solvent to methanol (250 ml)A solution of 1 g of p-toluenesulfonic acid in 10 ml of methanol was added at room temperature.The mixture was stirred at naturally occurring temperature for 1 hour,Followed by heating under reflux.While methanol was distilled off,400 g of 10% strength sodium hydroxide solution was added dropwise,Finally 60 ml of water was added.This methanol was added drop-Until the bottom temperature reached 97 C,And distilled.After cooling to 55 C., 190 ml of MTB was added and the mixture was acidified to pH 2 with about 77 ml of concentrated hydrochloric acid.After cooling to room temperature, the aqueous phase was distilled off, and the organic phase was concentrated by distilling 60 ml of MTB.The product was crystallized by adding 500 ml of heptane and gradually cooled to room temperature.The crude crystalline solid was filtered off with suction, washed with heptane and dried in a vacuum oven at 40 C. to constant weight.Yield: 108.9 g (80%), HPLC> 99.5% area.
3-methoxy-2-((p-toluenesulfonyl)oxy)-3,3-diphenylpropionic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85%
With triethylamine; In tert-butyl methyl ether; at 0 - 5℃;
27.2 g of <strong>[178306-51-9]2-hydroxy-3-methoxy-3,3-diphenylpropionic acid</strong> (0.10 mol, 81% of R configuration content) was added to 200 ml of methyl tert-butyl ether, 10 ml of triethylamine was added, and ice 21.0 g (0.11 mol) of p-toluenesulfonyl chloride was added dropwise at a temperature of 0-5 C in a water bath. After the reaction was completed, 200 ml of water was added to stir and the layers were separated. The organic layer was washed with 0.05 mol / L of dilute hydrochloric acid 100 ml and saturated saline 100 ml. Concentration to dryness gave 3-methoxy-2-((p-toluenesulfonyl) oxy) -3,3-diphenylpropionic acid 36.2 in a yield of 85.0%
3-methoxy-2-((methanesulfonyl)oxy)-3,3-diphenylpropionic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
94.8%
With triethylamine; In tert-butyl methyl ether; at 0 - 5℃;
27.2 g of <strong>[178306-51-9]2-hydroxy-3-methoxy-3,3-diphenylpropionic acid</strong> (0.10 mol, 70% of R configuration content) was added to 200 ml of methyl tert-butyl ether, 10 ml of triethylamine was added, and ice 12.6 g (0.11 mol) of methanesulfonyl chloride was added dropwise at a temperature of 0-5 C in a water bath. After the reaction was completed, 200 ml of water was added to stir and the layers were separated. The organic layer was washed with 0.05 mol / L diluted hydrochloric acid 100 ml and saturated saline 100 ml, and concentrated under reduced pressure. 33.2 g of 3-methoxy-2-((methanesulfonyl) oxy) -3,3-diphenylpropionic acid was obtained until the yield was 94.8%.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping