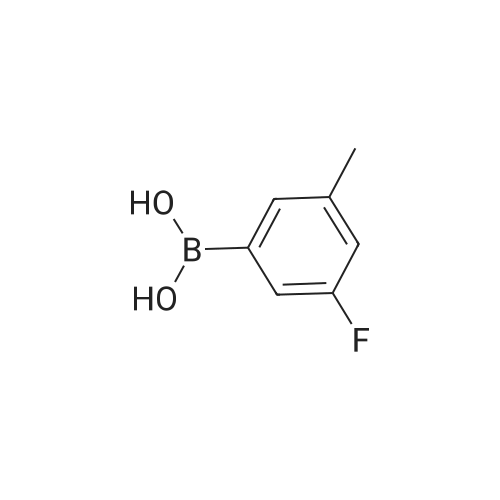
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
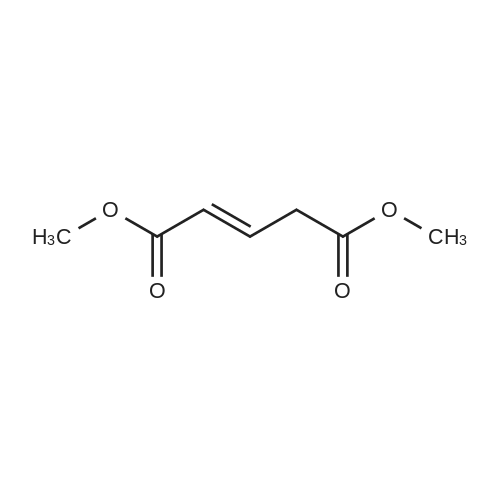
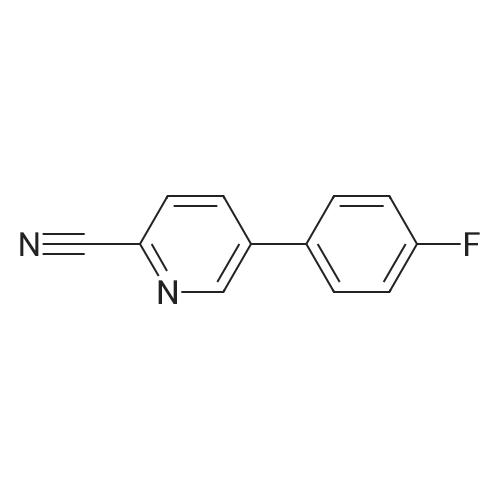
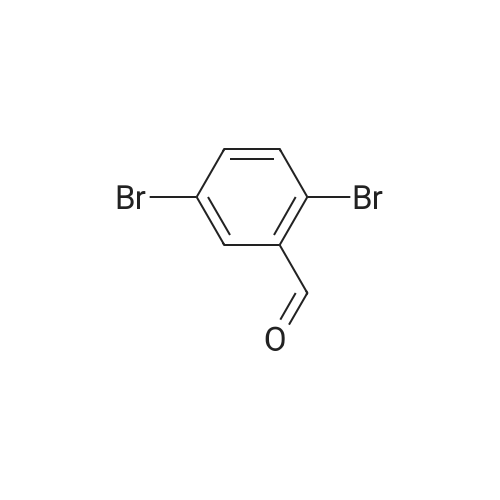
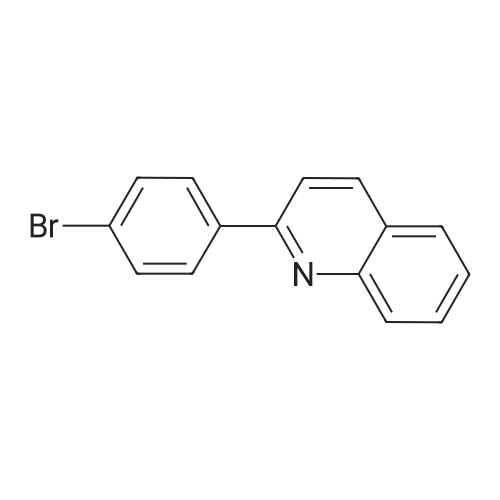
[1] Synlett, 2000, # 10, p. 1485 - 1487
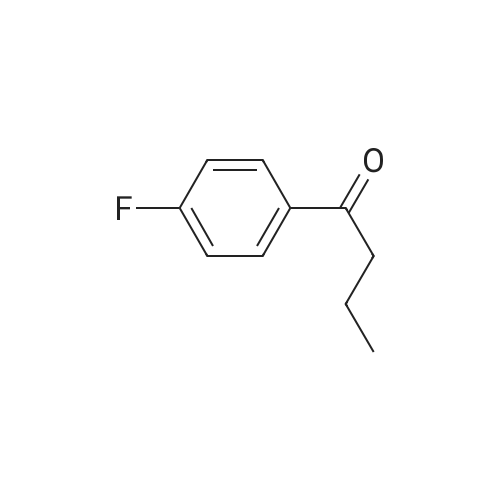
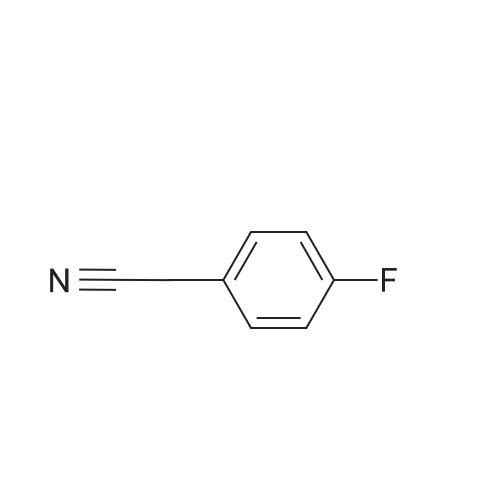
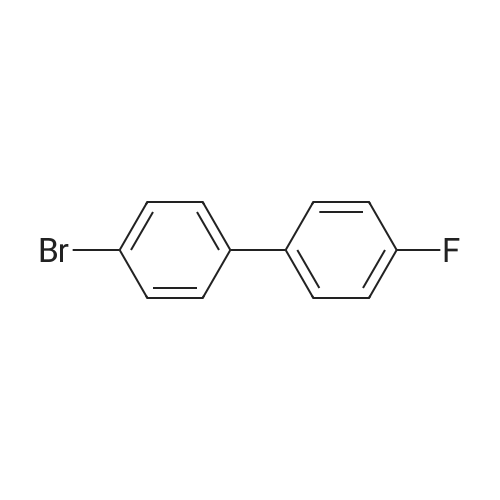
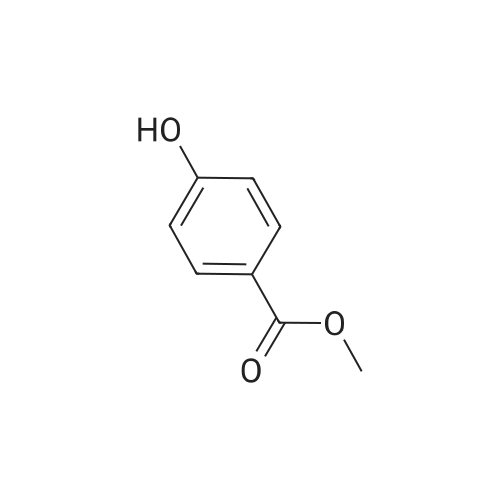
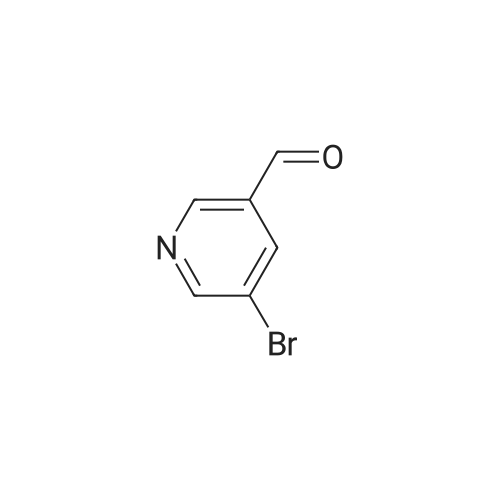
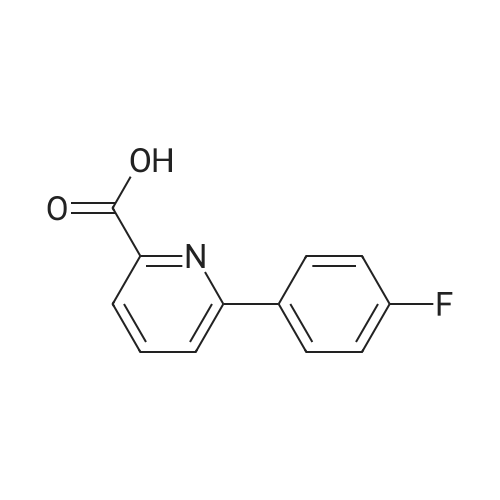
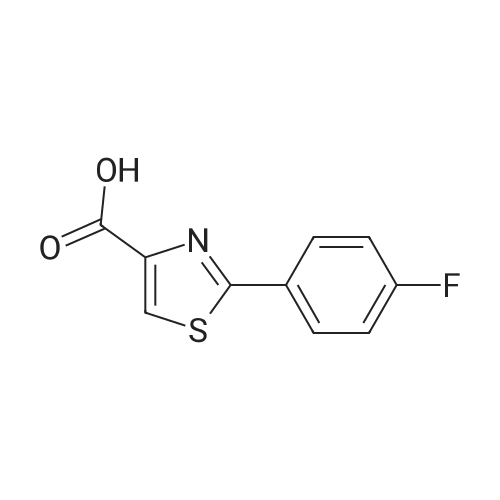
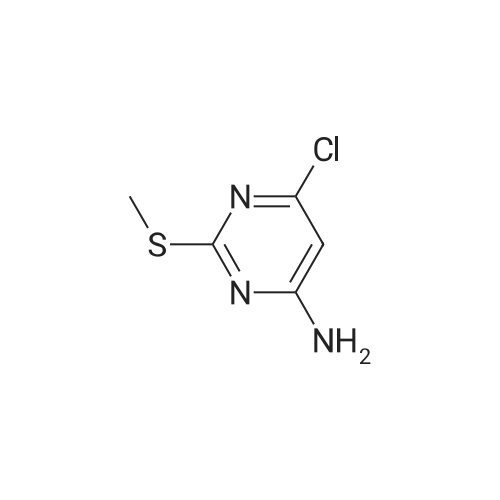
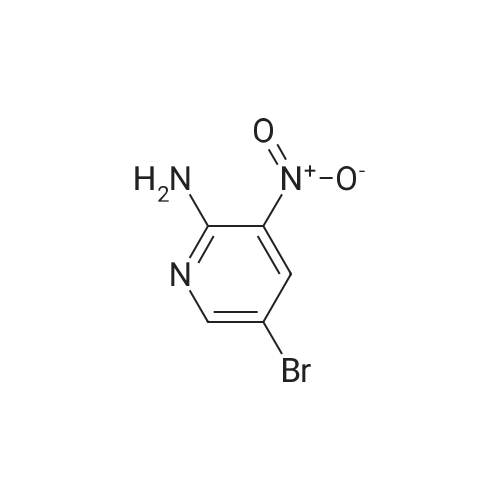
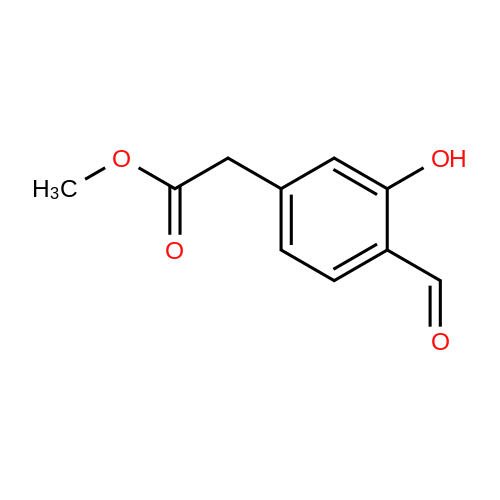
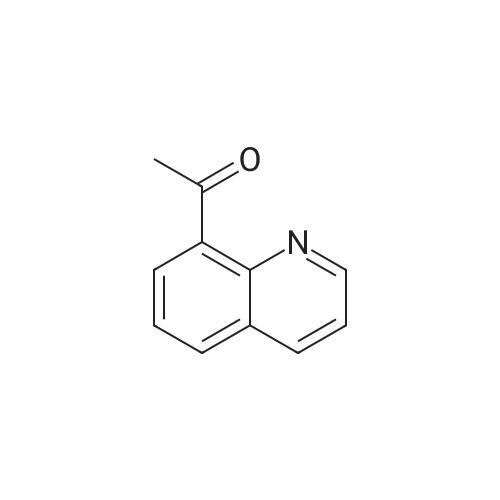
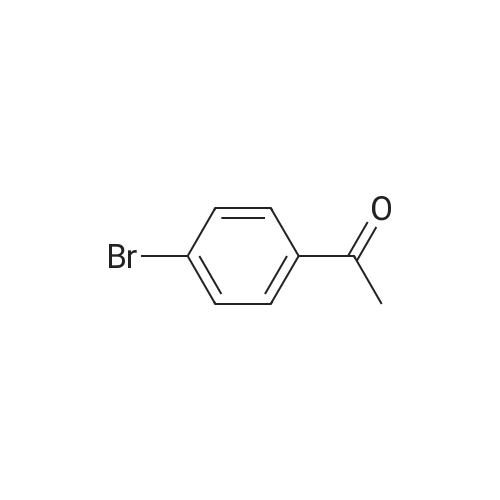
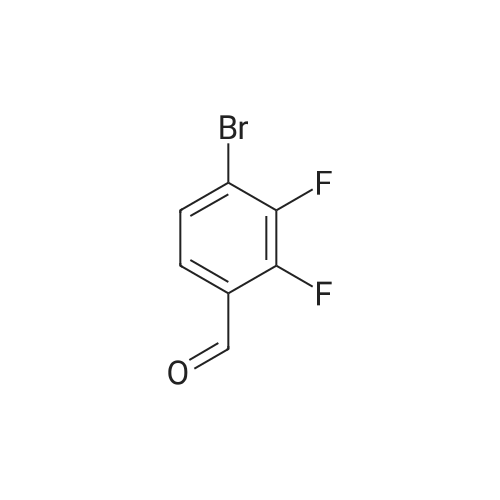
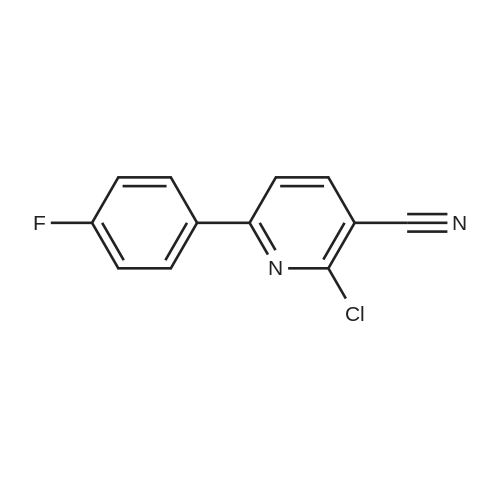
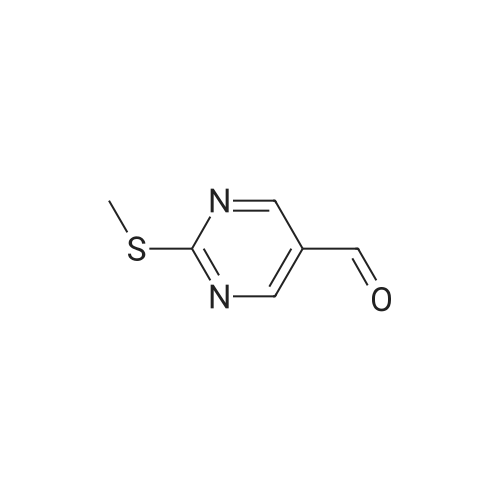
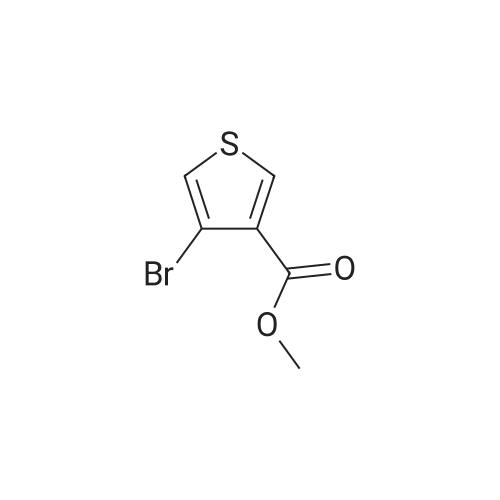
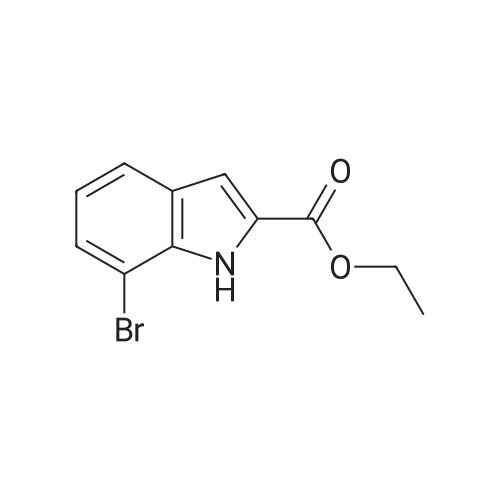
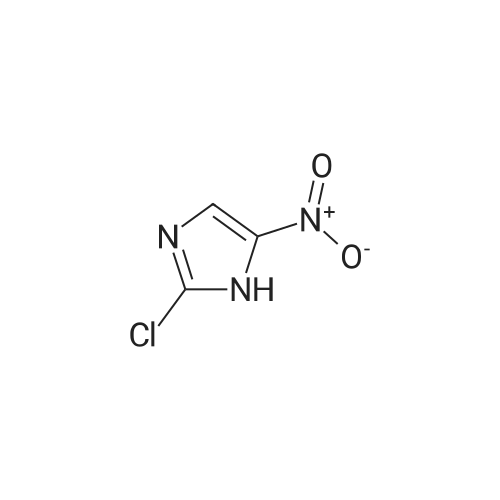
3
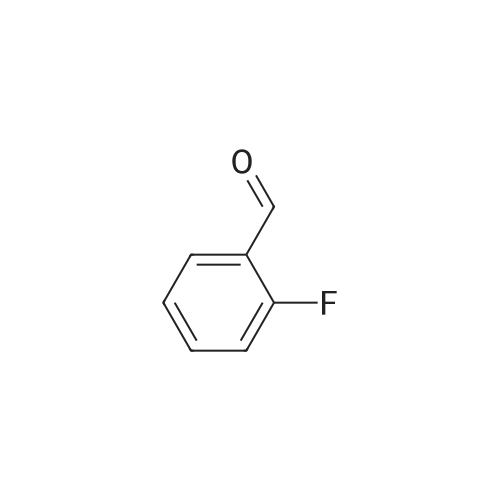
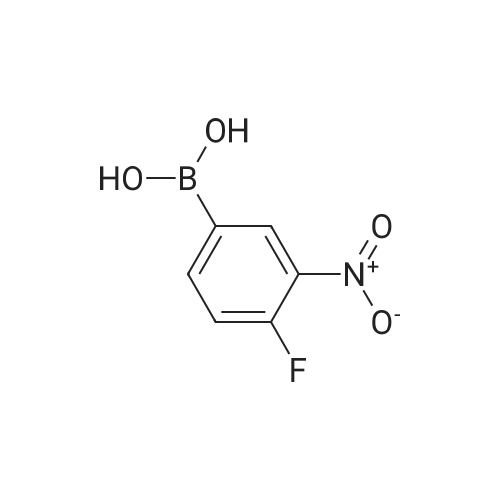
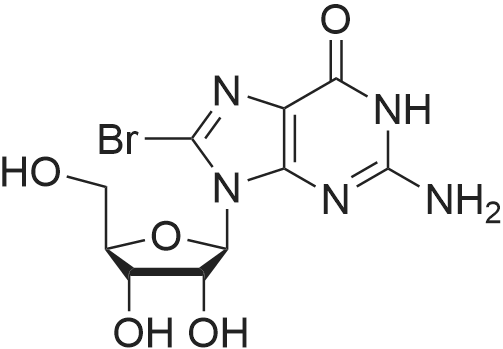
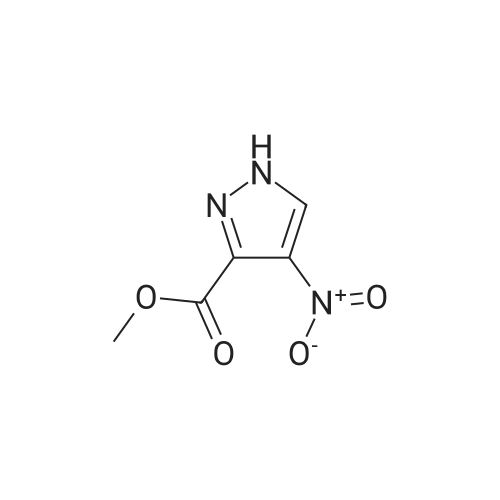
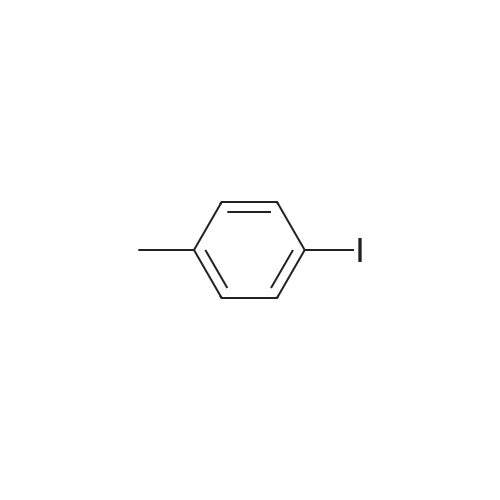
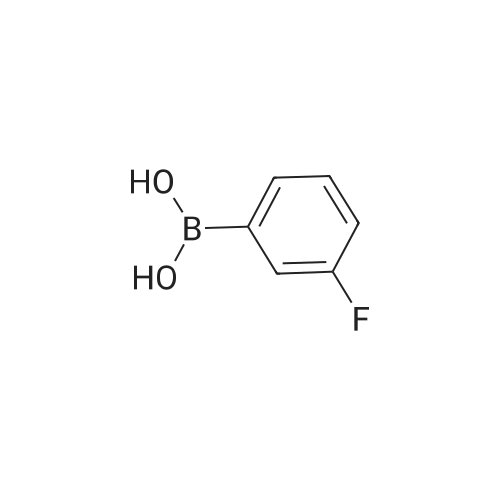
[ 6940-76-7 ]
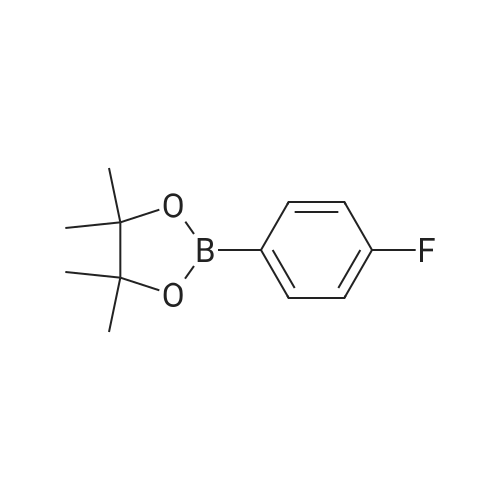
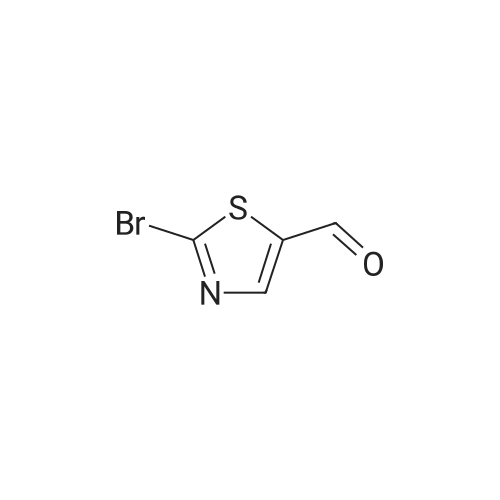
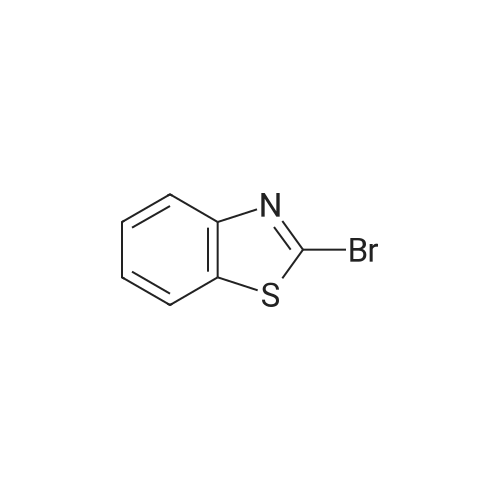
[ 1765-93-1 ]
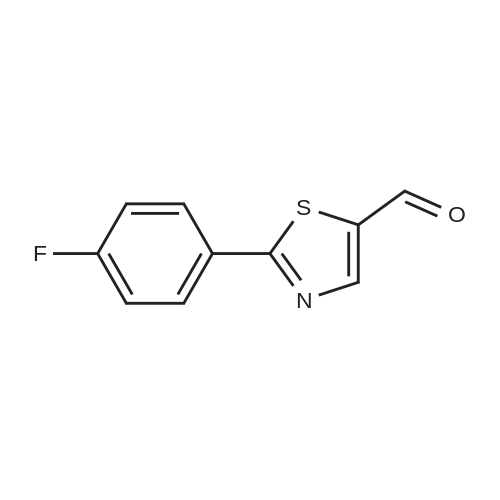
[ 13939-06-5 ]
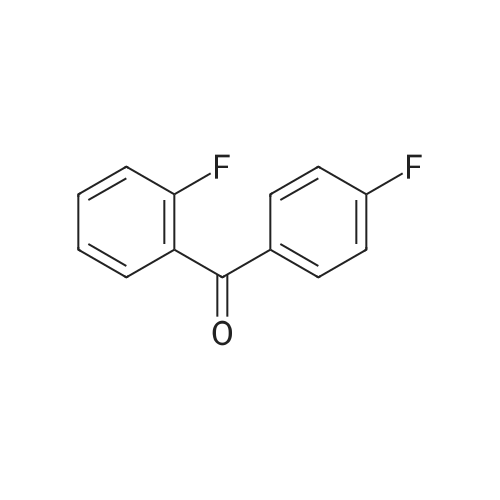
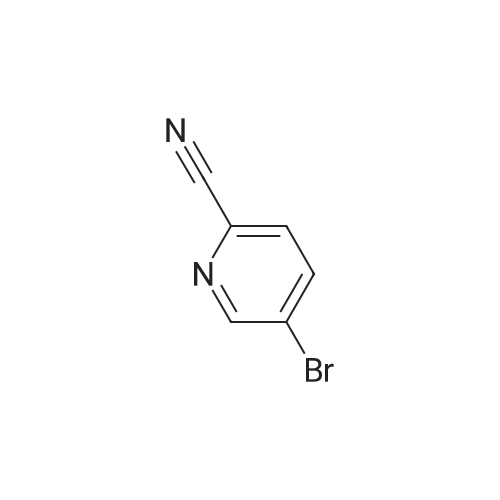
[ 3874-54-2 ]
Reference:
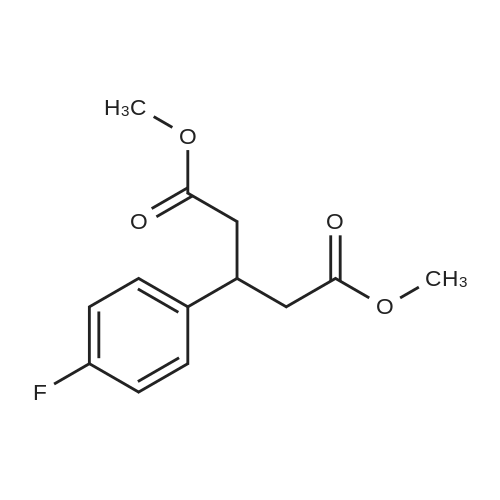
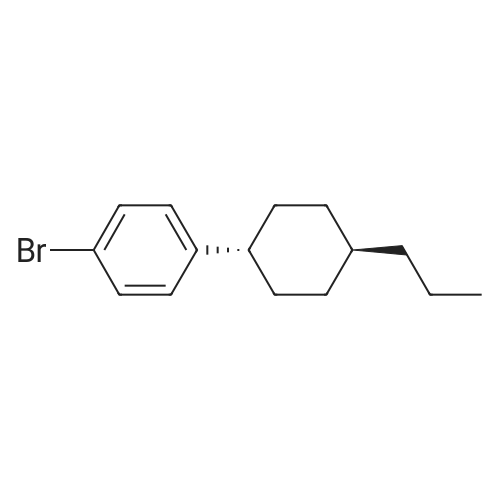
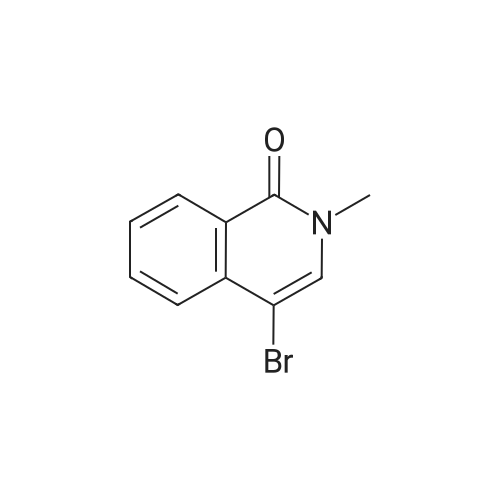
[1] Chemical Communications, 2017, vol. 53, # 51, p. 6895 - 6898
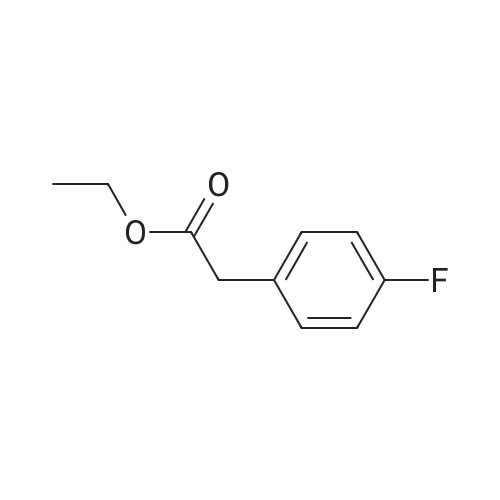
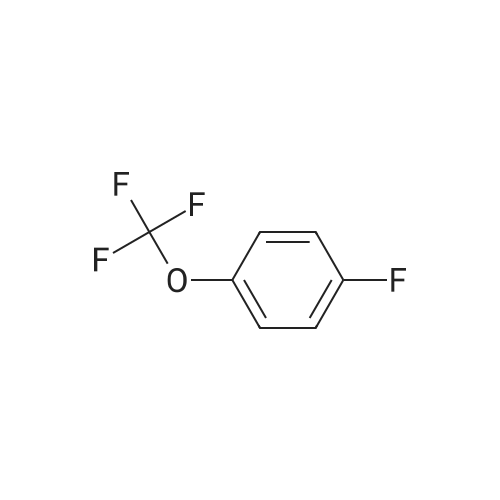
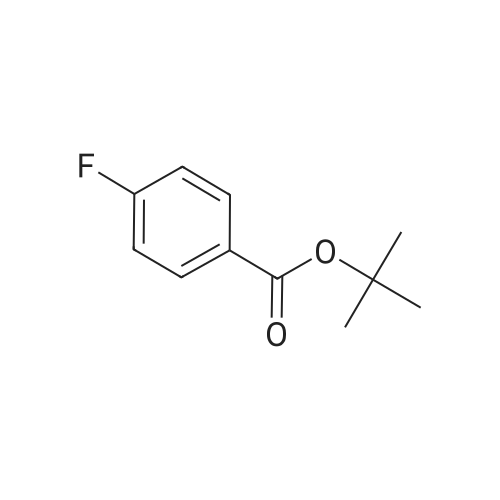
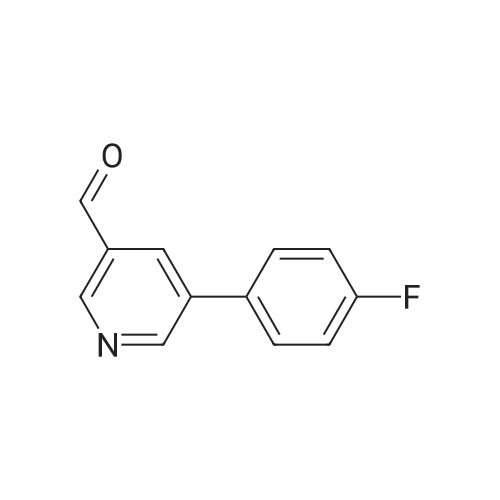
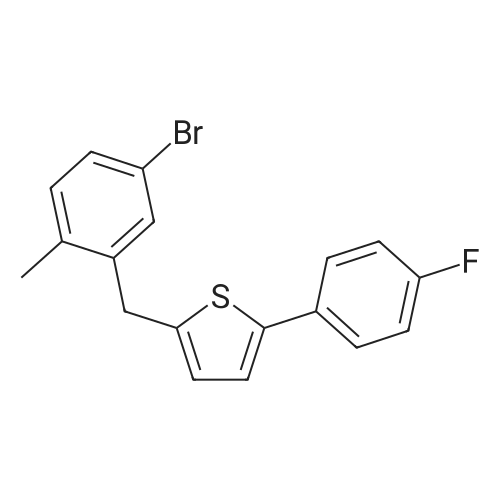
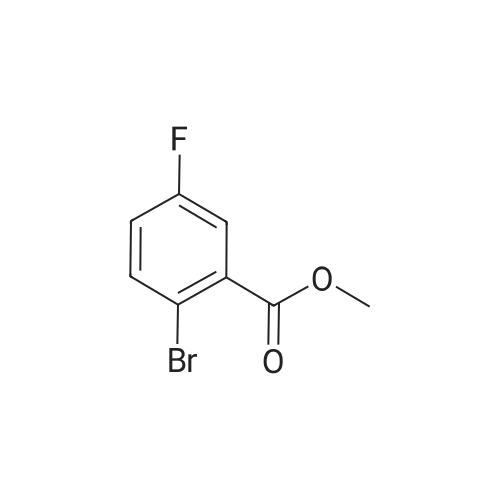
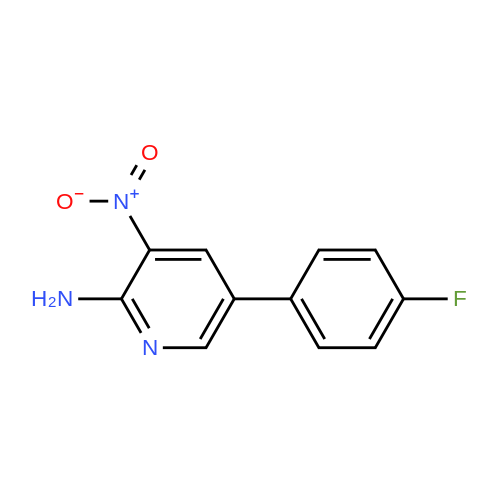
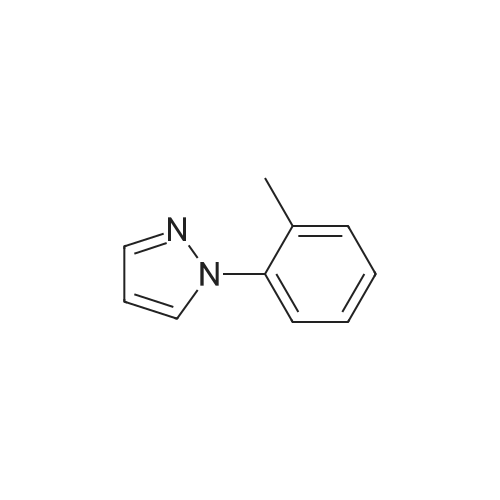
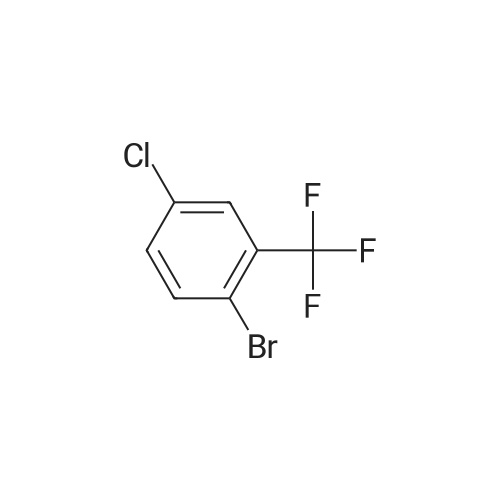
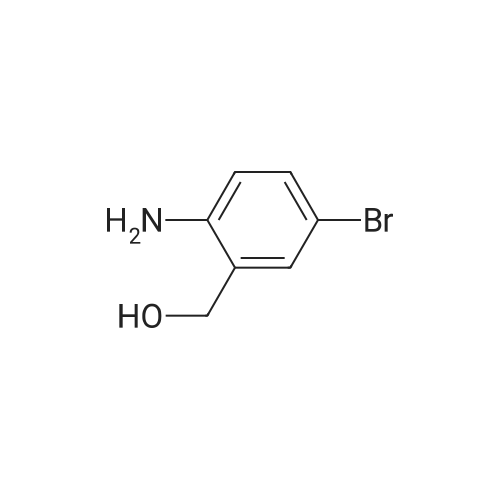
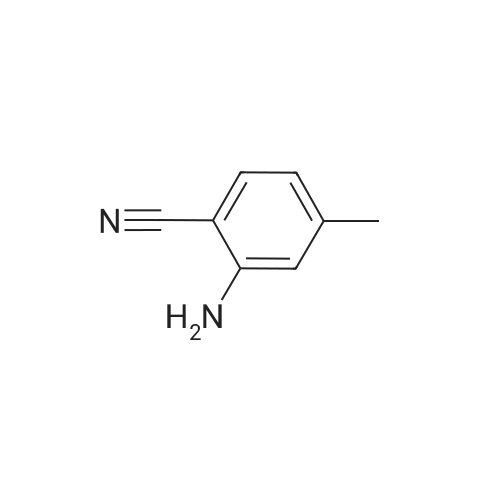
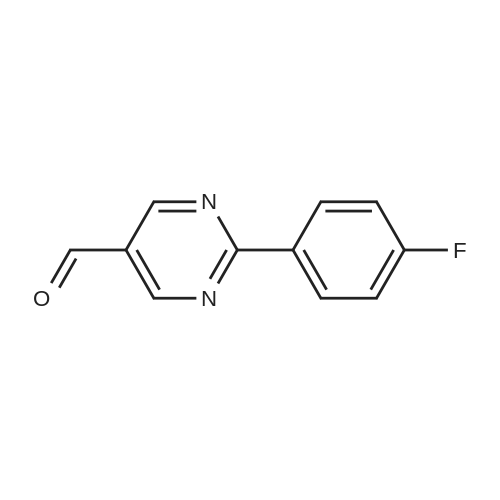
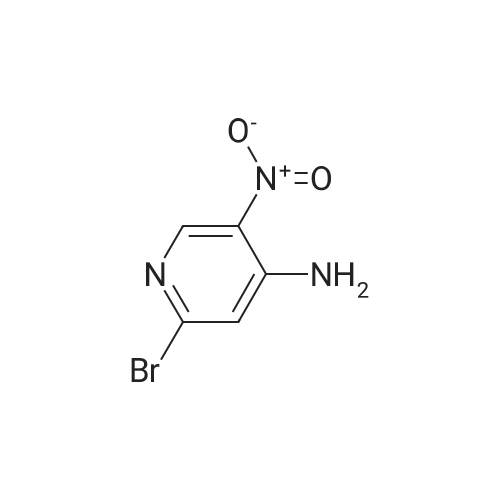
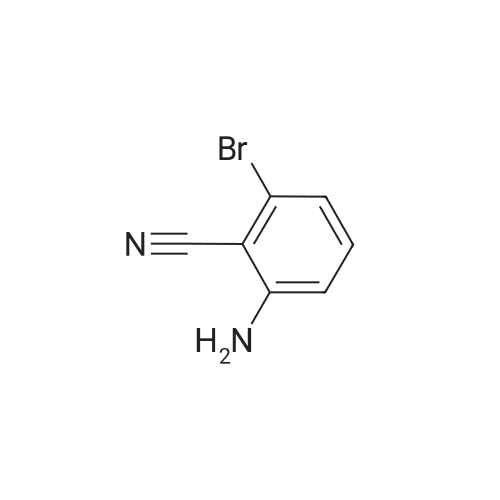
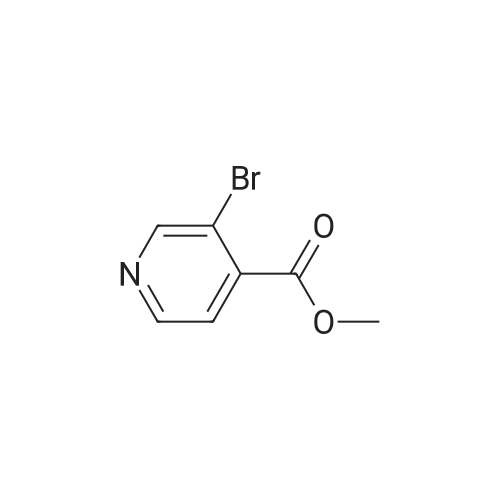
4
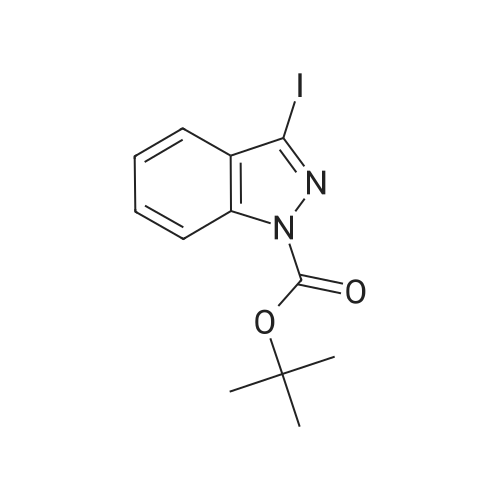
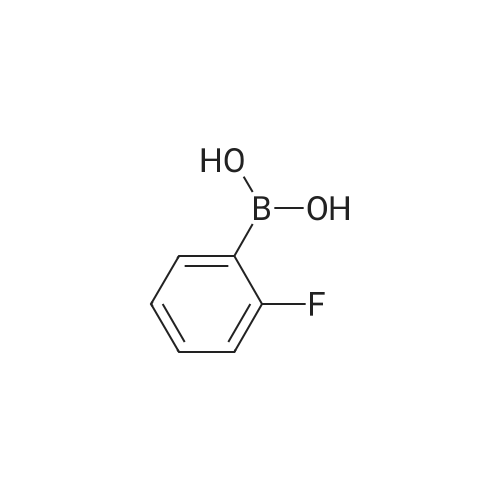
[ 123-73-9 ]
[ 1765-93-1 ]
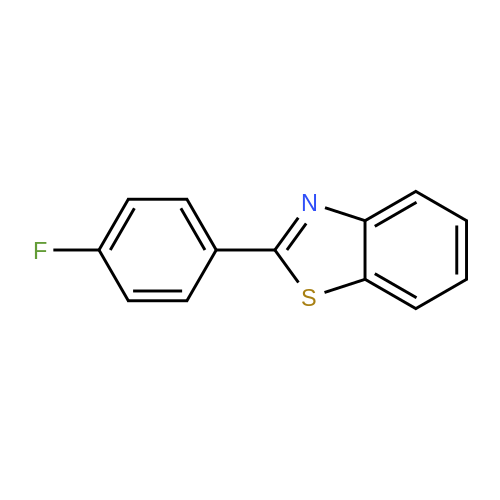
[ 582-83-2 ]
Reference:
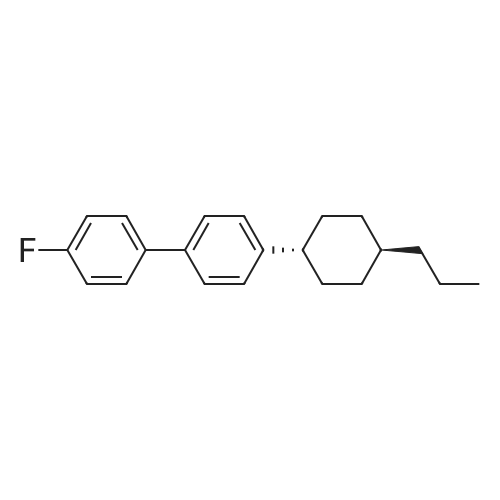
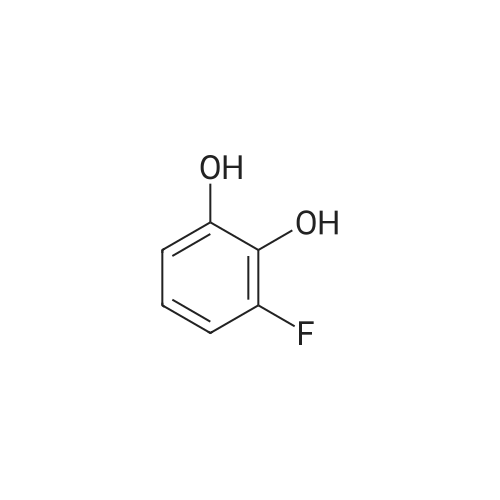
[1] Organic and Biomolecular Chemistry, 2018, vol. 16, # 40, p. 7470 - 7476
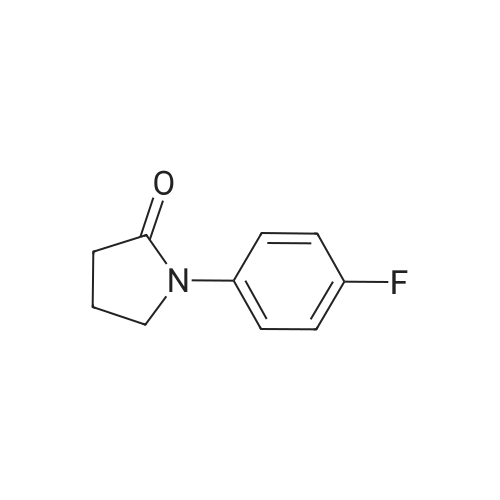
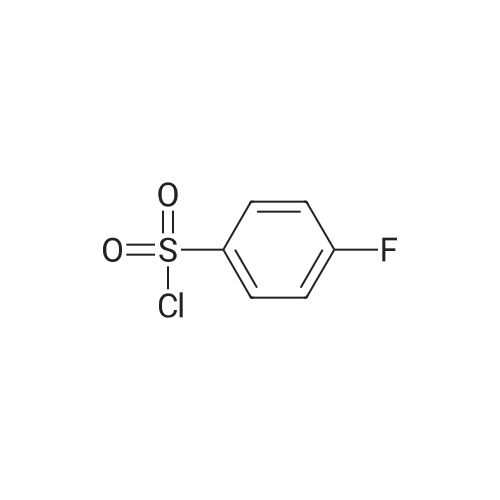
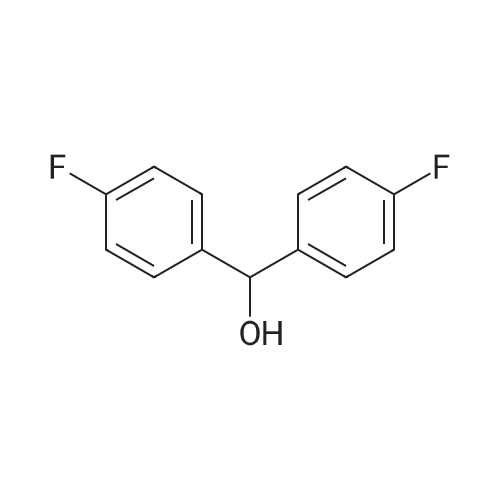
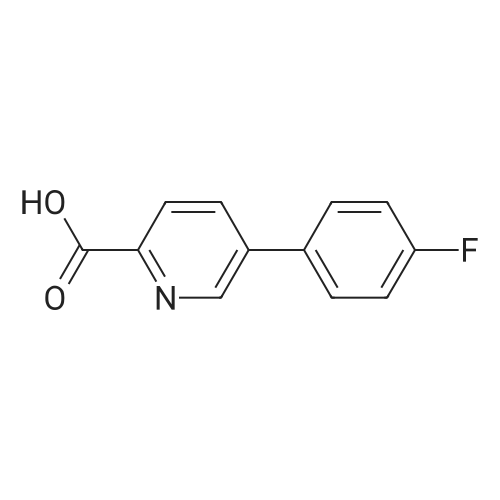
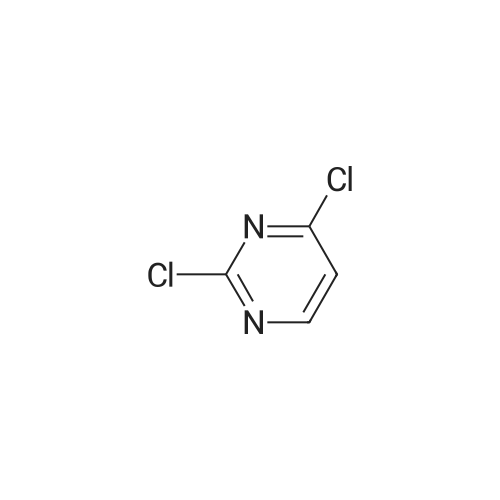
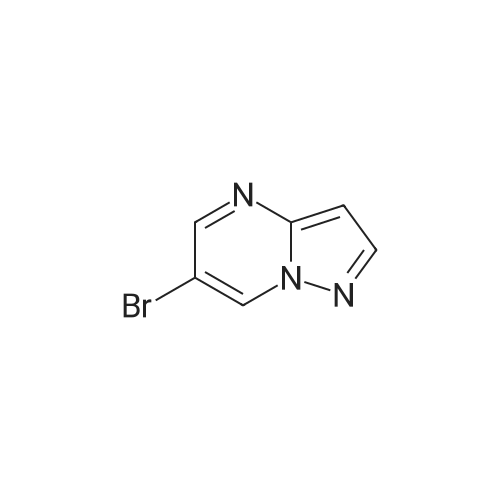
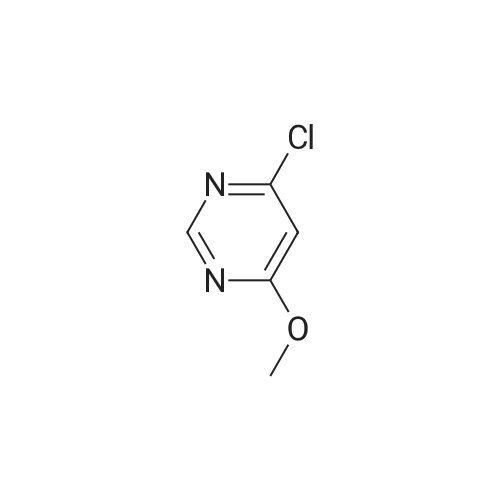
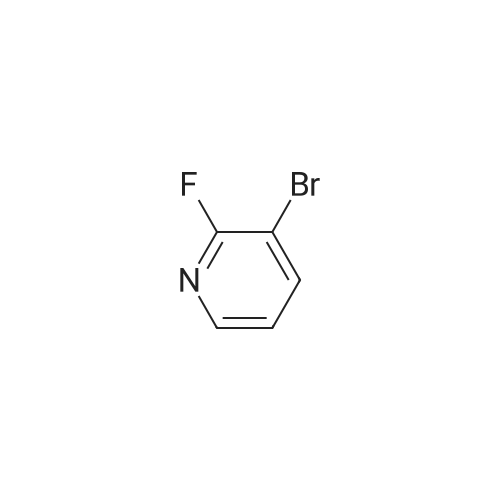
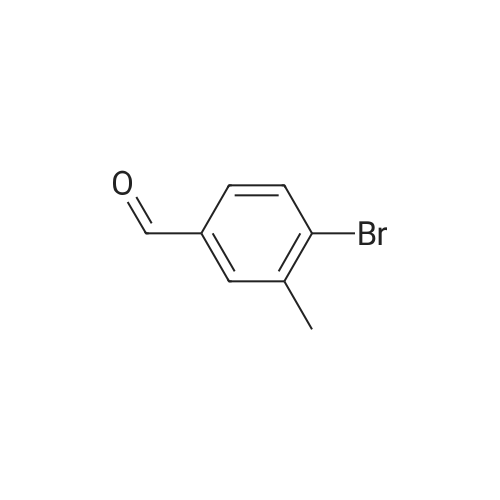
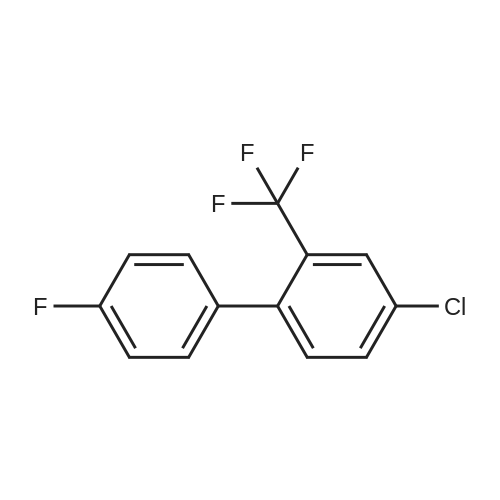
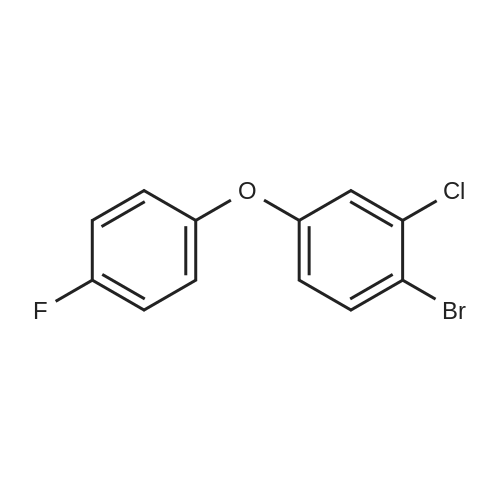
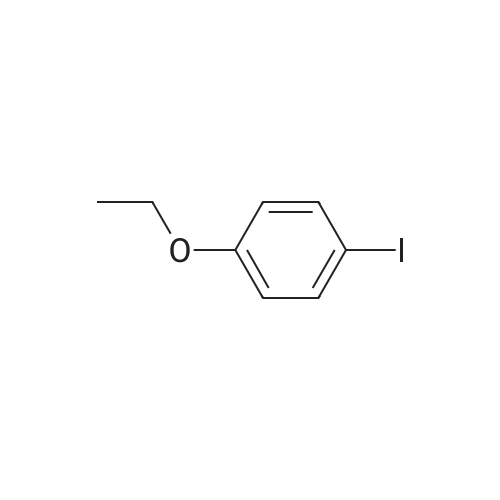
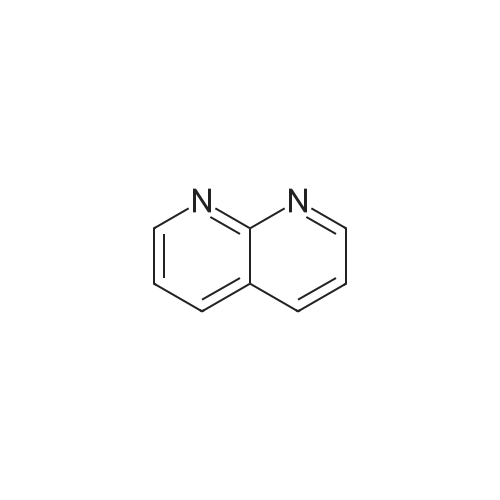
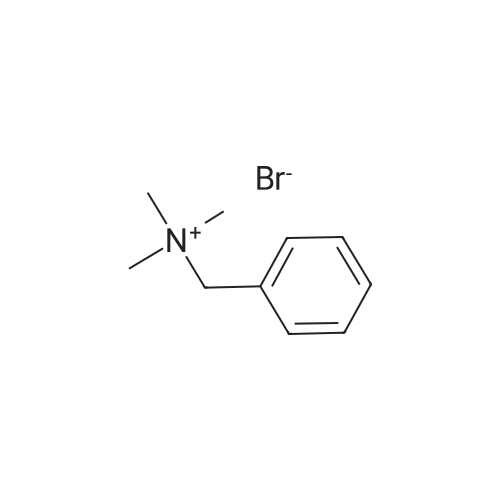
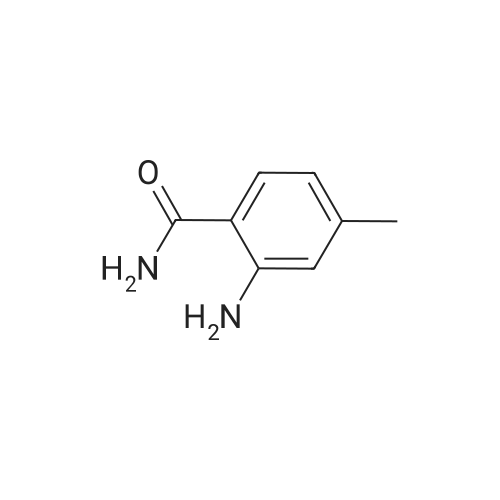
5
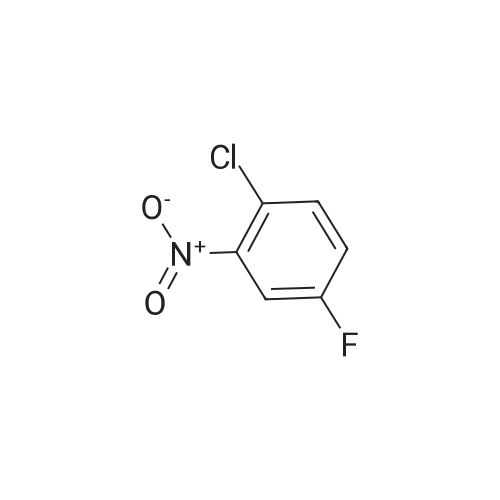
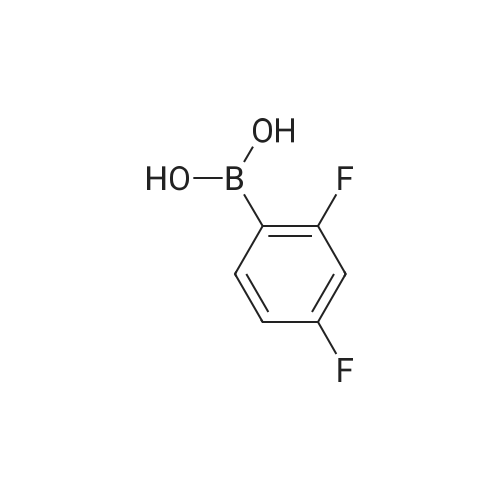
[ 1765-93-1 ]
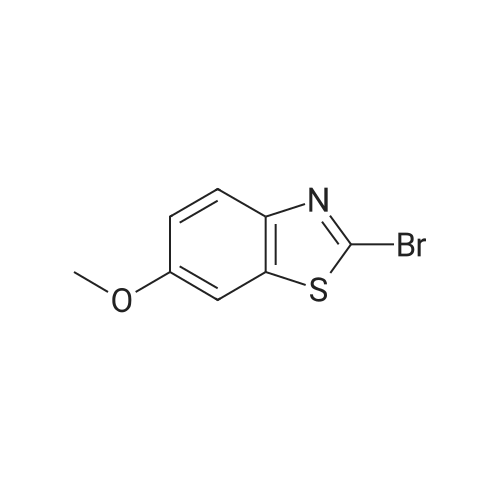
[ 105-36-2 ]
[ 587-88-2 ]
Reference:
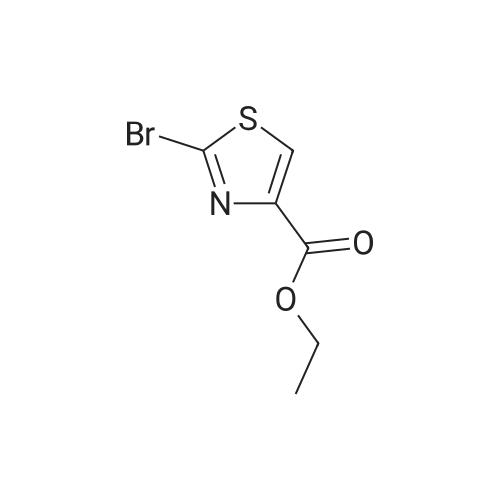
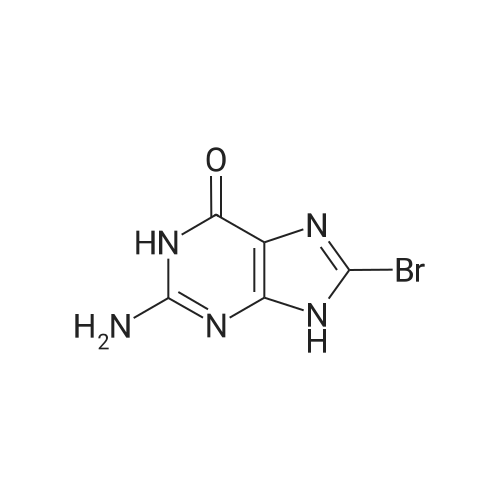
[1] Journal of Organic Chemistry, 2011, vol. 76, # 19, p. 8107 - 8112
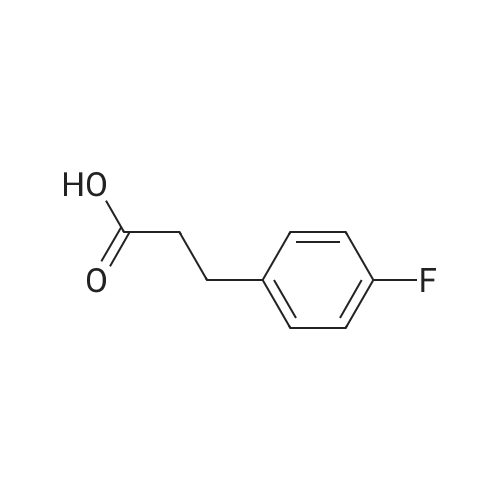
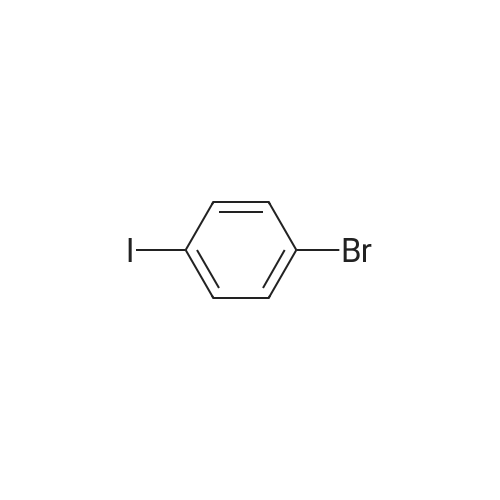
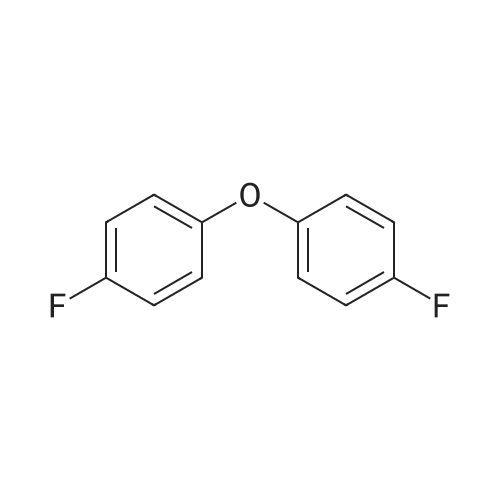
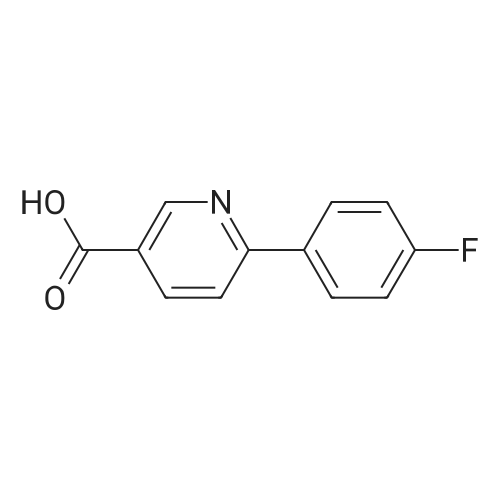
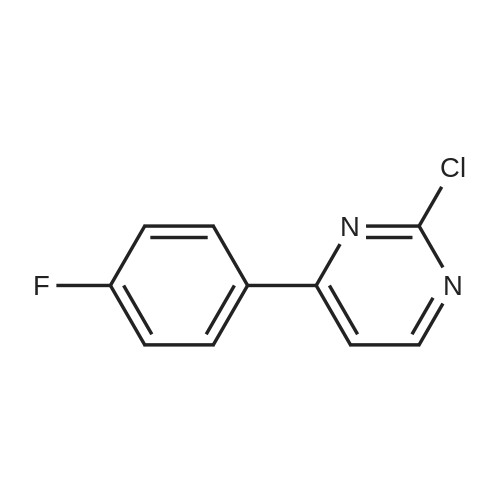
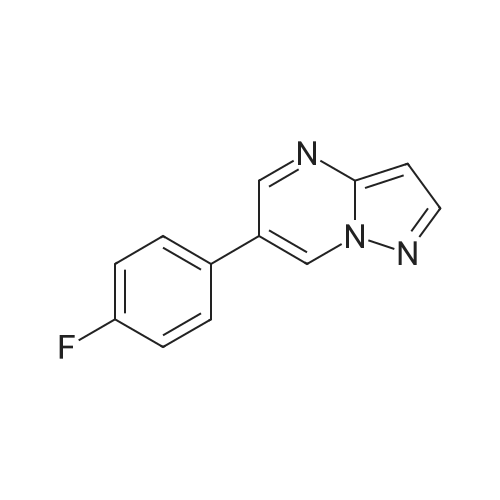
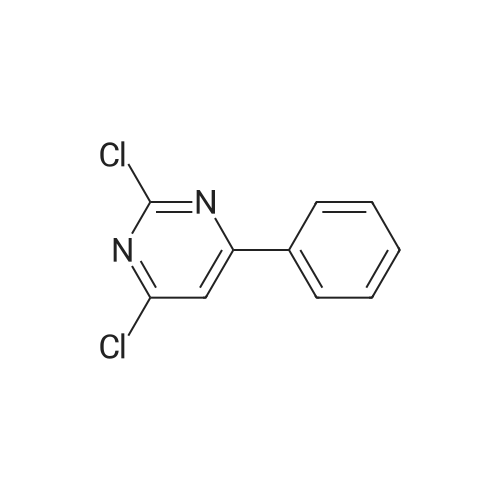
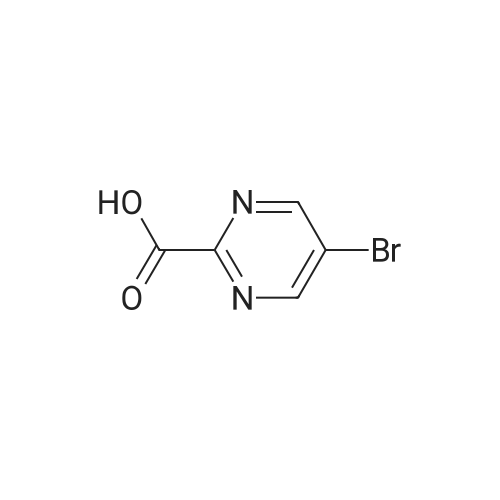
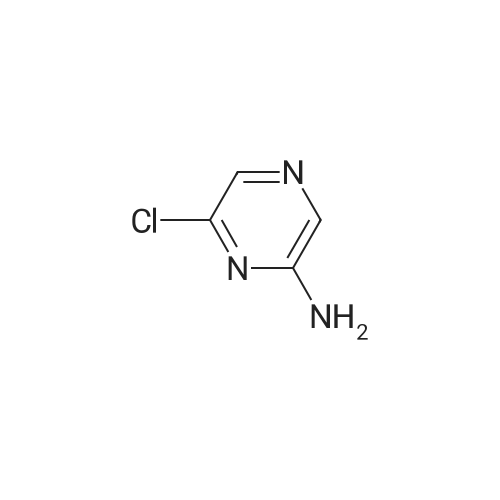
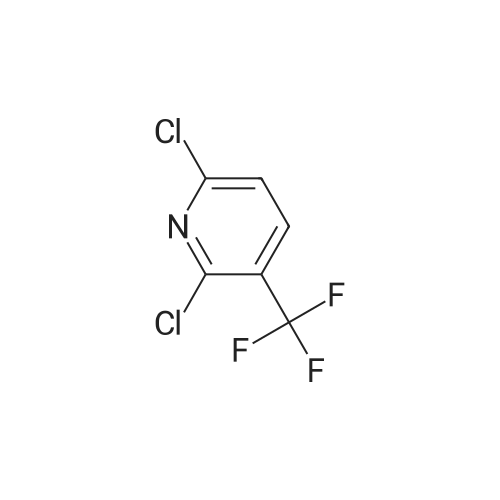
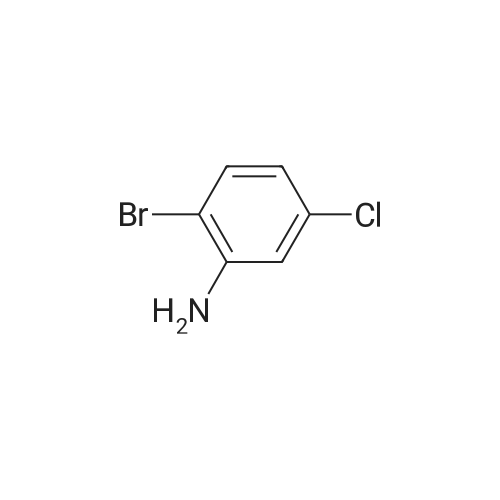
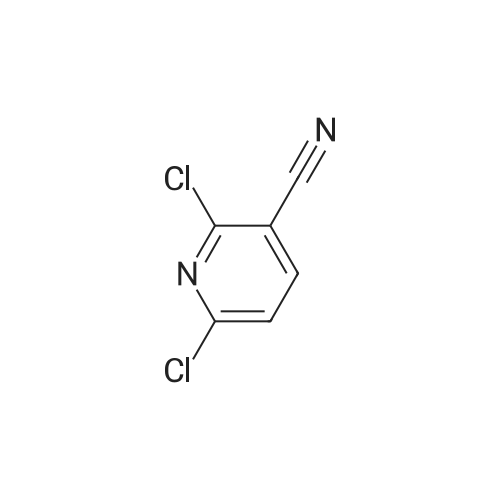
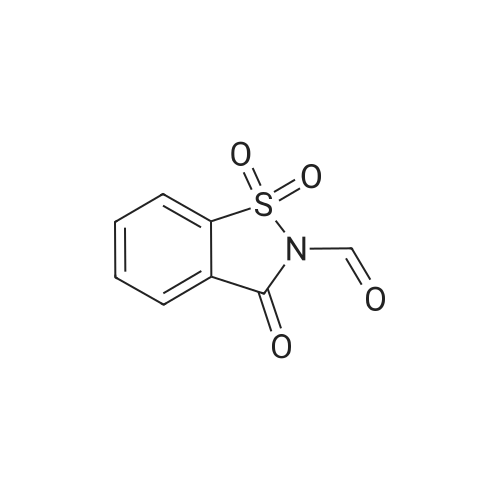
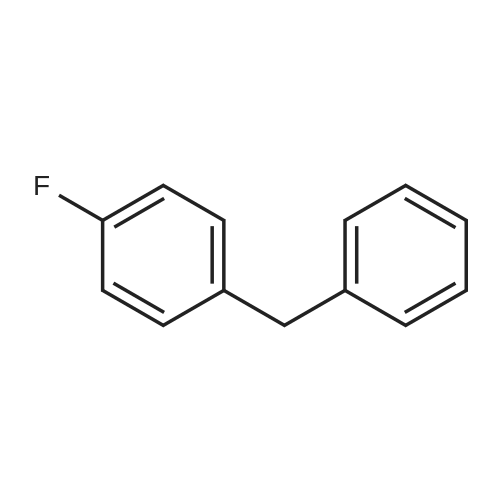
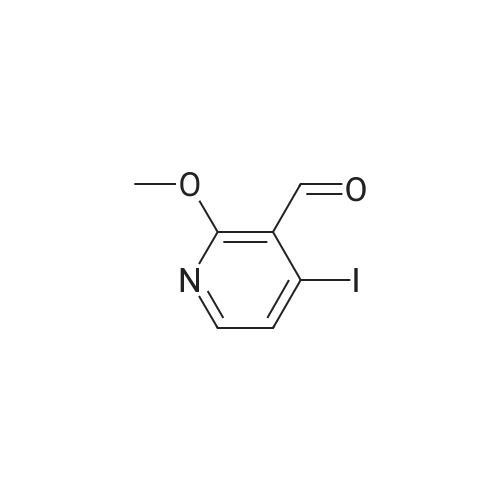
6
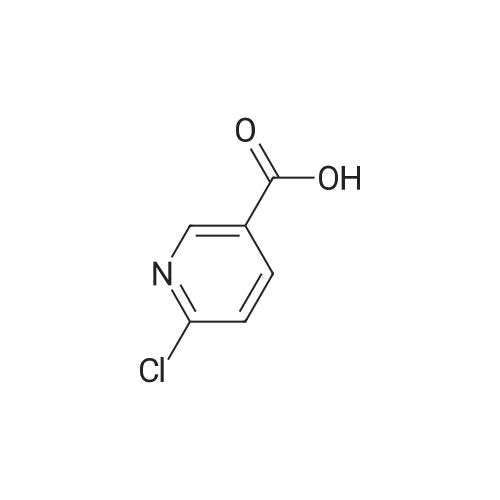
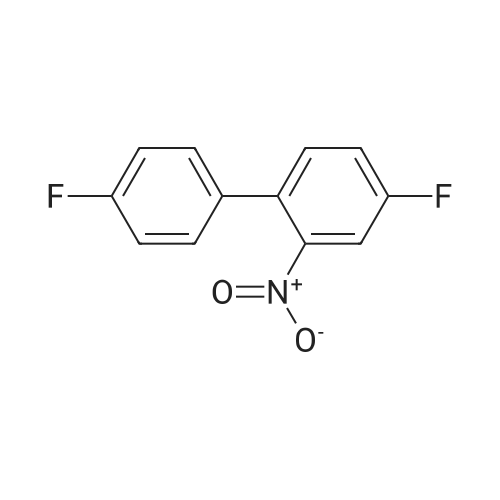
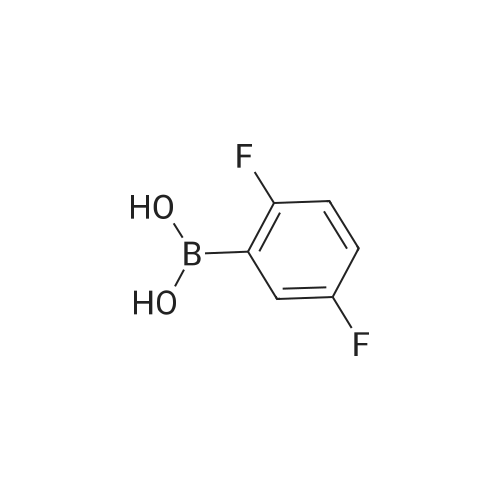
[ 616-45-5 ]
[ 1765-93-1 ]
[ 54660-08-1 ]
Yield
Reaction Conditions
Operation in experiment
97%
Stage #1: With copper(l) iodide In dimethyl sulfoxide at 20℃; for 0.166667 h; Stage #2: With tert.-butylhydroperoxide In water; dimethyl sulfoxide at 60℃; for 1 h;
General procedure: Aryl boronic acid (1.0 mmol), CuI (5 molpercent),amide (3.0 mmol), and DMSO (1.0 mL) were added to a reactionvial, and the mixture was stirred at room temperature for10 min. A 70percent aqueous solution of TBHP (1.1 mmol) was addedto the reaction mixture dropwise over 5 min. The reaction vialwas then immersed in a preheated oil bath and the progress ofreaction was followed by TLC. Upon completion of reaction, thecooled mixture was partitioned between water and ethyl acetate.The aqueous layer was further extracted with ethyl acetate,and the combined organic layers were washed with brine,dried over Na2SO4, filtered, and concentrated in vacuo. Theresidue was purified by column chromatography on silica gel(hexane–ethyl acetate) to give the desired N-aryl lactam
With C38H28O4P2Pd; dihydrogen peroxide; potassium carbonate In water at 20℃; for 4 h;
To the reaction flask was added 1 mmol of 4-fluorobenzyl bromide, 1.2 mmol of 4-fluorobenzeneboronic acid, and anhydrous potassium carbonate2 mmol, PEG 2000 2.5 mmol, 0.01 mmol catalyst, 5 ml pure water, air at room temperatureThe reaction was stopped for 4 h, and the reaction was quenched with about 40 ml of ethyl acetate to give ethyl acetateMachine phase, add 2-3 tablespoons of anhydrous sodium sulfate for 4h in addition to water, filter steaming steam, using TLC methodThe product was isolated using (n-hexane: dichloromethane = 7: 1, v: v) as a developing solvent.Ethanol to give bis (4-fluorophenyl) methanol in a yield of 75percent.
Reference:
[1] Patent: CN105801350, 2016, A, . Location in patent: Paragraph 0044-0046
14
[ 1765-93-1 ]
[ 330-93-8 ]
Reference:
[1] Tetrahedron Letters, 2003, vol. 44, # 37, p. 7061 - 7063
[2] Journal of Organic Chemistry, 2001, vol. 66, # 2, p. 633 - 634
15
[ 1765-93-1 ]
[ 446-52-6 ]
[ 342-25-6 ]
Yield
Reaction Conditions
Operation in experiment
98%
With potassium phosphate; palladium diacetate In water; methyl cyclohexane for 8 h; Reflux
Method of the present embodiment provides a 2,4 ′-difluorobenzophenone and preparation method thereof, as follows: in a 1000 ml round bottom flask The bottle was added sequentially o-fluoro benzaldehyde 124 g of 4-fluorophenylboronic acid and 140 g of potassium phosphate, 106 grams, 1.12 g of palladium acetate, then adding 500 ml of methyl cyclohexane and 100 ml of water and heated to reflux for 8 hours., sequentially passes through filtering, layering, washing with water, removing the solvent to obtain the product., 2,4 '-difluorobenzophenone 214 g, 98percent yield.
Reference:
[1] Chemical Communications, 2016, vol. 52, # 14, p. 2980 - 2983
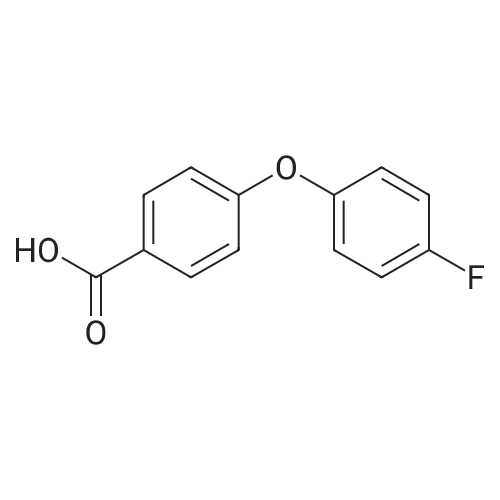
18
[ 1765-93-1 ]
[ 99-76-3 ]
[ 129623-61-6 ]
Yield
Reaction Conditions
Operation in experiment
90%
Stage #1: With molecular sieve; copper diacetate; triethylamine In dichloromethane at 20℃; for 16 h; Stage #2: With hydrogenchloride; lithium hydroxide In 1,4-dioxane; water at 20℃; for 18 h;
To a flask with 4-fluorophenyl boronic acid (2.1 g, 15 mmol), copper(II) acetate (1.4 g, 7.5 mmol), activated powdered molecular sieves (approximately 2 g), and methyl 4-hydroxybenzoate (1.2 g, 7.5 mmol) is added TEA (5.2 mL, 38 mmol) followed by CH2Cl2 (75 mL). The reaction is stirred for 16 hours at room temperature with air bubbling through it. The reaction mixture is diluted with CH2Cl2 and filtered through silica gel. The silica gel is washed with EtOAc-heptane. The solution is concentrated in vacuo and dissolved in dioxane (15 mL). To this solution is added aqueous LiOH (1N, 15 mL) and stirred for 18 hours at room temperature. To this reaction mixture is added aqueous HCl (IN) until acidic, having a pH less than 6. The resulting precipitate is collected by filtration and rinsed with water, and dried in vacuo to give 4-(4-fluorophenoxy)benzoic acid (1.6 g, 90percent). MS for C13H9FO3 (ESI) (M-H)- m/z 231.
With hydrogenchloride; triethanolamine In 1,4-dioxane; dichloromethane
Step F. Preparation of 4-(4-fluorophenoxy)benzoic acid. To a flask with 4-fluorophenyl boronic acid (2.1 g, 15 mmol), copper(II) acetate (1.4 g, 7.5 mmol), activated powdered molecular sieves (approximately 2 g), and methyl 4-hydroxybenzoate (1.2 g, 7.5 mmol) is added TEA (5.2 mL, 38 mmol) followed by CH2Cl2 (75 mL). The reaction is stirred for 16 hours at room temperature with air bubbling through it. The reaction mixture is diluted with CH2Cl2 and filtered through silica gel. The silica gel is washed with EtOAc-heptane. The solution is concentrated in vacuo and dissolved in dioxane (15 mL). To this solution is added aqueous LiOH (1N, 15 mL) and stirred for 18 hours at room temperature. To this reaction mixture is added aqueous HCl (1N) until acidic, having a pH less than 6. The resulting precipitate is collected by filtration and rinsed with water, and dried in vacuo to give the desired product (1.6 g, 90percent). MS for C13H9FO3 (ESI) (M-H)- m/z 231.
Reference:
[1] Patent: US2002/40035, 2002, A1,
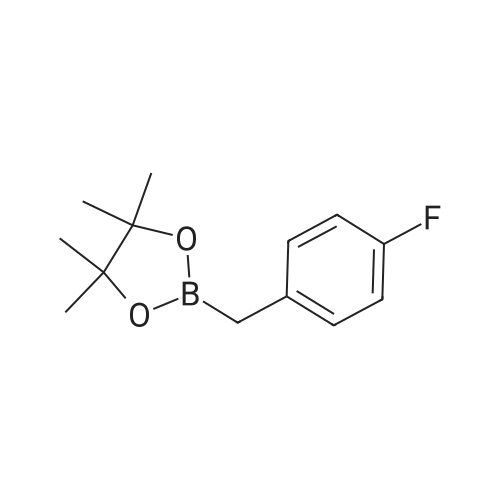
20
[ 76-09-5 ]
[ 1765-93-1 ]
[ 18107-18-1 ]
[ 243145-83-7 ]
Yield
Reaction Conditions
Operation in experiment
77%
Stage #1: at 50℃; for 1 h; Stage #2: With tetrabutyl ammonium fluoride In tetrahydrofuran; 1,4-dioxane; hexane; water at 50℃; for 1 h;
To a 10 mL reaction tube equipped with a magnet was added 0.4 mg (0.8 mmol) of 4-fluorophenylboronic acid (56 mmol), trimethylsilyl diazomethane (2M n-hexane solution), 1 mL of 1, 4-dioxane, plug the rubber stopper and react for 1 hour at 50 ° C on an electromagnetic heating stirrer.(0.4 mmol) of tetramethylammonium fluoride (1 M tetrahydrofuran solution) and 200 uL of water were added to an electromagnetic heating stirrer at 50 ° C, respectively, followed by the addition of 40 mg of pinacol (dissolved in 1 mL of 1,4-dioxane) To continue for 1 hour.After completion of the reaction, the organic solvent was removed by a rotary evaporator and purified by column chromatography4-fluorobenzyl boronic acid pinacol ester, its structure is as follows:The compound was a colorless liquid in a yield of 77percent with the following NMR data:
Reference:
[1] Patent: CN105884808, 2016, A, . Location in patent: Paragraph 0070; 0071; 0072; 0073; 0074; 0075
21
[ 5164-76-1 ]
[ 1765-93-1 ]
[ 216690-15-2 ]
Reference:
[1] Journal of the American Chemical Society, 2007, vol. 129, # 2, p. 290 - 291
22
[ 76-09-5 ]
[ 1765-93-1 ]
[ 214360-58-4 ]
Reference:
[1] Advanced Synthesis and Catalysis, 2013, vol. 355, # 18, p. 3553 - 3557
[2] Chemical Communications, 2015, vol. 51, # 21, p. 4406 - 4409
[3] Angewandte Chemie - International Edition, 2012, vol. 51, # 37, p. 9385 - 9388
[4] Journal of Organic Chemistry, 2014, vol. 79, # 21, p. 10568 - 10580
[5] Bulletin of the Chemical Society of Japan, 2009, vol. 82, # 7, p. 870 - 878
[6] Journal of the American Chemical Society, 2013, vol. 135, # 7, p. 2552 - 2559
[7] Organic Letters, 2018, vol. 20, # 20, p. 6573 - 6577
23
[ 1765-93-1 ]
[ 1321604-76-5 ]
[ 214360-58-4 ]
Reference:
[1] Chemistry - A European Journal, 2011, vol. 17, # 29, p. 8005 - 8008
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In 1,2-dimethoxyethane; water at 98℃; for 24 h; Inert atmosphere
Step A:5-bromopyridine-2-carboxylic acid (57, 1.0 g, 5.0 mmol)Dissolved in ethylene glycol dimethyl ether (12 mL) and water (4 mL),Add p-fluorophenylboronic acid (17, 1.0 g, 7.5 mmol)And anhydrous potassium carbonate (1.0 g, 7.5 mmol),Then tetrakis(triphenylphosphine)palladium (289 mg, 0.25 mmol) was added.The resulting mixture was stirred at 98 ° C for 24 hours under a nitrogen atmosphere.TLC analysis indicated that the reaction was over,The reaction solution was cooled to room temperature.Then add water (40 mL),The pH was adjusted to 2-3 with 6M hydrochloric acid.Filtered, the filter cake is dissolved in dichloromethane,The organic layer was washed with 20 mL of saturated sodium bicarbonate solution.Divide the water layer,The aqueous layer was adjusted to pH 2-3 with a 6M hydrochloric acid solution.Filter the solid,The filter cake is washed with water to neutrality.The filter cake was dried to give compound 61 (942 mg).Yield: 87.7percent.
Reference:
[1] Patent: CN108623532, 2018, A, . Location in patent: Paragraph 0206; 0207; 0208
[2] Patent: EP1657242, 2006, A1, . Location in patent: Page/Page column 31
[3] Bioorganic and Medicinal Chemistry, 2016, vol. 24, # 4, p. 812 - 819
[4] Medicinal Chemistry, 2017, vol. 13, # 2, p. 176 - 185
[5] Medicinal Chemistry Research, 2018, vol. 27, # 2, p. 374 - 387
27
[ 113118-81-3 ]
[ 1765-93-1 ]
[ 381684-96-4 ]
Reference:
[1] Zeitschrift fur Naturforschung - Section B Journal of Chemical Sciences, 2012, vol. 67, # 1, p. 75 - 84
[2] Patent: US2014/296528, 2014, A1, . Location in patent: Sheet 4
28
[ 3934-20-1 ]
[ 1765-93-1 ]
[ 85979-59-5 ]
Yield
Reaction Conditions
Operation in experiment
61%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In ethanol; water; toluene at 55℃; for 12 h;
To a stirred solution of 2,4-dichloropyrimidine (3.00 g, 20.1 mmol) in toluene (25 mL) was added 4-fluorophenylboronic acid (2.82 g, 20.1 mmol), potassium carbonate (8.32 g, 60.3 mmol), tetrakis(triphenylphosphine)palladium(0) (0.630 g, 0.545 mmol) and 1 : 1 (v/v) ethanol/water (36 mL). The mixture was heated at 55 °C for 12 hours and then concentrated. The residue was diluted with water and extracted with ethyl acetate. The combined extracts were washed with brine, dried (Na2S04) and concentrated. The crude material was purified by flash chromatography over silica using a hexane/ethyl acetate eluant to afford 2-chloro-4-(4-fluorophenyl)pyrimidine as a yellow solid (2.50 g, 61percent). To a stirred solution of this compound (1.27 g, 6.09 mmol) in N,N-dimethylformamide (8 mL) was added ethyl piperidine-4-carboxylate (0.959 g, 6.10 mmol) and cesium carbonate (2.10 g, 6.44 mmol). The mixture was heated at 100 °C for 12 hours and then concentrated. The residue was diluted with water and extracted with ethyl acetate. The combined extracts were washed with brine, dried (Na2S04) and concentrated. The crude material was purified by flash chromatography over silica using a hexane/ethyl acetate eluant to afford ethyl l-(4'-fluoro-[l, -biphenyl]-3-yl)piperidine-4-carboxylate as a yellow oil (1.60 g, 80percent>). To a stirred solution of this intermediate (1.60 g, 4.80 mmol) in 1 : 1 (v/v) methanol/water (20 mL) was added solid sodium hydroxide (0.968 g, 24.2 mmol). After 2 hours, the reaction was concentrated. The residue was dissolved in water, made acidic (pH ~6) with the addition of IN hydrochloric acid and extracted with ethyl acetate. The combined organic layers were washed with brine, dried (Na2S04) and concentrated to afford l-(4'-fluoro-[l, -biphenyl]-3-yl)piperidine-4-carboxylic acid as a white solid (1.40 g, 97percent). Using General Procedure D and Intermediate 5, this carboxylic acid was subjected to amide coupling to generate the title compound as a white solid (0.118 g, 27percent). FontWeight="Bold" FontSize="10" Η ΝΜΚ (500 MHz, CDC13) δ 8.37 (d, J= 5.0 Hz, 1H), 8.07-8.04 (m, 2H), 7.15 (t, J= 9.0 Hz, 2H), 6.89 (d, J= 10.0 Hz, 1H), 5.38 (s, 1H), 4.97-4.95 (m, 2H), 3.02-2.83 (m, 8H), 2.39-2.37 (m, 2H), 1.96-1.51 (m, 13H) ppm. 13C NMR (100 MHz, CDCI3) δ 174.1, 165.3, 163.3, 163.2, 161.7, 158.4, 133.8, 129.0, 128.9, 115.7, 115.5, 105.2, 59.4, 53.1, 47.6, 46.1, 44.6, 43.5, 39.3, 36.1, 28.9, 28.7, 25.1, 24.3, 24.2 ppm. Purity: >96percent (214 & 254 nm) LCMS; retention time: 1.44 min; (M+H+) 438.3.
fuming nitric acid (140 mL) was placed in a 250 mL four-necked flask and stirred at -35 ° C to -45 ° C with a dry ethanol bath. Fluorobenzene boric acid (20 g) was thoroughly dried and pulverized in batches and slowly added to a four-necked flask to maintain the reaction temperature between -35 ° C and -45 ° C. After the addition of p-fluorobenzene boronic acid, the reaction was complete by TLC. The reaction solution into 200g crushed ice, rapid and stirring, a yellow solid precipitation, Buchner funnel filter, ice water 20mL washing 2 times, drained. The resulting filtrate was adjusted to pH 6 with sodium bicarbonate and extracted with ethyl acetate. The oil phase was combined, dried over magnesium sulfate, dried to dryness, n-heptane beating, To give a pale yellow solid, 8.3 g, yield 31.5percent
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate In ethanol; toluene at 85℃; for 6 h; Inert atmosphere
To a solution of 2-bromothiazole-5-carbaldehyde (5.0 g, 26.04 mmol) in toluene (150 mL) and ethanol (75 mL) was added (4-fluorophenyl)boronic acid (7.29 g, 52.08 mmol), 2M sodium carbonate solution (73.58 mL), Pd(PPh3)4 (1.5 g, 1.3 mmol) under argon atmosphere. The resulting mixture was heated at 85°C for 6 h. The reaction mixture was concentrated under reduced pressure, the residue was diluted with water (150 mL), and extracted with ethyl acetate (5 x 500 mL). The organic layer was dried over Na2S04, filtered and concentrated under reduced pressures to obtain crude product. The crude product was purified by Combiflash® chromatography (Mobile phase: 10percent ethyl acetate in hexane) to give 2-(4- fluorophenyl)thiazole-5-carbaldehyde as off-white solid (200 mg, 88percent yield). IH NMR (400 MHz, DMSO-d6): δ 10.06 (s, IH), 8.74 (s, IH), 8.14-8.11 (m, 2H), 7.39 (t, 2H); LC-MS m/z calculated for [M+H]+ 208.02, found 207.9
Reference:
[1] Chemical Research in Toxicology, 2014, vol. 27, # 12, p. 2052 - 2061
[2] Bioorganic and Medicinal Chemistry Letters, 2016, vol. 26, # 18, p. 4403 - 4407
36
[ 1765-93-1 ]
[ 86579-53-5 ]
[ 87260-24-0 ]
Reference:
[1] European Journal of Organic Chemistry, 2013, # 20, p. 4345 - 4350
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; water; at 80℃; for 9h;
To a suspension of <strong>[6945-68-2]2-amino-5-bromo-3-nitropyridine</strong> [(2. 2] [G,] 10.1 mmol) in dimethoxyethane (20 mL) were added [4-FLUOROPHENYLBORONIC] acid (4.83 [G,] 13.1 mmol), tetrakis (triphenylphosphine) palladium (580 mg, 0.50 mmol) and 2N aqueous sodium carbonate solution (10 mL). The mixture was stirred at [80C] for 9 hours under a nitrogen atmosphere. After the reaction mixture was cooled to room temperature, it was diluted with ethyl acetate to give a precipitate, which was collected by filtration, washed with ethyl acetate and then dried to give the title compound (1.76 g, 76 [%).]
60%
With dichloro[1,1?-bis[bis(1,1-dimethylethyl)phosphino]ferrocene-P,P?]palladium; caesium carbonate; In 1,4-dioxane; water; at 80℃; for 4h;
A solution of <strong>[6945-68-2]2-amino-5-bromo-3-nitropyridine</strong> (623 mg, 2.86 mmol), 4-fluorophenylboronic acid (525 mg, 3.75 mmol), Cs2CO3 (1.87 g, 5.74 mmol), and PdCl2(dppf) (209 mg, 0.29 mmol) in 1,4-dioxane (14 mL) and water (3 mL) was heated to 80 C for 4 h and then concentrated. The residue was partitioned between DCM and water, upon which time the product crashed out. The heterogeneous mixture was redissolved with a solution of EtOAc containing 1.5% IPA. The aqueous layer was further extracted with EtOAc containing 1.5% IPA. The combined organic layers were dried, filtered and concentrated to yield the title compound (400 mg, 60%) as a solid, which was used in the next step without further purification.
With sodium carbonate;trans-bis(tricyclohexylphosphine)palladium(II) dichloride; In 1,4-dioxane; water; at 110℃; for 17h;Heating / reflux;
5.0 g of <strong>[6945-68-2]2-amino-5-bromo-3-nitropyridine</strong> are dissolved in 120 ml of anoxic dioxane under a nitrogen atmosphere. Subsequently, 69 mi of an aqueous sodium bicarbonate solution (2.0 M), 5.4 g of 4- fluorophenyl-boronic acid, and 1.0 g of trans-dichloro-bis (tricyclohexylphosphane) palladium- (Il) are added. The reaction-mixture is refluxed at 110C for 17 hours. Thereafter, the volatile components are removed in vacuo and the remaining residue is redissolved in 2.0 1 of a mixture of water/dichloromethane (1: 1). The aqueous phase is extracted four times each with 625 ml of dichloromethane. The organic layer is separated, dried using sodium sulfate, and evaporated to dryness to yield a colorless, crude solid. Subsequently, the residue is purified by flash chromatography on silica gel (eluent : toluene/ethyl acetate 20: 1) to afford 5.35 g of the title compound as a colorless solid of m. p. 232C. MS: 234.2 (MH+). TLC: Rf = 0.30 (toluene/ethyl acetate 20: 1).
With sodium carbonate; In ethanol; water; toluene; at 60 - 75℃; for 40h;
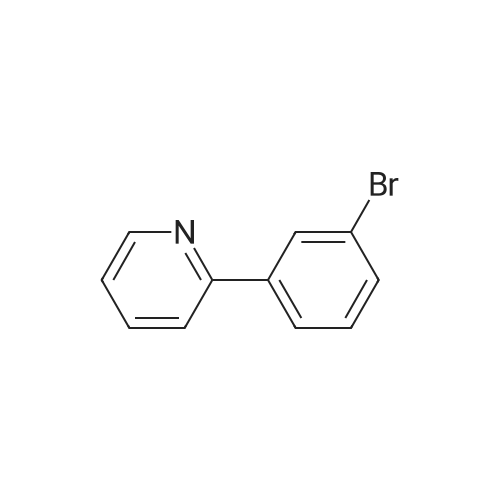
Example 2: (2S)-2-[(S)-[3-(4-fluorophenyl0pyridin-2-yl]thio}(phenyl)methyl]morpholine fumarate S, H J Fs / 0 H SH R Ph Fh C Ph Ph I Oh Fumarate salt i) To bis (benzonitrile) palladium (II) dichloride (0.054 g, 0.14 mmole) and 1,4- bis (diphenylphosphine) butane (0.091 g, 0.21 mmole) is added dry toluene (6 ml), under nitrogen, and the mixture stirred for half an hour. To this is added 3-bromo-2- fluoropyridine (0.50 g, 2.83 mmole) in ethanol (1.4 ml) followed by a solution of 4- fluorophenylboronic acid (0.793 g, 5.67 mmole) in ethanol (2.4 ml). To this is added an aqueous solution of sodium carbonate (1 M, 2.83 ML, 2.83 mmole). The mixture is heated at 60°C for 24 hours, then at 75°C for a further 16 hours. The organic layer is loaded directly onto a 40 g REDISEP SIO2 COLUMN and components isolated by automated flash chromatography (ISCO System, 0-30 percent ethyl acetate in cyclohexane gradient elution over 40 minutes). This gave 3- (4-fluorophenyl)-2- fluoropyridine as a white solid (0.387 g, 71 percent). LCMS 6 min gradient method, Rt = 3. 6 min, (M+H+) = 192
71%
With sodium carbonate;bis(benzonitrile)palladium(II) dichloride; 1,4-di(diphenylphosphino)-butane; In ethanol; water; toluene; at 60 - 75℃; for 40.5h;
To bis (benzonitrile) palladium (II) dichloride (0.054 g, 0.14 mmole) and 1,4- bis (diphenylphosphine) butane (0.091 g, 0.21 mmole) is added dry toluene (6 ml), under nitrogen, and the mixture stirred for half an hour. To this is added 3-bromo-2- fluoropyridine (0.50 g, 2. 83 mmole) in ethanol (1.4 ml) followed by a solution of 4- fluorophenylboronic acid (0.793 g, 5.67 mmole) in ethanol (2.4 ml). To this is added an aqueous solution of sodium carbonate (1 M, 2. 83 ml, 2.83 mmole). The mixture is heated at 60°C for 24 hours, then at 75°C for a further 16 hours. The organic layer is loaded directly onto a 40 g Redisep SI02 column and components isolated by automated flash chromatography (ISCO System, 0-30 percent ethyl acetate in cyclohexane gradient elution over 40 minutes). This gave 3- (4-FLUOROPHENYL)-2-FLUOROPYRIDINE as a white solid (0.387 g, 71 percent). LCMS 6 min gradient method, Rt = 3.6 min, (M+H+) = 192
71%
With sodium carbonate; 1,4-di(diphenylphosphino)-butane;bis(benzonitrile)palladium(II) dichloride; In ethanol; water; toluene; at 60 - 75℃; for 40.5h;
To bis (benzonitrile) palladium (II) dichloride (0.054 g, 0.14 mmole) and 1,4- bis (diphenylphosphine) butane (0.091 g, 0.21 mmole) is added dry toluene (6 ml), under nitrogen, and the mixture stirred for half an hour. To this is added 3-bromo-2- fluoropyridine (0.50 g, 2. 83 mmole) in ethanol (1.4 ml) followed by a solution of 4- fluorophenylboronic acid (0.793 g, 5.67 mmole) in ethanol (2.4 ml). To this is added an aqueous solution of sodium carbonate (1 M, 2. 83 ml, 2.83 mmole). The mixture is heated at 60°C for 24 hours, then at 75°C for a further 16 hours. The organic layer is loaded directly onto a 40 g Redisep SI02 column and components isolated by automated flash chromatography (ISCO System, 0-30 percent ethyl acetate in cyclohexane gradient elution over 40 minutes). This gave 3- (4-FLUOROPHENYL)-2-FLUOROPYRIDINE as a white solid (0.387 g, 71 percent). LCMS 6 min gradient method, Rt = 3.6 min, (M+H+) = 192
5-(4-Fluorophenyl)pyrimidine-2-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83.5%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In 1,2-dimethoxyethane; water; at 98℃; for 24h;Inert atmosphere;
Step A:<strong>[37131-87-6]5-bromopyrimidine-2-carboxylic acid</strong> (41, 1.0 g, 5.0 mmol)Dissolved in ethylene glycol dimethyl ether (12 mL) and water (4 mL),Add p-fluorophenylboronic acid (17, 1.0 g, 7.5 mmol)And anhydrous potassium carbonate (1.0 g, 7.5 mmol),Then tetrakis(triphenylphosphine)palladium (289 mg, 0.25 mmol) was added.The resulting mixture was heated to 98 C under nitrogen for 24 hours.TLC analysis indicated that the reaction was over,The reaction solution was cooled to room temperature.Then add water (40 mL),The pH was adjusted to 2-3 with 6M hydrochloric acid.Filtered, the filter cake is dissolved in dichloromethane,The organic layer was washed with 20 mL of saturated sodium bicarbonate solution.Divide the water layer,The aqueous layer was adjusted to pH 2-3 with a 6M hydrochloric acid solution.Filter the solid,The filter cake is washed with water to neutrality.The filter cake was dried to give compound 42 (910 mg).Yield: 83.5%.
Production Example 34 5-(4-Fluorophenyl)-2-pyrimidinecarboxylic acid Conducting the operations similar to those of Production Example 14 using <strong>[37131-87-6]5-bromopyrimidine-2-carboxylic acid</strong> and 4-fluorophenylboronic acid, the title compound was obtained as white solid. ESI-MS Found:m/z 219[M+H]+ ESI-MS Found:m/z 217[M-H]-
With sodium carbonate; In 1,2-dimethoxyethane; hexane; ethyl acetate;
Preparation 63 To a suspension of <strong>[89488-29-9]2-bromo-4-methoxypyridine</strong> (0.94 g), 4-fluorophenylboronic acid (909 mg) and tetrakis(triphenylphosphine)-palladium (289 mg) in 1,2-dimethoxyethane (20 ml) was added 2M aqueous solution of sodium carbonate (6.5 ml). The mixture was stirred at 80 C. for 4 hours under a nitrogen atmosphere, then cooled to room temperature and diluted with ethyl acetate. The organic layer was separated, washed with water and brine and dried over sodium sulfate. The solvent was evaporated under reduced pressure. The residue was purified by column chromatography (silica gel 50 g, 30% ethyl acetate in n-hexane) to give 4-(4-methoxypyridin-2-yl)fluorobenzene (0.866 g). 1H-NMR (CDCl3): delta3.91(3H,s), 6.7-6.9(1H,m), 7.1-7.2(2H,m), 7.9-8.1(1H,m), 8.50(1H,d,J=5.7 Hz)
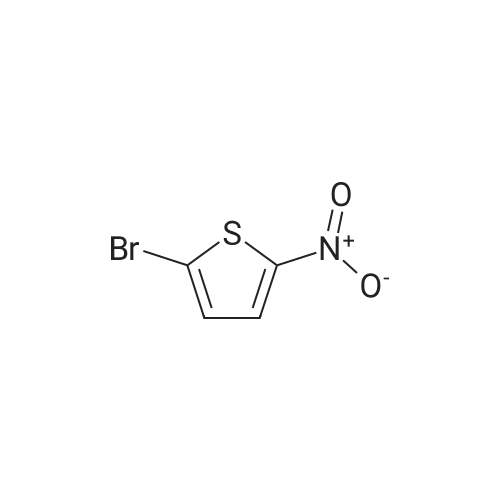
Example 32 A specific procedure for the synthesis of: N-5-[(4-fluoro)phenyl-thien-2-yl]methanesulfonamide (32) STR20 <strong>[13195-50-1]2-Bromo-5-nitrothiophene</strong> (3.50 g, 16.8 mmol) was dissolved in toluene (50 mL). To this solution was added 4-fluorophenyl boronic acid (2.52 g, 18.0 mmol), tetrakis(triphenylphosphine)paJladium (0) (0.58 g, 3 molepercent), potassium carbonate (4.56 g, 33.0 mmol) and water (25 mL) and the resulting mixture refluxed for 18 hours after which time the layers were separated. The organic layer was evaporated to a crude solid which was recrystallized from ethanol-water to give 2-nitro-5-(4-fluoro)phenylthiophene (2.99 grams (80percent)1. Mp 129°-130° C.
5-bromo-6-(4-fluorophenyl)-1,3-benzodioxole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
3.9 g (71%)
In ethanol; ethyl acetate; toluene;
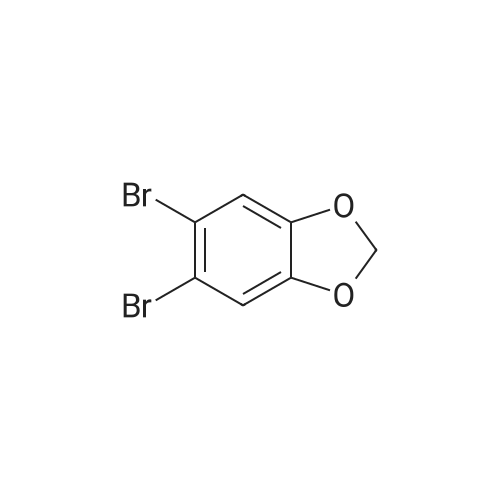
Step 1 Preparation of 5-bromo-6-(4-fluorophenyl)-1,3-benzodioxole Under nitrogen, 1.1 g of Pd(PPh3)4 was added to a stirred solution of 10.4 g (37.2 mmol) of <strong>[5279-32-3]5,6-dibromo-1,3-benzodioxole</strong> and 2.6 g (18.6 mmol) of 4-fluorophenylboronic acid in 100 mL of toluene, 60 mL of ethanol, and 40 mL of M Na2 CO3. After vigorous stirring at reflux overnight, the solvent was removed in vacuo. The residue was dissolved in ethyl acetate, washed with water, and dried over Na2 SO4. Purification by silica gel chromatography (Waters Prep-500A) with hexane as the eluent gave 3.9 g (71%) of 5-bromo-6 (4-fluorophenyl)-1,3-benzodioxole as a colorless solid: mp 86.0-87.5 C.; NMR (CDCl3) delta 6.02 (s, H), 6.77 (s, 1H), 7.04-7.13 (m, 3H), 7.28-7.35 (m, 2H).
5-(4-fluorophenyl)-6-[4-(methylthio)phenyl]-1,3-benzodioxole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In ethanol; ethyl acetate; toluene;
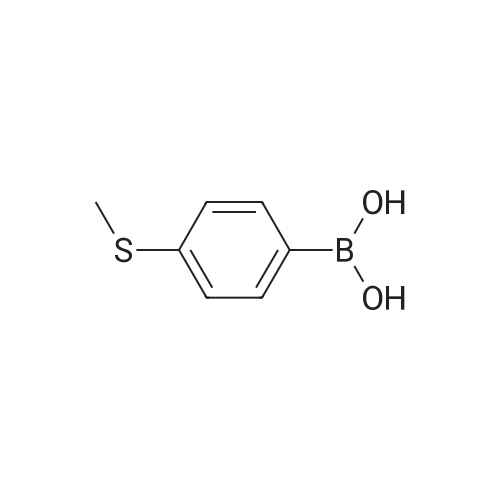
Step 1 5-(4-fluorophenyl)-6-[4-(methylthio)phenyl]-1,3-benzodioxole Under nitrogen, 1 g of Pd(PPh3)4 was added to a stirred solution of 4 g (14.3 mmol) of <strong>[5279-32-3]5,6-dibromo-1,3-benzodioxole</strong> (Lancaster), 2.4 g (17.2 mmol) of 4-fluorophenyl boronic acid, and 2.87 g (17.2 mmol) of 4-methylthiophenylboronic acid (Example 1, Step 2) in 70 mL of toluene, 40 mL of ethanol, and 30 mL of 2M Na2 CO3. After vigorous stirring at reflux overnight, the solvent was removed in vacuo. The residue was dissolved in ethyl acetate, washed with water, and dried over Na2 SO4. Concentration in vacuo gave 6.9 g of 5-(4-fluorophenyl)-6-[4-(methylthio)phenyl]-1,3-benzodioxole as a semi-solid which was used without further purification.
With potassium carbonate;tetrakis(triphenylphosphine) palladium(0); tris-(o-tolyl)phosphine; In 1,2-dimethoxyethane; water; at 80℃; for 16h;
Step 2: (4'-fluorobiphenyl-4-yl)fphenyl)methanol (12). To a solution of 11 (Ig, 3.80 mmol) and 4-fluorophenyl boronic acid (585 mg, 4.18 mmol) in a 2 : 1 mixture of DME : water (30 mL), was added Pd(PPh3J4 (307 mg, 0.27 mmol), tri-o-toly phosphine (81 mg, 0.27 mmol) and potassium carbonate (2.63g, 19.0 mmol). The solution was heated to 80 0C and stirred for 16 hours. Then water (50 mL) was added and the organic residue was extracted with ethyl acetate (2 x 40 mL). The organic layer was dried over Na2SO4, filtered and concentrated. The residue was purified via prep-HPLC (ISCO) in silica gel column with a gradient of ethyl acetate (2-50percent) in hexane to afford compound 12 as a white solid (600 mg, 57percent). 1H NMR (DMSO-d6) delta (ppm): 7.67 - 7.63 (m, 2H), 7.56 (d, J = 8.4 Hz, 2H), 7.45 - 7.38 (m, 4H), 7.32 - 7.17 (m, 5H), 5.93 (d, J = 3.9 Hz, IH), 5.73 (d, J = 3.8 Hz, IH).
With potassium phosphate; palladium diacetate; tricyclohexylphosphine; In water; toluene; at 90℃;Inert atmosphere;
Palladium acetate (1.98 g, 8.80 mmol) was added to a mixture of compound 30 (55.8 g, 176 mmol), (4-fluorophenyl)boronic acid (43.1 g, 308 mmol), tricyclohexylphosphine (20percent toluene solution, 31.2 mL, 17.6 mmol) and K3PO4 (112 g, 528 mmol) in toluene (300 mL) and water (150 mL) at room temperature. The mixture was stirred at 90 °C overnight under Ar atmosphere. The mixture was quenched with water at room temperature and extracted with AcOEt. The organic layer was separated, washed with water and brine, dried over MgSO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane/AcOEt = 95/5 to 70/30) to give the title compound as a green oil (50.7 g, 153 mmol, 87percent). MS (ESI/APCI) m/z 333.1 [M+H]+. 1H NMR (300 MHz, CDCl3) delta 1.27 (6H, t, J = 6.9 Hz), 1.41 (3H, t, J = 7.1 Hz), 4.03 (4H, q, J = 6.9 Hz), 4.40 (2H, q, J = 7.1 Hz), 6.99-7.14 (2H, m), 7.28-7.41 (4H, m).
With cesium fluoride; tri tert-butylphosphoniumtetrafluoroborate;tris-(dibenzylideneacetone)dipalladium(0); In 1,4-dioxane; at 90℃; for 20.0h;Inert atmosphere;
Ethyl 2,6-diethoxy-4'-fluorobiphenyl-4-carboxylate (Compound 2)Dioxane (120 mL) was added to a degassed mix of tri-t-butylphosphonium tetrafluoroborate (0.73 g, 2.5 mmol), 4-flurorphenylboronic acid (11.8 g, 84 mmol), Tris(dibenzylideneacetone)dipalladium(0) (0.77 g, 0.84 mmol), CsF (23.7 g, 156 mmol) and Compoundl (13.4 g, 42 mmol). The mixture was stirred at 900C under nitrogen for 2Oh, and then was partitioned between EtOAc and water. The aqueous phase was filtered and extracted with EtOAc. The combined organic phases were washed with water, brine, dried (MgSO^5 and concentrated. The residue was chromatographed on silica gel columns, eluting with EtOAc/Hexane. Product fractions were combined and concentrated to give Compound2. LC- MS, M+l-333.1; HNMR (50OMHz5 CDC13, ppm): 1.3 (t, 6H), 1.45 (t, 3H), 4.1 (q, 4H), 4.4 (q, 2H), 7.1 (m, 2H), 7.2 (s, 2H), 7.4 (m, 2H).
With cesium fluoride;tris-(dibenzylideneacetone)dipalladium(0); tri tert-butylphosphoniumtetrafluoroborate; In 1,4-dioxane; at 90℃; for 20.0h;Inert atmosphere;
Step B: Synthesis of Ethyl 2,6-diethoxy-4'-fluorobiphenyl-4-carboxylateDioxane (120 mL) was added to a degassed mixture of tri-t-butylphosphonium tetrafluoroborate (0.73 g, 2.5 mmol) 4-fluorophenylboronic acid (11.8 g, 84 mmol) tris(dibenzylideneacetone)dipalladium(0) (0.77 g, 0.84 mmol), CsF (23.7 g, 156 mmol) and <strong>[149517-92-0]ethyl 4-bromo-3,5-diethoxybenzoate</strong> (Step A, 13.4 g, 42 mmol). The mixture was stirred at 90°C under nitrogen for 20 hours, and then partitioned between EtOAc and water. The aqueous phase was filtered and extracted with EtOAc. The combined organic phases were washed with water, brine, dried (MgSO4), and concentrated. The resulting residue was chromatographed on silica gel columns by eluting with EtOAc/hexane. Product fractions were combined and concentrated to give the title compound. LC-MS, M+l=333.1; H NMR (500MHz, CDCB9 ppm): 1.3 (t, 6H), 1.45 (t, 3H), 4.1 (q, 4H), 4.4 (q, 2H), 7.1 (m, 2H), 7.2 (s, 2H), 7.4 (m, 2H).
50.7 g
With potassium phosphate; palladium diacetate; tricyclohexylphosphine; In water; toluene; at 90℃;Inert atmosphere;
[0377] Palladium acetate (1.98 g), tripotassium phosphate (112 g), (4-fluorophenyl)boronic acid (43. lg) and tricyclohexylphosphine (20percent toluene solution, 31.2 mL) were added to a mixture of <strong>[149517-92-0]ethyl 4-bromo-3,5-diethoxybenzoate</strong> (55.8 g), toluene (300 mL) and water (150 mL), and the resultant was heated and stirred overnight at 90°C in an argon atmosphere. After the reaction mixture was cooled to room temperature, water was added thereto, extraction thereof was performed using ethyl acetate, and the resultant was sequentially washed with water and a saturated saline solution. After the obtained organic layer was dried over anhydrous magnesium sulfate, the solvent was distilled off under reduced pressure. The obtained residue was purified by a silica gel column chromatography (hexane/ethyl acetate), thereby obtaining the title compound (50.7 g). 1H NMR (300 MHz, CDC13) delta 1.27 (6H, t, J = 6.9 Hz), 1.41 (3H, t, J = 7.1 Hz), 4.03 (4H, q, J = 6.9 Hz), 4.40 (2H, q, J = 7.1 Hz), 6.99-7.14 (2H, m), 7.28-7.41 (4H, m).
50.7 g
With potassium phosphate; palladium diacetate; tricyclohexylphosphine; In water; toluene; at 90℃;Inert atmosphere;
Palladium acetate (1.98 g), tripotassium phosphate (112 g), (4-fluorophenyl)boronic acid (43.1 g), and tricyclohexylphosphine (20percent toluene solution, 31.2 mL) were added to a mixture of <strong>[149517-92-0]ethyl 4-bromo-3,5-diethoxybenzoate</strong> (55.8 g) in toluene (300 mL) and water (150 mL), and the resultant mixture was heated with stirring overnight at 90° C. in an argon atmosphere. The reaction mixture was allowed to cool to room temperature, and then, water was added thereto, followed by extraction with ethyl acetate. The organic layer was washed with water and saturated saline in this order. The organic layer was dried over anhydrous magnesium sulfate, and then, the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane/ethyl acetate) to obtain the title compound (50.7 g). 1H NMR (300 MHz, CDCl3) delta 1.27 (6H, t, J=6.9 Hz), 1.41 (3H, t, J=7.1 Hz), 4.03 (4H, q, J=6.9 Hz), 4.40 (2H, q, J=7.1 Hz), 6.99-7.14 (2H, m), 7.28-7.41 (4H, m).
With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; sodium carbonate; In 1,4-dioxane; water; at 20℃; for 20h;Reflux;
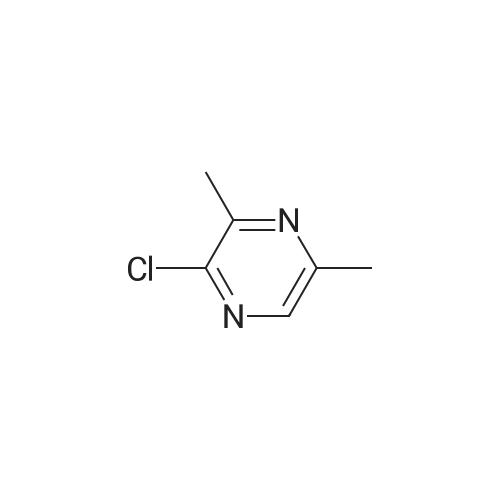
6-Chloropyrazin-2-amine (0.96 g, 7.41 mmol) was added to a degassed suspension of Na2CO3 (2.0 M aqueous solution, 7.4 mL, 14.82 mmol), Pd(dppf)Cl2·CH2Cl2 (0.49 g, 0.59 mmol) and (4-fluorophenyl)boronic acid (1.55 g, 11.11 mmol) in 1,4-dioxane (20 mL). The reaction was refluxed for 5 h and stirred at room temperature for 15 h. The mixture was poured into water (40 mL) and extracted with Et2O. The combined organic layers were dried over Na2SO4 (anhydrous), filtered and concentrated. The crude residue was purified by flash chromatography on SiO2 (20percent to 40percent EtOAc/hexanes) to give the title compound (off-white solid, 1.5 g, quantitative yield). HPLC-MS (Method A): Ret, 8.01 min; ESI+-MS m/z: 190 (M+1).
With sodium hydroxide; In toluene; at 100℃; for 2h;Schlenk technique;
General procedure: An oven-dried Schlenk flask, equipped with a magnetic stir bar, septum and a condenser was charged with acyl chloride (1.0 mmol), arylboronic acid (1.0 mmol), NaOH (4 mmol) and 5.0 mL of toluene. The flask was immersed and stirred in an oil bath at 100 °C. Upon complete consumption of starting materials as determined by GC analysis, the water (10.0 mL) was added. The reaction mixture was extracted with diethyl ether (3 × 5.0 mL). The combined organic layer was collected, dried over anhydrous Na2SO4 and concentrated in vacuum to afford product which was purified by silica gel column chromatography (eluent: n-hexane/ethyl acetate = 9:1 or 8:2).
45%
With C30H38Cl2N8Pd(2+)*2Cl(1-); sodium carbonate; In water; acetone; at 60℃; for 12h;
General procedure: The catalyst (3.8 mg, 1.0 molpercent), Na2CO3 (170 mg, 1.6 mmol), H2O (1.5 mL) and acetone (1.5 mL) was added into a 25 mL schlenk flask, then the reactor was stirred at room temperature for several minutes to dissolve the catalyst and base homogenously. Subsequently, benzoyl chloride (1.0 mmol) and boronic acid (1.2 mmol) were introduced. Then the flask was immersed in an oil bath preheated at 60 °C for 12 h. After the reaction was completed, the mixture was extracted with diethyl ether, and the combined organic layer was dried over anhydrous Na2SO4 and was subsequently purified by flash chromatography using silica gel (petroleum ether/ethyl acetate = 20:1) yielding the desired products.
With caesium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,4-dioxane; water; at 80℃; for 18h;Inert atmosphere;
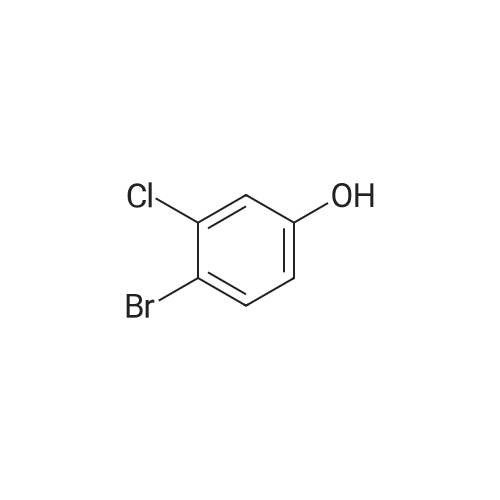
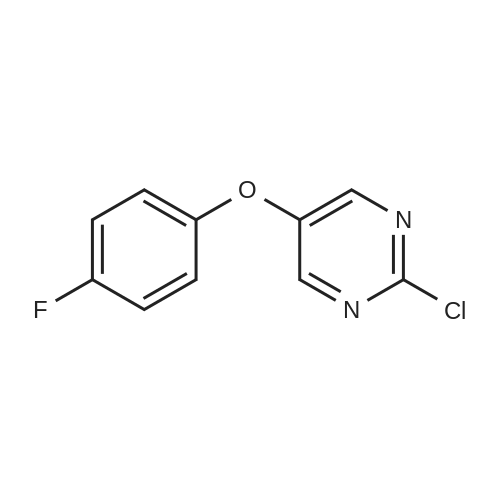
4-Fluorobenzeneboronic acid (405 mg, 2.89 mmol) and <strong>[13631-21-5]4-<strong>[13631-21-5]bromo-3-chlorophenol</strong></strong> (500 mg, 2.41 mmol) were dissolved in dioxane (15 mL) and water (3 mL) under nitrogen. The solution was degassed before tetrakis(triphenylphosphine) palladium (0) (278 mg, 0.24 mmol) was added followed by caesium carbonate (2.36 g, 7.23 mmol) and the reaction was stirred at 80 C. for 18 hours. The cooled reaction mixture was partitioned between ethyl acetate and water. The aqueous layer was separated and extracted with ethyl acetate. The combined organic extracts were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (20% ethyl acetate in heptane) to afford the title compound (503 mg, 94%) as a tan oil which solidified on standing.1HNMR (400 MHz, CDCl3): delta 6.78 (d, 1H), 6.98 (s, 1H), 7.08 (dd, 2H), 7.18 (d, 1H), 7.38 (dd, 2H).LCMS Rt=2.93 minutes MS m/z 221 [M-H]
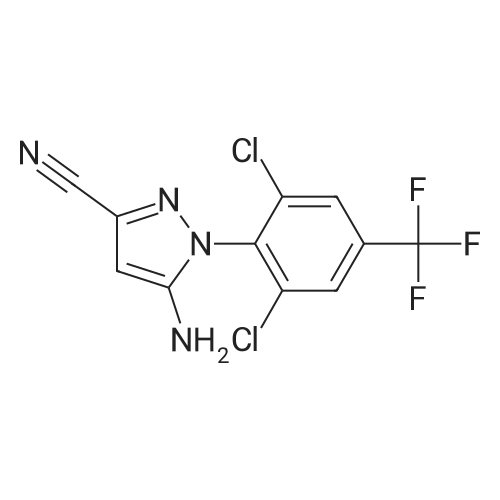
5-amino-1-(2,6-dichloro-4-(trifluoromethyl)phenyl)-4-(4-fluorophenyl)-1H-pyrazole-3-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
71%
With bis-triphenylphosphine-palladium(II) chloride; N-iodo-succinimide; sodium hydrogencarbonate; In ethanol; water; at 80℃; for 18h;Inert atmosphere;
General procedure: Phenylpyrazole (0.2 mmol), arylboronic acid (0.4 mmol), NIS (0.2 mmol), [Pd] (10 mol%), NaHCO3 (0.4 mmol) and C2H5OH:H2O (3:1, 10 mL), were added to a Schlenk tube. Then the tube was charged with N2 and the reaction mixture was stirred at 80 C for 18 h. After the completion of the reaction, as monitored by TLC, the mixture was cooled and filtrated. The filtrate was extracted with ethyl acetate and washed with brine. Then the combined organic extracts were dried over Na2SO4, concentrated under vacuum and the resulting residue was purified by silica gel column chromatography to afford the desired products.
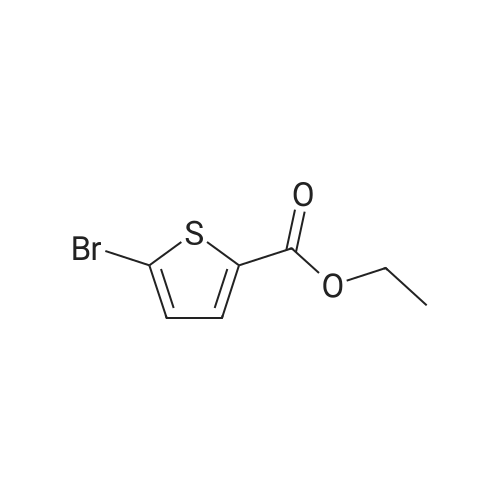
ethyl 5-(4-flurophenyl)thiophene-2-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
67.9%
With potassium fluoride;palladium diacetate; In ethanol; at 110℃; for 2h;Microwave irradiation;
A mixture of 4-fluorophenylboronic acid (214 mg, 1.531 mmol), ethyl 5-bromothiophene-2- carboxylate (300 mg, 1.276 mmol), potassium fluoride (185 mg, 3.19 mmol) and Pd(OAc)2 (28.6 mg, 0.128 mmol) in 10 ml of EtOH is microwave-heated at 110C for 2 h, until the reaction is complete. The mixture is diluted with AcOEt and washed with water and brine, then dried and evaporated; the residue is purified by flash chromatography (Hexane/Et20 95:5 to 9: 1) to obtain ethyl 5-(4-fluorophenyl)thiophene-2-carboxylate (217 mg, 0.867 mmol, 67.9% yield).
With NHC-Pd(II)-Im; sodium hydrogencarbonate; In water; at 50℃; for 12h;Inert atmosphere;
General procedure: Under N2 atmosphere, NaHCO3 (2.4 mmol), benzoic anhydride 3a (1.5 mmol), phenylboronic acid 2a (0.75 mmol), and H2O were successively added into a Schlenk reaction tube. The mixture was stirred at room temperature for about 10 min. Then NHC-Pd(II)-Im complex 1 (1.0 mol percent) was added. The mixture was stirred at room temperature for 12 h and then was diluted with CH2Cl2, washed with saturated brine, dried over anhydrous Na2SO4. The dried organic phase was then filtered, concentrated under reduced pressure and purified by flash column chromatography on silica gel to give the pure product 4a.
With potassium phosphate; tetrakis(triphenylphosphine) palladium(0); In 1,4-dioxane; at 75℃; for 4.0h;Inert atmosphere;
General procedure: A 1,4-Dioxane solution (8 mL) of 10 (1.0 mmol), arylboronic acid 2 (1.0 equiv.), K3PO4, and Pd(PPh3)4 (5 molpercent) was heated at 75 C for 4 h. After cooling to room temperature, H2O was added and the reaction mixture was extracted with CH2Cl2. The organic layer was dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by column chromatography (pure n-heptane).
With potassium phosphate; palladium diacetate; In water; N,N-dimethyl-formamide; at 20℃; for 12h;
General procedure: A DMF/water solution (1:1, 2 mL per 1 mmol of 1) of K3 PO4 (1.5 mmol),Pd(OAc)2 (2 mol %), and arylboronic acid 2a-n (0.9 mmol) was stirred atroom temperature for 8-12 h (tlc control). After completion of the reaction, themixture was extracted with CH2Cl2 and the combined organic layers weredried (Na2SO4), filtered and the filtrate was concentrated in vacuo. The residuewas purified by column chromatography (silica gel, EtOAc/heptane = 1:4).Starting with 1 (216 mg, 1.00 mmol), K3PO4 (165 mg, 1.50 mmol), Pd(OAc)2(2 mol %), arylboronic acid (149 mg, 0.90 mmol), and a solution ofDMF and water (1:1, 5 mL), 3a-3n was isolated
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In 1,4-dioxane; water; at 120℃; for 1h;Microwave irradiation;
General procedure: <strong>[290368-00-2]ter<strong>[290368-00-2]t-butyl 3-iodo-1H-indazole-1-carboxylate</strong></strong> (S2, 100 mg, 0.29 mmol) was placed in a microwave vial and dissolved in 1,4-dioxane (11.5 mL). 3-Methoxyphenylboronic acid (88 mg, 0.58 mmol, 2.0 equiv) and tetrakis(triphenylphosphine)palladium (20 mg, 0.017 mmol, 0.06 equiv) were added, and the resulting mixture was sparged thoroughly with nitrogen. An aqueous solution of sodium carbonate (2.0 M, 0.65 mL, 1.3 mmol, 4.5 equiv) was then added. The biphasic mixture was microwaved for 1 hour at a reaction temperature of 120 C. After cooling to room temperature, the reaction was diluted with ethyl acetate (2 mL), and then filtered through a celite pad with additional ethyl acetate. The filtrate was concentrated under reduced pressure to give an oil. The crude material was purified by column chromatography over silica gel (hexanes/ethyl acetate: 100/0 to 30/70) to give the title compound as an oil (58.0 mg, 89%).
With tetrabutylammomium bromide; palladium diacetate; sodium carbonate; In water; at 165℃; for 0.166667h;Microwave irradiation;
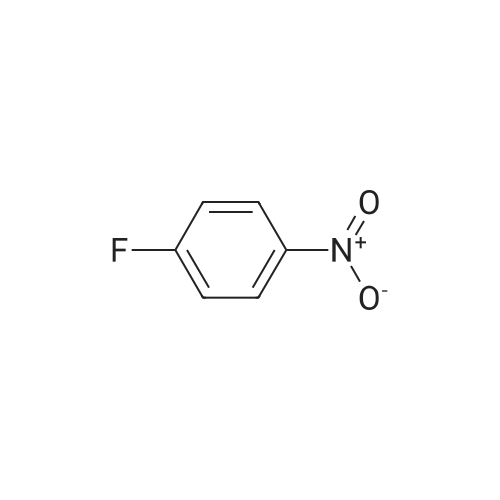
Preparation 41 4,4'-Difluoro-2-nitro-1,1'-biphenyl A microwave reaction vessel was charged with <strong>[345-17-5]2-chloro-5-fluoronitrobenzene</strong> (0.878 g, 5.0 mmol), 4-fluorophenylboronic acid (0.770 g, 5.5 mmol), sodium carbonate (1.590 g, 15.0 mmol), palladium diacetate (0.045 g, 0.2 mmol), tetrabutylammonium bromide (1.612 g, 5.0 mmol) in water (10 mL). The mixture was heated to 165 C. in a microwave reactor for 10 mins. The reaction was cooled to room temperature and poured into diethyl ether and 0.1N aqueous sodium hydroxide. The organic layer was washed with saturated aqueous sodium chloride, dried (anhydrous sodium sulfate), filtered, and concentrated in vacuo. The residue was purified by silica gel chromatography (10-30% methylene chloride/hexanes) to give the desired product as a yellow solid (0.61 g, 52%). 1H NMR (CDCl3, 300 MHz) delta 7.61 (dd, 1H, J=8.1, 2.7 Hz), 7.44-7.30 (m, 2H), 7.30-7.21 (m, 2H), 7.16-7.07 (m, 2H).
With potassium phosphate; nickel dichloride; Trimethylacetic acid; at 80℃; under 760.051 Torr; for 20h;Green chemistry;
General procedure: Into a 25 ml reaction flask was successively added nickel chloride (0.01 mmol), R2 substituted aryl iodide (table 2) (0.5 mmol), R3 substituted arylboronic acid (0.75 mmol), potassium phosphate (1.0 mmol), pivalic acid (0.25 mmol) and polyethylene glycol 400 (2.0 g), and introduce one atmospheric pressure carbon monoxide. The reaction mixture at 80 °C react until starting material reaction complete and cool to room temperature, pressure reducing evaporate the solvent column chromatography separation to obtain the product. The experimental results are set out in table 2.
91%
With palladium diacetate; sodium carbonate; In water; at 100℃; under 760.051 Torr; for 6h;Sealed tube; Autoclave; Green chemistry;
General procedure: A 75 mL autoclave equipped with a Teflon liner and a magnetic stirrer bar was charged with Pd(OAc)2 (4.48 mg, 2.0 × 10?2 mmol), L (46.7 mg, 4.0 × 10?2 mmol) and H2O (6 mL) and the mixture was stirred at room temperatures for 0.5 h under N2. Then iodobenzene (113 muL, 1 mmol), phenylboronic acid (134 mg, 1.1 mmol), Na2CO3(106 mg, 1 mmol), and n-decane (0.1 mL, GC internal standard) were added. Once sealed, the autoclave was purged three times with CO, and pressurized to 1 atm of CO. The reaction mixture was stirred at 100 °C for 2 h. After reaction, the mixture was extracted with diethyl ether (3 × 5 mL). The combined organic layer was concentrated in vacuo and the product was purified by column chromatography. In the recycling experiment, the aqueous phase containing the catalyst was subjected to a second run by charging it with the same substrates as mentioned above, and the reaction performed under the same conditions.
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In 1,4-dioxane; water; for 19.0h;Reflux; Inert atmosphere;
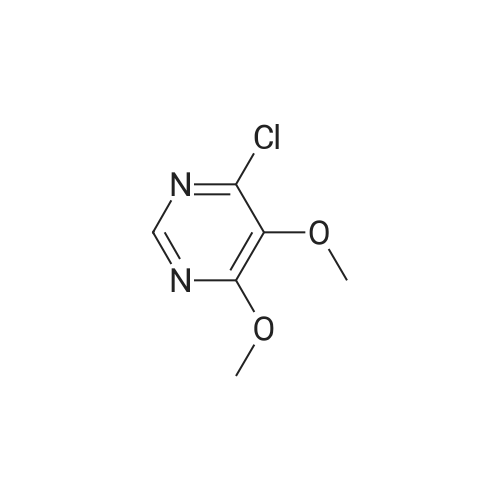
b. Preparation of Compound 4-(4-Fluorophenyl)-5,6-dimethoxypyrimidine 4-Chloro-4,5-dimethoxypyrimidine (165 mg, 0.95 mmol), (4-fluorophenyl)boronic acid (99 mg, 1.42 mmol), Pd(PPh3)4 (110 mg, 0.095 mmol) and Na2CO3(300 mg, 2.84 mmol) were dissolved in a mixture of dioxane (9 mL) and water (3 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 19 hours. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with sat. NH4CI followed by brine. The organic layer was dried over Na2S04 and concentrated under reduced pressure and the resulting residue was flash chromatographed on silica gel eluting with 0 to 10% EtOAc/Hexane. This afforded 4-(4-fluorophenyl)-5,6-dimethoxypyrimidine as a white solid (181 mg, 82%); m.p.62-64 C; 1H NMR (400 MHz, CDC13) delta 8.53 (s, 1H), 8.07 (dd, J= 9 Hz, J= 6 Hz, 2H), 7.15 - 7. 1 1 (m, 2H), 4.07 (s, 3H), 3.73 (s, 3H); 13C NMR (100 MHz, CDCI3) delta 163.7 (Jc,F= 249 Hz), 164.0, 154.3, 152.0,139.4, 131.4 (JCF= 8 Hz), 131.3, 115.3 (JC,F = 21 Hz), 60.2, 54.2; 19F NMR (376 MHz, CDC13) 6 -1 1 1.0
With copper diacetate; triethylamine; In dichloromethane;
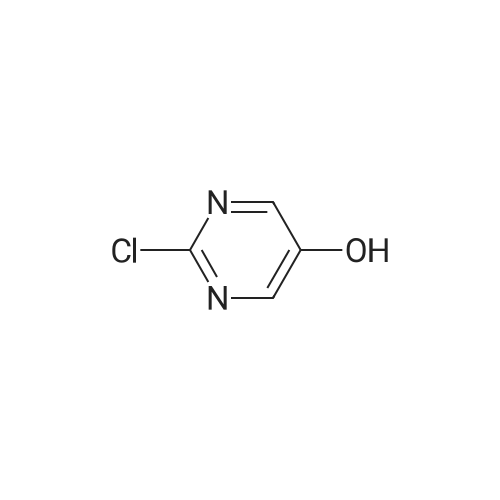
To a stirred solution of <strong>[4983-28-2]2-chloropyrimidin-5-ol</strong> (1.50 g, 1 1.6 mmol) in dichloromethane (20 mL) was added 4-fluorophenylboronic acid (3.30 g, 23.2 mmol), copper(II) acetate (2.49 g, 13.9 mmol) and triethylamine (8.0 mL, 57 mmol). The mixture was left open to the air and stirred overnight. The suspension was then filtered through a pad of Celite and concentrated. The residue was purified by flash chromatography over silica using a hexane/ethyl acetate eluant to afford 2-chloro-5-(4-fluorophenoxy)pyrimidine as a light yellow solid (0.400 g, 17percent). Exchanging 2-chloro-4-(4-fluorophenyl)pyrimidine for this intermediate, ethyl piperidine-4-carboxylate for ethyl 4-fluoropiperidine-4-carboxylate hydrochloride and Intermediate 5 for Intermediate 1 , the final three steps of Example 41 were used to prepare the title compound. 1H NMR (400 MHz, CD3OD) delta 8.20 (s, 2H), 7.09-6.99 (m, 4H), 4.70-4.66 (m, 2H), 3.33-3.16 (m, 3H), 2.90-2.83 (m, 5H), 2.30-2.01 (m, 3H), 1.92-1.89 (m, 4H), 1.70-1.50 (m, 5H) ppm. 13C NMR (100 MHz, CD3OD) delta 172.4, 172.2, 159.8, 158.5, 157.4, 154.4, 150.0, 143.3, 1 18.2, 1 18.1 , 1 16.1 , 1 15.8, 95.7, 93.8, 60.7, 52.8, 45.6, 45.5, 39.6, 31.5, 31.4, 31.3, 31.2, 29.3, 23.0, 21.8, 21.3 ppm. Purity: > 99percent LCMS (214 nm & 254 nm); retention time 1.39 min; (M+H+) 458.0.
With copper diacetate; triethylamine; In dichloromethane; at 20℃; for 16.5h;Molecular sieve;
Step 1: preparation of 1-bromo-2-chloro-4-(4-fluorophenoxy)benzene. To amixture of <strong>[13631-21-5]4-<strong>[13631-21-5]bromo-3-chlorophenol</strong></strong> (1.2 g, 5.8 mmol), copper (II) acetate (1.79 g, 9.83 mmol), triethylamine (4.82 mL, 34.7 mmol), and 1.5 g activated 4A molecular sieves in anhydrous dichloromethane (80 mL) at 00 C was added (4-fluorophenyl)boronic acid (2.43 g, 17.4 mmol, 3.0 equiv) portion-wise over 30 mm. The reaction mixture was allowed to warm to ambient temperature over 16 h, after which it was filtered. Thefiltrate concentrated in vacuo and purified by silica gel column chromatography to afford the title compound as a light yellow oil (0.60 g, 35%). MS (M+H) m/z 302.
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; caesium carbonate; In tetrahydrofuran; water; at 80℃; for 1h;Inert atmosphere; Sealed tube;
General procedure: Synthesis of Intermediates Intermediate 129 4-Chloro-4'-fluorobiphenyl-2-amine 2-Bromo-5-chloroaniline (CAS 823-57-4; 5 g), 4-fluorophenylboronic acid (3.56 g) and cesium carbonate (31.6 g) were combined in THF (70 ml) and water (35 ml). The mixture was degassed by bubbling argon through the solution. After addition of [Iota,Gamma- bis(diphenylphosphino)ferrocene]dichloropalladium(II) (886 mg) the reaction mixture was stirred 1 h at 80 °C in a sealed tube. The reaction mixture was diluted with EtOAc, washed with water, dried over Na2S04, evaporated and purified by chromatography (silica gel, 0percent to 30percent EtOAc in heptane). The product was finally bulb-to-bulb distilled at 0.3 mbar and 120-130 °C oven temperature to give the title compound (5.07 g) as a light yellow liquid. In analogy to the synthesis of Intermediate 129, the following intermediate was prepared:
5.07 g
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; caesium carbonate; In tetrahydrofuran; water; at 80℃; for 1h;Inert atmosphere; Sealed tube;
Intermediate 129 4-Chloro-4'-fluorobiphenyl-2-amine 2-Bromo-5-chloroaniline (CAS 823-57-4; 5 g), 4-fluorophenylboronic acid (3.56 g) and cesium carbonate (31.6 g) were combined in THF (70 ml) and water (35 ml). The mixture was degassed by bubbling argon through the solution. After addition of [Iota, - bis(diphenylphosphino)ferrocene]dichloropalladium(II) (886 mg) the reaction mixture was stirred 1 h at 80 °C in a sealed tube. The reaction mixture was diluted with EtOAc, washed with water, dried over Na2S04, evaporated and purified by chromatography (silica gel, 0percent to 30percent EtOAc in heptane). The product was finally bulb-to-bulb distilled at 0.3 mbar and 120-130 °C oven temperature to give the title compound (5.07 g) as a light yellow liquid. In analogy to the synthesis of Intermediate 129, the following intermediates were prepared:
With dicyclohexyl-(2',6'-dimethoxybiphenyl-2-yl)-phosphane; tris-(dibenzylideneacetone)dipalladium(0); sodium carbonate; In water; toluene; at 100℃; for 16h;Inert atmosphere;
[1526] (4-Fluorophenyl)boronic acid (17.3 g), dicyclohexyl (2^6'-dimethoxybiphenyl-2-yl)phosphine (5.09 g), 2 M aqueous sodium carbonate solution (124 mL) and tris(diber.zyUdeneacetone)dipalladium(0) (5.29 g) were added to a solution of 4-bramo-2,3-difluorobenzaldehyde (18.3 g) in toluene (300 mL) at room temperature, and the resultant was stirred 16 hours at 100C in an argon atmosphere. Water was added to the reaction mixture at room temperature, the resultant was filtered using celite, and the filtrate was extracted using ethyl acetate. The obtained organic layer was sequentially washed with water and a saturated saline solution, dried over anhydrous magnesium sulfate, and then the solvent was distilled off under reduced pressure. After the obtained residue was passed through a short column of a silica gel (hexane/ethyl acetate), the solvent was distilled off under reduced pressure. The obtained residue was crystallized (hexane/ethyl acetate), and further recrystallized (hexane/ethyl acetate), thereby obtaining the title compound (15.1 g). 1H NMR (300 MHz, CDC13) delta 7.15-7.24 (2H, m), 7.27-7.35 (1H, m), 7.53-7.61 (2H, m), 7.69 (1H, ddd, J = 8.2, 6.3, 1.7 Hz), 10.37 (1H, d, J = 0.6 Hz).
6.01 g
With dicyclohexyl-(2',6'-dimethoxybiphenyl-2-yl)-phosphane; tris-(dibenzylideneacetone)dipalladium(0); sodium carbonate; In water; toluene; at 100℃; for 16h;Inert atmosphere;
(4-Fluorophenyl)boronic acid (6.67 g), dicyclohexyl(2',6'-dimethoxybiphenyl-2-yl)phosphine (1.96 g), a 2 M aqueous sodium carbonate solution (47.6 mL), and tris(dibenzylideneacetone)dipalladium(0) (2.04 g) were added at room temperature to a toluene (200 mL) solution of <strong>[644985-24-0]4-bromo-2,3-difluorobenzaldehyde</strong> (7.02 g), and the mixture was stirred at 100 C. for 16 hours in an argon atmosphere. Water was added to the reaction mixture at room temperature, and the mixture was filtered through celite. Then, the filtrate was subjected to extraction with ethyl acetate. The obtained organic layer was washed with water and saturated saline in this order and dried over anhydrous magnesium sulfate, and then, the solvent was distilled off under reduced pressure. The obtained residue was passed through a short silica gel column (hexane/ethyl acetate), and the solvent was distilled off under reduced pressure. The obtained residue was crystallized (hexane/ethyl acetate) and further recrystallized (hexane/ethyl acetate) to obtain the title compound (6.01 g). 1H NMR (300 MHz, CDCl3) delta 7.15-7.23 (2H, m), 7.28-7.34 (1H, m), 7.53-7.61 (2H, m), 7.69 (1H, ddd, J=8.2, 6.2, 1.8 Hz), 10.37 (1H, d, J=0.7 Hz).
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In water; N,N-dimethyl-formamide; at 110℃;Inert atmosphere; Sealed tube;
General procedure: To a solution of 4-Methylphenylboronic acid (2.8g,21mmol) in DMF/water (v/v, 1:1) was added 1-bromo-4-iodobenzene (4g, 14mmol) , Tetrakis (triphenylphosphine)palladium (0.8g, 0.7mmol) and K2CO3 (5.8g, 42mmol), themixture was stirred at 110oC in a sealed tube under N2 over night. The reaction mixture was cooled and poured into an ice-wate rmixture solution, filtering to get solid material. The residue was purified by column chromatography on silica gel (petroleum ether) to yield the title compound
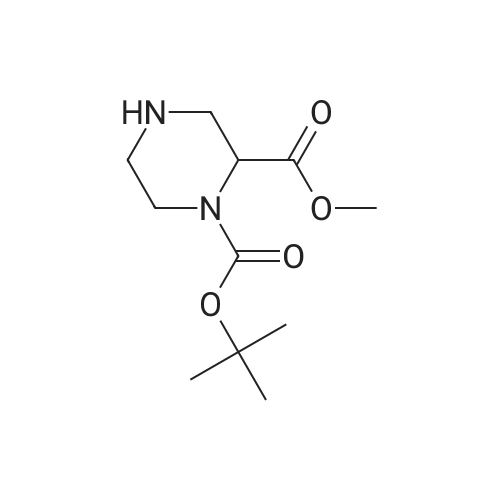
1-tert-butyl 2-methyl 4-(4-fluorophenyl)piperazine-1,2-dicarboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
With pyridine; copper diacetate; In dichloromethane; at 25℃; for 16h;
Step 1 : 1-tert-Butyl 2-methyl 4-(4-fluorophenyl)piperazine-l,2-dicarboxylate (1113) [00386] To a solution of A29-1 (200.00 mg, 0.819 mmol, 1.00 eq) in DCM (5 mL) were added A29-1A (229. 11 mg, 1.64 mmol, 2.00 eq), Cu(OAc)2 (148.70 mg, 0.819 mmol, 1.00 eq), 4A MS (50 mg) and pyridine (0. 13 mL, 1.64 mmol, 2.00 eq). The reaction mixture was stirred at 25 C for 16 hours under oxygen atmosphere. The reaction mixture was concentrated under reduced pressure. The residue was purified by column chromatography over silica gel (petroleum ether: ethyl acetate = 1 :0 to 10: 1) to afford the title compound (250 mg, 90% yield). LCMS (ESI): RT = 0.800 min, mass calcd. for C17H23FN2O4 338.16, m/z found 338.9 [M+H] , NMR (400MHz, COCh-d) delta 7.00 - 6.93 (m, 2H), 6.91 - 6.84 (m, 2H), 4.92 - 4.85 (m, 0.5H), 4.73 - 4.66 (m, 0.5H), 4.09 - 3.88 (m, 2H), 3.78 (s, 3H), 3.40 - 3.18 (m, 2H), 2.94 - 2.83 (m, 1H), 2.79 - 2.66 (m, 1H), 1.49 (d, J=16.8 Hz, 9H).
79%
With pyridine; oxygen; copper diacetate; sodium hydrogencarbonate; In dichloromethane; at 20℃; for 60h;
In 9 parallel batches, A mixture of tert-butyl 2-methyl piperazine-l,2-dicarboxylate (1.0 g, 4.09 mmol), (4-fluorophenyl)boronic acid (1.72 g, 12.28 mmol), Copper(II) acetate (1.5 g, 8.19 mmol), pyridine (647 mg, 8.19 mmol) and sodium bicarbonate (688 mg, 8.19 mmol) in dichloromethane (50 ml) was stirred at ambient temperature under 02 (balloon) for 60 h, at which time LCMS indicated the reaction had gone to completion. The combined solutions were concentrated under reduced pressure. The residue was dissolved in ethyl acetate (50 mL), washed with water (2 x 40 mL) and concentrated. The crude product was purified by silica gel chromatography column (Hexanes/ethyl acetate = 5: 1) to give the title compound (9.8 g, yield 79%) as a brown oil. NMR (400 MHz, CDC13): delta 6.91 -6.80 (m, 2 H), 6.80-6.62 (m, 2 H), 4.86-4.68 (m, 1 H), 4.05-3.88 (m, 2 H), 3.77 (s, 3 H), 3.37-3.15 (m, 2 H), 2.90-2.86 (m, 1 H), 2,77-2.68 (m, 1 H), 1.49-1.40 (m, 9 H). LCMS M/Z (M+H) 338.
General procedure: A one dram vial equipped with a stir bar was charged with <strong>[54198-88-8]2-(chloromethyl)pyrimidine</strong> (12.mg,0.070 mmol), 4-bromothiophenol (13.06 mg, 0.070 mmol), and cesium carbonate (95.37 mg, 0.290 mmol). The solids were then dissolved in 1,4-Dioxane (0.327 mL) and stirred at 90 °C for 20 min before the vial was removed from the heat. At this time pyridine-3-boronic acid (11.62 mg, 0.090 mmol), water (36 lL), and [1,10-bis(diphenylphosphino)ferrocene]dichloropalladium (II) (5.34 mg, 0.010 mmol) were added to the reaction mixture followed by a thirty second purge with argon. The reaction vessel was allowed to stir at 90 °C for anadditional two hours. After being allowed to cool to room temperature the crude reaction mixture was filtered over a thin celite plug in a phase separator with EtOAc (10 mL) and the resulting organics concentrated before crude product was purified using Gilson reverse phase automated system (30 50 mm column, 5?50percent MeCN in 0.1percent Aq. TFA, 5 min gradient). Fractions containing product by LC/MS were pooled and quenched with sat. sodium bicarbonate before being extracted with DCM (5 mL) and dried to afford 2-[[4-(3-pyridyl)phenyl]sulfanylmethyl]pyrimidine (12d,17.6 mg,0.063 mmol, 87percent) as a yellow solid.
With potassium phosphate; palladium diacetate; In water; methyl cyclohexane; for 8h;Reflux;
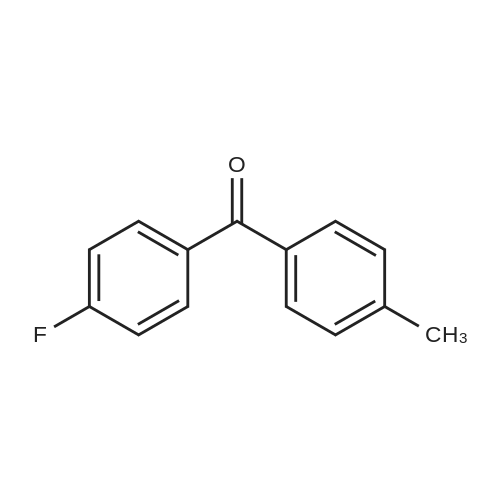
Method of the present embodiment provides a 2,4 ?-difluorobenzophenone and preparation method thereof, as follows: in a 1000 ml round bottom flask The bottle was added sequentially o-fluoro benzaldehyde 124 g of 4-fluorophenylboronic acid and 140 g of potassium phosphate, 106 grams, 1.12 g of palladium acetate, then adding 500 ml of methyl cyclohexane and 100 ml of water and heated to reflux for 8 hours., sequentially passes through filtering, layering, washing with water, removing the solvent to obtain the product., 2,4 '-difluorobenzophenone 214 g, 98percent yield.
With tetrakis(triphenylphosphine) palladium(0); caesium carbonate; In 1,4-dioxane; water; at 95℃; for 2h;Inert atmosphere;
Synthesis of 323-1. To a mixture of 323-0 (1.00 g, 4.6 mmol), 4-fluorophenylboronic acid (773 mg, 5.5 mmol) and Cs2CO3 (3.00 g, 9.2 mmol) in dioxane/H2O (20 mL/4 mL) was added Pd(PPh3)4 (531 mg, 0.5 mmol) under N2 atmosphere. The mixture was stirred at 95 C for 2 hours and then concentrated in vacuo. The residue was dissolved with EtOAc (60 mL) and the resulting solution was washed with brine (20 mL × 3). The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (DCM : MeOH = 100 : 1 ~ 50 : 1) to give 323-1 (1.00 g, 93 %) as a yellow solid.
With tetrakis(triphenylphosphine) palladium(0); sodium hydrogencarbonate; In ethanol; water; toluene; at 110℃;
One neck r.b.f4-fluorophenyl boronic acid (36.5 g, 261 mmol), <strong>[63860-31-1]2-bromo-4-chloro-1-nitrobenzene</strong> (47.5 g,10 mmol), Tetrakis (triphenylphosphine) palladium (0) (11.5 g, 10 mmol), NaHCO3 (32.4 g, 400 mmol)toluene / ethanol / water (800 ml / 160 ml / 160 ml) was refluxed at 110 C.Extraction with MC and drying over MgSO4. After column packing HX: MC = 5: 1 packing, go down to 4: 1, remove upper side, and lower to HX: MC = 3: 15-chloro-4'-fluoro-2-nitro-1,1'-biphenyl was obtained (42.5 g, 84%).
With pyridine; oxygen; copper diacetate; In N,N-dimethyl-formamide; at 20℃; for 2h;
General procedure: To a solution of <strong>[461-89-2]6-azauracil</strong> (100 mg, 0.88 mmol) inDMF (10.0 mL) was added base (1.76 mmol) and Cu(OAc) 2(159 mg, 0.88 mmol) at room temperature. The resulting reationmixture was degassed with oxygen for 10 min and then addedarylboronic acids (0.96 mmol) at room temperature and stirredat appropriate temperature (Table-1) under oxygen atmosphere.The reaction mixture was diluted with water (15 mL) andextracted with dichloromethane (3 × 15 mL). The organic layerwashed with H 2 O (15 mL), brine solution (15 mL), dried overNa 2 SO 4 and concentrated. The obtained crude product waspurified by column chromatography (0 to 10 % CH 3 OH/CH 2 Cl 2 )to afford the title compounds.
With tetrakis(triphenylphosphine) palladium(0); sodium hydrogencarbonate; In ethanol; water; toluene; at 100℃;
The mixture of <strong>[16732-69-7]ethyl 7-bromo-1H-indole-2-carboxylate</strong> (536 mg, 2 mmol), (4- fluorophenyl)boronic acid (420 mg, 3 mmol) in a mixture of toluene, ethanol and sat. NaHCO3 solution (10/4/4 mL) was degassed and Pd(PPh3)4 (100 mg, 0.09 mmol) was added. The reaction mixture was heated at 100 oC overnight and extracted with EtOAc, then washed with brine and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel to give the desired product (320 mg, 57% yield) as an off-white powder. 1H NMR (300 MHz, CDCl3) delta 8.89 (br, 1H), 7.69 (d, J = 8.4 Hz, 1H), 7.60 (m, 1H), 7.58 (m, 1H), 7.23 (m, 5H), 4.40 (q, J = 6.9 Hz, 2H), 1.41 (t, J = 6.9 Hz, 3H).
5-benzyloxy-2-(4-fluorophenyl)pyrimidine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
78%
4-Fluorophenylboronic acid (0.72 g, 5.1 mmol) and [1,1'-bis(diphenylphosphino)ferrocene]palladium dichloride (0.26 g, 0.35 mmol) were dissolved in toluene (42 mL) and a mixed solvent of ethanol (21 mL),The resulting mixture isAn aqueous solution (7.5 mL) of sodium carbonate (1.7 g, 16 mmol) was added under nitrogen.After stirring for 10 minutes, 5-benzyloxy-2-chloro-pyrimidine 34b (0.76 g, 3.4 mmol) was added, and the mixture was reacted at 80 ° C for 2 hours.The reaction solution was cooled to room temperature and water (20 mL) was added.Extracted with ethyl acetate (30 mL).Dry over anhydrous sodium sulfate, concentrate by suction filtration, and the residue obtained was purified by silica gel column chromatography[ethyl acetate / petroleum ether (v / v) = 1/10] purified to give the title compound34c (0.75 g, yield 78percent) as a white solid.
methyl 3-(3,5-difluorophenyl)isonicotinate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83.3%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In toluene; at 100℃;Inert atmosphere;
Compound 52a. To a solution of ethyl 3-bromoisonicotinate (0.2 g, 0.93 mmol, 1.0 equiv) in Toluene (15 mL) was added (4-fluorophenyl)boronic acid (0.21 g, 1.85 mmol, 2.0 equiv), K2CO3 (0.38 g, 2.78 mmol, 3.0 equiv). The resulting reaction mixture purged with N2 gas for 10 minute, followed by the addition of Pd(PPh3)4 (0.054 g, 0.046 mmol. 0.05 equiv). The reaction mixture was heated at 100 C. for overnight. Product formation was confirmed by LCMS. After the completion of reaction, the mixture was filtered through celite bed, washed with ethyl acetate (100 mL). Filtrate was concentrated under reduced pressure. The crude product obtained was purified by flash chromatography (0-20% ethyl acetate in hexane as an eluent) to obtain methyl 3-(4-fluorophenyl)isonicotinate (0.185 g, 83.3%) as an off white solid. (0682) LCMS 232.2 [M+H]+ (0683) 1H NMR (400 MHz, DMSO-d6) delta 8.74 (d, J=5.26 Hz, 1H), 8.70 (s, 1H), 7.70 (d, J=4.82 Hz, 1H), 7.42 (dd, J=5.70, 8.33 Hz, 2H), 7.31 (t, J=8.77 Hz, 2H), 3.68 (s, 3H).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping