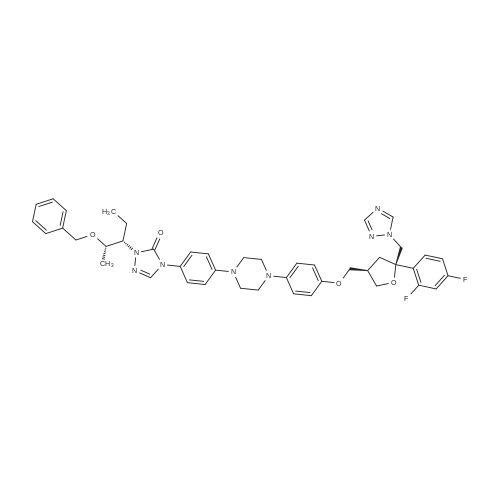
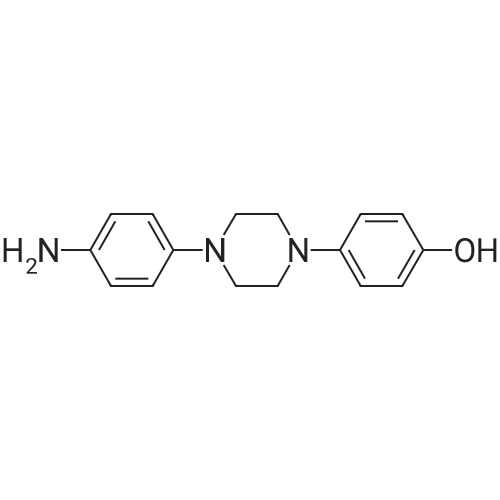
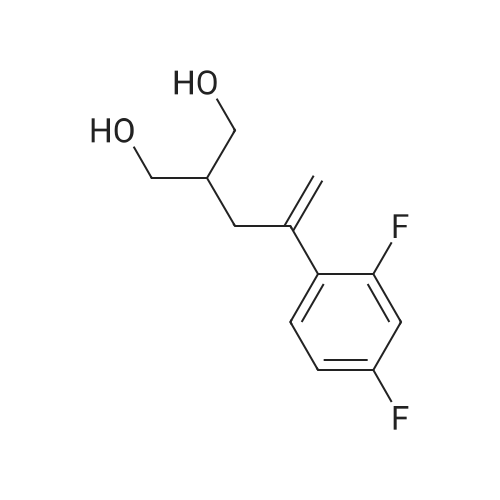
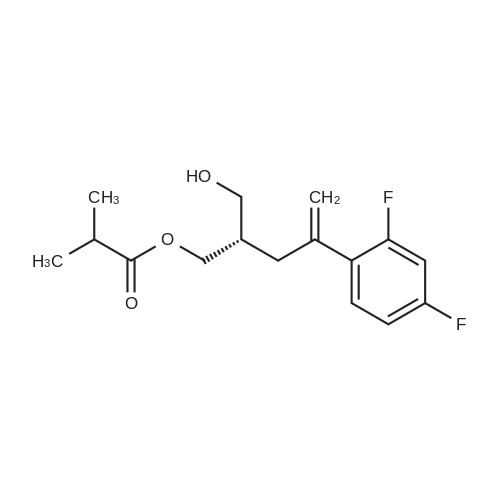
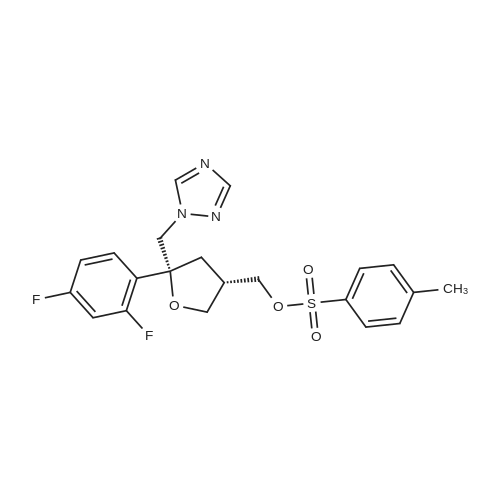
| 77.42% |
With ammonium formate; palladium(II) hydroxide; In isopropyl alcohol; at 70 - 75℃; |
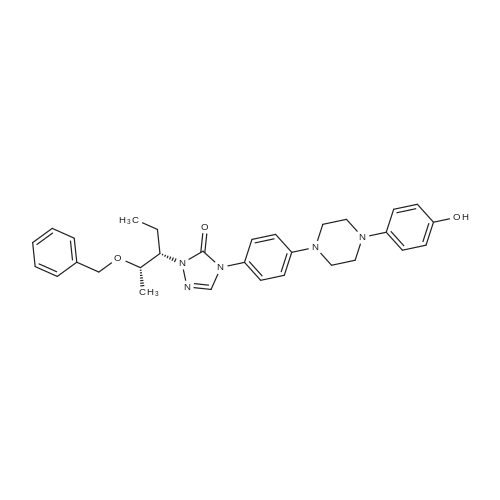
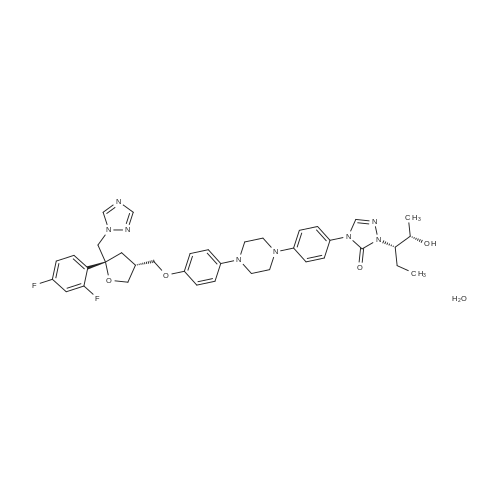
5 g of the compound of formula I is added to 80 ml of isopropanol and 1 g of palladium hydroxide is added.4.8 g of ammonium formate (12 eq), 70-75 C reaction until the starting reaction is complete,The purity of posaconazole in the reaction solution was determined by HPLC to be 96.8%. The reaction solution was filtered under hot water at 40-50 C, and the filtrate was concentrated to dryness. The obtained solid was dissolved in ethyl acetate. The organic phase was washed with brine and concentrated to give a crude product of 4.0 g, yield 90.23%, HPLC purity 98.63 %. The obtained crude product was heated to a clear solution with a mixture of 32 ml of methanol and 8 ml of acetone, and then slowly cooled to room temperature, and filtered to obtain 3.83 g of posaconazole in a yield of 77.42%.The purity by HPLC was 99.68%. Its nuclear magnetic data is basically the same as in the first embodiment. |
| 75% |
With hydrogenchloride; palladium 10% on activated carbon; hydrogen; In methanol; water; at 50℃; for 5h; |
Example-16 Preparation of Posaconazole (Formula-1) 5N hydrochloric acid (72 ml) and 10% Pd-C(10 g) were added to a solution of 4-(4-(4-(4-(((3R,5R)-5-((1H-1,2,4-triazol-1-yl)methyl)-5-(2,4-difluorophenyl)tetrahydro furan-3-yl)methoxy)phenyl)piperazin-1-yl)phenyl)-1-((2S,3R)-2-(benzyloxy)pentan-3-yl)-1H-1,2,4-triazol-5(4H)-one compound of formula-21 (42 g) in methanol (420 ml). The reaction mixture was hydrogenated for 5 hours under a hydrogen gas pressure of 4-5 kg/cm2 at 50. After completion of reaction, the catalyst was filtered off and washed with methanol. pH of the filtrate was adjusted to ?7.0 using 4N sodium hydroxide. Water was added to the reaction mixture and stirred for 2 hours at 25-35 C. Filtered the separated solid and washed with water. The obtained solid was dissolved in acetone (320 ml) and stirred at reflux temperature for 30 minutes. Filtered the undissolved product and added water to the filtrate and stirred the reaction mixture for 4 hours at 25-35 C. Filtered the separated solid and washed with water. Further the solid was recrystallized from isopropyl alcohol to get the title compound. Purity by HPLC: 99.85%; Yield: 75.0%: Chiral purity by HPLC: 99.82%. |
| 56.4% |
With hydrogen bromide; for 4h;Large scale; |
6.00 kg of compound (II) and 41.37 kg of mass concentration of 40%Hydrobromic acid(The mass concentration referred to as the mass of hydrogen bromide as a percentage of the total mass of hydrobromide) was added to a 100 L glass reactor,Stirring, heating to 50 ~ 60 , reaction 4 ± 0.5 hours. TLC monitoring reaction, after the reaction is completed. Cooled to 15 to 25 C, 32.27 kg of ethyl acetate was added,Stirring for 0.5 to 1 hour, suction filtration.The filter cake was washed twice with 21.51 kg of ethyl acetate. The filter cake was dissolved in 47.8 kg of dichloromethane,A mass concentration of 10% aqueous sodium hydroxide solution (sodium hydroxide 3.00 kg dissolved in pure water 27.00 kg, said mass concentration means that the mass of sodium hydroxide is the percentage of the total mass of the aqueous sodium hydroxide solution) to pH = 10 ~ 11 (sodium hydroxide aqueous solution to the amount of pH).The layers were allowed to stand, the lower organic phase was collected and the reactor was purged with purified water.The organic phase was added to the reaction vessel and 29.98 kg of purified water was added. Stirring for 10 to 20 minutes, standing and stratifying, collecting the organic phase,The organic phase repeats the above water wash step once. The organic phase was transferred to a 20 L rotary vial, and at a vacuum of -0.08 to -0.1 MPa,Control temperature 40 ~ 50 under reduced pressure to solvent-free distillation, a white solid.The resulting white solid was transferred to a reaction kettle, and 11.69 kg of acetone and 2.36 kg of methanol were added. Temperature rise.0.29 kg of activated carbon was added, and the mixture was stirred for 2 ± 0.5 hours. Filtration by hot, the filtrate transferred to the reactor, heated to 50 ~ 60 , adding purified water 3.00kg, natural cooling to 15 ~ 25 .Stir for 2 to 3 hours. Centrifuge the filter to the solvent-free effluent, filter cake with a mixed solvent (acetone 2.36kg methanol 0.46kg) washing, centrifugal filtration to solvent-free effluent. Will be centrifuged to get the whole batch of wet goods in the vacuum -0.08 ~ -0.1MPa, the control temperature of 40 ~ 50 vacuum drying 6 to 10 hours.A white solid of 3.00 kg was obtained. HPLC purity 99.92%, maximum single product 0.06%, chiral HPLC did not detect isomers, yield: 56.4%. |
| 12.9% |
With palladium 10% on activated carbon; hydrogen; In ethanol; at 25 - 30℃; under 1500.15 Torr; for 5h;Large scale; |
Add 192 kg of absolute ethanol, 1 kg of palladium on carbon (10%) to a 500 L hydrogenator. 19.2kg POC, the replacement system N2 is replaced five times, H2 is replaced five times, The pressure of the control system H2 is not higher than 0.2 MPa. After incubating for 5 hours at 25~30 C, the monitoring was carried out until the raw materials were ?0.1%. After replacing the system with N2 for five times, the material is pressed out to filter out palladium carbon. The filtrate is controlled to a temperature of T ? 40 C and concentrated under reduced pressure to about 30 L. After adding 40 kg of n-heptane and stirring and crystallization for 3 hours, the temperature was lowered to 0 to 10 C at a rate of 10 C / h, and the filter cake was rinsed with pre-cooled 5 kg of n-heptane. Drain and transfer to the drying room for 40h at 40~45C. Received 15.2 kg of posaconazole.The HPLC purity was 99.8%, the diastereomer content was 0.01%, and the total yield was 12.9%. |
|
|
Benzyl ether of posaconazole (12.5g) was taken in 5N hydrochloric acid (25ml) in methanol (125ml), and was hydrogenated for 4-5 hours under a hydrogen gas pressure of 4 kg/cm2 at 50 C in the presence of palladium on carbon (10%, 1.2g). After completion of hydrogenation (monitored by TLC), the catalyst was filtered off and washed with methanol (25ml). The combined filtrate was concentrated to obtain a residue. Residue was dissolved in tert. butanol (250ml) and pH of the reaction mixture was adjusted to 6-7 with 4N sodium hydroxide solution to give 1Og of crude title compound having purity of 93.5% by HPLC.Purification: Crude posaconazole obtained above was taken in methanol (100ml) and was stirred at reflux temperature for 30 minutes followed by cooling at ambient temperature. The precipitated product was filtered and washed with chilled methanol (20ml) and dried at 50 C to obtain 7g of the title compound having purity of 99.5% by HPLC |
|
|
EXAMPLE 3PREPARATION OF POSACONAZOLE OF FORMULA (I); In a clean, dry Pari shaker bottle 5 gm of benzyl posaconazole, 50 ml of methanol were charged and 3.1 ml of methane sulfonic acid and 0.5 gm of 10% Pd/C were added at about room temperature. 5 kg/cm2 of hydrogen gas pressure was applied and stirred for about 4 hours at about room temperature. After completion of the reaction, the pressure was released and the reaction solution was filtered and the catalyst was washed with 1 5 ml of methanol. The methanol solution was concentrated completely under vacuum. Then 15 ml of isopropyl alcohol and 25 ml of water were added to the residue and stirred for about 15 minutes and cooled to about 5C. The pH of the solution was adjusted to 7.5 using 4N NaOH solution and stirred for about 1 hour at about 5C. The solid obtained was filtered and washed with 30 ml of water. The wet material was dried in a vacuum oven at about 50 C for about 12 hours to yield 4.3 gm of the title compound.Purity by HPLC: 95% |
| 41 g |
With hydrogenchloride; In water; at 25 - 63℃; for 11h; |
Example-21: Preparation of 4-(4-(4-(4-(((3R,5R)-5-((1H-1,2,4-triazol-1-yl)methyl)-5-(2,4- difluorophenyl)tetrahydrofuran-3-yl)methoxy)phenyl)piperazin-1-yI)phenyl)-1-((2S,3S)-2-hydroxypentan -3-yI)-1H-1,2,4-triazol-5(411)-one (Posaconazole)4-(4-(4-(4-(((3R,5R)-5-(( I H-i ,2,4-triazol- 1 -yl)methyl)-5-(2,4-difluorophenyl) tetrahydrofuran-3-yl)methoxy)phenyl)piperazin- 1 -yl)phenyl)- 1 -((2S,3 S)-2-(benzyloxy) pentan-3-yl)- 1 Hl,2,4-triazol-5 (4H)-one (50 gms) was added to hydrochloric acid (150 ml) at 25-30C. Heated the reaction mixture to 60-63C and stirred for 11 hours at the same temperature. A mixture of water (500 ml), methanol (400 ml) and acetone (200 ml) was added to the reaction mixture at 25- 30C. Cooled the reaction mixture to 10-15C. Adjusted the pH of the reaction mixture to 7.0using aqueous sodium hydroxide solution below 15C. Raised the temperature of the reaction mixture to 25-30C and stirred for 1 hour at the same temperature. Filtered the precipitated solid, washed with water. To the obtained wet compound, acetone (500 ml) was added at 25-30C. Heated the reaction mixture to 55-60C and stirred for 30 minutes at the same temperature. Carbon (5.0 gins) was added to the reaction mixture at 55-60C and stirred for 30 minutes at thesame temperature. Filtered the reaction mixture through hy-flow bed and washed with hot acetone. Distilled off the solvent from the filtrate under reduced pressure. Acetone (500 ml) was added to the obtained compound at 25-30C. Heated the reaction mixture to 55-60C and stirred for 30 minutes at the same temperature. Cooled the reaction mixture to 25-30C. Water was slowly added to the reaction mixture at 25-30C and stirred for 2 hours at the same temperature.Filtered the precipitated solid, washed with water and dried to get the title compound.26 Yield: 41 gms. |
| 9.4 g |
|
10 g of compound of formula B, 100 mL methanol and 10 g refined hydrochloric acid were added to a reaction flask at room temperature, and stirred for 1 h. Then, 1 g of 5% Pd/C was added thereto. Then, hydrogen was introduced thereto. The mixture was warmed up to 40 C., and stirred and reacted for 10-16 h. After the reaction was completed, the mixture was filtered under suction. The resultant solid was washed with 10 mL methanol. The filtrate was warmed up to 45 C. And an alkaline solution prepared from 2.7 g sodium hydroxide and 17 g water was dropwise added thereto to adjust the pH of the solution to 7-8. Then, the mixture was warmed up to 60-65 C. And 60 mL purified water was slowly dropwise added thereto. After that, the mixture was cooled to 45 C., crystallized while keeping the temperature for 1 h, further cooled to 15-20 C., crystallized while keeping the temperature for 1 h, and filtered under suction. The resultant solid was first drip washed with 5 mL of 50% aqueous methanol solution, and then drip washed with 4×50 mL purified water. The solid was collected, placed in a ventilated oven at 50-55 C. and dried for 16 h to obtain 9.4 g of product. |
| 0.68 kg |
With hydrogenchloride; palladium 10% on activated carbon; hydrogen; In methanol; water; at 50℃;Large scale; |
4-(4-(4-(4-(((3R)-5-((lH-l,2,4-triazol-l-yl)methyl)-5-(2,4-difluorophenyl)- tetrahydrofuran-3-yl)methoxy)phenyl)piperazin-l-yl)phenyl)-2-((2S,3S)-2- (benzyloxy)pentan-3-yl)-2H-l,2,4-triazol-3(4H)-one (1 kg) was added to the flask along with Methanol (7 lit), followed by the addition of 10% Pd / C (0.24 kg) and Hydrochloric acid (1.78 lit) at room temperature. Hydrogen gas (5 kg) was applied and temperature was raised to 50C. The resultant material was cooled to room temperature, filtered through the Hyflow bed and washed with Methanol (1 lit). The resultant filtration mass was cooled to 0-5C and pH was adjusted to 7.0-7.5 with 16% Sodium hydroxide solution, maintained for 2 hrs and filtered the solid and washed with Methanol.The resultant material was added to the Flask along with Methanol (10 lit) and heated to 60-65C and maintained for 1 hr. Activated Carbon was added, stirred for 30 min and filtered through Hyflow bed and washed with methanol (1 lit). The resultant mass was cooled to room temperature and maintained for 1-2 hrs, filtered the solid and washed with Methanol (1 lit) to get the title compound.Yield: 0.68Kg; HPLC: 99.4%. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping