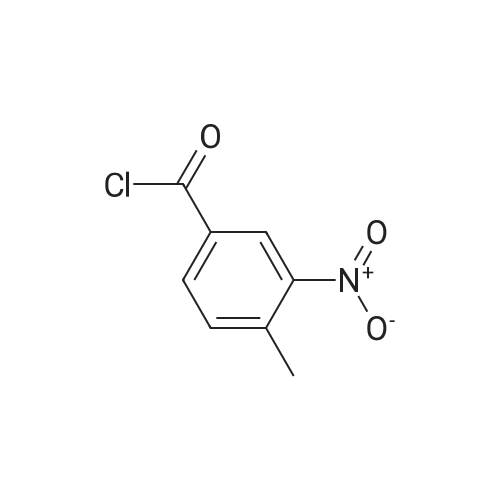
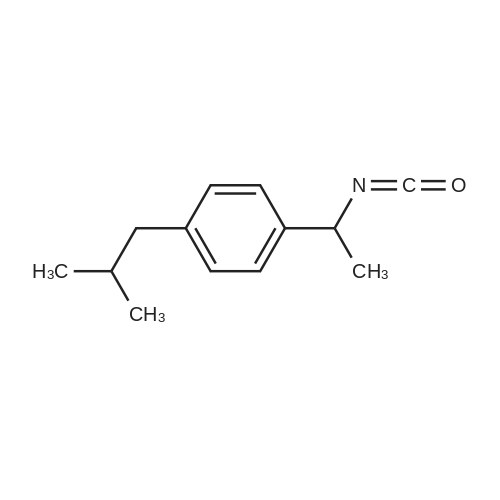
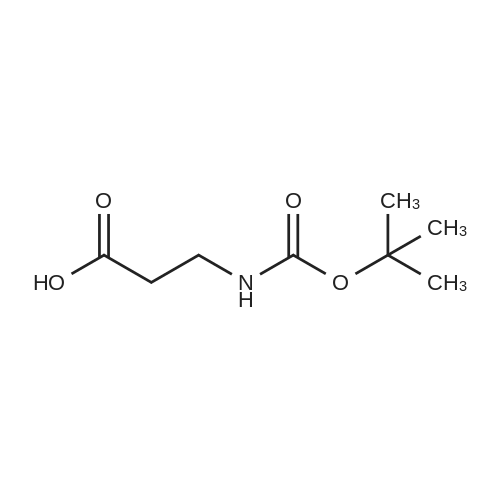
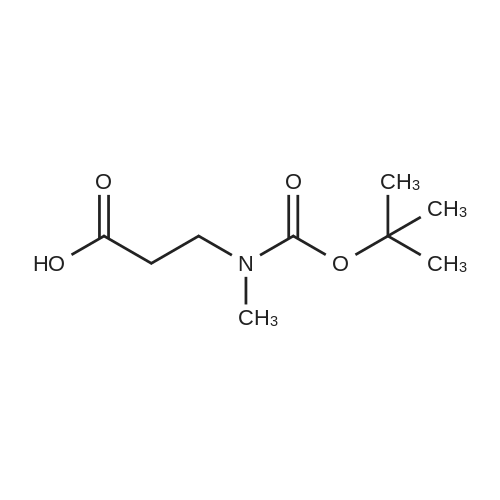
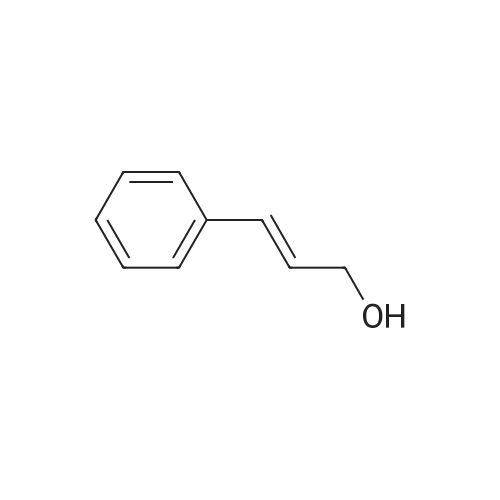
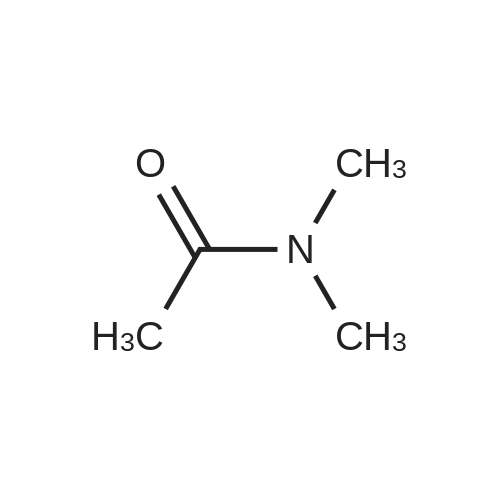
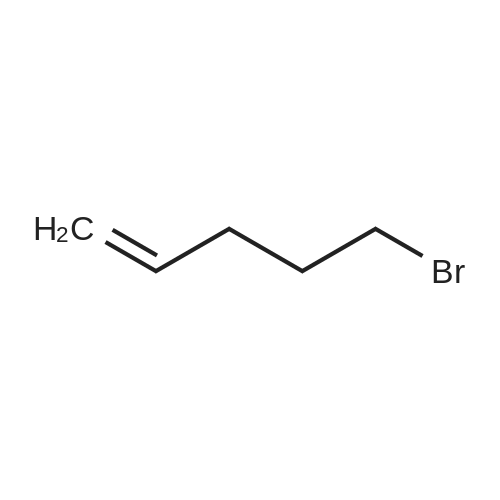
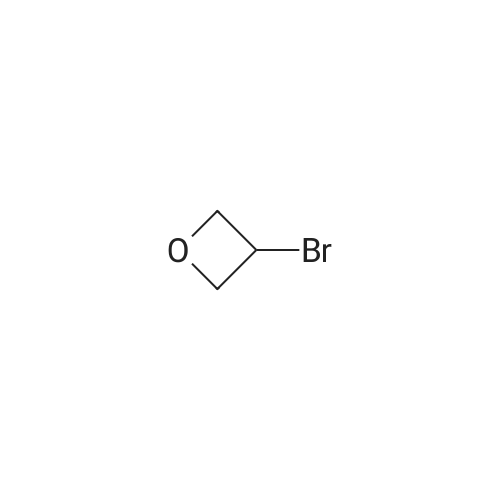
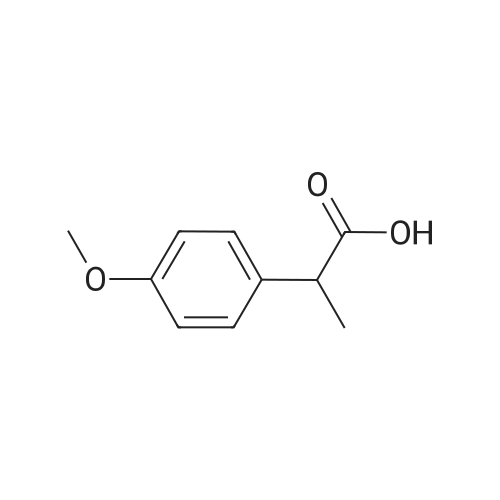
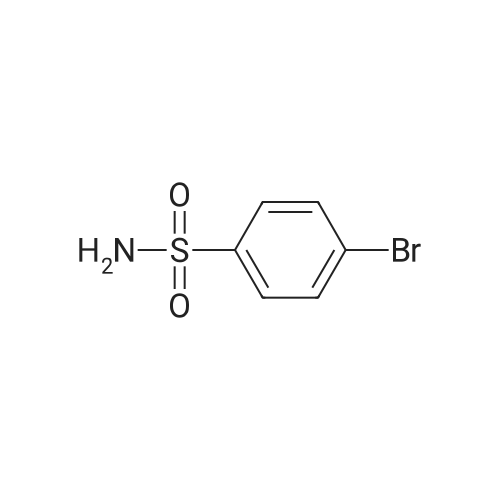
| 46% |
In ethanol; for 24h;Heating / reflux; |
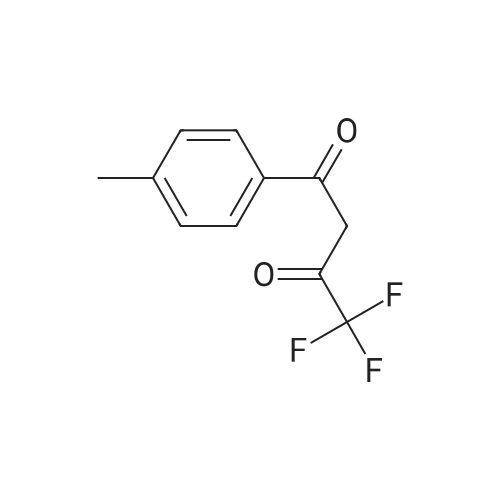
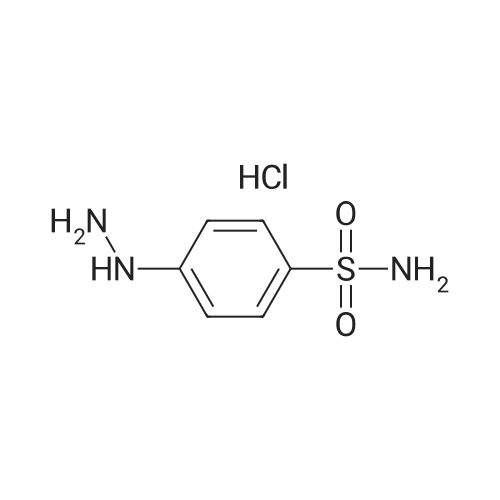
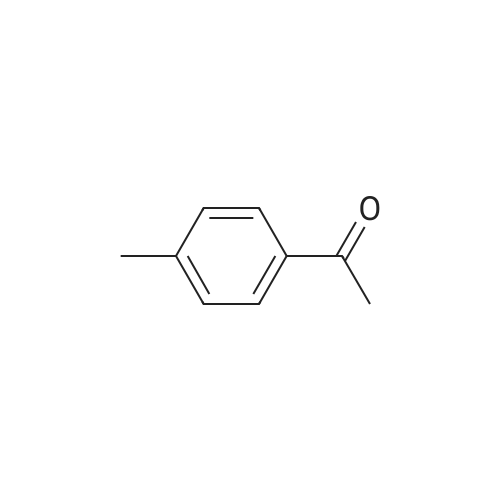
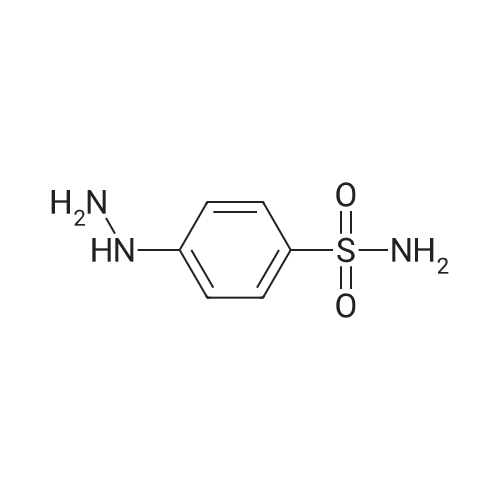
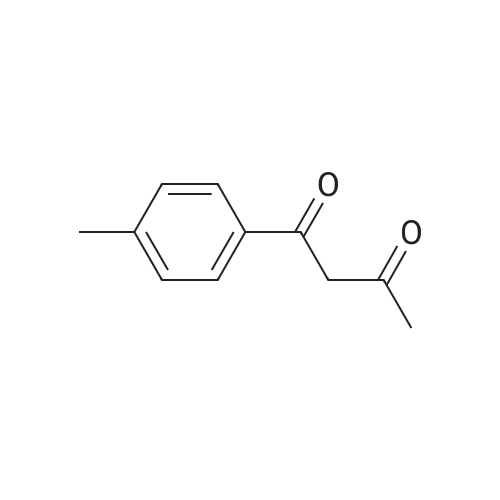
To the dione from Step 1 (4. 14 g, 18. 0 mmol) in 75 mL absolute ethanol, 4.26 g (19.0 mmol) 4-sulphonamidophenylhydrazine hydrochloride was added. The reaction was refluxed under argon for 24 hours. After cooling to room temperature and filtering, the reaction mixture was concentrated to afford 6.13 g of an orange solid. The solid was recrystallized from methylene chloride/hexane to give 3.11 g (8.2 mmol, 46%) of the product as a pale yellow solid, having a melting point (mp) of 157 -159 C ; and a calculated composition of C17 H14 N3 2 SF3 ; C, 53.54 ; H, 3.70 ; N, 11.02. The composition that was found by analysis was: C, 53.17 ; H, 3.81 ; N, 10.90. |
| 46% |
In ethanol; for 24h;Heating / reflux; |
To the dione from Step 1 (4. 14 g, 18.0 mmol) in 75 mL absolute ethanol, 4.26 g (19.0 mmol) 4-sulphonamidophenylhydrazine hydrochloride was added. The reaction was refluxed under argon for 24 hours. After cooling to room temperature PC26183 46 and filtering, the reaction mixture was concentrated to afford 6.13 g of an orange solid. The solid was recrystallized from methylene chloride/hexane to give 3. 11 g (8. 2 mmol, 46%) of the product as a pale yellow solid, having a melting point (mp) of 157-159C ; and a calculated composition of C17 H14 N3 02 SF3 ; C, 53.54 ; H, 3. 70 ; N, 11.02. The composition that was found by analysis was: C, 53.17 ; H, 3.81 ; N, 10.90. |
| 46% |
In ethanol; for 24h;Heating / reflux; |
Step 2: Preparation of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide. To the dione from Step 1 (4.14 g, 18.0 mmol) in 75 mL absolute ethanol, 4.26 g (19.0 mmol) 4-sulphonamidophenylhydrazine hydrochloride was added. The reaction was refluxed under argon for 24 hours. After cooling to room temperature and filtering, the reaction mixture was concentrated to afford 6.13 g of an orange solid. The solid was recrystallized from methylene chloride/hexane to give 3.11 g (8.2 mmol, 46%) of the product as a pale yellow solid, having a melting point (mp) of 157-159 C.; and a calculated composition of C17H14N3O2SF3; C, 53.54; H, 3.70; N, 11.02. The composition that was found by analysis was: C, 53.17;H, 3.81; N, 10.90. |
| 46% |
In ethanol; for 24h;Heating / reflux; |
Step 2: Preparation of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide. To the dione from Step 1 (4.14 g, 18.0 mmol) in 75 mL absolute ethanol, 4.26 g (19.0 mmol) 4-sulphonamido-phenylhydrazine hydrochloride was added. The reaction was refluxed under argon for 24 hours. After cooling to room temperature and filtering, the reaction mixture was concentrated to afford 6.13 g of an orange solid. The solid was recrystallized from methylene chloride/hexane to give 3.11 g (8.2 mmol, 46%) of the product as a pale yellow solid, having a melting point (mp) of 157-159 C.; and a calculated composition of C17H14N3O2SF3; C, 53.54; H, 3.70; N, 11.02. The composition that was found by analysis was: C, 53.17; H, 3.81; N, 10.90. |
| 46% |
In ethanol; for 24h;Heating / reflux; |
To the dione from Step 1 (4.14 g, 18.0 mmol) in 75 mL absolute ethanol, 4.26 g (19.0 mmol) 4-sulphonamidophenylhydrazine hydrochloride was added. The reaction was refluxed under argon for 24 hours. After cooling to room temperature and filtering, the reaction mixture was concentrated to afford 6.13 g of an orange solid. The solid was recrystallized from methylene chloride/hexane to give 3.11 g (8.2 mmol, 46%) of the product as a pale yellow solid, having a melting point (mp) of 157-159C ; and a calculated composition of C17 H14 N3 02 SF3 ; C, 53.54 ; H, 3.70 ; N, 11.02. The composition that was found by analysis was: C, 53.17 ; H, 3.81 ; N, 10.90. |
| 46% |
In ethanol; for 24h;Heating / reflux; |
Step 2: Preparation of 4- [5- (4-methylphenyl)-3- (trifluoromethyl)-1 H-pyrazol-1-yl] benzenesulfonamide. [000203] To the dione from Step 1 (4.14 g, 18.0 MMOL) in 75 mL absolute ethanol, 4.26 g (19.0 MMOL) 4-SULPHONAMIDOPHENYLHYDRAZINE hydrochloride was added. The reaction was refluxed under argon for 24 hours. After cooling to room temperature and filtering, the reaction mixture was concentrated to afford 6.13 g of an orange solid. The solid was RECRYSTALLIZED from methylene chloride/hexane to give 3.11 g (8. 2 MMOL, 46%) of the product as a pale yellow solid, having a melting point (mp) of 157-159C |
| 46% |
In ethanol; for 24h;Heating / reflux; |
To the dione from Step 1 (4.14 g, 18.0 MMOL) in 75 mL absolute ethanol, 4.26 g (19.0 MMOL) 4-sulphonamidophenylhydrazine hydrochloride was added. The reaction was refluxed under argon for 24 hours. After cooling to room temperature and filtering, the reaction mixture was concentrated to afford 6.13 g of an orange solid. The solid was recrystallized from methylene CHLORIDE/HEXANE to give 3.11 g (8.2 mmol, 46%) of the product as a pale yellow solid, having a melting point (mp) of 157-159C ; and a calculated composition of C17 H14 N3 02 SF3 ; C, 53.54 ; H, 3.70 ; N, 11.02. The composition that was found by analysis was: C, 53.17 ; H, 3.81 ; N, 10.90. |
| 46% |
In ethanol; for 24h;Heating / reflux; |
To the dione from Step 1 (4.14 g, 18.0 MMOL) in 75 mL absolute ethanol, 4.26 g (19.0 MMOL) 4-SULPHONAMIDOPHENYLHYDRAZINE hydrochloride was added. The reaction was refluxed under argon for 24 hours. After cooling to room temperature and filtering, the reaction mixture was concentrated to afford 6.13 g of an orange solid. The solid was RECRYSTALLIZED from methylene chloride/hexane to give 3.11 g (8.2 MMOL, 46%) of the product as a pale yellow solid, having a melting point (mp) of 157-159C ; and a calculated composition of C17 HR4 N3 02 SF3 ; C, 53. 54 ; H, 3.70 ; N, 11.02. The composition that was found by analysis was: C, 53.17 ; H, 3.81 ; N, 10.90. |
| 46% |
In ethanol; for 24h;Heating / reflux; |
To the digne from Step 1 (4.14 g, 18.0 mmol) in 75 mL absolute ethanol, 4.26 g (19.0 mmol) 4-sulphonamidophenylhydrazine hydrochloride was added. The reaction was refluxed under argon for 24 hours. After cooling to room temperature and filtering, the reaction mixture was concentrated to afford 6.13 g of an orange solid. The solid was recrystallized from methylene chloride/hexane to give 3.11 g (8.2 mmol, 46%) of the product as a pale yellow solid, having a melting point (mp) of 157-159C ; and a calculated composition of C17 H14 N3 2 SF3 ; C, 53.54 ; H, 3.70 ; N, 11.02. The composition that was found by analysis was: C, 53.17 ; H, 3.81 ; N, 10.90. |
|
|
To a 100 mL Morton flask was charged [1G 4-SAPH-HCI] and 20 mL THF. To this slurry, under nitrogen at room temperature, was added 4.5 mL [NAOH] [(1 M] in [H20)] to pH 11.2. The solution turned homogeneous. After 30 minutes the pH of the solution was adjusted to 0.9 with [TRIFLUOROACETIC] acid. At room temperature, 105 mg diketone, in 10 mL THF, was added. Aliquots were taken periodically as the reaction proceeded and analyzed by HPLC on a 4.6 mm, 5 micron Supelco [SUPELCOSILE] LC-DP column. HPLC analysis in all of the following examples was done by the same method. Results are given in table 1 a below. |
|
|
A 50 mL [3-NECK] Morton flask was fitted with a thermometer, a reflux condenser and a nitrogen inlet. To this reactor was charged 25 mL isopropanol and [1G] (4.5 [MMOL)] [4-SAPH-HCI,] followed by the addition of 0.5 g triethylamine (4.4 [MMOL)] to free base the 4-SAPH. The reaction mixture was allowed to stir for 15 minutes at room temperature. To the resultant slurry was added 0.87g (9.2 [MMOL)] chloroacetic acid. The mixture was allowed to stir at room temperature for 15 minutes. After this time period, [1 G] (4.3 [MMOL)] diketone was added and the reaction mixture was heated to 60 [C] and held at this temperature for 2.5 hr. An aliquot was taken and analyzed via HPLC. The reaction was repeated with each of the following acids: acetic, [DICHLOROACETIC,] trichoroacetic, and [TRIFLUOROACETIC.] Comparative results are given in table 2 below. |
|
|
A 50 mL [3-NECK] Morton flask was fitted with a thermometer, a reflux condenser and a nitrogen inlet. To this reactor was charged 25 mL isopropanol and [1G] (4.5 [MMOL)] [4-SAPH-HCI,] followed by the addition of 0.5 g triethylamine (4.4 [MMOL)] to free base the 4-SAPH. The reaction mixture was allowed to stir for 15 minutes at room temperature. To the resultant slurry was added 0.87g (9.2 [MMOL)] chloroacetic acid. The mixture was allowed to stir at room temperature for 15 minutes. After this time period, [1 G] (4.3 [MMOL)] diketone was added and the reaction mixture was heated to 60 [C] and held at this temperature for 2.5 hr. An aliquot was taken and analyzed via HPLC. The reaction was repeated with each of the following acids: acetic, [DICHLOROACETIC,] trichoroacetic, and [TRIFLUOROACETIC.] Comparative results are given in table 2 below. |
|
|
A 50 mL [3-NECK] Morton flask was fitted with a thermometer, a reflux condenser and a nitrogen inlet. To this reactor was charged 25 mL isopropanol and [1G] (4.5 [MMOL)] [4-SAPH-HCI,] followed by the addition of 0.5 g triethylamine (4.4 [MMOL)] to free base the 4-SAPH. The reaction mixture was allowed to stir for 15 minutes at room temperature. To the resultant slurry was added 0.87g (9.2 [MMOL)] chloroacetic acid. The mixture was allowed to stir at room temperature for 15 minutes. After this time period, [1 G] (4.3 [MMOL)] diketone was added and the reaction mixture was heated to 60 [C] and held at this temperature for 2.5 hr. An aliquot was taken and analyzed via HPLC. The reaction was repeated with each of the following acids: acetic, [DICHLOROACETIC,] trichoroacetic, and [TRIFLUOROACETIC.] Comparative results are given in table 2 below. |
|
|
A 50 mL [3-NECK] Morton flask was fitted with a thermometer, a reflux condenser and a nitrogen inlet. To this reactor was charged 25 mL isopropanol and [1G] (4.5 [MMOL)] [4-SAPH-HCI,] followed by the addition of 0.5 g triethylamine (4.4 [MMOL)] to free base the 4-SAPH. The reaction mixture was allowed to stir for 15 minutes at room temperature. To the resultant slurry was added 0.87g (9.2 [MMOL)] chloroacetic acid. The mixture was allowed to stir at room temperature for 15 minutes. After this time period, [1 G] (4.3 [MMOL)] diketone was added and the reaction mixture was heated to 60 [C] and held at this temperature for 2.5 hr. An aliquot was taken and analyzed via HPLC. The reaction was repeated with each of the following acids: acetic, [DICHLOROACETIC,] trichoroacetic, and [TRIFLUOROACETIC.] Comparative results are given in table 2 below. |
|
|
A 50 mL [3-NECK] Morton flask was fitted with a thermometer, a reflux condenser and a nitrogen inlet. To this reactor was charged 25 mL isopropanol and [1G] (4.5 [MMOL)] [4-SAPH-HCI,] followed by the addition of 0.5 g triethylamine (4.4 [MMOL)] to free base the 4-SAPH. The reaction mixture was allowed to stir for 15 minutes at room temperature. To the resultant slurry was added 0.87g (9.2 [MMOL)] chloroacetic acid. The mixture was allowed to stir at room temperature for 15 minutes. After this time period, [1 G] (4.3 [MMOL)] diketone was added and the reaction mixture was heated to 60 [C] and held at this temperature for 2.5 hr. An aliquot was taken and analyzed via HPLC. The reaction was repeated with each of the following acids: acetic, [DICHLOROACETIC,] trichoroacetic, and [TRIFLUOROACETIC.] Comparative results are given in table 2 below. |
|
|
A 100 mL 3-neck Morton flask was fitted with a reflux condenser, a nitrogen inlet, a thermometer and a glass stopper. This setup was purged with nitrogen for 15 min after which time 1 g (4.47 [MMOL)] [4-SAPH-HCI] and 25 mL anhydrous methyl alcohol was charged to the flask. To the slurry was added 0.5 g (4.95 [MMOL)] triethylamine at room temperature. After 15 minutes, 1 g (8.77 [MMOL)] [TRIFLUOROACETIC] acid was added at room temperature. After 15 minutes at room temperature, the reaction mixture was heated to 55 [C,] 1 g solid diketone (4.3 [MMOL)] was added all at once followed by 5 mL methyl alcohol to wash the addition funnel. Aliquots from the reaction mixture were taken and analyzed by HPLC. Results in mole% by-product formed based on celecoxib at reaction completion are show in table 3 below. The reaction was repeated for the following solvents: Ethyl alcohol, n- Propyl alcohol, i-Propyl alcohol, Trifluoroethanol, and t-Butyl alcohol. |
|
|
To a 100 mL Morton flask was charged 1.03g 4-SAPH and 20 mL THF. To this slurry, under nitrogen at room temperature, was added 1g NaOMe (25 wt% in [MEOH)] to pH 10.4. The solution turned homogeneous. After 30 minutes the pH of the solution was adjusted to 1.1 with [TRIFLUOROACETIC] acid. At room temperature, 104 mg diketone, in 10 mL THF, was added. Aliquots were taken periodically as the reaction proceeded and analyzed by HPLC. Results are given in table [1B] below. |
|
In ethanol; for 20h;Inert atmosphere; Reflux; |
General procedure: 4-Hydrazinylbenzenesulfonamide hydrochloride (3?HCl) (982 mg, 4.4 mmol) was added to the stirred solution of diketone 2 (4.0 mmol) in EtOH (60 mL). If free base 3 was used to replace 3?HCl, additional HCl (3 N, 2.0 mL) is needed. The mixture was heated to reflux for 20 h. Then the reaction mixture was concentrated in vacuo. The residue was added EtOAc, washed with water, dried, filtered, and concentrated. The crude product was purified by column chromatography on silica gel (12% MeOH/CH2Cl2) to give a white solid 4 in 85 +/- 5% (n = 3) yield, Rf = 0.78-0.82 (10% MeOH/CH2Cl2). |
| 24.06g |
With trifluoroacetic acid; In water; isopropyl alcohol; at 55 - 65℃; for 1h; |
Take another 500 mL three-necked glass vial, add 50 mL of isopropanol, to a solution of hydrazinobenzenesulfonamide hydrochloride (17.4 g, 0.078, 1101) and trifluoroacetic acid (0.428, 0.0037111001), and the temperature was raised to 55 (after adding reaction solution 3, reaction 111, adding50mL water, the temperature rose to 65 C full solution, cooling 10 C per hour, down to 15 C, pumping, 45 C under reduced pressure drying to get celecoxib eight crystal 24.068, the yield of 85% Ie ^: detection purity of 99.7%. |
|
In ethanol; for 24h;Heating / reflux; |
To the dione from Step 1 (4.14 g, 18.0 mmol) in 75 mL absolute ethanol, 4.26 g (19.0 mmol) 4-sulphonamidophenylhydrazine hydrochloride was added. The reaction was refluxed under argon for 24 hours. After cooling to room temperature and filtering, the reaction mixture was concentrated to afford 6.13 g of an orange solid. The solid was recrystallized from methylene chloride/hexane to give 3.11 g (8.2 mmol, 46%) of the product as a pale yellow solid, having a melting point of 157-159C ; and a calculated composition of C17Hl4N302SF3 ; C, 53.54 ; H, 3.70 ; N, 11.02. The composition that was found by analysis was: C, 53.17 ; H, 3. 81 ; N, 10.90. |
|
In ethanol; for 24h;Heating / reflux; |
To the dione from Step 1 (4.14 g, 18.0 mmol) in 75 mL absolute ethanol, 4.26 g (19.0 mmol) 4-sulphonamidophenylhydrazine hydrochloride was added. The reaction was refluxed under argon for 24 hours. After cooling to room temperature and filtering, the reaction mixture was concentrated to afford 6.13 g of an orange solid. The solid was recrystallized from methylene chloride/hexane to give 3.11 g (8.2 mmol, 46%) of the product as a pale yellow solid, having a melting point of 157-159C ; and a calculated composition of C17Hl4N302SF3 ; C, 53.54 ; H, 3.70 ; N, 11.02. The composition that was found by analysis was: C, 53.17 ; H, 3. 81 ; N, 10.90. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +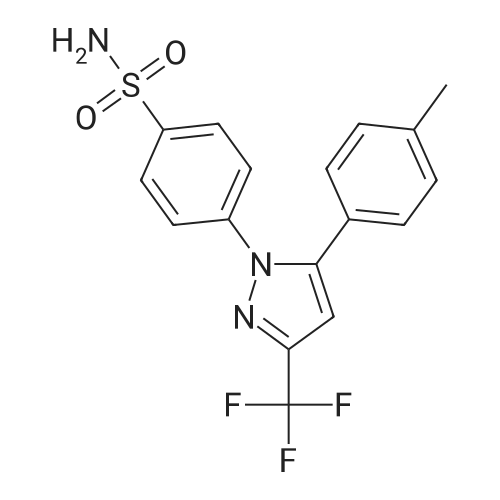

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping