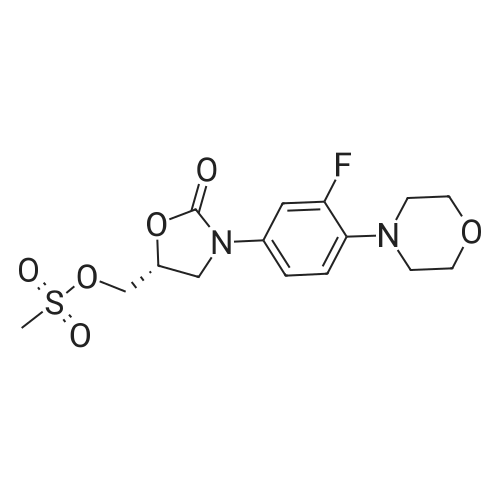
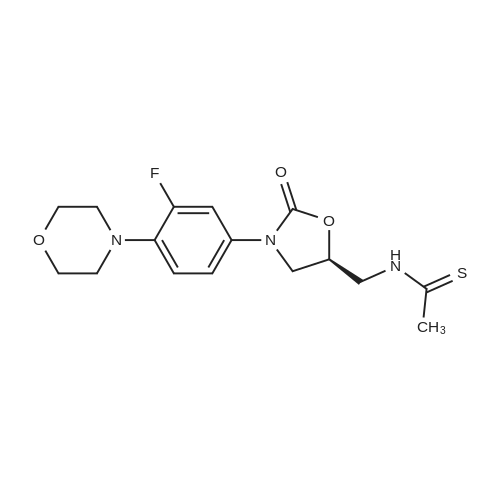
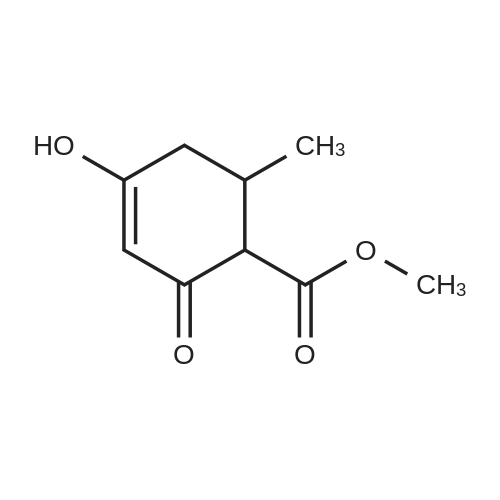
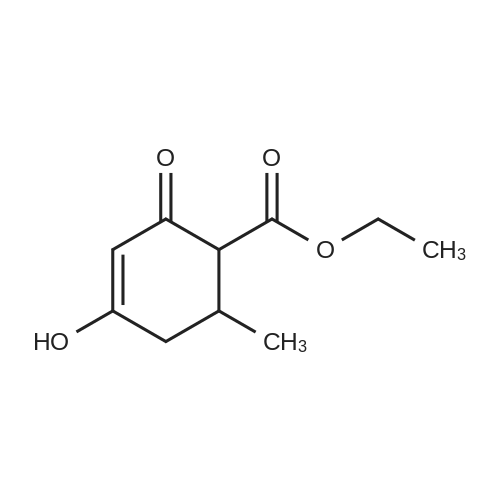
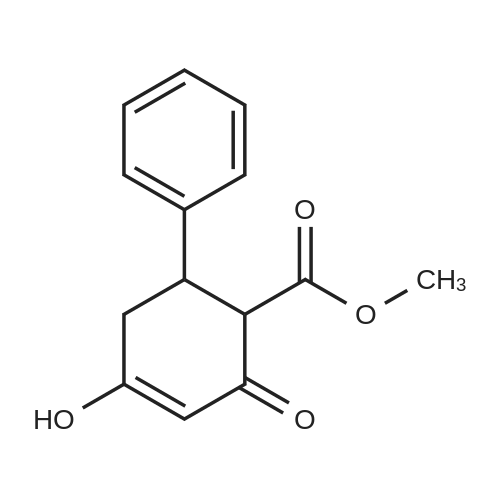
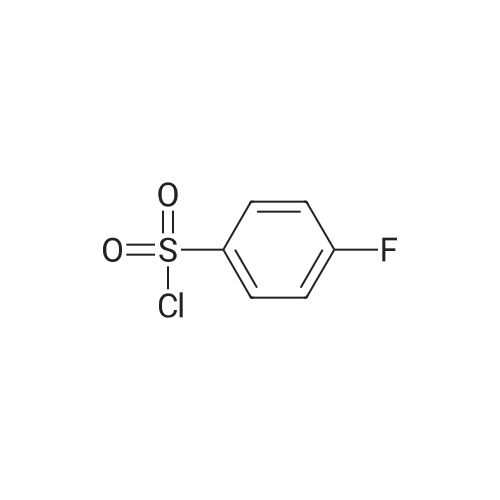
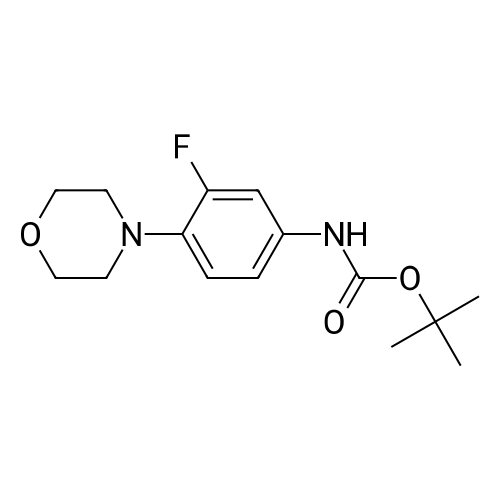
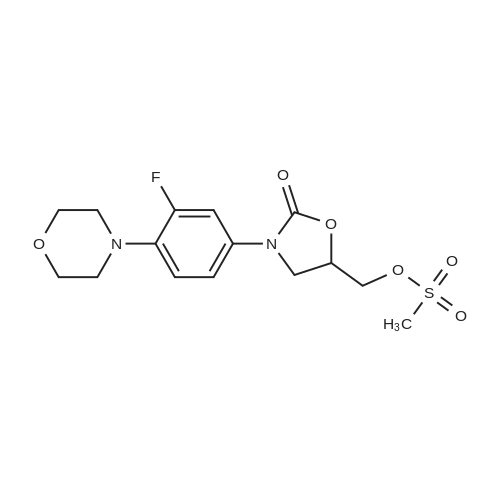
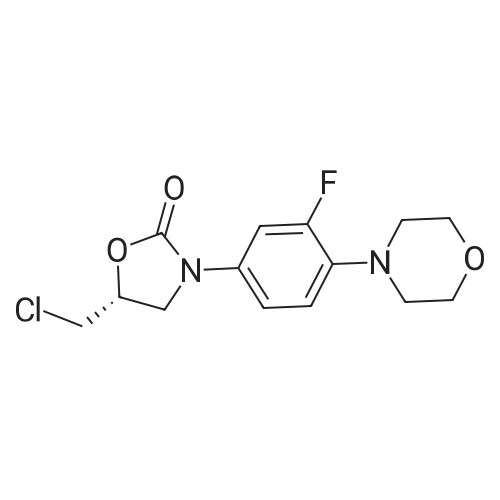
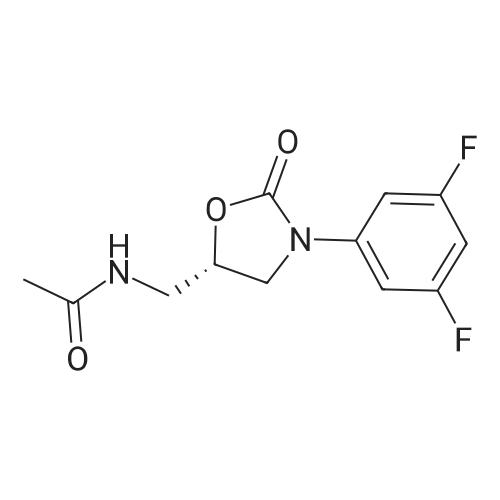
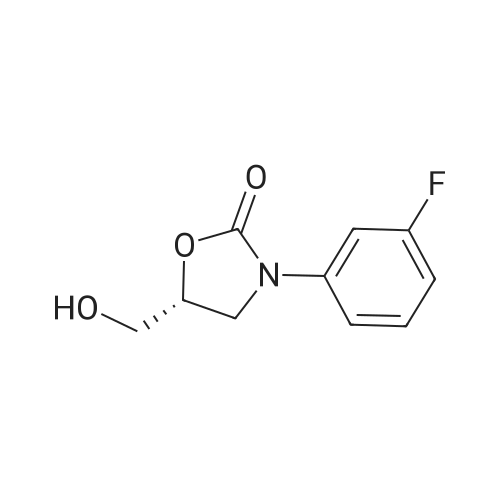
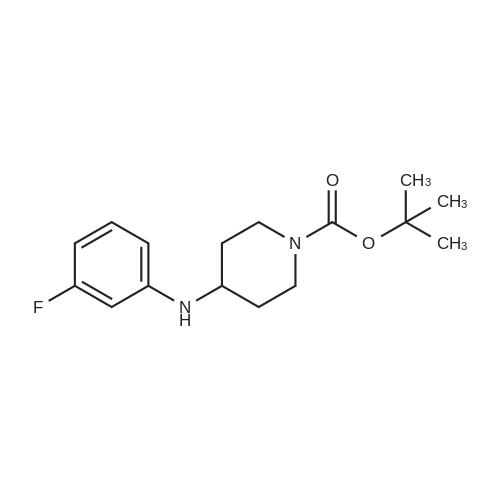
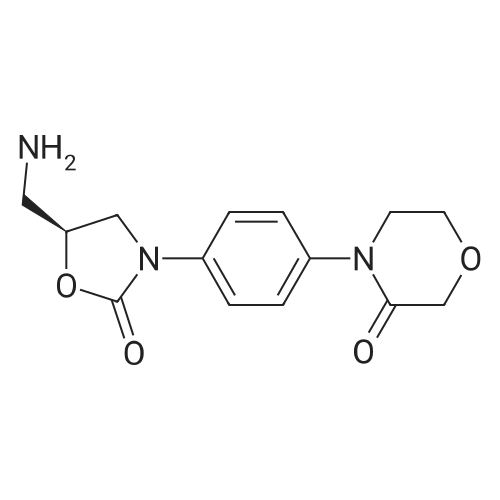
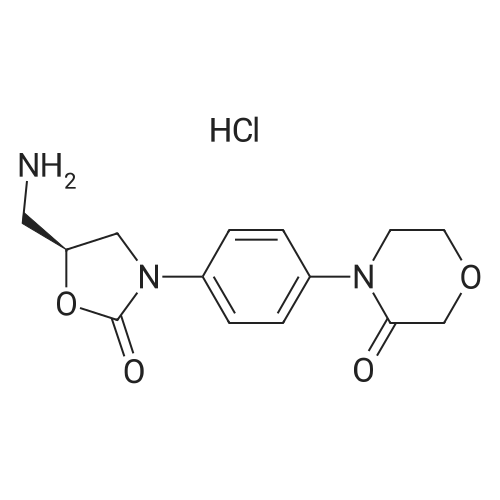
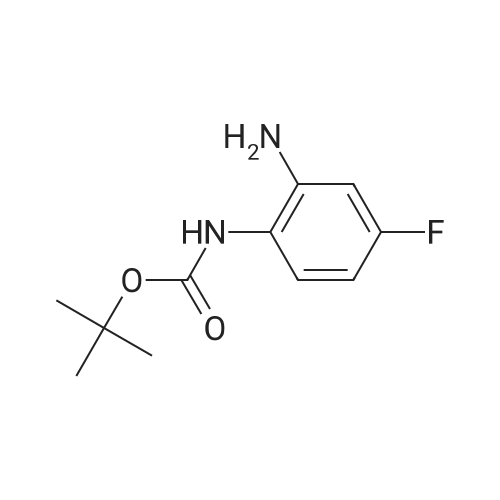
| 88.4% |
With triethylamine; In dichloromethane; at 20℃; for 1h; |
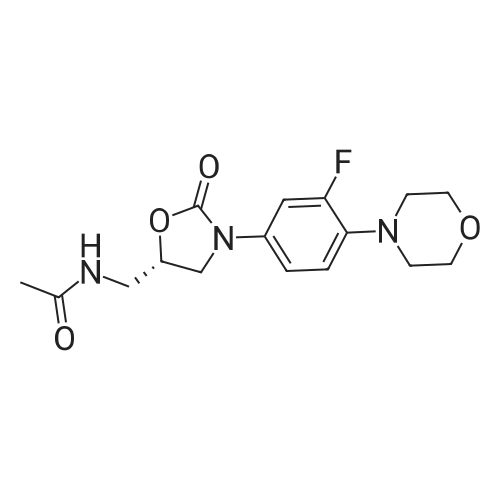
(S) - [3- (3-fluoro-4-morpholinophenyl) -2-oxo-5-oxazolidinyl] methylamine (7.2 g, 24.4 mmol) In dichloromethane, acetic anhydride (3.0 g, 29.2 mmol) was added,Triethylamine (4.1 mL, 29.2 mmol), reacted at room temperature for 1 hour, and monitored by TLC.After the reaction was complete, the mixture was washed with water, saturated sodium chloride solution, dried over anhydrous sodium sulfate, concentrated,To give the crude product as a milky white which was recrystallized from ethyl acetate,To give linezolid 7.27g,Yield: 88.4%, chemical purity: 99.9%; optical purity: 99.96%. |
| 87% |
In ethyl acetate; at 20℃; for 1h; |
General procedure: A solution of acetic anhydride andpropionic anhydride (0.020 mol) was added dropwise to a stirred solution of (S)-N-[[3-[3-Fluoro-4-[4-morpholinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]amine (6) (2.5 g, 0.008 mol) inethyl acetate (20 mL) at ambient temperature andthe reaction mixture was allowed to stirred for 1 h.The reaction mixture was cooled to 0-5C. Theprecipitated solid was filtered off and re-crystallized from methanol (20 ml-) to give the correspondingcompounds (7a-b). |
| 86.9% |
With triethylamine; In ethyl acetate; at 20℃; for 2h; |
Amino-methyl-3- (3-fluoro-4-morpholinophenyl) oxazolin-2-one obtained in Example 4-1 was added to a reaction flask (7,23.7 g, 0.08 mol), acetic anhydride (10.2 g, 0.1 mol), triethylamine (40.5 g, 0.4 mol) was added 500 mL of ethyl acetate at room temperature for 2 hours until the reaction was complete. Add water, the organic layer, saturated brine washing, anhydrous magnesium sulfate drying, filtration, evaporation of most of the solvent, frozen crystals, filtered crude. The crude product was recrystallized from ethyl acetate to give nicotinamide 23.5 g, molar yield 86.9%, HPLC chemical purity 99.8%, optical purity 99.9%. |
| 80% |
With triethylamine; at 25℃; for 0.5h; |
2) The compound (intermediate 3) in dichloromethane was suction filtered to take the filtrate, which was added to the reaction vessel and cooled to 0 C, and added.570g of triethylamine, slowly adding 345g acetic anhydride; after the addition is completed, the system is heated to 25 C for 0.5h, then added 2.4L hydrochloric acid aqueous solution, stirred for 10min;Wash again with water 3 times, combine the organic phases, dry over anhydrous sodium sulfate,The mixture was suction filtered, and the solvent was evaporated under reduced pressure.Cool down to 10 C for suction filtration, dry at 40 C,Obtained 758.85g of white solid.The yield was 80%. |
| 77% |
With triethylamine; In toluene; at 20℃; for 5.5h;Product distribution / selectivity; |
To a suspension of (S)-5-(aminomethyl)-3-(3-fluoro-4-morpholinophenyl)oxazolidin-2-one (III) (452 mg, 1.53 mmol) in toluene (2.3 mL) was added triethylamine (215 muL, 1.54 mmol, 1 eq) and acetic anhydride (146 muL, 1.54 mL, 1 eq) and the mixture was stirred at room temperature for 5.5 h. The suspension was cooled in an ice bath for 30 min, was filtered and the collected solids were washed with cold toluene (2 x 0.5 mL) and were dried in vacuo to give N-(((S)-3-(3-fluoro-4-morpholinophenyl)-2-oxazolidin-5-yl)methyl)acetamide, linezolid (390 mg, 77%, HPLC 99.1%, 99.9% ee) as an off-white solid. For characterization, see Example 11. |
| 75% |
In dichloromethane; at 25 - 30℃; for 1h; |
Example-9: (0148) Preparation of N-({(5S)-3-[3-fluoro-4-(morpholin-4-yl) phenyl]-2-oxo-l, 3- oxazolidin-5-yl} methyl) acetamide (Linezolid) (0149) To the mixture of Methanol (100 ml) ,DM water (400 ml) and (S) 2-[3-(3-Fluoro-4- morpholin-4-yl-phenyl)-2-oxo-oxazolidin-5-ylmethyl] -isoindole-1, 3-Dione (100 g 0.212 moles ) were added Methyl amine solution (47 g) to the reaction mixture at 25- 30C, stirred and the temperature was slowly raised to 80-85C and stirred for 2-3 hours at 80-85C. The reaction mixture was cooled to 25-30C and dichloromethane (500 ml) was added to it and stirred the reaction mixture for 15 min and separated the two layers. MDC was distilled out by atmospheric pressure completely to get the residual product (5S)-5-(amino methyl)-3-[3-fluoro-4-(morpholin-4-yl) phenyl]-l,3-oxazolidin-2-one. Dichloromethane (400 ml) was added to the residue and acetic anhydride (25 g) was slowly added at 25-30C over a period of 60 min. After completion, 5% aqueous sodium bicarbonate solution was slowly added to reaction mixture, stirred for 15 min and the two layers were separated. The dichloromethane layer was washed with DM Water (200 ml). The dichloromethane layer was filtered through hyflo and distilled out dichloromethane completely under vacuum below 40C. Cyclohexane (500 ml) was added to the residue and heated to 45-50C. The slurry obtained was cooled to 20-25C, stirred for 60 min, filtered the solid, washed with cyclohexane (200 ml) and dried the solid at 45-55C to furnish pure crystalline N-({(5S)-3-[3-fluoro-4-(morpholin-4- yl)phenyl]-2-oxo-l,3-oxazolidin-5-yl}methyl)acetamide (Linezolid) (53 g 75% ). |
| 68% |
|
Example- 7: Preparation of (5S)-(N)-[[3-fluoro-(4-morpholinylphenyl)-2-oxo-5-oxazolidinyl] methyl acetamide (Form II)(5S)-(N)-[[3- fluoro-(4-mophiholinylphenyl)-2-oxo-5-oxazolidinyl]methyl]amine (20 gms) was added in water (200 ml) and ethyl acetate (80 ml) and the mixture was stirred at 25C-30C followed by the addition of acetic anhydride (19.2 ml, 3 eq.) at the same time. The reaction mixture was neutralized to 6-8 pH with aqueous ammonia and stirred for 2 hours. The reaction mixture was then filtered, to give (5S)-(N)-[3-fluoro-(4-morpholinylphenyl)-2-oxo-5-oxazolidinyl]methyl acetamide (Yield: 68%) |
| 60% |
With triethylamine; In dichloromethane; at 5 - 20℃; for 4h; |
Example-14: Preparation of (S)-N-[[3-(3-fluoro-4-morpholinylphenyl)-2-oxo-5- oxazolidinyljmethyl acetamide, Linezolid (I) starting from (S)-[[N-3-(3-fluoro-4- morpholinylphenyl)-2-oxo-5-oxazolidinyl]methyl]amine(S)-[[N-3-(3-fluoro-4-morpholinylphenyl)-2-oxo-5-oxazolidinyl]methyl]amine (3 g) obtained in example 11 and triethylamine (1.54 g) were dissolved in DCM (15 ml). The solution was cooled to 5C and acetic anhydride (1.25 g) was added. The resulting reaction mixture was warmed to room temperature and stirred for 4h. After completion of reaction, water (50 ml) was added in reaction mixture and stirred for 30 min. Separation of DCM and aqueous layers was carried out. Aqueous layer was extracted with DCM. The combined DCM layers were washed with 10% sodium bicarbonate solution followed by washing with water. DCM layer was concentrated under vacuum at 35-40C. Toluene (20 ml) was added into the residue and heated to 70C. The suspended solution was stirred at 65-70C for 30 min and further at room temperature for lh. The solid mass was filtered, washed with toluene and dried to obtain the titled compound (2.1 g) with 60% yield. |
|
In ethyl acetate; at 20℃; for 1h;Product distribution / selectivity; |
Example 5; To the mixture of (S)-N-[[3-[3-fluoro-4-[4-morpholinyl]phenyl]-2-oxo-5- oxazolidinyl] methyl] amine (10 gm) and ethylacetate (100 ml), acetic anhydride (10 ml) is slowly added at ambient temperature, then stirred at ambient temperature for 1 hour. The separated solid is filtered and dried under reduced pressure at 50C to give 9.5 gm of linezolid form 111. |
|
With ammonium hydroxide; triethylamine; In water; toluene; at 3 - 25℃;Product distribution / selectivity; |
Example 1 - preparation of crystalline linezolid Form IVA flask containing 5.5 g of (S)-N-(4-morpholinyl-3-fluorophenyl)-2-oxo-5-oxazolidinyl-methyl amine and 20 ml ammonium hydroxide in 200 ml toluene wasmixed at 25 C. Triethyl amine (2 equivalents) was added. The mixture was cooled to3 C and acetic anhydride (2.5 equivalents) was added dropwise. The reaction mixturewas brought to RT. Linezolid that precipitated from the reaction mixture was filtered.The wet crystals were analyzed by PXRD and shown to be crystalline linezolid FormIV.Example 2 - Procedure for the preparation of crystalline linezolid Form IVThis example was carried out on a Buchi Mini Spray Dryer B-290. 5 g of linezolid was dissolved in methanol (300 ml) at room temperature. Theobtained solution was pumped into the spray dryer. The nitrogen was at an inlettemperature of 50 C. The evaporated solvent and nitrogen exited the spray dryer at35 C. The obtained sample was analyzed by PXRD and shown to be linezolid FormIV. |
|
In toluene; at 20℃; for 1h; |
S-N-[[3-[3-Fluoro4-[4-morpholinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl] amine (20 gm) is stirred in toluene (200 ml) for 15 minutes, acetic anhydride (20 gm) is added drop wise at ambient temperature and stirred for 1 hour. The reaction mixture is cooled to 0-5C, filtered the solid and re-crystallized from methanol (200 ml) to give 16 gm of N-[[(5S)-3-[3-fluoro-4-(4-morphblinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide. |
|
In ethyl acetate; at 20℃; for 1h; |
Example 5; S-N-[[3-[3-Fluoro4-[morpholinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]amine (20 gm) is dissolved in Ethyl acetate (200 ml), Acetic anhydride (20 gm) is added drop wise at ambient temperature and stirred for 1 hour. The reaction mixture is then cooled to 0-5 C., filtered the solid and re-crystallized from Isopropyl alcohol (400 ml) to give 16 gm of N-[[(5S)-3-[3-fluoro-4-(4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide. |
|
With triethylamine; In ethyl acetate; at 25 - 30℃;Product distribution / selectivity; |
[139] Example 6: Synthesis of Linezolid Crude.[140] Ethyl acetate (3500ml) and 10% palladium on carbon catalyst (6.0g) are added in autoclave having (R)- [N- 3 - (3 -Fluoro-4-morpholinylphenyl) -2-oxo- 5 -oxazolidinyl] methyl azide (lOOg) at 20-30C. Cool the reaction mass & maintain 2-3kg hydrogen pressure at 15-20C for 6-7 hrs. Filter it & wash the hyflo bed by Ethyl acetate(100mlx2). Then add the Triethyl amine (35. lg) & Acetic anhydride (29.9g) slowly at 25-30C under stirring. Cool the mix, filter it and wash the solid with chilled (0-5C) Ethyl acetate (100 ml) followed by water (100mlx2). Finally product is dried at 55-60 C. Yield: 0.85.: Percentage 81%w/w. |
|
|
EXAMPLE 25. Scale-up of synthesis of (S)-N-[{ 3-[3-fluoro-4- (morpholinyl)phenyl]-oxo-5-oxazolidyl]methyl]acetamide: Linezolid (9). To a 2 L 3 neck round bottom flask with overhead stirrer was added water (425 ml), 12 N hydrochloric acid (34 ml, 408 mmol), methylene chloride (340 ml) and (S)-(E,Z)-5-((4- chlorobenzylideneamino)methyl)-3-(3-fluoro-4-morpholinophenyl)oxazolidin-2-one (17) (85 g, 203.4 mmol) rinsed in with methylene chloride (85 ml). The mixture was rapidly stirred at room temp and within 30 minutes a two-phase clear homogenous solution formed. The reaction was stirred for 1 hour and the orange yellow lower organic layer was discarded. The lemon yellow aqueous layer was washed with additional methylene chloride (200 ml) and the methylene chloride was discarded. Methylene chloride (425 ml) was added to the aqueous layer and the two phase solution was transferred to a 2 L Erlenmeyer flask, cooled in an ice bath and neutralized to ca. pH 7 with ice cold 6 N NaOH (ca. 45 ml) while stirring the reaction in the ice bath. The reaction changed in color from yellow to colorless and a white precipitate formed. The ice bath was removed from the reaction flask and acetic anhydride (72 ml, 720 mmol) was added all at once to the rapidly stirring mixture. The mixture was vigorously stirred at room temp for 1 hour yielding a light yellow clear 2-layer solution. The solution was cooled in an ice bath and made basic (ca. pH 9) with 6 N sodium hydroxide. The lower organic layer was separated and the aqueous layer was extracted with additional methylene chloride (3 x 100 ml). The combined organic layers were dried (MgS04) and evaporated (bath temp 25 C) to a volume of ca. 400 ml. The light yellow solution |
|
In ethyl acetate; at 20℃; for 1h; |
(S)-3-(3-fluoro-4-morpholinophenyl)-2-oxo-5-oxazolidinyl methylamine ( 15 gr) is dissolved in ethylacetate (1 50 ml); acetic anhydride ( 1 5 gr) is added dropwise at ambient temperature and stirred for 1 hr. The reaction mixture is then cooled to 0-5 C. Filtered the solid to give 12 gr of (S)-3-(3-fluoro-4-morpholinophenyl) -2-oxo-5-oxazolidinyl methyl acetamide. |
| 60 g |
at 0 - 25℃; |
ExampIe-5: ; (SMN-[3-(3-fluoro-4-morpholinylphenyl)-2-oxo-5- oxazolidinyllmethyll- acetamide (Linezolid)Methanol (500 mL) and aqueous methylamine (500 mL of 40%) were added to a flask containing the 100 g phthalimido oxazolidinone of Example-4. The suspension was heated at 65C for 3 hours and cooled to room temperature. 500 mL methylene dichloride and 500 mL water were added to the reaction mixture and stirred for 30 minutes. The separated aqueous layer was extracted with 1 L of methylene dichloride and allowed to settle. The combined organic layer was washed with brine solution and distilled to remove 2 times of methylene dichloride under vacuum below 50C. The reaction mixture was cooled to 0C and 72 g acetic anhydride was added to it. The reaction mixture was stirred at 25C and treated with 10% sodium bicarbonate solution (280 mL, 28g). The separated organic layer was washed with water and allowed to settle for 30 minutes. The organic layer was treated with 5 g activated carbon and stirred for 30 minutes. The reaction mixture was filtered and methylene dichloride was completely removed under vacuum below 50C. The residue (linezolid oil) was co- distilled with 100 mL toluene to obtain 83 g Linezolid as a residue. The residue was cooled to room temperature and 300 mL toluene was added to it. The reaction mixture was heated at 50C to 55C for 3 hours and cooled to room temperature. The solid was filtered and washed with 100 mL toluene to obtain 64.8 g wet-cake. The wet-cake was taken in another RBF and 1.5 L ethyl acetate was added. The reaction mixture was heated below 78C and 5 g activated carbon was added to the clear solution. The reaction mixture was fine filtered and washed with 100 mL ethyl acetate. Approx. 700 mL ethyl acetate was distilled below 78C and reaction mixture was cooled to room temperature. Finally, the reaction mixture was cooled to 18C to 20C and stirred for 1 hour. The product was filtered and washed with 100 mL ethyl acetate. The wet-cake was dried at 65C to 70C to obtain 60 g (69%) Linezolid. |
| 21.5 g |
With triethylamine; In dichloromethane; at 10 - 30℃; |
Triethyl amine (12.32 ml) and methylene chloride (220 ml) were added to (S)-N-[[3-[3- fluoro-4-[4-morpholinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]amine (22 g) at 25-30C, the resulting solution was cooled to 10-15C, followed by slow addition of acetic anhydride at 12-14C for 10 to 15 minutes. During the addition process, the temperature of the reaction mass raised up to 22C. The resulting mass was stirred for 10 to 15 minutes at 10-15C, followed by raising the temperature to 25-30C and then stirring the mass for 1 hour at the same temperature. The reaction mass was washed with water (50 ml x 2), followed by separation of the organic layer and subsequently stirring the organic layer with activated carbon (2.5 g) for 5 minutes at 25-30C. The resulting mixture was filtered through hyflo bed and the bed was washed with methylene chloride (20 ml), followed by removal of solvent by ordinary distillation at 55-60C to produce a solid. The resulting solid was co-distilled two times with ethyl acetate (35 ml x 2) under vacuum at 65-75C. Water (1 10 ml) was added to the resulting solid, followed by heating the mixture at 60- 65C and then stirring for 20 to 25 minutes at the same temperature. The resulting mass was cooled to 25-30C and then stirred for 10 minutes at the same temperature, followed by cooling the mass to 10C and then stirring for 10 to 15 minutes at the same temperature. The separated solid was filtered, washed with water (50 ml) and then dried the material at 80-85C for 4 hours to produce 21.5 g of highly pure linezolid crystalline Form III [Purity by HPLC: 99.95%; Chiral Purity by HPLC: (S)-isomer = 99.989%; (R)-isomer = 0.01 1%; and Melting Point: 177.8C - 178.5C]. |
| 12 g |
In ethyl acetate; at 20℃; for 1h; |
(S)-3-(3-fluoro-4-morpholinophenyl)-2-oxo-5-oxazolidinyl methylamine (15 gr) is dissolved in ethylacetate (150 ml); acetic anhydride (15 gr) is added dropwise at ambient temperature and stirred for 1 hr. The reaction mixture is then cooled to 0-5 C. Filtered the solid to give 12 gr of (S)-3-(3-fluoro-4-morpholinophenyl)-2-oxo-5-oxazolidinyl methyl acetamide. |
| 8.5 g |
In dichloromethane; at 0 - 5℃;Reflux; |
To a solution of (5)-5-aminomethyl-3-(3-fluoro-4-morpholin-4-yl-phenyl)-oxazolidin-2-one (compound 6a) ( 10.0 gr, 0.033 mol) in methylene dichloride (25.0 mL), acetic anhydride (compound 7a) (6.0 mL) was added at 0-5C. Slowly temperature was raised to room temperature and heated reflux temperature and stirred for 1 -2 hrs. The reaction mass was distilled and add methanol (20.0 mL) then stirred at room temperature for 10- 1 min, filtered and wash with methanol to get crude Linezolid. The crude compound was crystallized in methanol to get pure Linezolid (compound 8a) (8.5 gr). |
| 50 g |
With sodium hydroxide; In water; at 20 - 25℃; for 1h;pH 6.2-6.4; |
Example 4: Preparations of linezolid form-I from the reaction To a solution of azide of formula II (100 gm), ethanol (800 ml) and water (200 ml), NH4CI (55.8gm) was added at 25-30 C under stirring. Zinc powder (39.0 gm) was charged in one-lot to the reaction mixture, raised the temperature to reflux and stirred for 2 hours. The reaction mixture was cooled to 25-30 C and 23% of aqueous ammonia (200ml) was charged. The reaction mixture was filtered through hyflow bed and washed the bed with water (100 ml) and dichloromethane (100 ml). Dichloromethane (900 ml) and water (300 ml) were charged to the filtrate and stirred for 10-15 minutes. The layers were separated and the aqueous layer was stirred with dichloromethane (2x500ml). The total organic layer was collected and combined with water (500 ml) and subjected for pH adjustment of 1.5-2.0 with 15% aqueous HC1 solution (180-200.0ml) at 25-30C. The resultant two layers were separated and then the aqueous layer was stirred with dichloromethane (2x300 ml). The aqueous layer was separated, treated with ceca carbon (10 gm) at 25-30 C, filtered through hyflow bed and then washed the bed with water (200 ml). The filtrate was cooled to 20-25 C and then adjusted pH 6.5-6.8 by 25% aqueous NaOH solution (5-7 ml). Acetic anhydride (68.4gm) was added in single lot (pH drops to 2.0-3.0) at 20-25 C followed by 25% aqueous NaOH solution (-180.0-190.0 ml) was added to adjust pH 6.2- 6.4 at 20-25 C to the reaction solution and stirred for 1 hour. After the completion of the reaction, the reaction mixture was subjected for pH adjustment to 7.5-8.0 with 25% aqueous solution of sodium hydroxide at 25-30C to extract the reaction mixture into dichloromethane. The organic layer was charcoalized, filtered through hyflow bed and washed with dichloromethane (200 ml). The filtrate was distilled out completely to obtain crude Linezolid at 30 C under vacuum.Water (350 ml) was added to the crude and raised the temperature to 95-100 C. Dimethyl formamide (8.4 ml) was added drop wise to the reaction mixture at 98-100C and maintained for 30 minutes at 100C. The reaction mixture was cooled slowly up to 90C and maintained for 5 hours at the same temperature. The resultant obtained solid was filtered and dried in oven at 75-80 C for 10 hours to afford titled compound.Dried weight: 50 gm.XRPD pattern: Matches with the Fig. 1.Purity by HPLC: 99.9%; Chiral purity: greater than 99.9% and R-isomer: not detected. Residual content: DMF (Below detection limit); ethanol (below detection limit); dichloromethane (below detection limit).Loss on Drying (%w/w):0.36. |
| 62.1 g |
at 20℃; for 0.25h; |
General procedure: To a 2L 3 neck round bottom flask with overhead stirrer was added water (500 ml), 12N hydrochloric acid (40 ml, 480 mmol), methylene chloride (400 ml) and(S)-(E,Z)-5-((4-chlorobenzylideneamino)methyl)-3-(3-fluoro-4-morpholinophenyl)oxazolidin-2-one(12) (100 g, 239 mmol) rinsed inwith methylene chloride (100 ml) The mixture was rapidly stirred at room tempand within 30 minutes a two-phase clear homogenous solution formed. Thereaction was stirred for 1 hour and the orange yellow lower organic layer wasdiscarded. The lemon yellow aqueous layer was washed with additional methylenechloride (250 ml) and the methylene chloride was discarded. Methylene chloride(500 ml) was added to the aqueous layer and the two phase solution wastransferred to a 2 L Erlenmeyer flask, cooled in an ice bath and neutralized toca. pH 7 with ice cold 6 NNaOH (ca. 50 ml) while stirring the reaction in an ice bath. Thereaction changed in color from yellow to colorless and a whiteprecipitateformed that went back into solution with continued strirring. The ice bath wasremovedfrom the reaction flask and acetic anhydride (72 ml, 720 mmol) was added all atonceto the rapidly stirring solution. The mixture was stirred at room temp for 15minutes, cooled in an ice bath and made basic (ca. pH 9) with 6 N sodiumhydroxide (ca. 350 ml). The lower organic layer was separated and the aqueouslayer was extracted with additional methylene chloride (3 x 150 ml). Thecombined organic layers were dried (MgSO4) and evaporated (bath temp 25oC) to a volume of ca. 400 ml. The light yellow solution was slowly added to astirring refluxing solution of ethyl acetate (800 ml) and refluxed down to avolume of 800 ml. Hoy ethyl acetate was added to a volume of 1800 ml and themilky solution was treated with Celite (caution to avoid foaming) filtered andconcentrated by refluxing to a volume of 1100 ml with stirring. Crystalsstarted forming in the refluxing solution at a volume of ca. 1250 ml. The flaskwas stirred at room temp overnight and cooled in an ice bath for 30 minutes beforethe white crystals of 1 werecollected by filtration, air dried and dried in vacuo at room temp (62.1 g, 77%) homogenous by TLC (CH2Cl2:MeOH 9:1, Rf = 0.54; 1HNMR ( 500 MHz, CDCl3) delta 7.44 (dd, 1H, J = 13.8 Hz, J = 2.6 Hz), 7.08 (dd, 1H, J= 8.8Hz, J = 2.6 Hz), 6.93 ( t, 1H, J = 9 Hz), 6.1 (bt, 1H, J = 6.1 Hz), 4.8(m, 1H), 4.0 (t, 1H. J =8.9 Hz), 3.87 (t, 4H, J = 4.5 Hz), 3.75 ( dd, 1H, J =9.1 Hz, 6.8 Hz), 3.70 (ddd, 1H, J = 11.6Hz, J = 5.9 Hz, J = 3.1 Hz), 3.62 (dt,1H, J = 14.7. J = 6.0 Hz), 3.05 (t, 4H, 5 J = 4.5 Hz), 2.02(s, 3H); [alpha]25D-13.6o (c = 1.00, EtOH). Lit.3i [alpha]25D-16o (c = 1.05, EtOH). |
| 2.25 g |
In methanol; dichloromethane; water; |
25 g of (S)-2-((3-(3-fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl methyl) - isoindoline-1, 3- dione (III)was taken into a clean round bottom flask. To this 100 mL ofdemineralized water, 25 mL methanol and 20 g of mono -methylamine were added at 35C and the temperature wasraised to 85C, stirred for 3 h at 85C and washed withmethylene dichloride 3 times. The compound was extractedwith methylene dichloride and to this acetic anhydride wasadded slowly drop-wise over the period of 45 minutes. Thenmethylene dichloride layer was washed 3 times with demineralizedwater (70mL x 3) and the layer was separated.Methylene dichloride layer was dried over Na2SO4 and distilledout completely at 50C and then stripping with 35 mLof methanol at 60C to remove methylene dichloride traces.70 mL of methanol was added and the temperature wasraised to 65C for refluxing it to get a clear solution. To this3 g of activated carbon was added and filtered through thehyflowbed and washed with 5 mL of methanol. The filtratewas cooled to 35C and stirred for 30 minutes at the sametemperature to obtain (S)-N-((3-(3-fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl) methyl) acetamide (or) Linezolid(formula- I) product. The product was filtered and purifiedby recrystallization from methanol to obtain 22.5 g ofpure Linezolid. Yield: 90%. 13C NMR (CDCl3): delta 23.4 (C1), 42.1 (C3), 45.7 (C5), 46.3 (C13, C16), 66.4 (C14, C15), 84.3(C4), 111 (C8), 116.1 (C11), 118.1 (C12), 130.2 (C7, C-N),132.9 (C10), 153 (C6, C=O), 155.9 (C9, C-F); 170.8(C2,C=O); 1H NMR (DMSO) ppm: 9.52 (s, 1H), 7.49-7.53(dd, 1H), 7.484-7.490 (d, 1H), 7.20-7.23 (dd, 1H), 7.04-7.15(t, 1H), 4.85-4.95 (m, 1H), 4.00-4.05 (t, 1H :diastereotopicproton), 3.80-3.82 (q, 1H: diastereotopic proton), 3.72-3.75(t, 4H), 2.95-2.97 (t, 2H), 2.85-2.87 (t, 4H), 2.70-2.83(s,3H). IR (KBr, cm-1): 3393 (N-H stretching), 3014, 2994(aromatic C-H stretching), 2915, 2883, 2821 (aliphatic C-Hstretching), 1749, 1720 (C=O stretching), 1580, 1528 (aromaticC=C stretching), 1455, 1407 (N-H bending), 1300,1323 (aliphatic C-H bending), 1203 (C-N stretching), 1156(C-F stretching), 1124, 1112 (C-O stretching), 1012, 958 (CCstretching). MS: 338 (M++H). |
| 55 g |
In dichloromethane; at 25 - 30℃; for 1h; |
To a mixture of methanol (100 ml), DM water (400 ml) and (S) 2-[3-(3-fluoro-4-morpholin-4-yl-phenyl)-2-oxo-oxazolidin-5-ylmethyl]-isoindole-1,3-dione (100 g, 0.212 moles) a methyl amine solution (47 g) was added at a temperature of 25-30 C. The reaction mixture was stirred and the temperature was slowly raised to 80-85 C. and maintained for 2-3 hours. The reaction mixture was cooled to 25-30 C. and dichloromethane (500 ml) was added. The reaction mixture was stirred for 15 min and the layers were separated. MDC was distilled out completely under atmospheric pressure to get the residual product <strong>[168828-90-8](5S)-5-(amino methyl)-3-[3-fluoro-4-(morpholin-4-yl) phenyl]-1,3-oxazolidin-2-one</strong>. Dichloromethane (400 ml) was added to the residue and acetic anhydride (25 g) was slowly added at a temperature of 25-30 C. over a period of 60 min. After completion, 5% aqueous sodium bicarbonate solution was slowly added to the reaction mixture. After stirring for 15 min the two layers were separated. The dichloromethane layer was washed with DM Water (200 ml). The dichloromethane layer was filtered through hyflo and the solvent was distilled off completely under vacuum below 40 C. Cyclohexane (500 ml) was added to the residue and heated to 45-50 C. The obtained slurry was cooled to 20-25 C. and stirred for 60 min. filtered the solid, washed with cyclohexane (200 ml) and dried the solid at 45-55 C. to furnish pure crystalline N-({(5S)-3-[3-fluoro-4-(morpholin-4-yl)phenyl]-2-oxo-1,3-oxazolidin-5-yl}methyl)acetamide (Linezolid) |
| 83.6 g |
In dichloromethane; for 0.5h; |
Add to a 2000ml four-necked flask(S,E)-5-((benzylimino)methyl)-3-(3-fluoro-4-morpholinyl)oxazolidin-2-one (100.0 g, 260 mmol) andDichloromethane (600ml), stirring to warm up to 30 C,After adding water, add water(600 ml), then concentrated hydrochloric acid (30 wt%, 63.5 g, 520 mmol) was added for hydrolysis, and the reaction was incubated at 30C for 3 hours. After the reaction was completed, the aqueous layer was separated and the aqueous layer was washed with dichloromethane (100 ml * 2).The aqueous phase was added to dichloromethane (400 mL) and the pH was adjusted to 9 with 2M NaOH solution. The phases were separated, the aqueous layer was discarded, and acetic anhydride (39.8 g, 390 mmol) was added dropwise to the organic layer. After 30 minutes from the completion of the addition, the 2M NaOH solution was adjusted to pH 7, the phases were separated, the organic phase was concentrated to 150 ml, isopropanol (200 ml) was added, and the mixture was concentrated under reduced pressure to 150 ml, and isopropyl alcohol (400 ml) was added and the mixture was stirred and cooled. To 0C, drying under vacuum at a temperature of 50C to obtain the target compound linezolid 83.6g, the yield was 95.3% |
|
With triethylamine; In ethyl acetate; at 20℃; for 3h; |
Add 484.8 mg of triethylamine and 1.26 g of acetic anhydride to the ethyl acetate solution of the compound VI, stir at room temperature for 3 h, dilute with ethyl acetate, and wash it with water and saturated brine several times, anhydrous sulfuric acid Sodium is dried, petroleum ether-ethyl acetate is recrystallized,The white solid was 1.12 g, and the total yield of catalytic hydrogenation and acetylation was 83%, mp 178.8 -179.0 C. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping