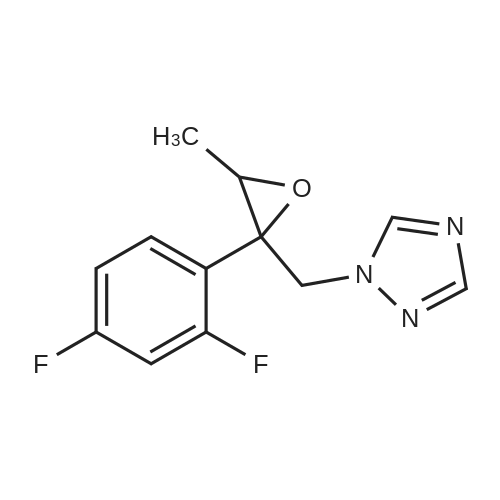
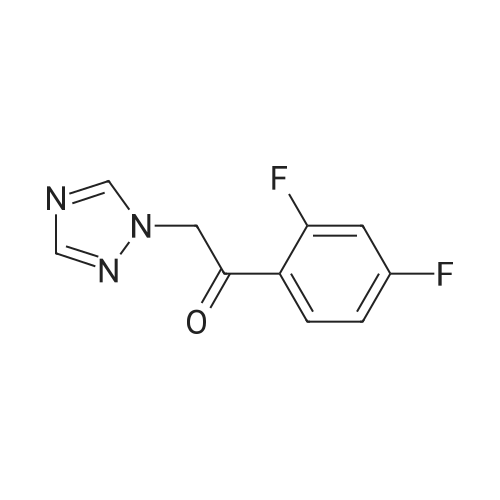
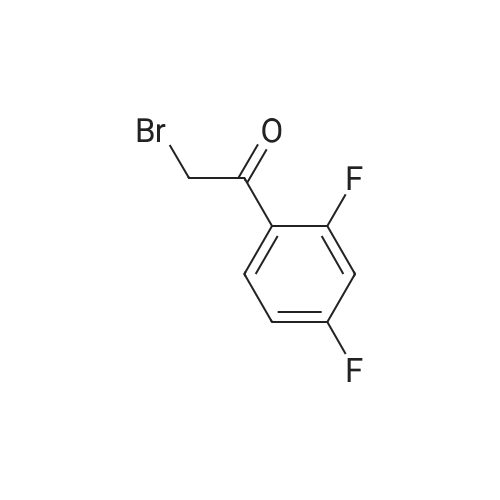
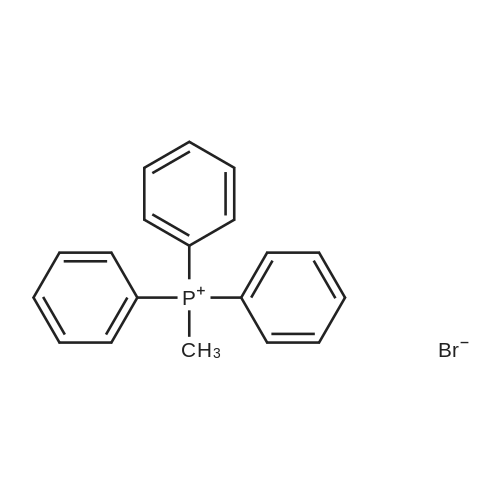
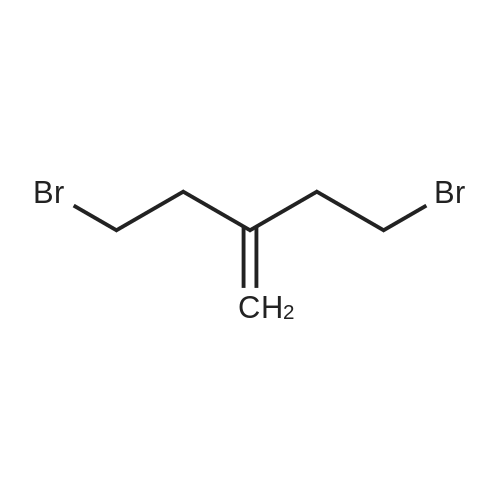
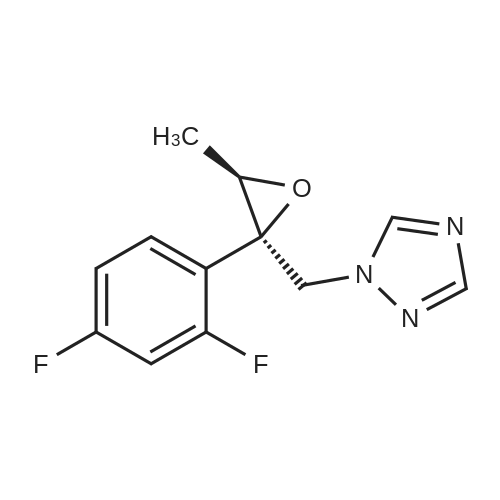
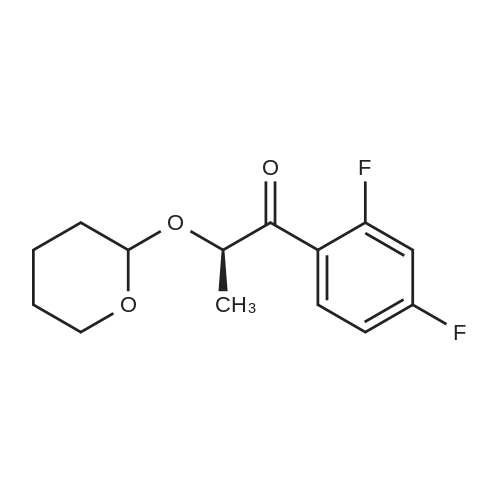
| 84% |
With N-ethyl-N,N-diisopropylamine; magnesium chloride In acetonitrile at 0 - 75℃; for 16h; |
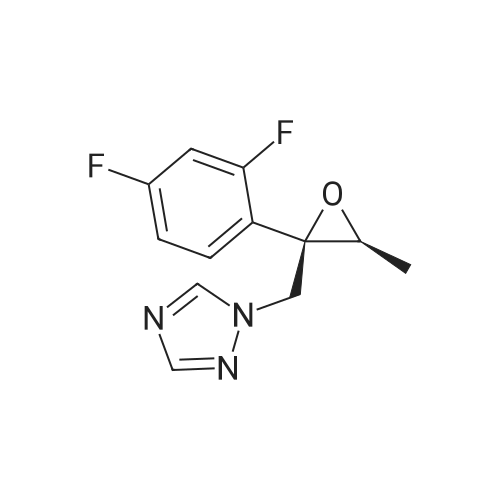
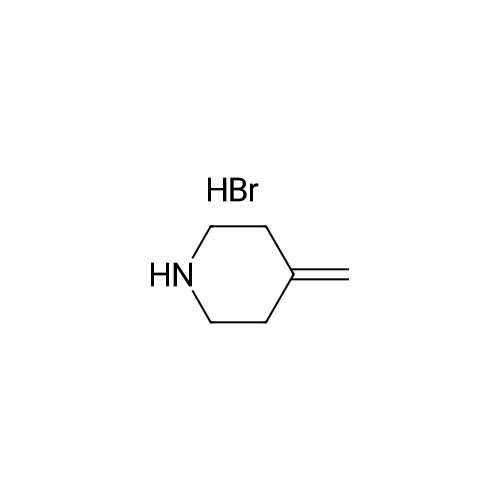
1 Example 1: synthesis of efmaconazole in the presence of methylenepiperidine hydrochloride and N,N-diisopropylethylamine
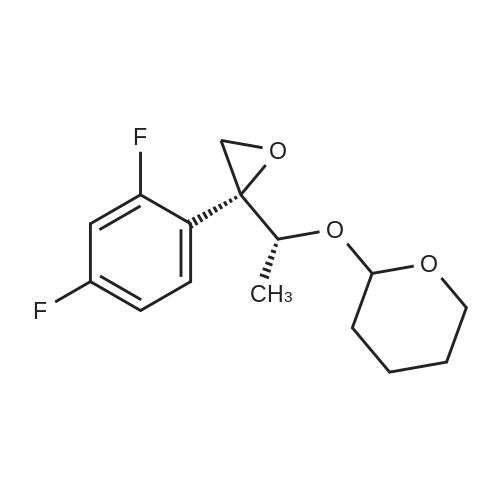
Methylenepiperidine hydrochloride (1 19.27 g, 0.8926 moles) is added to a solution of 1 -[[(2 ,3S)-2-(2,4-difluorophenyl)-3-methyloxiranyl]methyl]- 1 H- 1 ,2,4-triazole (172.5 g, 0.6866 moles) in acetonitrile (690 ml. Diisopropylethylamine (124.2 g, 0.961 moles) is added to the resulting solution. The resulting solution is then cooled to 0-5 °C and anhydrous magnesium chloride (a total of 130.74 g, 1.373 moles) is added in about 4 portions, monitoring the exothermy. The reaction mixture is then heated to 70-75 °C and maintained at that temperature for 16 h. The reaction is then monitored by UPLC. After completion of the reaction, the mixture is concentrated to a small volume and taken up with ethyl acetate. Ethyl acetate (720 ml) is then added to the resulting residue, and water (720 ml) is slowly dropped therein, monitoring the exothermy. After phase separation the organic phase is filtered and concentrated, taking up with ethanol up to about 2 volumes relative to the expected product. Water (335 ml) is dropped into the resulting ethanol solution at room temperature. After the start of precipitation of the product the suspension is cooled to 0-5°C and filtered, washing the panel with a 45:55 mixture of water/ethanol (409 ml). The resulting solid is then dried under vacuum at the temperature of 50°C. The yield obtained from the starting intermediate epoxytriazole (II) is about 84%. |
| 80% |
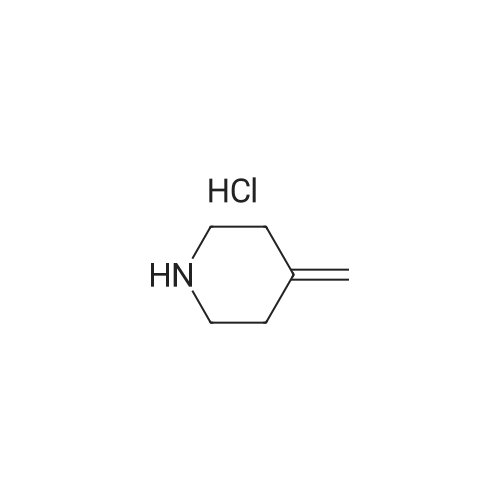
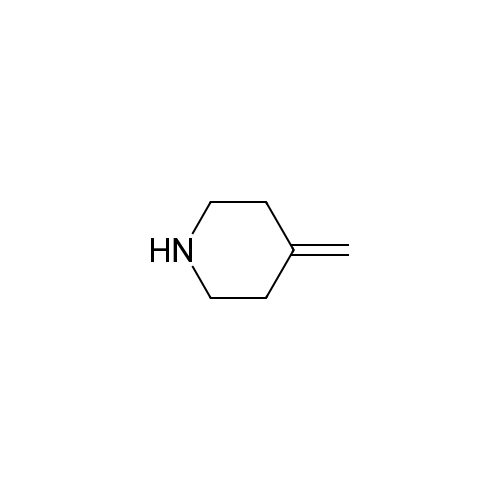
Stage #1: 4-methylenepiperidine monohydrochloride With potassium hydroxide In water at 23 - 28℃; for 2h;
Stage #2: 1-(((2R,3S)-2-(2,4-difluorophenyl)-3-methyloxiran-2-yl)-methyl)-1H-1,2,4-triazole With 1-ethyl-3-methylimidazol-3-ium ethyl sulfate at 100℃; for 6h; |
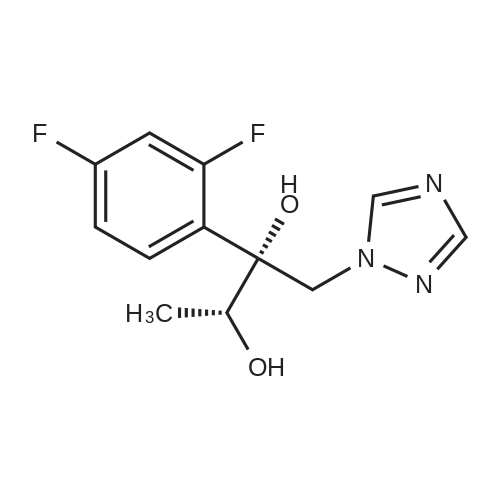
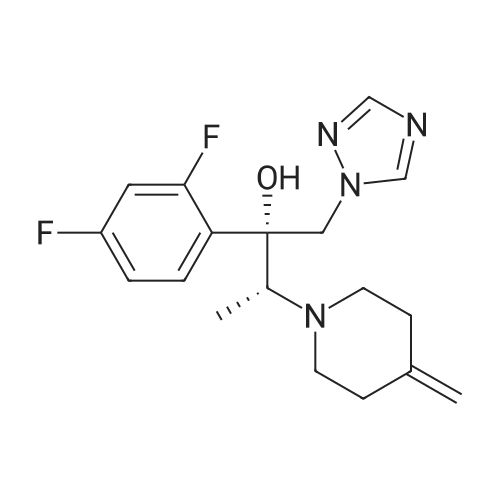
Synthesis of (2R,3R)-2-(2,4-difluorophenyl)-3-(4-methylenepiperidin-1-yl)-1-(1H-1,2,4-triazol-1-yl)butan-2-ol
4-methylenepiperidine hydrochloride42.44 g, potassium hydroxideAfter adding 354.01 ml of 50% aqueous solution,It was stirred at room temperature (23 ~ 28 ) for 2 hours.After adding 638.4 ml of ethyl ether to the reaction solution and stirring for 30 minutes, The layers were separated.After adding 638.4 ml of purified water to the organic layer and stirring for 30 minutes, the organic layer obtained by layer separation was concentrated.To the concentrate, 1-[[(2R,3S)-2-(2,4-difluorophenyl)-3-methyloxiranyl]methyl]-1H-1,2,4-triazole 7.98 g, purified water95.76 ml,Ionic liquidAfter adding 40 ml of 1-ethyl-3-methyl imidazolium ethyl sulfate,The reaction solution was stirred at 100° C. for 6 hours. After confirmation of completion of the reaction, the mixture was cooled to room temperature, 638.4 ml of ethyl acetate and 638.4 ml of purified water were added, stirred, and the layers were separated.After adding 638.4 ml of saturated aqueous sodium chloride solution to the organic layer and stirring for 30 minutes, the organic layer was separated.Anhydrous sodium sulfate was added to the separated organic layer for dehydration, followed by filtration and the organic solvent was concentrated under reduced pressure. The obtained compound was crystallized to give 8.85 g (80%). |
| 80% |
Stage #1: 4-methylenepiperidine monohydrochloride With potassium hydroxide In water at 23 - 28℃; for 2h;
Stage #2: 1-(((2R,3S)-2-(2,4-difluorophenyl)-3-methyloxiran-2-yl)-methyl)-1H-1,2,4-triazole With water; 1-ethyl-3-methylimidazol-3-ium ethyl sulfate at 100℃; for 6h; |
1 Cpd1a: manufacture of ((2R,3R)-2-(2,4-difluorophenyl)-3-(4-methylenepiperidin-1-yl)-1-(1H-1,2,4-triazole- 1-yl) butan-2-ol)
After adding 42.44 g of 4-methylenepiperidine hydrochloride and 354.01 ml of a 50% aqueous potassium hydroxide solution, the mixture was stirred at room temperature (23-28° C.) for 2 hours. After adding 638.4 ml of ethyl ether to the reaction solution and stirring for 30 minutes, the layers were separated. After adding 638.4 ml of purified water to the organic layer and stirring for 30 minutes, the organic layer obtained by layer separation was concentrated.In the concentrate, 1-[[(2R,3S)-2-(2,4-difluorophenyl)-3-methyloxiranyl]methyl]-1H-1,2,4-triazole 7.98g, purified water 95.76ml , After adding 40 ml of 1-ethyl-3-methyl imidazolium ethyl sulfate as an ionic liquid, the reaction solution was stirred at 100° C. for 6 hours. After confirming the completion of the reaction, it was cooled to room temperature, 638.4 ml of ethyl acetate and 638.4 ml of purified water were added, stirred, and the layers were separated. After adding 638.4 ml of saturated sodium chloride aqueous solution to the organic layer and stirring for 30 minutes, the organic layer was separated. Anhydrous sodium sulfate was added to the separated organic layer for dehydration, filtered, and the organic solvent was concentrated under reduced pressure. The HPLC purity of the obtained cpd1 was 86%, and it was crystallized to obtain 8.85 g (80%). |
|
With potassium hydroxide; sodium chloride In ethanol; hexane; ethyl acetate |
1 (2R,3R)-2-(2,4-difluorophenyl)-3-(4-methylenepiperdine-1-yl)-1-(1H-1,2,4-triazole-1-yl)butane-2-ol
EXAMPLE 1 (2R,3R)-2-(2,4-difluorophenyl)-3-(4-methylenepiperdine-1-yl)-1-(1H-1,2,4-triazole-1-yl)butane-2-ol There was added 11.2 ml of 50% aqueous solution of potassium hydroxide to 1.336 g of 4-methylenepiperidine hydrochloride and, after dissolved under stirring, the resulting solution was extracted with 20 ml of ethyl ether. Then the aqueous phase was further extracted with 10 ml of ethyl ether, and the organic phases were combined and ethyl ether was removed therefrom. To the residue there were added 3 ml of ethanol, 251 mg of (2R,3S)-2-(2,4-difluorophenyl)-3-methyl-2-[(1H-1,2,4-triazole-1-yl)methyl]oxirane and 3 ml of distilled water in order, and the mixture was refluxed with heating for 24 hours in the oil bath at 85° C. After the reaction, the reaction solution was cooled to room temperature, and thereto were added 20 ml of ethyl acetate and 20 ml of distilled water, and the organic phase was separated. The aqueous phase was further extracted with 10 ml of ethyl acetate, and the organic phase was combined with the above-separated organic phase, and the mixture was washed with a saturated aqueous solution of sodium chloride, and dried over anhydrous magnesium sulfate and then the solvent was removed. The residue was subjected to HPLC using 8 g of silica gel and was eluted with a mixed solvent of ethyl acetate/hexane (4:1 to 3:1) to obtain 188 mg of the titled compound. Yield: 54.0%. Upon recrystallization from a mixed solvent of ether/hexane, a pure product having a melting point of 86°-87° C. was obtained. HPLC: The analysis was carried out using hexane/isopropyl alcohol of 9/1 as a mobile phase, at a flow rate of 1.0 ml/min at room temperature under the conditions capable of detecting with UV (254 nm), and then a single peak appeared at a retention time of 6.6 minutes. |
|
Stage #1: 4-methylenepiperidine monohydrochloride With water; lithium carbonate; lithium bromide In ethanol at 0.25 - 30℃; for 72h; Reflux;
Stage #2: 1-(((2R,3S)-2-(2,4-difluorophenyl)-3-methyloxiran-2-yl)-methyl)-1H-1,2,4-triazole In ethanol for 72h; Reflux; |
1 Example 1: Preparation of (2R,3R)-2-(2,4-Difluorophenyl)-3-(4-methylene- 1-piperidinyl)- 1 -(1-1 ,2,4-triazol- 1 -yl)-2-butanol
To a suspension of 4-methylene-piperidine hydrochloride (1.59gm) in ethanol(2.5m1), lithiumcarbonate( 1.47gm) was added at about 25-30°C. To this reaction mixture aqueous lithiumbromide(0.84gm) was added and the reaction mass was stined. To this 1-[2-(2,4- dimethylphenyl)oxiran-2-yl]methyl}-1H-1,2,4-triazole(0.5gm) was added and heated to reflux for about 72 hrs. On completion, the reaction mass was concentrated under vacuum and the residue was taken up in a mixture of ethyl acetate and water. The layers were separated. Theethyl acetate layer was concentrated under vacuum. The residue was stined in n-heptane (1.5m1) and at about 5-10°C. The precipitated solid was filtered and dried under vacuum to obtain (2R,3R)-2-(2,4-Difluorophenyl)-3-(4-methylene- 1 -piperidinyl)- 1 -( 1H- 1 ,2,4-triazol- 1 -yl)-2-butanol. |
|
Stage #1: 4-methylenepiperidine monohydrochloride With sodium hydroxide In dichloromethane at 0 - 5℃; for 1h;
Stage #2: 1-(((2R,3S)-2-(2,4-difluorophenyl)-3-methyloxiran-2-yl)-methyl)-1H-1,2,4-triazole With lithium bromide In acetonitrile at 20 - 100℃; |
2 Example-2: Preparation of Efinaconazole
Example-2: Preparation of Efinaconazole Stage-01: (0092) MDC solvent (5 vol) was taken in a clean and dry RBF and added the 4-Methylene piperidine hydrochloride (150 gm) and cooled to 0-5° C., 48% NaOH solution (2 eq) was charged and stirred for 1 hr. The R.M. was quenched with DM water and separated the MDC layer and distilled out completely to get the 4-Methylenepiperidine (100 gm). Stage-02: (0093) 4-Methylenepiperidine (100 gm), triazole compound of Formula III (100 gm) and 3 Volume of acetonitrile solvent (ACN) were taken in to a clean and dry R.B.F and added the Lithium Bromide (70 gm) at RT stirred for 30 min and at RT. (0094) The R.M. temp was raised up to 95-100° C. and stirred for 18-20 hrs and the R.M. was cooled to RT and poured in to IPA+DM water mixture to get the crude Efinaconazole compound. (0095) This crude Efinaconazole compound is purified with ethyl acetate and silica gel to get the pure Efinaconazole compound |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping