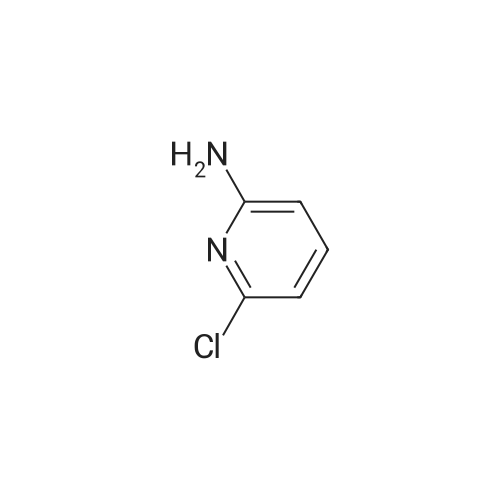
| 88% |
With sodium hexamethyldisilazane; In tetrahydrofuran; at 0 - 20℃; for 0.5h; |
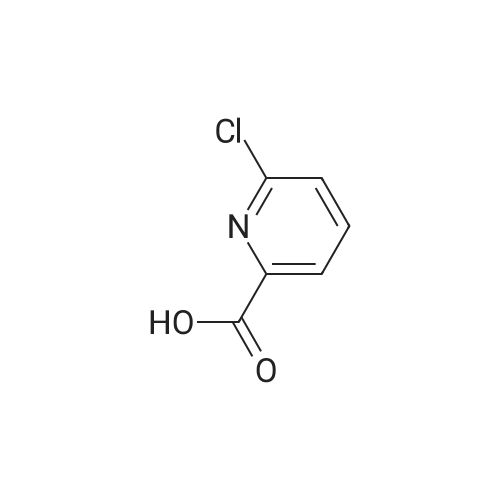
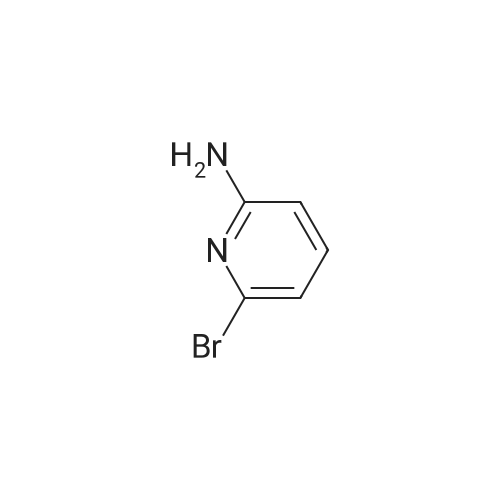
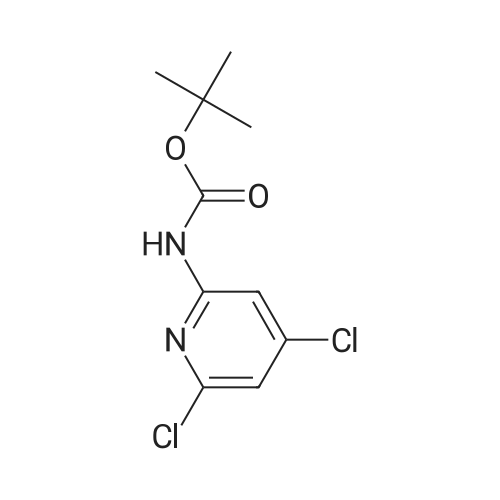
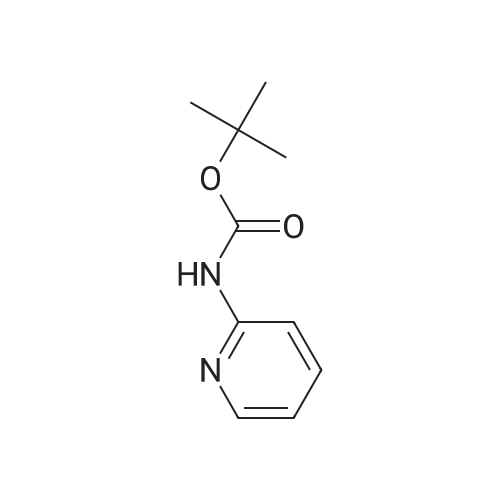
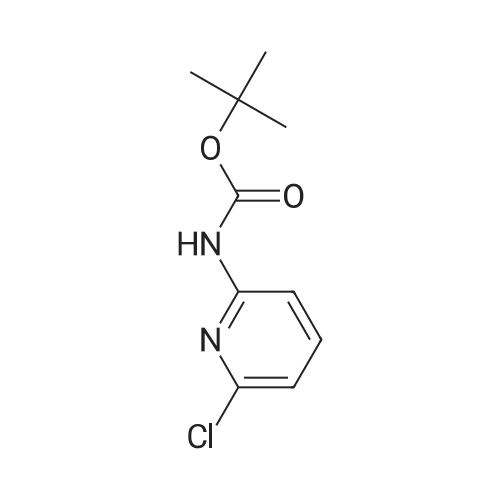
Example 11A tert-Butyl (6-chloropyridin-2-yl)carbamate Under argon, 150 ml of THF were added to 23.4 g (181.8 mmol) of 2-chloro-5-aminopyridine, and the mixture was cooled to 0 C. 73.3 g (400 mmol) of sodium bis(trimethylsilyl)amide and 43.65 g (200 mmol) of di-tert-butyl dicarbonate, dissolved in 150 ml of THF, were added dropwise. After 15 min, the cooling bath was removed, and stirring was continued at RT for 15 min. The THF was removed using a rotary evaporator, ethyl acetate and 0.5 N hydrochloric acid were added to the residue and the mixture was extracted. The organic phase was separated off, dried over magnesium sulphate and concentrated using a rotary evaporator. The reaction mixture was chromatographed on silica gel (mobile phase dichloromethane/methanol 100%?100:3). This gave 36.54 g (88% of theory) of the product as a solid. LCMS (method 3): Rt=2.41 min. (m/z=175 (M+H)+). 1H-NMR (400 MHz, DMSO-d6): delta=10.11 (s, 1H), 7.78 (d, 2H), 7.1 (t, 1H), 1.47 (s, 9H). |
| 88% |
With sodium hexamethyldisilazane; In tetrahydrofuran; at 0 - 20℃;Inert atmosphere; |
Example 105Atert-Butyl (6-chloropyridin-2-yl)carbamate 23.4 g (181.8 mmol) of 2-chloro-5-aminopyridine are mixed with 150 ml of THF under argon and cooled to 0 C. 73.3 g (400 mmol) of bis(trimethylsilyl)sodium amide and 43.65 g (200 mmol) of di-tert-butyl dicarbonate, dissolved in 150 ml of THF, are added dropwise. After 15 min, the cooling bath is removed and stirring is continued at RT for 15 min. The THF is removed in a rotary evaporator, and the residue is mixed and extracted with ethyl acetate and 0.5 N hydrochloric acid. The organic phase is separated off, dried over magnesium sulphate and concentrated in a rotary evaporator. The reaction mixture is chromatographed on silica gel (mobile phase dichloromethane/methanol 100%?100:3). 36.54 g (88% of theory) of the product are obtained as a solid.LCMS (method 3): Rt=2.41 min. (m/z=175 (M+H)+).1H-NMR (400 MHz, DMSO-d6): d=10.11 (s, 1H), 7.78 (d, 2H), 7.1 (t, 1H), 1.47 (s, 9H). |
| 88% |
With sodium hexamethyldisilazane; In tetrahydrofuran; at 0 - 20℃; for 0.5h;Inert atmosphere; |
Example 3Atert-Butyl(6-chloropyridin-2-yl)carbamate; Under argon, 150 ml of THF were added to 23.4 g (181.8 mmol) of 2-chloro-5-aminopyridine, and the mixture was cooled to 0 C. 73.3 g (400 mmol) of sodium bis(trimethylsilyl)amide and 43.65 g (200 mmol) of di-tert-butyl dicarbonate, dissolved in 150 ml of THF, were added dropwise. After 15 min, the cooling bath was removed and stirring was continued at RT for 15 min. The THF was removed on a rotary evaporator, ethyl acetate and 0.5N hydrochloric acid were added to the residue and the mixture was extracted. The organic phase was separated off, dried over magnesium sulfate and concentrated on a rotary evaporator. The reaction mixture was chromatographed on silica gel (mobile phase dichloromethane/methanol 100%?100:3). This gave 36.54 g (88% of theory) of the product as a solid.LCMS (Method 3): Rt=2.41 min. (m/z=175 (M+H)+).1H-NMR (400 MHz, DMSO-d6): delta=10.11 (s, 1H), 7.78 (d, 2H), 7.1 (t, 1H), 1.47 (s, 9H). |
| 88% |
With sodium hexamethyldisilazane; In tetrahydrofuran; at 0 - 20℃; for 0.5h;Inert atmosphere; |
Example 1A tert-Butyl (6-chloropyridin-2-yl)carbamate 23.4 g (181.8 mmol) of 2-chloro-5-aminopyridine were admixed with 150 ml of THF under argon and cooled to 0 C. 73.3 g (400 mmol) of sodium bis(trimethylsilyl)amide and 43.65 g (200 mmol) of di-tert-butyl dicarbonate, dissolved in 150 ml of THF, were added dropwise. After 15 min, the cooling bath was removed and the mixture was stirred at RT for a further 15 min. The THF was removed by rotary evaporation, and the residue was admixed and extracted with ethyl acetate and 0.5 N hydrochloric acid. The organic phase was removed, dried over magnesium sulphate and concentrated on a rotary evaporator. The reaction mixture was chromatographed on silica gel (eluent: dichloromethane/methanol 100%?100:3). 36.54 g (88% of theory) of the product were obtained in solid form. LCMS (method 3): Rt=2.41 min (m/z=175 (M+H)+). 1H NMR (400 MHz, DMSO-d6): delta=10.11 (s, 1H), 7.78 (d, 2H), 7.1 (t, 1H), 1.47 (s, 9H). |
| 87.4% |
|
Sodium hexamethyl disilazane (63.03 g, 343 mmol) was added portion-wise to a stirred solution of <strong>[45644-21-1]2-chloro-6-aminopyridine</strong> (20.0 g, 156.25 mmol) in THF (100 mL) at 0 C. under argon. After 5 minutes of stirring at the same temperature, di-tertiary-butyl dicarbonate (36.77 mL, 171 mmol) was added drop-wise into the reaction mixture. After 15 minutes of additional stirring at the same temperature, the reaction temperature was brought to room temperature and stirred until starting material was consumed completely (1 hour, monitored by silica gel TLC using ethyl acetate-hexanes, 1:9 as mobile phase). THF was distilled off under reduced pressure, the obtained residue was taken up in ethyl acetate (300 mL), washed with an aqueous solution of 0.5 M HCl (100 mL), water (2*75 mL), dried over anhydrous sodium sulfate, filtered and concentrated on the rotary evaporator to obtain a gummy mass, which was purified over silica gel column chromatography (eluant:EtOAc-hexanes, 1:19) to give (6-chloro-pyridin-2-yl)-carbamic acid tert-butyl ester as a white solid. (Yield 31.0 g, 87.4%). |
| 63.4% |
|
NaHMDS (351 ml, 0.7mol) in THF (300 ml) was cooled to 0 C, a solution of 2-amino-6- chloropyridine (107; 40 g, 0.3 1 1 mol) in THF (300 ml) was added, followed by a solution of di- tert-butyl dicarbonate(68 g, 0.3 1 1 mol) in THF, ensuring the internal temperature remained below 0 C. The resulting reaction mixture was aged for 1 h at room temp and then careful ly acidified to pH 3 by addition of 1 M hydrochloric acid, extracted with EtOAc, the combined organic layers were then washed sequentially with saturated aqueous NaHCCh and brine, dried over Na2S04, filtered, concentrated to afford crude product, Triturated with ether to afforded desired product tert-butyl 6-chloropyridin-2-ylcarbamate (108; 45g, yield 63.4%). M S (ESI) calcd for C,oH,3CIN202 (m/z) 228.69. |
| 56% |
With lithium hexamethyldisilazane; In tetrahydrofuran; at 20℃; |
Drop in a solution of <strong>[45644-21-1]2-amino-6-chloropyridine</strong> (40.0 g, 311 mmol) and bistrimethylsilylamide lithium (685 mL, 685 mmol, 1 M in tetrahydrofuran) in tetrahydrofuran (400 mL) Di-tert-butyl dicarbonate (74.7 g, 342 mmol) was added. The reaction system was stirred at room temperature overnight, then concentrated, diluted with ethyl acetate (400 mL), and the organic phase was washed with hydrochloric acid (1M), saturated aqueous sodium hydrogen carbonate and brine, The sodium was dried, filtered and concentrated, and the obtained residue was recrystallized from ethanol and filtered. The filter cake was dried to give compound 1.1 (39.5 g, yield: 56%) as a yellow solid. |
|
With hydrogenchloride; In dichloromethane; water; |
a) 2-tert-Butoxycarbonylamino-6-chloropyridine. A solution of 4.36 g (0.02 mol) of di-tert-butyl dicarbonate in 10 ml of methylene chloride is added dropwise to a solution of 2.57 g (0.02 mol) of <strong>[45644-21-1]2-amino-6-chloropyridine</strong> in 25 ml of methylene chloride. It is left at ambient temperature overnight, washed with a 1N solution of HCl and with water, and is dried over sodium sulphate and concentrated in vacuum to obtain 4.0 g of the compound referred to above, as a yellow oil. |
|
With sodium hexamethyldisilazane; In tetrahydrofuran; for 24h; |
To a solution of <strong>[45644-21-1]2-amino-6-chloropyridine</strong> (5.24 g, 40.8 mmol) and sodium hexamethyldisilazide (1.0 M, 89.8 mL, 89.8 mmol) in TEtaF (35 mL) was added a solution of di-tert-butyl dicarbonate (9.80 g, 44.9 mmol) in TEtaF (35 mL). After 24 h the reaction was concentrated and the residue was partitioned between EtOAc (30 mL) and IN HCl (100 mL). The aqueous layer was extracted further with EtOAc (2x). The combined organic layers were washed with NaEtaCOs, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel chromatography, eluting with a gradient of 20 to 100% dichloromethane: hexane to give the title compound (7.73 g). MS: m/z = 173.0 (M - 1Bu). |
|
With sodium hexamethyldisilazane; In tetrahydrofuran; for 24h; |
INTERMEDIATE 2; Spirorpiperidine-4.4'-pyridor2.3-6?ri.31oxazin1-2'd'H)-one; Step A. tot-Butyl (6-chloropyridin-2-yl)carbamate; To a solution of <strong>[45644-21-1]2-amino-6-chloropyridine</strong> (5.24 g, 40.8 mmol) and sodium hexamethyldisilazide (1.0 M, 89.8 mL, 89.8 mmol) in TEtaF (35 mL) was added a solution of di-tert-butyl dicarbonate (9.80 g, 44.9 mmol) in TEtaF (35 mL). After 24 h the reaction was concentrated and the residue was partitioned between EtOAc (30 mL) and IN HCl (100 mL). The aqueous layer was extracted further with EtOAc (2x). The combined organic layers were washed with NaHCO3, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel EPO <DP n="56"/>chromatography, eluting with a gradient of 20 to 100% dichloromethane: hexane to give the title compound (7.73 g). MS: mlz = 173.0 (M - 'Bu). |
|
With sodium hexamethyldisilazane; In tetrahydrofuran; for 48h; |
INTERMEDIATE 12 EPO <DP n="80"/> Spirorpirhoeridine-4.4'-pyrido|"2.3-Jiri31oxazin]-2'(rH)-one Step A. tert-Butyl f-chloropyridin^-yPcarbamate; To a solution of <strong>[45644-21-1]2-amino-6-chloropyridine</strong> (25.0 g, 194.5 mmol) and sodium hexamethyldisilazide (1.0 M; 427.8 mL, 427.8 mmol) in tetrahydrofuran (175 mL) was added a solution of di-tert-butyl dicarbonate (46.69 g, 213.9 mmol) in tetrahydrofuran (175 mL). After 48 h, the reaction mixture was concentrated and the residue was partitioned between ethyl acetate (150 mL) and IN HCl (500 mL). The aqueous layer was extracted further with ethyl acetate (2x). The combined organic layers were washed with saturated aqueous sodium bicarbonate, dried over magnesium sulfate, filtered, and concentrated. Recrystalization was accomplished by dissolving the crude residue in a minimal amount of ethanol at 60 0C. The solution was allowed to cool to ambient temperature and water was added. Precipitated solid was filtered and dried to give the title compound (33.45 g). MS: mlz = 173.0 (M - 1Bu). |
|
With sodium hexamethyldisilazane; In tetrahydrofuran; for 24h; |
To a solution of <strong>[45644-21-1]2-amino-6-chloropyridine</strong> (5.24 g, 40.8 mmol) and sodium hexamethyldisilazide (1.0 M, 89.8 mL, 89.8 mmol) in TEtaF (35 mL) was added a solution of di-tert-butyl dicarbonate (9.80 g, 44.9 mmol) in TEtaF (35 mL). After 24 h the reaction was concentrated and the residue was partitioned between EtOAc (30 mL) and IN HCl (100 mL). The aqueous layer was extracted further with EtOAc (2x). The combined organic layers were washed with NaHCO3, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel chromatography, eluting with a gradient of 20 to 100% dichloromethane: hexane to give the title compound (7.73 g). MS: mlz = 173.0 (M - 1Bu). |
|
In tert-butyl methyl ether; at 35℃; for 6h;Green chemistry; |
Starting with <strong>[45644-21-1]6-chloro-2-aminopyridine</strong> in methyl tert-butyl etherDi-tert-butyl dicarbonate is subjected to a condensation reaction in a stirred state,<strong>[45644-21-1]6-chloro-2-aminopyridine</strong>,Methyl tert-butyl ether and di-tert-butyl dicarbonate in a molar ratio of 1: 20:The condensation reaction temperature was 35 C,The condensation reaction time was 6 h,TLC plate to determine the reaction is completed,The reaction solution was distilled to dryness,To give 6-chloro-2-tert-butoxycarbonylaminopyridine, 6-chloro-2-tert-butoxycarbonylaminopyridine was added to a sodium hydroxide solution having a mass percentage of 4.2%Tetrabutylammonium bromide,P-xylene and water, and the hydrolysis reaction is carried out in a stirred state,6-chloro-2-tert-butoxycarbonylaminopyridine,The concentration of 4.2% sodium hydroxide solution,Tetrabutylammonium bromide,The molar ratio of p-xylene to water is 1: 1: 0.05: 0.06: 25: 50,The temperature of the hydrolysis reaction was 90 C,The hydrolysis time was 8 h,TLC plate to determine the reaction is completed,Down to room temperature,Adding methylene chloride and water,The organic phase is separated,First with water,And then washed with salt,And then dried with magnesium sulfate,Steamed to dry,The residue was purified by recrystallization from a methanol-isopropanol mixed solvent,To give 6-hydroxy-2-tert-butoxycarbonylaminopyridine as an off-white solid,Yield 88.7%The reaction of this step is the same as in Example 1; |
|
With sodium hexamethyldisilazane; In tetrahydrofuran; at 20℃; for 20h; |
<strong>[45644-21-1]2-amino-6-chloropyridine</strong> and sodium bis(trimethylsilyl)amide are dissolved in tetrahydrofuran. A solution of di-tert-butyl dicarbonate in tetrahydrofuran is added dropwise. The reaction mixture is stirred at room temperature for twenty hours. Water and ethyl acetate are added, and the product is extracted with ethyl acetate. The organic phases are combined, washed once with water and once with a saturated aqueous solution of sodium chloride, then dried over magnesium sulphate, filtered and concentrated under vacuum. (6-Chloro-pyridin-2-yl)-tert-butyl carbamate is obtained in the form of a brown solid. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping