* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of the American Chemical Society, 2010, vol. 132, # 50, p. 17933 - 17944
2
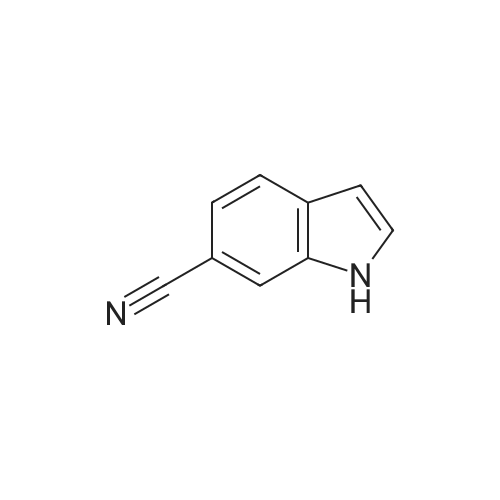
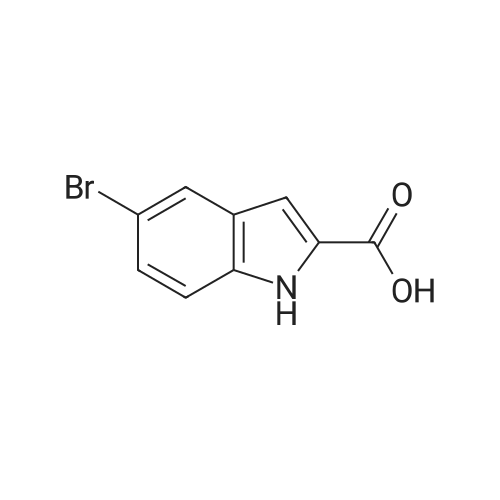
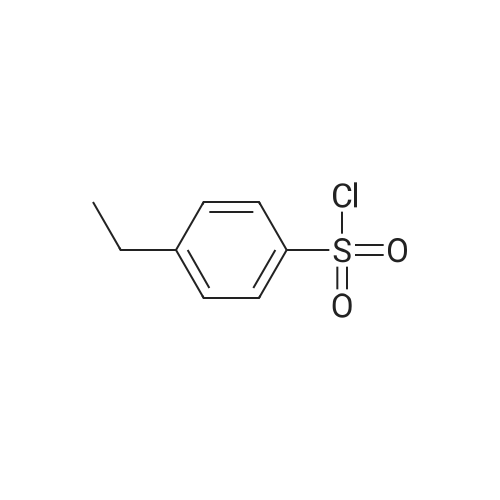
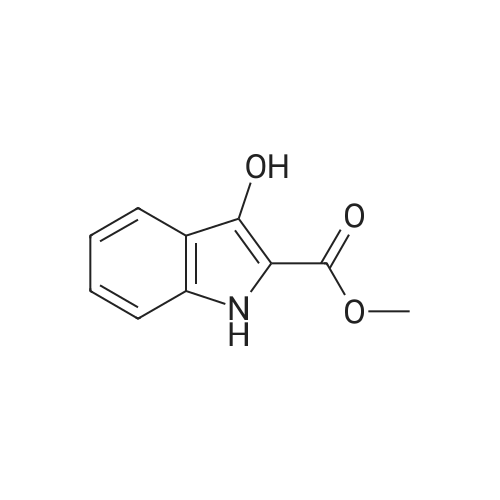
[ 15861-24-2 ]
[ 1670-81-1 ]
Reference:
[1] Journal of Organic Chemistry, 1955, vol. 20, p. 1458
[2] Chinese Chemical Letters, 2010, vol. 21, # 12, p. 1407 - 1410
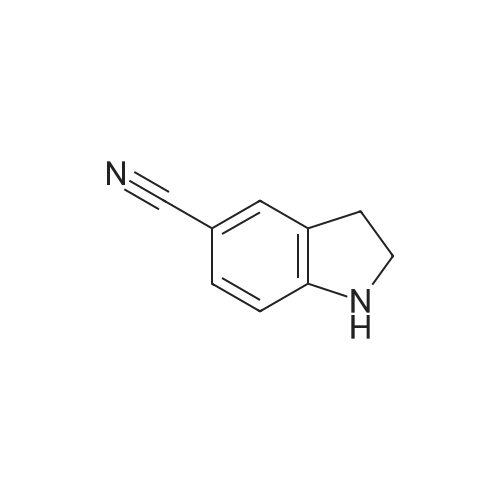
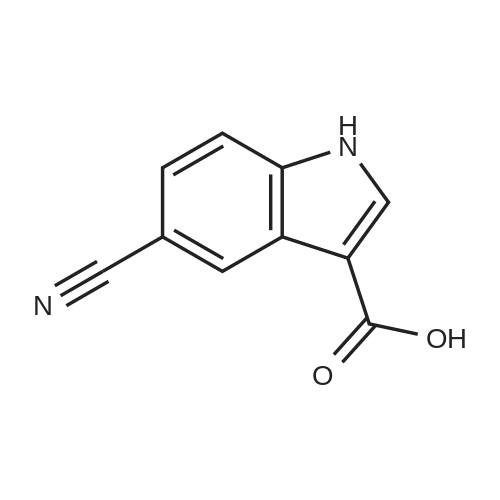
3
[ 15861-24-2 ]
[ 15861-23-1 ]
Yield
Reaction Conditions
Operation in experiment
77%
With triethylsilane In trifluoroacetic acid at 0℃; for 4 h;
A solution of 5-cyanoindole (1 g, 7 mmol) in 10 mL TFA was cooled to 0° C. and then triethylsilane (1.6 g, 2 eq.) was added. The reaction mixture was stirred at 0° C. for 4 h then diluted with EtOAc and washed with 1M HCl solution. The aqueous layers were combined and neutralized with 50percent NaOH to pH10 then extracted 3.x. with EtOAc. These latter extracts were combined, washed with brine, dried and evaporated to yield the indoline (77percent).
67%
With boron trifluoride diethyl etherate; sodium cyanoborohydride In methanol at 25℃;
Dissolve 5-cyanoindole (142 mg, 1 mmol) and sodium cyanoborohydride (63 mg, 1 mmol) in methanol (2 ml). Boron trifluoride etherate (152 drops, 1.2 mmol) was added dropwise and allowed to react at room temperature for 2-3 hours. After quenching, add 25percent ammonia to quench. The reaction was followed by extraction with ethyl acetate (10 mL x 3 times). The organic layers were combined, dried over anhydrous magnesium sulfate, concentrated and subjected to column chromatography (oilEther: ethyl acetate = 10:1) was isolated to give 97 mg of the desired product in 67percent yield.
56%
at 0 - 20℃; for 2 h;
Triethylsilane (2.0 eq.) was slowly added to a solution of indole (1 .0 eq.) in trifluoroacetic acid (1 .4 M), cooled to 0 °C. The reaction mixture was stirred at 0 °C for 1 h, then at r.t. for 1 h. Upon completion (monitored by TLC), the reaction was basified to pH 1 1 with NaOH (5.0 M) and extracted with EtOAc (x3). The organic layers were combined, washed with brine, dried over Na2S04 and concentrated under reduced pressure. The crude product was purified by flash chromatography to give the desired indoline. Following general procedure M, 5-cyanoindole (600.0 mg, 4.22 mmol) afforded indoline-5-carbonitrile (340.0 mg, 2.36 mmol, 56percent yield) as a light yellow solid. UPLC-MS (ES+, Short acidic): 1 .30 min, m/z 289.0 [2M+H]+.
40%
With sodium cyanoborohydride In acetic acid at 20℃;
A solution of 5-cyanoindole (3 g, 0.021 mol) in glacial acetic acid (25 mL) was treated portionwise with sodium cyanoborohydride (4 g, 0.063 mol) over 20-30 min then the solution was stirred overnight at rt under N2. The reaction was quenched by addition of water, and most of acetic acid was removed in vacuo. The residue was diluted with water and adjusted to pH>8 with 1M NaOH then extracted 3.x. with ethyl acetate. Extracts were combined and back extracted 2.x. with 1M HCl then set aside. (Starting indole can be recovered from these initial EtOAc extracts if desired.) Aqueous acid extracts were combined and rebasified with 5N NaOH, and then re-extracted with EtOAc. The latter extracts were combined, washed with brine, dried over anh. Na2SO4, filtered and evaporated to provide the indoline product (1.22 g, 40percent) as an off-white crystalline solid. 1H NMR (CDCl3, 300 MHz) δ 7.29-7.31 (m, 2H), 6.54 (d, 1H, J=8.4 Hz), 4.20 (bs, 1H), 3.67 (t, 2H, J=8.6 Hz), 3.06 (t, 2H, J=8.6 Hz)
33%
Stage #1: at 0 - 20℃; for 5 h; Stage #2: With sodium hydroxide In water
Reference Example 7 2,3-dihydro-1H-indole-5-carbonitrile; To a solution (5 mL) cooled to 0°C in an ice bath of 1H-indole-5-carbonitrile (640 mg, 4.50 mmol) in acetic acid was added sodium cyanotrihydroborate (848 mg, 13.5 mmol). The reaction mixture was stirred at room temperature for 5 hr and diluted with water. The mixture was basified with 8N aqueous sodium hydroxide solution and extracted with ethyl acetate. The organic layer was extracted with 1N hydrochloric acid and the aqueous layer was again basified with 8N aqueous sodium hydroxide solution and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate and filtered. The filtrate was concentrated. The residue was crystallization from ethyl acetate to give the title compound as pale-yellow crystals (213 mg, yield 33percent). 1H-NMR (300 MHz, CDCl3)δ:3.06 (t, J = 8.5 Hz, 2 H), 3.67 (t, J = 8.7 Hz, 2 H), 3.83 (br s, 1 H), 6.45 - 6.59 (m, 1 H), 7.24 - 7.33 (m, 2 H).
Reference:
[1] Patent: US2004/220206, 2004, A1, . Location in patent: Page 25-26
[2] Journal of Materials Chemistry A, 2015, vol. 3, # 34, p. 17704 - 17712
[3] Patent: CN105085504, 2018, B, . Location in patent: Paragraph 0139; 0147-0150
[4] Patent: WO2014/188173, 2014, A1, . Location in patent: Paragraph 00239; 00521
[5] Patent: US2004/220206, 2004, A1, . Location in patent: Page 25
[6] Patent: EP2399914, 2011, A1, . Location in patent: Page/Page column 92
[7] Patent: US2012/252853, 2012, A1, . Location in patent: Page/Page column 22
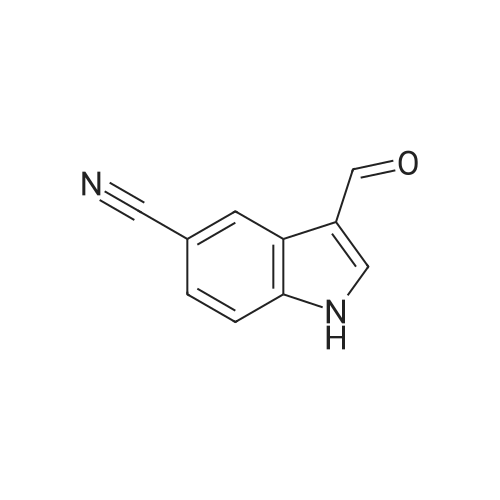
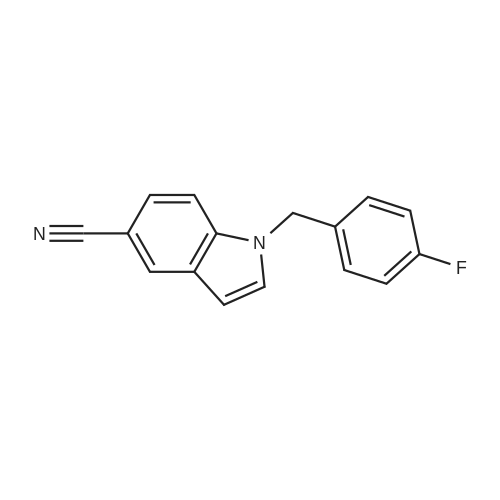
4
[ 15861-24-2 ]
[ 17380-18-6 ]
Yield
Reaction Conditions
Operation in experiment
84%
With trichlorophosphate In <i>N</i>-methyl-acetamide; water
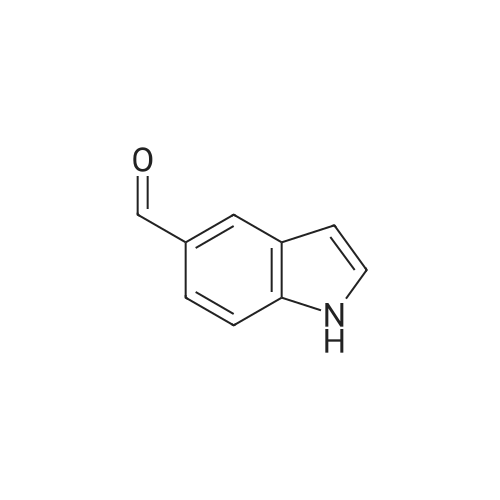
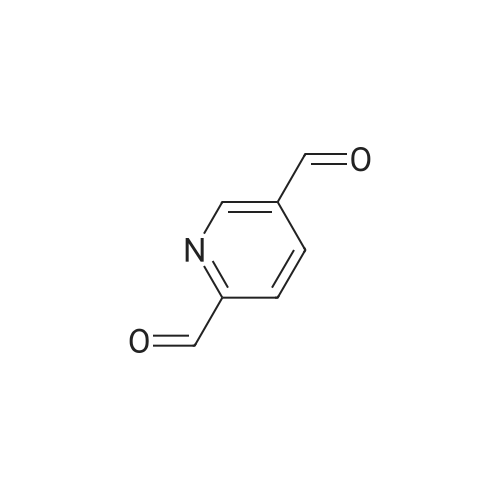
Preparation 35 3-Formyl-1 H -indole-5-carbonitrile Phosphoryl chloride (4.24ml, 45.48mmol) was added dropwise to dimethylformamide (3.52ml, 45.48mmol) and stirred for 30mins at room temperature. A solution of 1H-indole-5-carbonitrile (5.39g, 37.9mmol) in dimethylformamide (10ml) was added dropwise. A solid precipitated, further dimethylformamide (10ml) was added to aid stirring and the reaction mixture was then stirred at room temperature for 3hr. Water was added to quench the reaction mixture which was then stirred for 18hr. The stirring was stopped and the reaction mixture was left to stand, after 24hr a pink solid had precipitated in the organic layer. The layers were separated and organic layer filtered, washed with water and dried to give the desired product (5.44g, 84percent). 1H NMR (DMSO)δ 7.60-7.80 (m, 2H), 8.20-8.30 (m, 2H), 10.00 (s, 1H), 12.20-12.35 (s, br, 1H).
75%
With sodium hydroxide; trichlorophosphate In <i>N</i>-methyl-acetamide
EXAMPLE 1 Trans-2-[5-Cyanoindol-3-yl]-1-(N,N-dimethylaminomethyl)cyclopropane Phosphorus oxychloride (10.9 ml, 117 mmol) was added dropwise over 30 min to anhydrous dimethylformamide (50 ml) that was maintained at 10-20° C. (internal temperature). The resulting mixture was stirred for 30 min and then chilled to 0° C. A solution of commercially available 5-cyanoindole (15 g, 106 mmol) in anhydrous dimethylformamide (30 ml) was added over 10 min. The ice bath was removed and the solution was allowed to warm to room temperature. After 2 h, a very thick paste resulted. The off-white paste was carefully quenched with ice chips. An aqueous solution of sodium hydroxide (2.12 g NaOH/100 ml H2O) was added. After a mild exotherm, a clear yellow solution resulted. The solution was poured into water (~400 ml) and a fine solid immediately precipitated. The mixture was filtered through a 600 ml glass fritted funnel of medium porosity. The yellow filtrate was diluted with an equal volume of water and left to stand for 16 h. A yellow precipitate was collected by vacuum filtration. The solid was dried overnight under vacuum to afford 13.6 g (75percent yield) of (5-cyanoindol-3-yl)carboxaldehyde: 1H NMR (500 MHz, DMSO-d6) 12.58 (1 H, br s,), 10.00 (1 H, s), 8.51 (1 H, d, J=3.1 Hz), 8.46 (1 H, d, J=0.6 Hz), 7.22 (1 H, dd, J=8.6, 0.5 Hz), 7.64 (1 H, dd, J=8.5, 1.6 Hz); MS m/e 171 (M+H)+.
Reference:
[1] Patent: EP997474, 2000, A1,
[2] Patent: US6180627, 2001, B2,
[3] Patent: US2003/73849, 2003, A1,
[4] Organic Process Research and Development, 2008, vol. 12, # 2, p. 168 - 177
5
[ 15861-24-2 ]
[ 110-18-9 ]
[ 17380-18-6 ]
Yield
Reaction Conditions
Operation in experiment
52%
With water; iodine; oxygen; sodium carbonate In 1,4-dioxane at 100℃; for 36 h; Schlenk technique; Sealed tube
General procedure: Under air, a 20 mL of Schlenk tube equipped with a stir bar was charged with indole 1 (0.2 mmol, 1 equiv),TMEDA (75 µL, 0.5 mmol, 2.5 equiv), Na2CO3 (42.4 mg, 0.4mmol, 2.0 equiv), 1,4-dioxane (0.5 mL) and H2O (100 µL). Then I2 (101.5 mg, 0.4 mmol, 2.0 equiv) was added and the tube was sealed with a rubber plug and charged with O2. The reaction mixture was stirred at 100 °C for 36 h in oil bath. After cooling to room temperature, the resultant mixture was evaporated with EtOAc (20 mL) under reduced pressure and the residue was purified by flash column chromatography on a silica gel to give the products.
POCI3 (3.6 mL, 38.68 mmol) was added to DMF (l 6.5 mL) dropwise at 0 °C- 10 °C. The resulting mixture was stirred for 30 minutes, cooled to 0 °C, and a solution of Intermediate Z (5.0 g, 35.17 mmol) in DMF (10.0 mL) was added over15 minutes. After the addition was complete, the reaction mixture was stirred at ambient temperature for 2 hours. The reaction mixture was quenched with ice (25 g), poured into water (50 mL), and NaOH (1.5g) was added. The mixture was filtered, and the yellow colored filtrate was diluted with water (100 mL) and left to stand at room temperature for 20 hours. The solid was then filtered and dried to affordIntermediate AA (1.5 g, 62percent) as a yellow solid. 1H NMR (DMSO-c3/4: δ 12.59 (bs, 1H), 10.00 (s, 1H), 8.52 (s, 1 H), 8.51 (s, 1H), 7.71 (s, J = 8.29 Hz; 1H), 7.65 (d, J = 8.70 Hz; 1H). Mass (M-H): 169.1
Reference:
[1] Organic Letters, 2013, vol. 15, # 17, p. 4330 - 4333
[2] Chemical Biology and Drug Design, 2011, vol. 78, # 5, p. 864 - 868
[3] Journal of Agricultural and Food Chemistry, 2013, vol. 61, # 24, p. 5696 - 5705
[4] MedChemComm, 2018, vol. 9, # 11, p. 1882 - 1890
[5] Organic Letters, 2005, vol. 7, # 13, p. 2651 - 2654
[6] Journal of Medicinal Chemistry, 2005, vol. 48, # 19, p. 6023 - 6034
[7] Patent: WO2011/47156, 2011, A1, . Location in patent: Page/Page column 57-58
[8] Farmaco, 1994, vol. 49, # 6, p. 443 - 448
[9] Bioorganic and Medicinal Chemistry Letters, 1996, vol. 6, # 1, p. 81 - 86
[10] Journal of Medicinal Chemistry, 1997, vol. 40, # 18, p. 2843 - 2857
[11] Bioorganic and Medicinal Chemistry Letters, 2007, vol. 17, # 6, p. 1793 - 1798
[12] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 24, p. 7274 - 7277
[13] Bulletin of the Korean Chemical Society, 2011, vol. 32, # 1, p. 307 - 310
[14] Patent: EP1859798, 2015, B1, . Location in patent: Paragraph 0160
[15] Organic and Biomolecular Chemistry, 2018, vol. 16, # 36, p. 6647 - 6651
7
[ 15861-24-2 ]
[ 298-12-4 ]
[ 17380-18-6 ]
Reference:
[1] Organic and Biomolecular Chemistry, 2018, vol. 16, # 20, p. 3707 - 3710
8
[ 15861-24-2 ]
[ 1196-69-6 ]
Yield
Reaction Conditions
Operation in experiment
55%
With acetic acid In pyridine; water; ethyl acetate
PREPARATION 6 Indole-5-carboxaldehyde To a solution of 5-cyanoindole (5 g, 32.2 mmol) in pyridine (70 ml) was added acetic acid (35 ml), an aqueous solution of sodium hypophosphite (10 g in 35 ml H2 O) followed by the addition of Ra-Ni. The resultant mixture was heated at 45° C. for 3 hours and then filtered through celite. The filtrate was combined with water (150 ml) and ethyl acetate (150 ml). The organic extract was separated, washed with aqueous cupric sulfate (3*1001 ml), water (2*100 ml), dried (MgSO4) and evaporated under reduced pressure to afford 5.1 g of a crude product (a beige solid). The crude product was purified by crystallization from chloroform (40 ml) yielding 2.8 g (55percent) of the title compound as a white solid. (Helv. Chim. Acta, 51, 1616 (1968)) 1 H NMR (CDCl3) δ=6.70 (t, J=2 Hz, 1H), 7.28 (t, J= 2 Hz, 1H), 7.46 (d, J=6 Hz, 1H), 7.74 (d, J=6 Hz, 1H), 8.16 (s, 1H). 8.58 (bs, 1H), 9.12 (s, 1H).
Reference:
[1] Patent: US5399574, 1995, A,
[2] Patent: US5409941, 1995, A,
9
[ 15861-24-2 ]
[ 56-45-1 ]
[ 139393-02-5 ]
Reference:
[1] Journal of the American Chemical Society, 2017, vol. 139, # 31, p. 10769 - 10776
[2] Angewandte Chemie - International Edition, 2016, vol. 55, # 38, p. 11577 - 11581[3] Angew. Chem., 2016, vol. 128, p. 11749 - 11753,5
10
[ 15861-24-2 ]
[ 81881-74-5 ]
Yield
Reaction Conditions
Operation in experiment
100%
With ammonia; hydrogen In methanol; water
5-Cyanoindole was hydrogenated over Raney Nickel in methanol with aqueous ammonia. The solution was concentrated under reduced pressure to a light yellow solid (C-(1H-indol-5-yl)-methylamine, 5.25 g, quantitative).
79%
With lithium aluminium tetrahydride In tetrahydrofuran at 0 - 45℃; for 16 h;
To a stirring suspension of LiA1H4 (452 mg, 12.0 mmol) in THF (10 mL) was added asolution of 1H-indole-5-carbonitrile SM (994 mg, 7.0 mmol) in THF (8 mL) at 0 °C. The mixturewas warmed to 45 °C and stirred for 16 h. The reaction mixture was quenched with water (0.5 mL), 15percent NaOH( 0.5 mL) and then water (1.5 mL). The mixture was filtered and concentrated to obtain the residue, which was diluted with EtOAc (30 mL) and washed with water (10 mL) and then brine (10 mL). The organic extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was washed with Et20 (15 mL) to afford (1H- indol-5-yl)methanamine compound 1 (790 mg, 79percent) as light yellow solid. ‘H NMR (300 MHz, DMSO-d6): 10.94 (s, 1H), 7.44 (s, 1H), 7.28 (d, J= 8.0 Hz, 1H), 7.04 (d, J 8.0 Hz, 1H), 6.34(s, 1H), 3.74 (s, 2H), 1.71 (s, 2H).
59%
With hydrogen In methanol for 20 h;
Cyano derivative (N.ident.C-R7) (0.3g, 1 equivalent) was dissolved in 100 ml of MeOH, then a 40 bar pression of hydrogen is applied in the presence of Ni/Raney for 20 h. The reaction mixture is filtered through celite and concentrated. The crude product was purified by flash chromatography to afford the amine. The amine (1 equivalent) was dissolved in DMF (0.4M), then the ethyl isocyanatoacetate (1 equivalent) was added in one portion and the reaction mixture was let 2h at room temperature. After the reaction was complete (TLC control), the reaction mixture was concentrated and purified by flash chromatography to afford the urea.; Example 47: Preparation of ethyl 2-(3-((indol-5-yl)methyl)ureido)acetate (F575)(47). 5-cyano-indole (0.3 g, 2.1 1 mmol) was reduced to obtain the 5- aminomethylindole (0.18g, 59percent) after purification by flash chromatography (AcOEt/MeOH 7/3 then MeOH) Rf=0.09 (MeOH). H NMR (DMSO): δ 2.40 (s, 2H), 3.78 (s, 2H), 6.38 (m, 1 H), 7.10 (d, 1 H, J = 8.3 Hz), 7.29 (m, 1 H), 7.33 (d, 1 H, J = 8.3 Hz), 7.49 (s, 1 H), 1 1 .00 (s, 1 H). The 5-aminomethylindole (57 mg, 0.39 mmol) was used to obtain urea 47 (63 mg, 66percent) after treatment of the crude product by EDP Rf=0.57 (AcOEt). H NMR (DMSO): δ 1 .21 (t, 3H, J= 7.1 Hz), 3.81 (d, 2H, J = 6.0 Hz), 4.1 1 (q, 2H, J = 7.1 Hz), 4.28 (d, 2H, J = 5.7 Hz), 6.24 (t, 1 H, J = 6.0 Hz), 6.39 (s, 1 H), 6.58 (t, 1 H, J = 5.7 Hz), 7.01 (d, 1 H, J= 8.3 Hz), 7.38 (m, 3H), 1 1 .03 (s, 1 H). HPLC method A tr= 8.37 mn (97.3percent). ESI-MS m/z: 276.2 [M + H]+.
45%
Stage #1: With lithium aluminium tetrahydride In tetrahydrofuran at 0 - 20℃; Stage #2: With sodium hydroxide; water In tetrahydrofuran at 0℃;
Example 6; Indole-5-methanamme (4).; To an ice-cold 1.0 M solution Of LiAlH4 in THF(18 mL, 0.018 mol) was added dropwise under N2 a solution of 5-cyanoindole (16, 1.56 g, 0.011 mol) in dry THF (25 mL). After the addition was complete, the mixture was allowed to warm to room temperature and was stirred overnight. The resulting mixture was cooled in an ice bath, and excess LiAlH4 was quenched with 10percent NaOH. The product was extracted with ethyl acetate and dried over anhydrous magnesium sulfate. The solvent was removed by rotary evaporation to give the crude product (1.1 g), which was recrystallized from ethyl acetate/hexanes to give crystalline 5 (0.7 g, 45percent). 1H NMR (400 MHz, DMSO-^) δ 11.0 (s, IH), 7.5 (s, IH), 7.3 (d, 2H, J= 8.4 Hz), 7.0 (d, IH, J= 8.4 Hz), 6.4 (s, IH), 3.8 (s, 2H), 2-3 (br, 2H). HRMS (EI): calculated for C9H10N2 (M+): 146.0838. Found: 146.0835.
32%
With ammonia; hydrogen In methanol at 20℃;
To a solution of lH-indole-5-carbonitrile (1.0 g, 7.0 mmol, 1.0 eq) in NLh/MeOH (10 mL) was added Raney Ni (about 100 mg). The suspension was hydrogenated at rt overnight. The mixture was filtered. The filtered cake was washed with MeOH (10 mL). The filtrate was concentrated under reduced pressure. The residue was purified via flash chromatography to afford (lH-indol-5-yl)methanamine as an off-white solid (330 mg, 32percent).
Reference:
[1] Patent: US2004/204455, 2004, A1, . Location in patent: Page/Page column 18
[2] Chemistry - A European Journal, 2016, vol. 22, # 14, p. 4991 - 5002
[3] Patent: US2003/153596, 2003, A1,
[4] Patent: WO2016/44770, 2016, A1, . Location in patent: Page/Page column 584; 585
[5] Patent: WO2011/76784, 2011, A2, . Location in patent: Page/Page column 66
[6] Bioorganic and Medicinal Chemistry, 2006, vol. 14, # 5, p. 1331 - 1338
[7] Patent: WO2007/35964, 2007, A2, . Location in patent: Page/Page column 6; 13
[8] Patent: WO2015/103317, 2015, A1, . Location in patent: Page/Page column 160
[9] Journal of Medicinal Chemistry, 2005, vol. 48, # 18, p. 5823 - 5836
[10] Patent: US6180627, 2001, B2,
[11] Chemistry - A European Journal, 2008, vol. 14, # 31, p. 9491 - 9494
[12] ChemSusChem, 2017, vol. 10, # 5, p. 842 - 846
[13] Enzyme and Microbial Technology, 2018, vol. 118, p. 83 - 91
11
[ 15861-24-2 ]
[ 24424-99-5 ]
[ 475102-10-4 ]
Yield
Reaction Conditions
Operation in experiment
96%
Stage #1: With dmap In tetrahydrofuran at 20℃; for 0.5 h; Stage #2: for 2 h;
In a 100 mL round-bottom flask was placed 1H-INDOLE-5-CARBONITRILE (2.0 g, 14.07 MMOL) in 20 mL of anhydrous THF. To this solution was added DMAP (0.86 g, 7.03 MMOL) and the mixture was allowed to stir for 0.5 h at rt. At this point, BOC20 (3.07 g, 14.07 MMOL) was added and the reaction stirred for an additional 2 h. The reaction was then quenched with water and extracted twice with ethyl ether. The combined organic layers were washed successively with 1 N HCI, water, and brine, then dried over MGS04 and concentrated to provide 3.26 g (96percent) of the desired product as a white SOLID.APOS;H- NMR (DMSO-d6) 8 8.20-8. 14 (m, 2H), 7.83 (d, 1H), 7.70 (d, 1H), 6.80 (d, 1H), 1.63 (s, 9H).
96%
Stage #1: With dmap In tetrahydrofuran at 20℃; for 0.5 h; Stage #2: for 2 h;
In a 100 ml round-bottom flask was placed 1W-indole-5-carbonitrile (2.0 g, 14.07mmol) in 20 ml of anhydrous THF. To this solution was added DMAP (0.86 g, 7.03mmol) and the mixture was allowed to stir for 0.5 h at rt. At this point, BocaO (3.07 g,14.07 mmol) was added and the reaction stirred for an additional 2 h. The reaction wasthen quenched with water and extracted twice with ethyl ether. The combined organiclayers were washed successively with 1N HCI, water, and brine, then dried over MgSO4and concentrated to provide 3.26 g (96percent) of the desired product as a white solid. 1H-NMR (DMSO-c/e) 5 8.20-8.14 (m, 2H), 7.83 (d, 1H), 7.70 (d, 1H), 6.80 (d, 1H), 1.63 (s,9H).
90%
With dmap In acetonitrile at 20℃; for 0.5 h;
[0486] tert-butyl 5-cyano-l H-indole-1 -carboxylate (INT-65) [0487] To a flask containing 5-cyanoindole (500 mg, 3.52 mmol) in CH3CN (5 mL) was added Boc20 (920 mg, 4.22 mmol) and DMAP (42 mg, 0.35 mmol) and the mixture was stirred at room temperature for 0.5 h. The mixture was concentrated, redissolved in DCM and chromatographed (EtOAc / hexanes) to provide 766 mg (90percent) of tert-butyl 5-cyano-lH- indole-l-carboxylate INT -65 as a white solid. LCMS-ESI (m/z) calculated for C14H14N20 :242.27; found 243.1 [M+H]+, tR = 3.93 min.
Reference:
[1] Synthesis, 2009, # 21, p. 3617 - 3632
[2] Journal of Organic Chemistry, 2002, vol. 67, # 21, p. 7551 - 7552
[3] Patent: WO2004/43950, 2004, A1, . Location in patent: Page 134
[4] Patent: WO2005/51957, 2005, A1, . Location in patent: Page/Page column 54-55
[5] Patent: WO2011/60389, 2011, A1, . Location in patent: Page/Page column 98-99
[6] Synthesis, 2008, # 5, p. 707 - 710
[7] Synlett, 2008, # 2, p. 294 - 296
[8] Journal of Organic Chemistry, 2005, vol. 70, # 1, p. 175 - 178
[9] Journal of Organic Chemistry, 2007, vol. 72, # 14, p. 5046 - 5055
[10] Patent: US2004/242559, 2004, A1, . Location in patent: Page 18
[11] European Journal of Medicinal Chemistry, 2017, vol. 128, p. 70 - 78
12
[ 15861-24-2 ]
[ 884507-17-9 ]
Reference:
[1] Journal of Medicinal Chemistry, 2007, vol. 50, # 15, p. 3651 - 3660
[2] Patent: WO2013/26914, 2013, A1,
13
[ 15861-24-2 ]
[ 676273-39-5 ]
Yield
Reaction Conditions
Operation in experiment
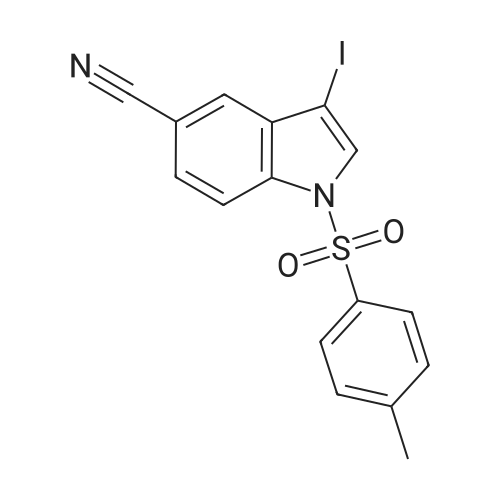
95%
With potassium penicillin V; sodium thiosulfate In N,N-dimethyl-formamide
Example 1 Synthesis of 3-iodo-1H-indole-5-carbonitrile (V) 1H-indole-5-carbonitrile (5 g, 35.2 mmol), KOH (7.90 g, 141 mmol) and I2 (8.90 g, 35.2 mmol) are suspended in 25 mL of DMF under inert atmosphere. The reaction is maintained under stirring in the dark for 30 min. at 10°C, and then treated with an 0.1M solution of Na2S2O3 (150 mL). The resulting suspension is maintained under stirring for 30 min, then filtered, and the resulting solid is washed with water and dried at 50°C under vacuum to constant weight. Product (V) (9.0 g) is obtained as a white solid with a yield of 95percent. 1H-NMR (400 MHz, CDCl3), δ: 8.78 (1H, bs); 7.80 (1H, s); 7.46-7.40 (3H, m).
With iodine; potassium hydroxide In N,N-dimethyl-formamide at 10℃; for 0.5 h; Inert atmosphere; Darkness
Example 1 Synthesis of 3-iodo-1H-indole-5-carbonitrile (V) 1H-indole-5-carbonitrile (5 g, 35.2 mmol), KOH (7.90 g, 141 mmol) and I2 (8.90 g, 35.2 mmol) are suspended in 25 mL of DMF under inert atmosphere. The reaction is maintained under stiffing in the dark for 30 min at 10° C., and then treated with an 0.1M solution of Na2S2O3 (150 mL) The resulting suspension is maintained under stirring for 30 min, then filtered, and the resulting solid is washed with water and dried at 50° C. under vacuum to constant weight. Product (V) (9.0 g) is obtained as a white solid with a yield of 95percent. 1H-NMR (400 MHz, CDCl3), δ: 8.78 (1H, bs); 7.80 (1H, s); 7.46-7.40 (3H, m).
94.5%
With iodine; potassium hydroxide In N,N-dimethyl-formamide at 20℃;
Preparation of compound 69a: 3-iodo-l/7-indole-5-carbonitrileTo a solution of l//-indole-5-carbonitrile (lg, 7.048 mmol) in DMF (10 mL) was added KOH pellets (1.15 g, 21.14 mmol), followed by I2 (3.57 g, 14.08 mmol) portionwise. The reaction was stirred for 1 h at RT. 10percent aq sodium bisulphite solution (10 mL) was added to the mixture and an off-white solid precipitate was formed. The suspension was filtered and the solid was washed with H20 (10 mL) and dried under vacuum to give 3-iodo-lH- indole-5-carbonitrile (1.7 g, 94.5percent) as an off white solid. .H NMR (400MHz, DMSO-d6): δ 12.18 (brs, 1H), 7.78 (s, 1H), 7.75 (d, J=1.2Hz, 1H), 7.59 (dd, J=8.4, 0.4Hz, 1H), 7.52 (dd, J=8.4, 1.6Hz, 1H).
75%
With iodine; sodium hydroxide In N,N-dimethyl-formamide at 20℃; for 0.416667 h;
5-Cyanoindole (0.500 g, 3.52 mmol) was dissolved in DMF (25 mL), and sodium hydroxide (0.493 g, 8.79 mmol) and iodine (0.902 g, 3.55 mmol) were added thereto, then the mixture was stirred at room temperature for 25 minutes. A saturated aqueous sodium thiosulfate solution was added to the mixture, and the precipitated solid was filtered off to give 5-cyano-3-iodoindole(0.707 g, 75percent). ESI-MS: m/z 269 [M + H]+.
With sodium hydride In N,N-dimethyl-formamide; mineral oil at 20 - 40℃; for 2h;
1.A Step A 1-ethyl-1H-indole-5-carbonitrile (II)
Add 100.2g (5.0mol) 60% NaH to 1300mLN,N-dimethylformamide at room temperature,Stir, slowly warm up to 40°C, add 5-cyanoindole 284.0g (2.0mol), after H2 escapes, cool to 25°C, add ethyl iodide 468.0g (3.0mol), react at room temperature for 2h. After the reaction was completed, 5000 mL of water was added, and after stirring for 30 min, 299.2 g of solid was obtained by suction filtration, with a yield of 88.0%.
88%
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide at 40℃;
Stage #2: ethyl iodide In N,N-dimethyl-formamide at 25℃; for 2h;
88%
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide at 25 - 40℃;
Stage #2: ethyl iodide In N,N-dimethyl-formamide at 25℃; for 2h;
4.1.1. Preparation of 1-ethyl-1H-indole-5-carbonitrile (a2)
To a solvent of DMF (800 mL) was added 60% NaH (200.2 g, 5.0 mol)at 25 C. 1H-indole-5-carbonitrile (284.0 g, 2.0 mol) was added whenthe temperature reached to 40 C. After the gas escaped, the solutionwas cooled to 25 C. Then, iodoethane (468.0 g, 3.0 mol) was added andthe reaction mixture was stirred for 2 h. When completed, the mixturewas diluted with plenty of water (1500 mL) under stirring for 0.5 h. Themixture was filtered to obtain the solid a3 in 88.0% yield. MS (ESI) m/z:171.4 [M+H]+.
81%
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide at 20 - 40℃;
Stage #2: ethyl iodide In N,N-dimethyl-formamide at 25℃; for 2h;
4.1.1. 1-ethyl-1H-indole-5-carbonitrile (2)
60% NaH (100.2 g, 5.0 mol) was suspended in DMF (800 mL) atambient temperature. 1H-indole-5-carbonitrile (284.0 g, 2.0 mol)was added when the temperature reached to 40 C. After the gasescaped, the solution was cooled to 25 C. Then iodoethane(468.0 g, 3.0 mol) was added and the reaction mixture was stirredat room temperature for 2 h. When completed, the mixture wasdiluted with water (500 mL). After stirring for 0.5 h, the mixturewas filtered to obtain the solid 2 in 88.1% yield.
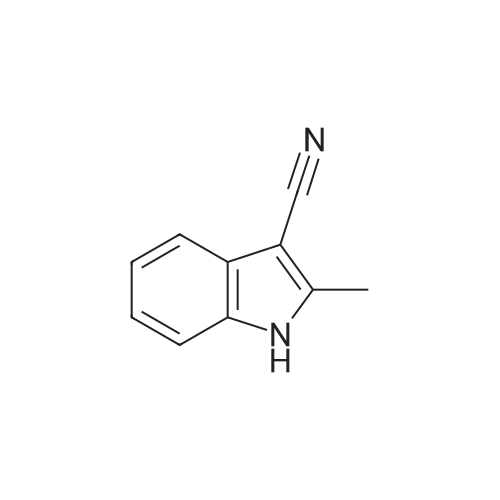
33%
With potassium hydroxide In acetone for 1h; Ambient temperature;
With sodium hydroxide; trichlorophosphate; In N,N-dimethyl-formamide;
EXAMPLE 1 Trans-2-[5-Cyanoindol-3-yl]-1-(N,N-dimethylaminomethyl)cyclopropane Phosphorus oxychloride (10.9 ml, 117 mmol) was added dropwise over 30 min to anhydrous dimethylformamide (50 ml) that was maintained at 10-20 C. (internal temperature). The resulting mixture was stirred for 30 min and then chilled to 0 C. A solution of commercially available 5-cyanoindole (15 g, 106 mmol) in anhydrous dimethylformamide (30 ml) was added over 10 min. The ice bath was removed and the solution was allowed to warm to room temperature. After 2 h, a very thick paste resulted. The off-white paste was carefully quenched with ice chips. An aqueous solution of sodium hydroxide (2.12 g NaOH/100 ml H2O) was added. After a mild exotherm, a clear yellow solution resulted. The solution was poured into water (~400 ml) and a fine solid immediately precipitated. The mixture was filtered through a 600 ml glass fritted funnel of medium porosity. The yellow filtrate was diluted with an equal volume of water and left to stand for 16 h. A yellow precipitate was collected by vacuum filtration. The solid was dried overnight under vacuum to afford 13.6 g (75% yield) of (5-cyanoindol-3-yl)carboxaldehyde: 1H NMR (500 MHz, DMSO-d6) 12.58 (1 H, br s,), 10.00 (1 H, s), 8.51 (1 H, d, J=3.1 Hz), 8.46 (1 H, d, J=0.6 Hz), 7.22 (1 H, dd, J=8.6, 0.5 Hz), 7.64 (1 H, dd, J=8.5, 1.6 Hz); MS m/e 171 (M+H)+.
62%
With trichlorophosphate; at 0 - 26℃; for 3h;
POCI3 (3.6 mL, 38.68 mmol) was added to DMF (l 6.5 mL) dropwise at 0 C- 10 C. The resulting mixture was stirred for 30 minutes, cooled to 0 C, and a solution of Intermediate Z (5.0 g, 35.17 mmol) in DMF (10.0 mL) was added over15 minutes. After the addition was complete, the reaction mixture was stirred at ambient temperature for 2 hours. The reaction mixture was quenched with ice (25 g), poured into water (50 mL), and NaOH (1.5g) was added. The mixture was filtered, and the yellow colored filtrate was diluted with water (100 mL) and left to stand at room temperature for 20 hours. The solid was then filtered and dried to affordIntermediate AA (1.5 g, 62%) as a yellow solid. 1H NMR (DMSO-c¾: delta 12.59 (bs, 1H), 10.00 (s, 1H), 8.52 (s, 1 H), 8.51 (s, 1H), 7.71 (s, J = 8.29 Hz; 1H), 7.65 (d, J = 8.70 Hz; 1H). Mass (M-H): 169.1
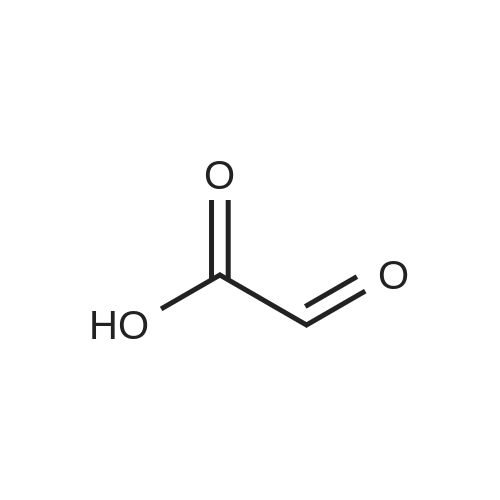
Example 23 3-Formyl-5-cyano-1H-indole Add phosphorous oxychloride (11.76 g, 76.67 mmol) dropwise to DMF (24.3 ml) wile maintaining the temperature at less than 10C. Warm to ambient temperature and stir for 15 minutes at ambient temperature. Add dropwise 5-cyanoindole (10.00 g, 70.34 mmol) as a solution in DMF (30 ml) while keeping the temperature below 35. After 1 hour, pour the reaction mixture into ice/water (300 ml) and then add 5N NaOH (54 ml) with stirring. Add slowly an additional amount of 5N NaOH (19.7 ml) and then heated to 90 for 1 minute and then cooled to ambient temperature to give a precipitate. Filter the precipitate and washed with water and dry to give the title compound: mp 248-250 C. MS (ACPI): m/e 171.0 (M+1). Analysis for C10H6N2O: Calcd: C, 70.58; H, 3.55; N, 16.46; found: C, 70.41; H, 3.53; N, 16.33.
With sodium azide; ChCl*2ZnCl2 at 140℃; for 7h; Green chemistry;
III. General process for synthesis of 5-substituted 1H-tetrazoles (3a-3k)
General procedure: NaN3 (0.975 g, 15 mmol) was dissolved in DES (10 mL) at room temperature by stirring until a clear solution was formed. Then benzonitrile (10 mmol) was added. The reaction mixture was constantly stirred at 140 °C and monitored by TLC. After completion of the reaction, the reaction mixture was cooled to room temperature and poured into the cold water (10 mL). The solid was obtained and filtered. The obtained solid is taken into cold water (10 mL). Then it was acidified carefully to pH 5 with 5 M HCl. The organic material was extracted thrice with ethyl acetate; the resultant organic layer was washed with distilled water, dried over anhydrous sodium sulfate, and concentrated to give the crude solid crystalline 5-substituted 1H-tetrazole. The resulting product, although evident as a single compound by TLC, was purified by simple recrystallization from aqueous ethanol giving pure 5-substituted 1H-tetrazoles.
82%
With trimethylsilylazide; tetrabutyl ammonium fluoride at 120℃; for 18h;
81%
With sodium azide; tetra(n-butyl)ammonium hydrogensulfate In water at 85℃; for 8h; Green chemistry;
General Procedure for Preparation of Tetrazoles in Water(Method II).
General procedure: General Procedure for Preparation of Tetrazoles in Water(Method II). TBAHS (0.25 mmol) was added to a mixture of nitrile (1 mmol), sodium azide (1.5 mmol), and 2 mL H2O in around-bottomed flask. The reaction mixture was heated to 85 °C. After completion of the reaction (as monitored by TLC), the crude reaction mixture was transferred into a separatory funnel, to which was added 1 N HCl (15 mL) extracted by ethylacetate (EtOAc, 10 mL × 5). The combined organic layers were washed with H2O and dried over anhydrous sodium sulfate, and were evaporated under reduced pressure to give pure 5-substituted-1H-tetrazole.
80%
Stage #1: 1H-indole-5-carbonitrile With sodium azide; tris(pentafluorophenyl)borate In N,N-dimethyl-formamide at 120℃; for 20h;
Stage #2: With hydrogenchloride In water; ethyl acetate; N,N-dimethyl-formamide for 0.25h;
Typical experimental procedure
General procedure: B(C6F5)3 (67.3 mg, 0.13 mmol, 5 mol %) was added to a stirred solution of 3,4-dichlorobenzaldehyde (172 mg, 1 mmol) and NaN3 (97.5 mg, 1.5 mmol) in DMF (5 mL) and was heated at 120 °C. After completion of reaction (as monitored by TLC), the reaction mixture was cooled to room temperature and was added 5 mL of cold water followed by 10 mL of 2 N HCl and 10 mL of ethyl acetate. The resulting mixture was stirred vigorously for 15 min. The organic layer was separated and aqueous layer was again extracted with ethyl acetate (3 × 15 mL). The combined organic layer was washed with water and dried over anhydrous sodium sulfate and was evaporated under reduced pressure. The crude product was purified by column chromatography (silica gel, EtOAc/hexane 9:1) to obtain pure 5-(3,4-dichlorophenyl)-1H-tetrazole. The known compounds were characterized and confirmed by comparison of their spectral data and physical properties with reported literature.
75%
With sodium azide; acetic acid; urea In water; N,N-dimethyl-formamide at 60 - 110℃; for 9h;
Typical procedure for preparation
General procedure: The procedure for the synthesis of the tetrazole 2a is representative. A mixture of sodium azide (0.39 g 0.0060 mol), urea (0.36 g, 0.0060 mol) and water (2.5 mL) was taken in a round-bottom flask and stirred at 60 °C for 1 h. Charged benzonitrile 1a (0.5 g 0.0048 mol), acetic acid(0.5 mL) and DMF (2.5mL) at 60 °C and heat to 110°C stirred for 8 h. After completion of the reaction (as indicated by TLC), the reaction mixture was cooled to room temperature and diluted the reaction mass with water (2.5 mL)and ethyl acetate (5.0 mL) at 25-35 °C. Add 5N aqueous hydrochloric acid (2.5 mL) at 25-35 °C. Stirred for 20- 30 min, the resultant organic layer was separated and the aqueous layer was extracted with ethyl acetate (2.5 mL). The combined organic layer was washed with 40 % aq.NaCl solution (2.5 mL) and dried over anhydrous Na2SO4 and concentrated to give a crude product, which was isolated using chilled water after 3-4 h maintenance, and eventually filtered off to give 0.67 g (95%) of an off-white solid.
With tris-(2-chloro-ethyl)-amine
With triethylamine hydrochloride In 1,2-dimethoxyethane; water; ethyl acetate
1 5-Tetrazol-5-yl-1H-indole (1a)
EXAMPLE 1 5-Tetrazol-5-yl-1H-indole (1a) A mixture of 5-cyano-1H-indole (88 g), triethylamine hydrochloride (225 g), and sodium azide (150 g) was heated in 1,2-dimethoxyethane (DME) at reflux temperature for 48 hours. The solvent was evaporated in vaciio and the residue was dissolved in ethyl acetate (500 mL) and water (500 mL). pH was adjusted to 10 by addition of concentrated aqueous NaOH solution. The aqueous phase was separated and pH was adjusted to 4 by addition of acetic acid. The precipitated crystalline title compound 1a was filtered off and subsequently dried in vacuo. Yield 82 g, mp 205° C.
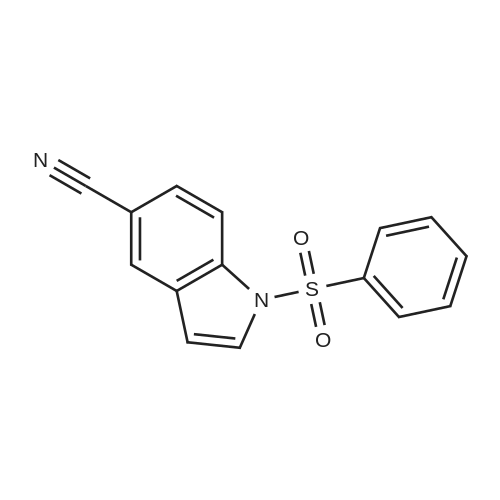
Stage #1: 1H-indole-5-carbonitrile With tetrabutylammomium bromide; sodium hydroxide In water; toluene for 0.5h; Inert atmosphere;
Stage #2: benzenesulfonyl chloride In water; toluene at 20℃; for 25h; Inert atmosphere;
3
Example 3; Preparation of 3-(2-Benzyl- lH-indol-5-yl)-2-ethoxy-proDanoic acid 3; l-Benzenesulfonyl-lH-indole-5-carbonitrile 24; To a vigorously stirred mixture of indole-5-carbonitrile 23 (864 mg, 6.08 mmol, 1.00 eq) and n- tetrabutylammonium bromide (197 mg, 0.609 mmol, 0.10 eq) in 50% aqueous NaOH (5.90 ml), toluene (5.00 ml) and water (9.00 ml) was added dropwise, over 30 min, benzenesulfonyl chloride (0.850 ml, 0.688 mmol, 1.10 eq) in toluene (4.00 ml) via a syringe pump. The reaction flask was covered with aluminium foil and the mixture stirred at RT for 25 hr. The solution was yellow. After removal of the aqueous phase, the organic phase was washed with 0.1 M NaHCO3 (20.0 ml), water (75.0 ml) and saturated brine (75.0 ml). After extractions with EtOAc (3x70.0 ml), the organic phase was dried over MgSO4 and concentrated in vacuo to give an off-white powder (1.69 g) which was purified by crystallisation from hexane/DCM 20:1 to yield 1.65 g (96%) of the product as white needles: mp 131.0-133.5 0C (hexane/DCM 20:1); Rf 0.35[hexane/EtOAc 6:4]; v;mx (nujoiycm 1 2222, 1458, 1375, 1080, 721; 4 (CDCl3) 8.10-8.08 (IH, d, /8.4, lxH2'), 7.90-7.89 (3H, m, lxH2 H4, W), 7.71-7.70 (IH, d, /3.6, H2), 7.62-7.56 (2H, m, 2xH3'), 7.51-7.47 (2H, t, /7.8, H4', H6), 6.74-6.73 (IH, d, J 3.6, H3); S0 (CDCl3) 137.73 (C), 136.43 (C), 134.42 (CH), 130.65 (C), 129.54 (CH), 128.37 (CH), 127.59 (CH), 126.77 (CH), 126.36 (CH), 119.18 (C), 114.27 (CH), 108.65 (CH), 107.03 (Q; m/z (EI+) 282 ([M]', 50), 141 ([M-BzSulfonyl group]"1", 66), 114 ([unsubstituted indole]"1", 12), 77 (100); found [M+H]+, 282.0474, Ci5H10N2O2S requires [M+Hf, 282.0463; Found: C, 63.7; H, 3.5; N, 9.9. Required: C, 63.8; H, 3.6; N, 9.9; Δ = 3.9 ppm.
96%
With tetrabutylammomium bromide; sodium hydroxide In water; toluene at 20℃; for 25h; Inert atmosphere;
86%
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In tetrahydrofuran at 0℃;
Stage #2: benzenesulfonyl chloride In tetrahydrofuran at 0 - 20℃;
69
Preparation 69: l-(Benzenesulfonyl)indole-5-carbonitrile; [812] To 30OmL of anhydrous tetrahydrofuran was dissolved sodium hydride (23.4g, 537mmol) at O0C. lH-indole-5-carbonitrile (25.43g, 179mmol) was dissolved in 10OmL of anhydrous tetrahydrofuran, which was then slowly added in drops at O0C. The mixture was stirred for 30 min. Benzenesulfonyl chloride (63.2g, 358mmol) was dissolved in 10OmL of anhydrous tetrahydrofuran, which was then slowly added in drops at O0C. The mixture was slowly warmed to room temperature while stirring for 15 h. After completion of the reaction, ethyl acetate was added. The mixture was washed with IN hydrochloric acid. The organic layer was concentrated under reduced pressure, and the resulting solid was washed with ethanol and dried to give 45.84g (157mmol, Yield 86%) of the title compound.[813] NMR 1H-NMR(CDCl3) δ 8.09(1H, d), 7.90(3H, m), 7.7O(1H, d), 7.55-7.62(2H, m), 7.49(2H, t), 6.73(1H, d)[814] Mass(EI): 283(M++1)
81%
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In tetrahydrofuran at 0℃; for 0.5h;
Stage #2: benzenesulfonyl chloride In tetrahydrofuran at 0 - 20℃; for 15h;
15.1
To a suspension of sodium hydride (60%, 42.0 g, 1.053 mol) THF (600 mL) at 0° C. was slowly added a solution of 5-cyanoindole (50.0 g, 0.351 mol) in THF (200 mL). The mixture was stirred for 30 min. A solution of benzene sulfonyl chloride (111.5 g, 0.633 mol) in THF (200 mL) was then slowly added at 0° C. The mixture was allowed to reach room temperature and was then stirred for 15 h. EtOAc was added and the mixture was washed with 1N hydrochloric acid. The aqueous layer was extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO, and concentrated to dryness. The resulting solid was washed with petroleum ether to give 1-benzenesulfonyl-1H-indole-5-carbonitrile as a yellow solid (80.0 g, 81%). 1H NMR (400 MHz, CDCl3) δ 8.09-8.07 (1H, d); 7.90-7.88 (3H, m), 7.70-7.69 (1H, d), 7.61 (1H, s), 7.59-7.55 (1H, t), 7.50-7.47 (2H, t), 6.73-6.72 (1H, d).
76%
With tetra(n-butyl)ammonium hydrogensulfate; sodium hydroxide In dichloromethane at 0 - 20℃; for 0.583333h;
With copper(l) iodide; potassium iodide; N,N`-dimethylethylenediamine In toluene at 110℃; for 24h;
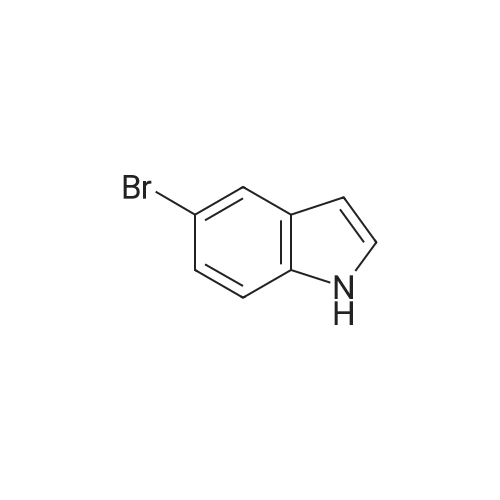
95%
Stage #1: 5-bromo-1H-indole; sodium cyanide With potassium iodide; N,N`-dimethylethylenediamine In toluene at 110℃; for 24h;
Stage #2: With ammonia In water; ethyl acetate; toluene at 25℃; for 0.166667h;
30 Example 30; [1H INDOLE-5-CARBONITRILE]
An oven dried screw cap test tube was charged with [NACN] (98 mg, 2.00 mmol) dried KI (55 mg, 0.331 mmol, 20 mol%) and CuI (32 mg, 0.168 mmol, 10 mol%), 5- Bromoindole (327 mg, 1.667 mmol), evacuated and backfilled with argon three times. Anhydrous toluene (1.1 mL) [ANDN, N'-DIMETHYLETHYLENEDIAMINE (180, UL,] 1.691 mmol) were added under argon. The tube was sealed and the reaction mixture was stirred magnetically at [110°C] for 24 h. The resulting yellow color suspension was cooled to room temperature, 2 mL of ethyl acetate, 1 mL of ammonium hydroxide 30% and 1 mL of water were added. The mixture was stirred at [25°C] for 10 min then the organic layer was separated and the aqueous layer was extracted three times with ethyl acetate (3 x 2 mL). The organic layers combined were washed with 5 mL of water and dried over [MGS04.] The GC analysis showed complete conversion of starting material with a desired product area of 99%. The solvent was removed at reduced pressure. Purification of the residue by flash chromatography on silica gel (2 x 15 cm ; hexane/ethyl acetate 10: 1) provided 225 mg (95% yield) of the title compound as a white solid.
76%
With copper(l) iodide In various solvent(s) at 115℃; for 2h;
With boron trifluoride diethyl etherate In dichloromethane at 20℃; for 0.0333333h; Green chemistry;
General procedure for the synthesis of 3-substituted indoles/ substituted arenes:
General procedure: Indole/ arene (1.0 mmol), α, β-unsaturated ketone (1.0 mmol) was dissolved in DCM (5mL) then BF3.OEt2 (1 equiv) was added. Immediately The progress of the reaction was monitored by TLC. After completion of the reaction which was indicated by TLC, reaction mixture was quenched with NaHCO3 (3 equiv) and concentrated in vacuo. The crude product was directly poured into silica gel column chromatography (100-200 mesh) using ethyl acetate: hexane (15:85 to 20:80) as eluent to afford corresponding 3-Substituted indoles and substituted arenes. The all obtained products were characterized by 1H NMR, 13C NMR, Mass and IR spectral data.
79%
at 20℃;
79%
With iodine at 20℃; for 0.333333h;
78%
In water at 20℃; for 2h;
70%
With hafnium(IV) trifluoromethanesulfonate In acetonitrile at 20℃; for 2h;
With boron trifluoride diethyl etherate In dichloromethane at 20℃; for 0.0333333h; Green chemistry;
General procedure for the synthesis of 3-substituted indoles/ substituted arenes:
General procedure: Indole/ arene (1.0 mmol), α, β-unsaturated ketone (1.0 mmol) was dissolved in DCM (5mL) then BF3.OEt2 (1 equiv) was added. Immediately The progress of the reaction was monitored by TLC. After completion of the reaction which was indicated by TLC, reaction mixture was quenched with NaHCO3 (3 equiv) and concentrated in vacuo. The crude product was directly poured into silica gel column chromatography (100-200 mesh) using ethyl acetate: hexane (15:85 to 20:80) as eluent to afford corresponding 3-Substituted indoles and substituted arenes. The all obtained products were characterized by 1H NMR, 13C NMR, Mass and IR spectral data.
85%
With scandium tris(trifluoromethanesulfonate) In acetonitrile at 20℃; for 0.5h;
85%
In water at 20℃; for 2h;
81%
With graphite oxide In tetrahydrofuran; water at 20℃; for 4h;
General procedure.
General procedure: Typical procedure for the Friedel-Crafts reaction of indoles to α,β-unsaturated ketones: To a stirred mixture of indole (117 mg, 1mmol), and methyl vinyl ketone (85 mg, 1.2 mmol) in H2O/THF (7:3, 2 mL ) solution, GO (20 mg) was added. The reaction mixture was allowed to stir at room temparature and progress of the reaction was monitored by TLC. After complete consumption of indole, water (5 mL) was added and the aqueous layer was extracted with ethyl acetate (2 X 5 mL). The combined organic extracts were dried with anhydrous Na2SO4. The solvent and volatiles were completely removed under vacuum to give the crude product, which was passed through a short pad of silica gel (petroleum ether/ethyl acetate) to afford the analytically pure product in 94% yield.The aqueous layer containing the catalyst was filtered and washed with acetone,water and dried in a dessicator and used for the next cyle.
76%
at 20℃;
76%
With iodine at 20℃; for 0.333333h;
68%
With zirconium(IV) chloride In dichloromethane at 20℃;
With iron(II) tetrafluoroborate hexahydrate; oxygen In water at 60℃; for 5h;
With aluminum (III) chloride In dichloromethane at 0 - 20℃;
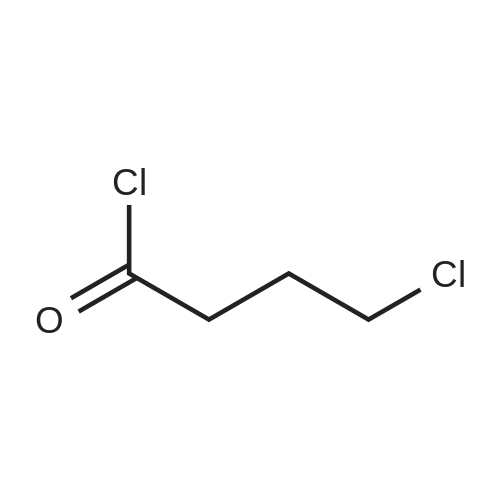
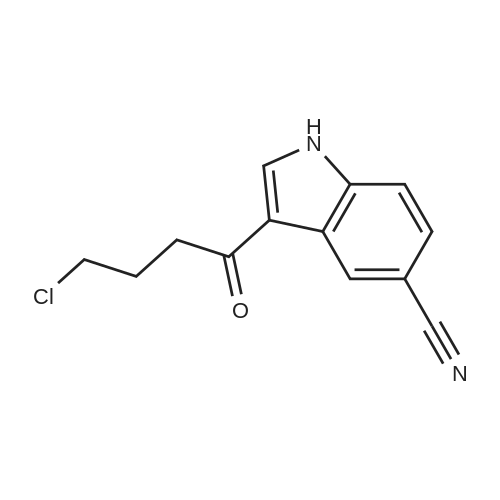
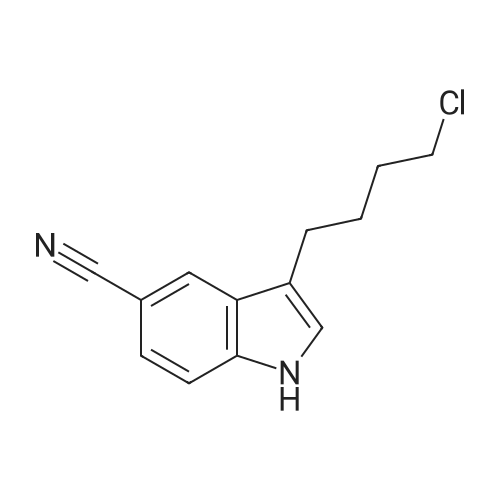
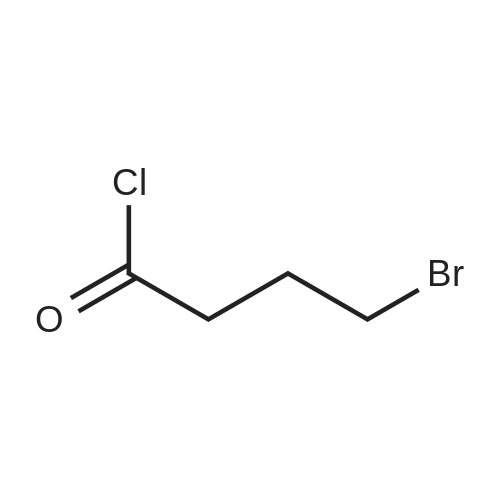
3-(4-Chlorobutanoyl)-1H-indole-5-carbonitrile (6b-1)
To the suspension of AlCl3 (60 mmol, 8.000 g) in CH2Cl2 (150 mL) was added 4-chlorobutyryl chloride (45 mmol, 5.04 mL) at 0 °C, the mixture solution was stirred for a moment and a solution of 5-cyanoindole (30 mmol, 4.265 g) in CH2Cl2 was added dropwise. Then return to room temperature and continue to react for 2 h. Detect the reaction process by TLC, pour the mixture solution to ice water when the reaction finished and a mass of solid was separated out, filter and collect the precipitate, then drying it to afford intermediate 6b-1 as white solid in 93% yield. 1H NMR (400 MHz, DMSO-d6) δ: 12.47 (s, 1H), 8.58 (d, J = 2.1 Hz, 1H), 8.55 (s, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 8.5 Hz, 1H), 3.73 (t, J = 6.6 Hz, 2H), 3.07 (t, J = 7.1 Hz, 2H), 2.11 (p, J = 6.9 Hz, 2H).
90%
With titanium tetrachloride In chloroform at 0℃; for 20h;
2 Example 1: Compounds of formula (3)
The 5-cyano-indole 28.4 g, chloroform was added to 284ml 500ml 4-neck flask, and dissolved with stirring, 40g of anhydrous titanium tetrachloride was added dropwise, maintaining 0 stirred for 20 hours, the reaction solution was poured into ice water 600g stirred, filtered and dried to give a solid 44g, yield of 90%, mp186 ~ 188 .
87.4%
Stage #1: 4-Chlorobutanoyl chloride With aluminum (III) chloride In 1,2-dichloro-ethane at 0℃; for 1h;
Stage #2: 1H-indole-5-carbonitrile In 1,2-dichloro-ethane at 0 - 20℃; for 2.5h;
1 Example 1: Preparation of 3- (4-chlorobutanoyl)indole-5-carbonitrile
At 0 , equipped with a mechanical stirrer three 2L flask was added 1,2-dichloroethane (400mL) and anhydrous aluminum chloride (40.0g, 0.3moL). At the same temperature was added dropwise 4-chlorobutyryl chloride (42.2g, 0.33moL), about half an hour the addition was complete, stirring was continued for 30 min. Was then added 5-cyano-indole (36.4g, 0.25moL) in 1,2-dichloroethane (400mL), keeping the temperature at 0 ~ 5 . The addition was complete within 30min. The ice bath was removed, brought to room temperature and then stirring was continued for 2h.Subsequently, the reaction mixture was added to 220g of ice and 220mL of concentrated hydrochloric acid. The reaction was stirred at room temperature overnight. Filtered and dried under vacuum to give a brown solid 3- (4-chlorobutanoyl) indole-5-carbonitrile 53.8 g of the, yield 87.4%.
83.6%
With aluminum (III) chloride In dichloromethane for 0.5h; Reflux;
2 Synthesis of 3-(4-Chlorobutyryl)-1 H-indol-5-carbonitrile
Aluminium trichloride 93 g, Methylene chloride 320 g, Heat under reflux, then add: 4-chlorobutyryle chloride 100 g, While maintaining the reflux add a separately prepared solution of: 5-Cyano-indole 40 g, Methylene chloride 160 g. Stir at reflux for 30 minutes then pour the reaction in a flask containing Distilled water400 g, Hydrochloric acid 32%, 32 g. On completion of casting distil up to 80° C. Add: Ethyl acetate 200 g. Cool at 5° C., filter and wash with:Ethyl acetate 60 g. Distilled water 120 g. DryThere are obtained 58 g; Yield: 83.6%.
83.6%
Stage #1: 4-Chlorobutanoyl chloride With aluminum (III) chloride In dichloromethane Reflux;
Stage #2: 1H-indole-5-carbonitrile In dichloromethane for 0.5h; Reflux;
2 Synthesis of 3-(4-Chlorobutyryl)-1H-indol-5-carbonitrile
Load into a flask: Aluminium trichloride 93 g; Methylene chloride 320 g. Heat under reflux, then add: 4-chlorobutyryle chloride 100 g. While maintaining the reflux add a separately prepared solution of: 5-Cyano-indole 40 g , Methylene chloride 160 g. Stir at reflux for 30 minutes then pour the reaction in a flask containing Distilled water 400 g, Hydrochloric acid 32% 32 g. On completion of casting distil up to 80°C. Add: Ethyl acetate 200 g. Cool at 5°C, filter and wash with: Ethyl acetate 60 g, Distilled water 120 g. Dry There are obtained 58 g. Yield: 83.6%
74.8%
Stage #1: 4-Chlorobutanoyl chloride With aluminum (III) chloride In dichloromethane at 0 - 30℃;
Stage #2: 1H-indole-5-carbonitrile In dichloromethane at 0 - 30℃;
3.1 Step-1: Preparation of 3-(4-Chlorobutyryl)-l/f-indole-5-carbonitrile
Aluminium chloride (82.44 g) was added to dichloromethane (350 ml) under stirring at 25- 30°C. The resulting mass was cooled to 0-5°C, followed by drop-wise addition of 4- chlorobutyryl chloride (87.5 g) for 30-45 minutes at 0°C and then stirring for 5 minutes at the same temperature. A solution of 5-cyanoindole (50 g) in dichloromethane (350ml) was added drop-wise to the resulting mass at 0-5°C within 1-2 hours, followed by stirring for 30 minutes at the same temperature. The temperature of the reaction mass was gradually increased to 25-30°C, followed by stirring for 12-16 hours at the same temperature. The reaction mass was slowly poured into crushed ice (420 g) with slow stirring, the resulting mixture was cooled to 0-5 °C and then stirred for 1 hour at the same temperature. The separated solid was filtered, washed the material subsequently with dichloromethane (2 x 85 ml) and water (2 x 85 ml) and then dried at 25-30°C to produce a solid material (Dry weight: 93 g). The resulting solid was dissolved in ethyl acetate (4000 ml) at 50-55°C, followed by the addition of activated carbon (5 g) and then stirring the mixture for 5 minutes at the same temperature. The resulting mass was filtered through celite bed and washed the bed with hot ethyl acetate (100 ml). The resulting filtrate was initially cooled to 25-30°C and further cooled to 0-5°C, followed by stirring the mass for 1 hour at the same temperature. The separated solid was filtered, washed with chilled ethyl acetate (100 ml) and then dried the material at 50-55°C until constant weight to produce 65 g of 3-(4- chlorobutyryl)-lH-indole-5-carbonitrile (Purity by HPLC: 99.2%; Yield: 74.8%).
73%
With isobutylaluminum dichloride In dichloromethane at 15 - 30℃;
64%
Stage #1: 4-Chlorobutanoyl chloride With aluminum (III) chloride In dichloromethane for 0.5h;
Stage #2: 1H-indole-5-carbonitrile In dichloromethane at 20℃; for 2h;
4.1 Step 1) 3-(4-Chlorobutyryl)-1H-indole-5-carbonitrile
At 0 ° C,4-Chlorobutyryl chloride (9.6 g, 68.0 mmol) was added dropwise to a solution containing aluminum chloride (9.0 g, 68.0 mmol)In the methyl chloride (90mL), the addition is completed.Continue to react for 0.5h,Then, a solution of 5-cyanoguanidine (8.1 g, 57.0 mmol) in dichloromethane (800 mL) was added dropwise.After the addition is completed,Move to room temperature for 2 h. Then, the reaction liquid was added to a mixed solvent of ice water (50 g) and concentrated hydrochloric acid (50 mL).Continue to react at room temperature for 20 h. The reaction is completed,Filtering,Wash the filter cake,Wash with ethyl acetate,Dry to a yellow solid (8.9 g, 64%).
63%
Stage #1: 4-Chlorobutanoyl chloride With aluminum (III) chloride In dichloromethane at 0℃; for 0.5h;
Stage #2: 1H-indole-5-carbonitrile In dichloromethane at 20℃; for 2h;
5.1 Step 1) 3 -(4-chlorobutanoyl)-lH-indole-5 -carbonitrile
Example 5 3 -(4-(4-(4,6-Dimethoxypyrimidin-2-yl)piperazin- 1 -yl)butyl)- lH-indole-5 -carbonitrile Step 1) 3 -(4-chlorobutanoyl)-lH-indole-5 -carbonitrile To a mixture of aluminium chloride (9.0 g, 68.0 mmol) in DCM (90 mL) was added 4-chlorobutyryl chloride (9.6 g, 68.0 mmol) dropwise at 0 °C. After the mixture was stirred at 0 °C for 30 mins, to it was added a mixture of 5-cyanoindole (8.1 g, 57.0 mmol) in DCM (800 mL) dropwise. The reaction mixture was heated to rt and stirred for 2 hours. Then the reaction mixture was poured into a mixture of ice water (50 g) in concentrated hydrochloric acid (50 mL), and stirred at rt for 20 hours. The resulting mixture was filtered, and the filter cake was washed with water (10 mL) and EtOAc (10 mL) in turn. Then the filter cake was dried to give the title compound as a yellow solid (8.9 g, 63%). The compound was characterized by the following spectroscopic data: LC-MS (ESI, pos. ion) m/z: 247.1 [M + H]+ and NMR (CD3OD, 400 MHz) δ (ppm): 8.63 (d, J= 0.7 Hz, 1H), 8.35 (s, 1H), 7.62-7.60 (m, 1H), 7.52 (dd, J = 8.4, 1.5 Hz, 1H), 3.69 (t, J = 6.5 Hz, 2H), 3.11 (t, J = 7.2 Hz, 2H), 2.24-2.17 (m, 2H).
63.6%
Stage #1: 4-Chlorobutanoyl chloride With aluminum (III) chloride In dichloromethane at 0℃; for 0.5h;
Stage #2: 1H-indole-5-carbonitrile In dichloromethane at 20℃; for 2h;
3.1 Step 1) 3 -(4-chlorobutanoyl)- 1H-indole-5 -carbonitrile
To a solution of aluminium chloride (9.10 g, 68.3 mmol) in DCM (90 mL) was added 4-chlorobutyryl chloride (9.6 g, 68.3 mmol) dropwise at 0 °C. After the mixture was stirred at 0°C for 30 minutes, to the mixture was added a solution of 5-cyanoindole (8.1 g, 57.0 mmol) in DCM (800 mL). Then the reaction mixture was stirred at rt for 2 hours and poured into a mixture of ice (50 g) and concentrated hydrochloric acid (50 mL), and thenfurther stirred for 20 hours. The resulting mixture was filtered, and the filter cake was washed with water (5 mL) and EtOAc (5 mL) in turn. The filter cake was then dried under vacuum to give the title compound as a yellow solid (8.9 g, 63.6%). The compound was characterized by the following spectroscopic data:LC-MS (ESI, pos. ion) m/z: 247.1 [M + H] and‘H NIVIR (400 MHz, CD3OD) (ppm):8.63 (d, J= 0.7 Hz, 1H), 8.35(s, 1H), 7.62-7.60 (m, 1H), 7.52 (dd, J 8.4, 1.5 Hz, 1H), 3.69 (t, J 6.5 Hz, 2H), 3.11 (t, J= 7.2 Hz, 2H), 2.24-2.17 (m, 2H).
63.1%
Stage #1: 4-Chlorobutanoyl chloride With aluminum (III) chloride In dichloromethane at 0℃; for 0.5h;
Stage #2: 1H-indole-5-carbonitrile With hydrogenchloride In dichloromethane; water at 20℃; for 22h; Cooling with ice;
1.1 Step 1’) Synthesis of 3 -(4-chlorobutanoyl)- 1H-indole-5-carbonitrile
To a solution of aluminium chloride (9.00 g, 68.00 mmol) in dichloromethane (90 mL) was added 4-chlorobutyryl chloride (9.60 g, 68.00 mmol) dropwise at 0 °C, the reaction mixture was stirred for 30 minutes, and then a soluton of 5-cyanoindole (8.10 g, 57.00 mmol) in dichloromethane (800 mL) was added dropwise. The reaction was warmed to room temperature, and after stirring 2 hours, the mixture was poured into a mixture of ice (50 g) and concentrated hydrochloric acid (50 mL), and then the stir was continued at room temperature for 20 hours. The resulting mixture was filtered by suction, and the filter cake was washed sequentially with water (10 mL) and ethyl acetate (10 mL), then dried to give the title product as a yellow solid (8.90 g, 63.1%). LC-MS (ESI, pos. ion) m/z: 247.1 [M + H].
63.1%
Stage #1: 4-Chlorobutanoyl chloride With aluminum (III) chloride In dichloromethane at 0℃; for 0.5h; Inert atmosphere;
Stage #2: 1H-indole-5-carbonitrile In dichloromethane at 20℃; for 2h;
Stage #3: With hydrogenchloride In dichloromethane; water at 20℃; for 20h; Cooling with ice;
1.1 Step 1) 3 - (4 - chloroprene acyl) - 1H - indole -5 - carbonitrile synthesis
0 °C lower, will be 4 - chlorobutyryl chloride (9.60g, 68 . 00mmol) drops containing aluminum chloride (9.00g, 68 . 00mmol) of dichloromethane (90 ml) solution, mixed solution stirring 30 minutes, continue to dropping 5 - cyano indole (8.10g, 57 . 00mmol) of methylene chloride (800 ml) solution. Heating the reaction to room temperature, stirring 2 hours, the reaction liquid slowly poured into ice/concentrated hydrochloric acid in a mixture of (50g/50 ml), then stirring continued at room temperature for 20 hours. The resulting mixture is filtered, and it with water (10 ml) and ethyl acetate (10 ml) washing the filter cake, the filter cake is dried to obtain the title compound as a yellow solid (8.90g, 63.1%).
With aluminum (III) chloride In dichloromethane
Stage #1: 1H-indole-5-carbonitrile With titanium tetrachloride In dichloromethane at 0 - 5℃; for 0.75h; Inert atmosphere;
Stage #2: 4-Chlorobutanoyl chloride In dichloromethane at 20℃; for 5h; Inert atmosphere;
12 Preparation of 3-(4-chlorobutanoyl)indole-5-carbonitrile
To a solution of 5-cyano indole (100 g) in dichloromethane (4 L) titanium tetrachloride (214 g) was added at 0-5 °C under nitrogen atmosphere. The resulting mixture was stirred for 45 minutes and then 4-chlorobutyryl chloride (168.8 g) was added to the mixture drop wise. The reaction mass was stirred at room temperature for 5 hours and again cooled to 0-5 °C. The reaction mass is quenched by ice-cold water (2 L) and stirred for 4 hours. The resulting solid material was filtered, washed with water (300 ml_). Water (1 L) was added to the wet material and the pH of the reaction mass was adjusted to 7.0 with aqueous solution of sodium bicarbonate (3 % w/v). The reaction mass was stirred for 10-15 minutes at room temperature, filtered, washed with water and dried to provide the desired compound.Yield: 120 g (69 %). Purity (by HPLC): 91 .96 %.
With aluminum (III) chloride In 1,2-dichloro-ethane
Stage #1: 1H-indole-5-carbonitrile With titanium tetrachloride In dichloromethane at 0 - 5℃; for 0.75h; Inert atmosphere;
Stage #2: 4-Chlorobutanoyl chloride In dichloromethane at 20℃; for 5h;
12 Preparation of 3-(4-chlorobutanoyl)indole-5-carbonitrile
To a solution of 5-cyano indole (100 g) in dichloromethane (4 L) titanium tetrachloride (214 g) was added at 0-5° C. under nitrogen atmosphere. The resulting mixture was stirred for 45 minutes and then 4-chlorobutyryl chloride (168.8 g) was added to the mixture drop wise. The reaction mass was stirred at room temperature for 5 hours and again cooled to 0-5° C. The reaction mass is quenched by ice-cold water (2 L) and stirred for 4 hours. The resulting solid material was filtered, washed with water (300 mL). Water (1 L) was added to the wet material and the pH of the reaction mass was adjusted to 7.0 with aqueous solution of sodium bicarbonate (3% w/v). The reaction mass was stirred for 10-15 minutes at room temperature, filtered, washed with water and dried to provide the desired compound. [0506] Yield: 120 g (69%) [0507] Purity (by HPLC): 91.96%.
With sodium hydride In N,N-dimethyl-formamide at 20℃; for 1h; Cooling with ice;
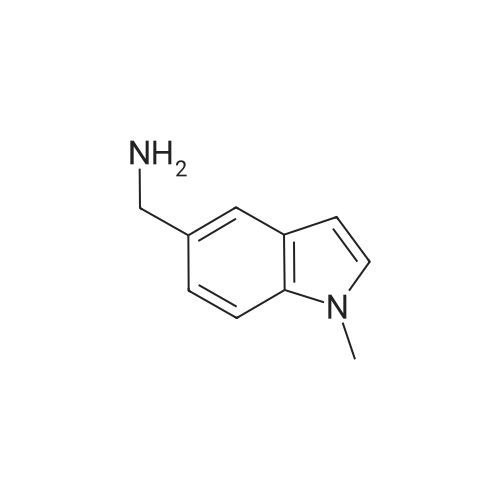
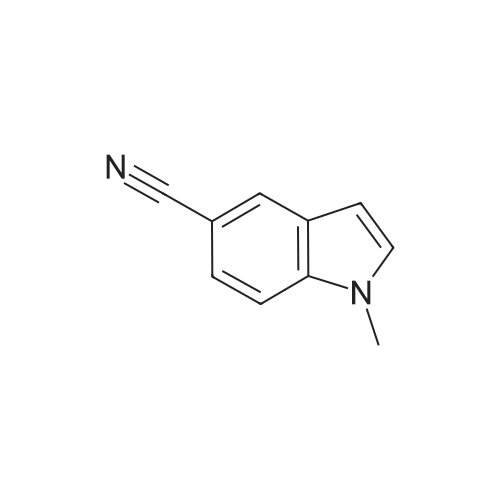
1.1.1 1.1 Preparation of 5-cyano-1-methylindole (502A, R1 = methyl)
In a 500mL three-necked bottle, add 5-cyanoindole (10g, 0.0703mol), DMF 50mL, under an ice bath.NaH (4.22 g, 0.1055 mol) was added in portions, iodomethane (12.0 g, 0.084 mol) was added dropwise, and the mixture was transferred to room temperature for 1 h.After the reaction was completed, 150 mL of saturated brine was added, and extracted three times with 80 mL of ethyl acetate, and the organic layers were combined.Add 50mL of saturated brine and wash three times. Separate the organic layer and add anhydrous sodium sulfate to dry overnight.Filter with suction and evaporate the solvent under reduced pressure to obtain 10.5g of light yellow solid. Yield: 93.6%.
89%
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide at 0℃;
Stage #2: methyl iodide In N,N-dimethyl-formamide at 0 - 20℃;
86%
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide at 0℃; Inert atmosphere;
Stage #2: methyl iodide In N,N-dimethyl-formamide at 0 - 20℃; Inert atmosphere;
85%
With sodium hydroxide In dimethyl sulfoxide at 20℃;
82%
With potassium hydroxide In acetone at 20℃; for 1h;
53%
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In acetonitrile at 0℃; for 0.5h;
Stage #2: methyl iodide In acetonitrile at 0 - 20℃; for 16h;
With sodium hydride In N,N-dimethyl-formamide; mineral oil at 20℃; Cooling with ice;
With potassium carbonate In N,N-dimethyl-formamide at 20℃; for 72h;
1
1 -methyl-1 H-indole-5-carbonitrileA mixture of 5-bromoindole (500 mg; 2.55 mmol) and cuprous cyanide (342 mg; 3.83 mmol) in NMP (10 ml_) was heated under microwave irradiations to 1000C for 30 minutes then at 2000C for 30 minutes. The reaction mixture was partitioned with water and DCM and the organic layer was washed with brine and concentrated in vacuo to give a pink solid. Purification by silica column chromatography (DCM) gave a white solid. It was dissolved in DMF (5 ml_) and K2CO3 (704 mg; 5.10 mmol) and iodomethane (543 mg; 3.83 mmol) were successively added. The reaction mixture was stirred at RT for 3 days and then partitioned between water and EtOAc. The organic layer was washed with brine, dried over MgSθ4 and concentrated under vacuum to give a slightly yellow oil which crystallized upon standing to give the title compound as an off-white solid (80 mg, 70%). LC/MS (Method A): 156.9 (M+H)+.
With sodium hydride In N,N-dimethyl-formamide for 0.5h; Cooling with ice;
57.1
5-Cyanoindole (3.0 g) was dissolved in dimethylformamide (20 ml), sodium hydride (1.27 g) and methyl iodide (1.97 ml) were added to the solution under ice cooling, and the resulting mixture was stirred at the same temperature for 30 minutes. Distilled water and ethyl acetate were added to the reaction mixture, the layers were separated, and the organic layer was dried over anhydrous magnesium sulfate, and filtered. By using a crude product obtained by concentrating the filtrate under reduced pressure as a starting material, a dimethyl compound (615.9 mg) was obtained in the same manner as that of Reference Example 17, (1)
With potassium <i>tert</i>-butylate In diethyl ether at 0℃; for 24h;
With potassium <i>tert</i>-butylate In diethyl ether at 0℃; for 24h;
800 mg
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide at 0℃; for 0.5h;
Stage #2: methyl iodide In N,N-dimethyl-formamide at 20℃; for 4h;
57.1
Example 57N4-(5-Cyclopropyl-lH-pyrazol-3-yl)-N2-((l -methyl-lH-indol-5-yl)methyl)pyrimidine-2,4-diamine (1-33) step 1 : To a solution of lH-indole-5-carbonitrile (600 mg, 4.2 mmol) in DMF (5 mL) at 0 °C was added NaH (201 mg, 60%> in oil, 8.4 mmol) with vigorous stirring. The solution was stirred 30 min, then iodomethane (1.8 g, 12.6 mmol) was added. The reaction mixture was stirred at RT for 4 h. The reaction was quenched with water (50 mL) and extracted with EtOAc (3x100 mL). The organic layers were combined, dried (MgSO i), filtered, and concentrated under reduced pressure to afford 800 mg of 1- methyl-lH-indole-5-carbonitrile (294) as white solid: MS (ESI) m/z = 157.3 [M+l]+.
With sodium hydride In N,N-dimethyl-formamide
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0 - 20℃; for 0.5h;
Stage #2: methyl iodide In N,N-dimethyl-formamide; mineral oil at 0 - 20℃;
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide at 0℃; for 0.5h; Inert atmosphere;
Stage #2: methyl iodide In N,N-dimethyl-formamide at 20℃; for 4h;
31.1 Step 1 : 1 -Methyl- l/T-indole-S-earbonitrile
To a solution of l//-indoie~5-carbonitrile (2.09 g, 14.7 mmol) in DMF (16 rnL), NaH (840 mg, 60 percent in oil, 21 mmol) was added with vigorous stirring at 0 °C under argon atmosphere. The solution was stirred for 30 minutes, then iodomethane (9.12 g, 4 mL, 63 mmol) was added. The reaction mixture was stirred at room temperature for 4 hours. The reaction w'as quenched with water (50 mL), and extracted with EtOAc (3x120 mL). The organic layers were combined, a hed with water (2x50 mL), dried over anhydrous NaaSO-y filtered and concentrated in vacuo , and dry toluene was evaporated from the residue to give 2.3 g of the title compound as an off white solid, used in the next step without any purification.
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0℃; for 0.5h; Inert atmosphere;
Stage #2: methyl iodide In N,N-dimethyl-formamide; mineral oil at 20℃; for 4h;
To a solution of l T-indole-5-carbonitrile (2.09 g, 14 7 mniol) in DMF (16 rnL), NaH (840 mg, 60 percent in oil, 21 mmol) was added with vigorous stirring at 0 °C under argon atmosphere. The solution was stirred for 30 minutes, then iodomethane (9 12 g, 4 rnL, 63 mmol) was added. The reaction mixture was stirred at room temperature for 4 hours. The reaction was quenched with water (50 rnL) and extracted with EtOAc (3x120 rnL). The organic layers were combined, washed with water (2x50 mL), dried over anhydrous NaiSCL, filtered and concentrated in vacuo, and dry toluene was evaporated from the residue to obtain 2.3 g of l-methyl-l -indole-5-carbonitrile as an off white solid used in the next step without any purification.
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide at 40℃;
Stage #2: methyl iodide In N,N-dimethyl-formamide at 25℃; for 2h;
With sodium hydride In N,N-dimethyl-formamide at 0 - 20℃; for 4h;
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In tetrahydrofuran; mineral oil at 0℃; for 0.5h; Inert atmosphere;
Stage #2: methyl iodide In tetrahydrofuran; mineral oil at 20℃; for 6h; Inert atmosphere;
Step 1. 1-Methyl-1H-indole-5-carbonitrile (32b)
To a solution oflH-indole-5-carbonitrile (32a, 356 mg, 2.5 mmol) in THF (16 mL)was added NaH (250 mg, 60% in oil, 6.25 mmol) with vigorous stirring at 0 °C under argon atmosphere. The solution was stirredfor 30 min, then iodomethane (710 mg, 312 mL, 5 mmol) was added. The reaction mixture was stirred at room temperature for 6 h. The reaction was quenched with water (50 mL) and extracted with EtOAc (3 120 mL). The organic layers were combined, washed with water (2 30 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo and the residue (390 mg of 32b) was used in the next step without further purification.
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0 - 20℃; for 1.25h;
Stage #2: methyl iodide In N,N-dimethyl-formamide; mineral oil at 0 - 20℃; for 0.5h;
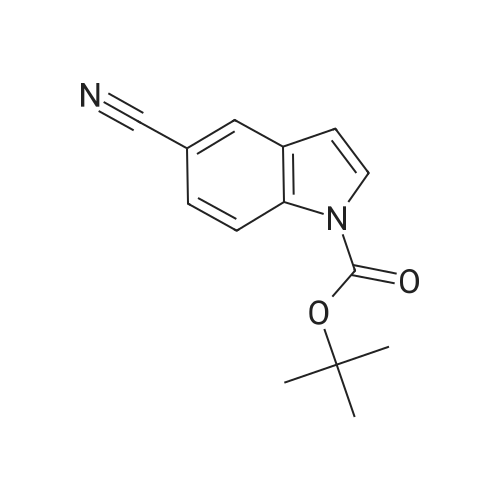
In a 100 mL round-bottom flask was placed 1H-INDOLE-5-CARBONITRILE (2.0 g, 14.07 MMOL) in 20 mL of anhydrous THF. To this solution was added DMAP (0.86 g, 7.03 MMOL) and the mixture was allowed to stir for 0.5 h at rt. At this point, BOC20 (3.07 g, 14.07 MMOL) was added and the reaction stirred for an additional 2 h. The reaction was then quenched with water and extracted twice with ethyl ether. The combined organic layers were washed successively with 1 N HCI, water, and brine, then dried over MGS04 and concentrated to provide 3.26 g (96%) of the desired product as a white SOLID.'H- NMR (DMSO-d6) 8 8.20-8. 14 (m, 2H), 7.83 (d, 1H), 7.70 (d, 1H), 6.80 (d, 1H), 1.63 (s, 9H).
96%
In a 100 ml round-bottom flask was placed 1W-indole-5-carbonitrile (2.0 g, 14.07mmol) in 20 ml of anhydrous THF. To this solution was added DMAP (0.86 g, 7.03mmol) and the mixture was allowed to stir for 0.5 h at rt. At this point, BocaO (3.07 g,14.07 mmol) was added and the reaction stirred for an additional 2 h. The reaction wasthen quenched with water and extracted twice with ethyl ether. The combined organiclayers were washed successively with 1N HCI, water, and brine, then dried over MgSO4and concentrated to provide 3.26 g (96%) of the desired product as a white solid. 1H-NMR (DMSO-c/e) 5 8.20-8.14 (m, 2H), 7.83 (d, 1H), 7.70 (d, 1H), 6.80 (d, 1H), 1.63 (s,9H).
90%
With dmap; In acetonitrile; at 20℃; for 0.5h;
[0486] tert-butyl 5-cyano-l H-indole-1 -carboxylate (INT-65) [0487] To a flask containing 5-cyanoindole (500 mg, 3.52 mmol) in CH3CN (5 mL) was added Boc20 (920 mg, 4.22 mmol) and DMAP (42 mg, 0.35 mmol) and the mixture was stirred at room temperature for 0.5 h. The mixture was concentrated, redissolved in DCM and chromatographed (EtOAc / hexanes) to provide 766 mg (90%) of tert-butyl 5-cyano-lH- indole-l-carboxylate INT -65 as a white solid. LCMS-ESI (m/z) calculated for C14H14N20 :242.27; found 243.1 [M+H]+, tR = 3.93 min.
With triethylamine;dmap; In dichloromethane; at 0℃; for 2h;
After introducing 5-cyanoindole, Boc2O, dichloromethane and DMAP into a 100 ml round-bottomed flask, the reaction medium is stirred at 0 C. under nitrogen for 2 hours. After disappearance of the starting cyanoindole, the reaction medium is poured into water and extracted with EtOAc. After drying and evaporating off the solvent, 1.7191 g of N-Boc-5-cyanoindole are obtained in the form of a yellowish powder. Rf (silica)=0.61; 7/3 cyclohexane/EtOAc. LC/MS m/z=242.
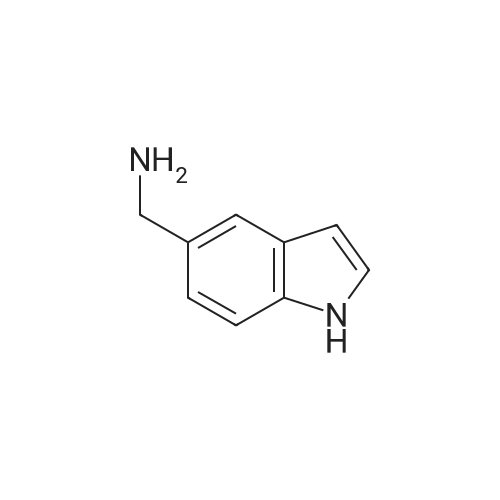
F
5-Cyanoindole was hydrogenated over Raney Nickel in methanol with aqueous ammonia. The solution was concentrated under reduced pressure to a light yellow solid (C-(1H-indol-5-yl)-methylamine, 5.25 g, quantitative).
98%
With [Ru(H)(BH4)(CO)(PPh3)(3-(di-tert-butylphosphino)-N-((1-methyl-1H-imidazol-2 yl)methyl)propylamine)]; hydrogen In isopropyl alcohol at 150℃; for 3h; Inert atmosphere; Autoclave;
94%
With lithium aluminium tetrahydride In tetrahydrofuran for 3h; Inert atmosphere; Reflux;
91%
With ammonia; hydrogen In water; isopropyl alcohol at 80℃; for 24h; Autoclave;
88%
1.1 Synthesis of 1-(1H-indol-5-ylmethyl)-3-phenethylthiourea (1-5)
Step 1: synthesis of (1H-indol-5-yl)methylamine To an ice cold suspension of aluminium chloride (126mg) in ether (1.5 ml) was added a suspension of lithium aluminium hydride (55 mg) in ether (1.5 ml), followed by stirring for 5 min. A solution of 5-cyanoindole (103 mg) in ether (5 ml) was added dropwise thereto. The mixture was stirred at room temperature for 6 hours, followed by adding aqueous Rochel solution thereto and then stirring for 5 hours. The resulting mixture was basified with 1M aqueous sodium hydroxide solution, extracted twice with ethyl acetate (50 ml), washed with saturated aqueous sodium chlroride solution, dried over magnesium sulfate and then filtered to yield (1H-indol-5-yl)methylamine (93 mg, 88%). 1H NMR(300 MHz, CD3OD): δ 7.46(d, 1H, J=l.OHz), 7.29(d, 1H, J=8.3 Hz), 7.14(d, 1H, J=3.2 Hz), 7.02(dd, 1H, J=1.7, 8.3 Hz), 6.34(dd, 1H, J=0.7, 3.2 Hz), 3.89(s,211)
79%
With lithium aluminium tetrahydride In tetrahydrofuran at 0 - 45℃; for 16h;
42 (1H-indol-5-yl)methanamine (compound 1):
To a stirring suspension of LiA1H4 (452 mg, 12.0 mmol) in THF (10 mL) was added asolution of 1H-indole-5-carbonitrile SM (994 mg, 7.0 mmol) in THF (8 mL) at 0 °C. The mixturewas warmed to 45 °C and stirred for 16 h. The reaction mixture was quenched with water (0.5 mL), 15% NaOH( 0.5 mL) and then water (1.5 mL). The mixture was filtered and concentrated to obtain the residue, which was diluted with EtOAc (30 mL) and washed with water (10 mL) and then brine (10 mL). The organic extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was washed with Et20 (15 mL) to afford (1H- indol-5-yl)methanamine compound 1 (790 mg, 79%) as light yellow solid. ‘H NMR (300 MHz, DMSO-d6): 10.94 (s, 1H), 7.44 (s, 1H), 7.28 (d, J= 8.0 Hz, 1H), 7.04 (d, J 8.0 Hz, 1H), 6.34(s, 1H), 3.74 (s, 2H), 1.71 (s, 2H).
59%
With hydrogen In methanol for 20h;
47
Cyano derivative (N≡C-R7) (0.3g, 1 equivalent) was dissolved in 100 ml of MeOH, then a 40 bar pression of hydrogen is applied in the presence of Ni/Raney for 20 h. The reaction mixture is filtered through celite and concentrated. The crude product was purified by flash chromatography to afford the amine. The amine (1 equivalent) was dissolved in DMF (0.4M), then the ethyl isocyanatoacetate (1 equivalent) was added in one portion and the reaction mixture was let 2h at room temperature. After the reaction was complete (TLC control), the reaction mixture was concentrated and purified by flash chromatography to afford the urea.; Example 47: Preparation of ethyl 2-(3-((indol-5-yl)methyl)ureido)acetate (F575)(47). 5-cyano-indole (0.3 g, 2.1 1 mmol) was reduced to obtain the 5- aminomethylindole (0.18g, 59%) after purification by flash chromatography (AcOEt/MeOH 7/3 then MeOH) Rf=0.09 (MeOH). H NMR (DMSO): δ 2.40 (s, 2H), 3.78 (s, 2H), 6.38 (m, 1 H), 7.10 (d, 1 H, J = 8.3 Hz), 7.29 (m, 1 H), 7.33 (d, 1 H, J = 8.3 Hz), 7.49 (s, 1 H), 1 1 .00 (s, 1 H). The 5-aminomethylindole (57 mg, 0.39 mmol) was used to obtain urea 47 (63 mg, 66%) after treatment of the crude product by EDP Rf=0.57 (AcOEt). H NMR (DMSO): δ 1 .21 (t, 3H, J= 7.1 Hz), 3.81 (d, 2H, J = 6.0 Hz), 4.1 1 (q, 2H, J = 7.1 Hz), 4.28 (d, 2H, J = 5.7 Hz), 6.24 (t, 1 H, J = 6.0 Hz), 6.39 (s, 1 H), 6.58 (t, 1 H, J = 5.7 Hz), 7.01 (d, 1 H, J= 8.3 Hz), 7.38 (m, 3H), 1 1 .03 (s, 1 H). HPLC method A tr= 8.37 mn (97.3%). ESI-MS m/z: 276.2 [M + H]+.
45%
With lithium aluminium tetrahydride In tetrahydrofuran at 20℃;
45%
Stage #1: 1H-indole-5-carbonitrile With lithium aluminium tetrahydride In tetrahydrofuran at 0 - 20℃;
Stage #2: With sodium hydroxide; water In tetrahydrofuran at 0℃;
6
Example 6; Indole-5-methanamme (4).; To an ice-cold 1.0 M solution Of LiAlH4 in THF(18 mL, 0.018 mol) was added dropwise under N2 a solution of 5-cyanoindole (16, 1.56 g, 0.011 mol) in dry THF (25 mL). After the addition was complete, the mixture was allowed to warm to room temperature and was stirred overnight. The resulting mixture was cooled in an ice bath, and excess LiAlH4 was quenched with 10% NaOH. The product was extracted with ethyl acetate and dried over anhydrous magnesium sulfate. The solvent was removed by rotary evaporation to give the crude product (1.1 g), which was recrystallized from ethyl acetate/hexanes to give crystalline 5 (0.7 g, 45%). 1H NMR (400 MHz, DMSO-^) δ 11.0 (s, IH), 7.5 (s, IH), 7.3 (d, 2H, J= 8.4 Hz), 7.0 (d, IH, J= 8.4 Hz), 6.4 (s, IH), 3.8 (s, 2H), 2-3 (br, 2H). HRMS (EI): calculated for C9H10N2 (M+): 146.0838. Found: 146.0835.
32%
With ammonia; hydrogen In methanol at 20℃;
79
To a solution of lH-indole-5-carbonitrile (1.0 g, 7.0 mmol, 1.0 eq) in NLh/MeOH (10 mL) was added Raney Ni (about 100 mg). The suspension was hydrogenated at rt overnight. The mixture was filtered. The filtered cake was washed with MeOH (10 mL). The filtrate was concentrated under reduced pressure. The residue was purified via flash chromatography to afford (lH-indol-5-yl)methanamine as an off-white solid (330 mg, 32%).
With lithium aluminium tetrahydride; aluminium trichloride In diethyl ether at 20℃; for 5h;
With sodium hydrogencarbonate In tetrahydrofuran
8 1H-Indol-5-ylmethylamine
Preparation 8 1H-Indol-5-ylmethylamine A stirred solution of 1H-indole-5-carbonitrile (4.0 g, 28.1 mmol) in THF (50 ml) at 0° C. was treated dropwise with lithium aluminum hydride in THF (1M, 98 ml, 98 mmol). The resultant mixture was stirred, warming to room temperature overnight. Sat. aq. NaHCO3 (60 ml) was added at 0° C. and resultant mixture was filtered through filter agent celite 521 and washed with THF. Evaporation of the solvent gave the crude product which was purified by chromatography (SiO2, gradient elution with 95:5 CH2Cl2:MeOH; 90:10 CH2Cl2:MeOH; 90:10:1 CH2Cl2:MeOH:NH3) to give the desired product (white powder, 12.9 g, 71%). 1H NMR (CD3OD) δ 4.10 (s, 2H), 6.50 (d, 1H), 7.15 (d, 1H), 7.30 (d, 1H), 7.40 (d, 1H), 7.60 (s, 1H). LRMS m/z=147.2 (M+1)+.
79 %Chromat.
With [bis(2-methylallyl)cycloocta-1,5-diene]ruthenium(II); 1,1'-bis(diphenylphosphino)ferrocene; potassium <i>tert</i>-butylate; hydrogen In toluene at 140℃; for 1h; Inert atmosphere; Autoclave; chemoselective reaction;
79 %Chromat.
Stage #1: 1H-indole-5-carbonitrile With cobalt(III) acetylacetonate; tris(2-(dicyclohexylphosphanyl)ethyl)phosphane In <i>tert</i>-butyl alcohol Sealed tube; Inert atmosphere;
Stage #2: With potassium <i>tert</i>-butylate In <i>tert</i>-butyl alcohol Sealed tube; Inert atmosphere;
Stage #3: With hydrogen In <i>tert</i>-butyl alcohol at 140℃; for 18h; Autoclave;
With nitrile reductase (Bac-2 over expressed in E.coli BL21) In aq. phosphate buffer; dimethyl sulfoxide at 37℃; for 24h; Microbiological reaction;
With N-iodo-succinimide; 1-hydroxy-3H-benz[d][1,2]iodoxole-1,3-dione In dimethyl sulfoxide at 15 - 25℃; for 6h;
Representative procedure for the synthesis of isatins from indoles
General procedure: A 5 mL round bottom flask was charged with 0.1 g of indole and 2.0 mL of DMSO. To the resultant solution was added NIS (1.2 equiv)/IBX (3.0 equiv), and the reaction mixture was stirred at 20 ± 5 C.Progress of the reaction was monitored by TLC (thin layer chromatography) analysis. After completion ofthe reaction, the reaction mixture was diluted with 20 mL of ethyl acetate, and the organic phase was washed with saturated solution of Na2S2O3 followed by brine, and then dried over anhyd. Na2SO4. Subsequently, the extract was stripped off the solvent under reduced pressure, and the product was isolated by silica gel column chromatography.
82%
With indium(III) chloride; 1-hydroxy-3H-benz[d][1,2]iodoxole-1,3-dione In water; acetonitrile at 80℃; for 2h;
Multi-step reaction with 2 steps
1: N-iodo-succinimide / dimethyl sulfoxide / 1 h / 30 °C
2: N-iodo-succinimide; 1-hydroxy-3H-benz[d][1,2]iodoxole-1,3-dione / dimethyl sulfoxide / 4 h / 25 - 35 °C
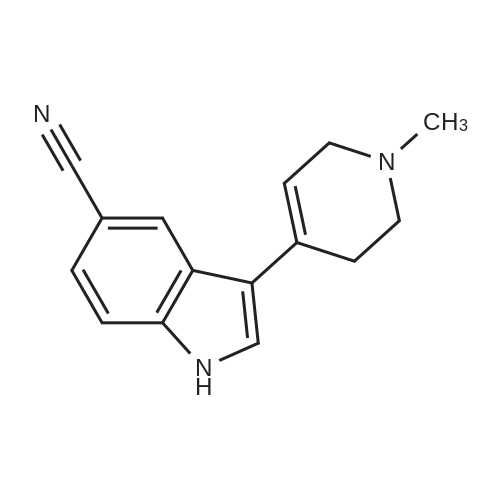
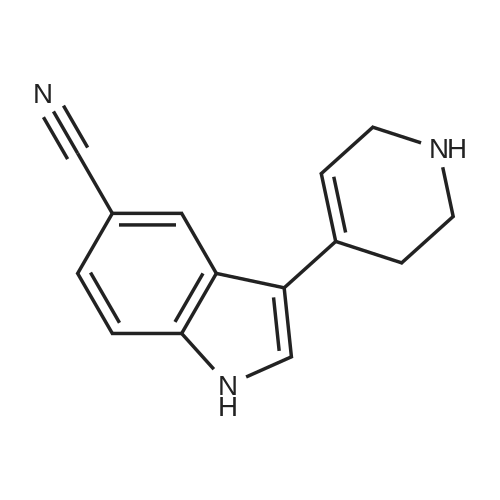
13 3-(1-Methyl-1,2,3,6-tetrahydropyridin-4-yl)-1H-indole-5-carbonitrile (49)
To an argon-purged round bottom flask fitted with a magnetic stirbar containing an orange solution of 5-cyanoindole (48) (250 mg, 1.76 mmol) dissolved in absolute ethanol (10 mL) were added 1-methyl-4-piperidone (0.43 mL, 3.50 mmol) and pyrrolidine (0.44 mL, 5.27 mmol). The reaction vessel was fitted with a condenser and transferred to an oil bath preheated to 80° C. The reaction was stirred at this temperature for 44 hrs. As no starting material remained (TLC 5% 2M NH3 in methanol/95% CH2Cl2) the reaction was cooled to room temperature followed by additional cooling in the fridge. As no precipitate formed, the reaction was concentrated under reduced pressure to afford an orange oil. The oil was redissolved in ethanol (20 mL) and the solvent removed under reduced pressure. This was repeated once more, and then the final residue was treated with ethanol and left in the fridge for 2 hrs. The precipitate which formed was collected by vacuum filtration and washed with hexanes (205 mg of pale yellow solid, compound 49, 48.7%) 1H NMR (DMSO) δ 11.90 (br s, NH), 8.51 (s, 1H), 7.80 (s, 1H), 7.77-7.74 (d, J=8.7 Hz, 1H), 7.68-7.65 (d, J=8.1 Hz, 1H), 6.41 (s, 1H), 3.53 (s, 2H), 3.27-3.26 (d, J=2.4 Hz, 2H), 2.79-2.77 (d, J=4.5 Hz, 2H), 2.72-2.71 (d, J=1.5 Hz, 3H).
With pyrrolidine In ethanol Heating;
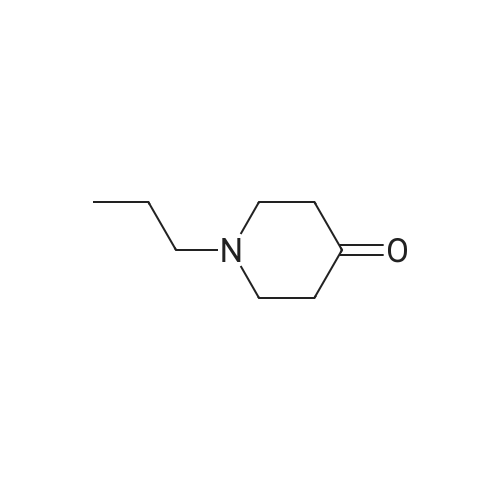
With potassium hydroxide In methanol
S.4.a (a)
(a) 3-(1-Methyl-1,2,3,6-tetrahydro-4-pyridyl)-1H-indole-5-carbonitrile 5-Cyanoindole (Aldrich, 20.0 g) was added to a solution of KOH (22.4 g) in methanol (200 ml). N-Methyl-4-piperidone (Aldrich, 40.4 g) was then added dropwise and the resulting mixture refluxed for 4 hours, then cooled and poured into water. The resulting precipitate was filtered off and dried to give the desired product as a pale pink crystalline solid (32.6 g).
14.1 g (80.5%)
With sodium In methanol
11 5-Cyano-3-(1-methyl-1,2,5,6-tetrahydropyrid-4-yl)indole
PREPARATION 11 5-Cyano-3-(1-methyl-1,2,5,6-tetrahydropyrid-4-yl)indole Procedure identical to Example 4. The reagents used include 5-cyanoindole (10 g, 70.4 mmol), 1-methyl-4-piperidone (8.65 ml, 70.4 mmol), sodium (3.45 g, 0.15 mmol) and methanol (200 ml). Reflux time was 48 hours. An orange solution was allowed to cool down to room temperature and concentrated under reduced pressure to ~100 ml volume. Product crystallized out of the methanolic solution as a beige solid, was collected by filtration and air-dried to afford 14.1 g (80.5%) of the title compound. 1 H NMR (CDCl3) δ=2.44 (s, 3H), 2.52 (bs, 2H), 2.68 (t, J=4 Hz, 2H), 3.12-3.22 (m, 2H), 6.08 (bs, 1H), 7.12 (s, 1H), 7.32 (bs, 2H), 8.12 (s, 1H).
3-(3-oxopentyl)-1H-indole-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
88%
With boron trifluoride diethyl etherate In dichloromethane at 20℃; for 0.0333333h; Green chemistry;
General procedure for the synthesis of 3-substituted indoles/ substituted arenes:
General procedure: Indole/ arene (1.0 mmol), α, β-unsaturated ketone (1.0 mmol) was dissolved in DCM (5mL) then BF3.OEt2 (1 equiv) was added. Immediately The progress of the reaction was monitored by TLC. After completion of the reaction which was indicated by TLC, reaction mixture was quenched with NaHCO3 (3 equiv) and concentrated in vacuo. The crude product was directly poured into silica gel column chromatography (100-200 mesh) using ethyl acetate: hexane (15:85 to 20:80) as eluent to afford corresponding 3-Substituted indoles and substituted arenes. The all obtained products were characterized by 1H NMR, 13C NMR, Mass and IR spectral data.
85%
In water at 20℃; for 2h;
65%
With ytterbium(III) triflate In acetonitrile at 20℃; for 14h; Inert atmosphere;
With triethylsilane; In trifluoroacetic acid; at 0℃; for 4h;
A solution of 5-cyanoindole (1 g, 7 mmol) in 10 mL TFA was cooled to 0 C. and then triethylsilane (1.6 g, 2 eq.) was added. The reaction mixture was stirred at 0 C. for 4 h then diluted with EtOAc and washed with 1M HCl solution. The aqueous layers were combined and neutralized with 50% NaOH to pH10 then extracted 3× with EtOAc. These latter extracts were combined, washed with brine, dried and evaporated to yield the indoline (77%).
67%
With boron trifluoride diethyl etherate; sodium cyanoborohydride; In methanol; at 25℃;
Dissolve 5-cyanoindole (142 mg, 1 mmol) and sodium cyanoborohydride (63 mg, 1 mmol) in methanol (2 ml). Boron trifluoride etherate (152 drops, 1.2 mmol) was added dropwise and allowed to react at room temperature for 2-3 hours. After quenching, add 25% ammonia to quench. The reaction was followed by extraction with ethyl acetate (10 mL x 3 times). The organic layers were combined, dried over anhydrous magnesium sulfate, concentrated and subjected to column chromatography (oilEther: ethyl acetate = 10:1) was isolated to give 97 mg of the desired product in 67% yield.
56%
With triethylsilane; trifluoroacetic acid; at 0 - 20℃; for 2h;
Triethylsilane (2.0 eq.) was slowly added to a solution of indole (1 .0 eq.) in trifluoroacetic acid (1 .4 M), cooled to 0 C. The reaction mixture was stirred at 0 C for 1 h, then at r.t. for 1 h. Upon completion (monitored by TLC), the reaction was basified to pH 1 1 with NaOH (5.0 M) and extracted with EtOAc (x3). The organic layers were combined, washed with brine, dried over Na2S04 and concentrated under reduced pressure. The crude product was purified by flash chromatography to give the desired indoline. Following general procedure M, 5-cyanoindole (600.0 mg, 4.22 mmol) afforded indoline-5-carbonitrile (340.0 mg, 2.36 mmol, 56% yield) as a light yellow solid. UPLC-MS (ES+, Short acidic): 1 .30 min, m/z 289.0 [2M+H]+.
40%
With sodium cyanoborohydride; In acetic acid; at 20℃;
A solution of 5-cyanoindole (3 g, 0.021 mol) in glacial acetic acid (25 mL) was treated portionwise with sodium cyanoborohydride (4 g, 0.063 mol) over 20-30 min then the solution was stirred overnight at rt under N2. The reaction was quenched by addition of water, and most of acetic acid was removed in vacuo. The residue was diluted with water and adjusted to pH>8 with 1M NaOH then extracted 3× with ethyl acetate. Extracts were combined and back extracted 2× with 1M HCl then set aside. (Starting indole can be recovered from these initial EtOAc extracts if desired.) Aqueous acid extracts were combined and rebasified with 5N NaOH, and then re-extracted with EtOAc. The latter extracts were combined, washed with brine, dried over anh. Na2SO4, filtered and evaporated to provide the indoline product (1.22 g, 40%) as an off-white crystalline solid. 1H NMR (CDCl3, 300 MHz) delta 7.29-7.31 (m, 2H), 6.54 (d, 1H, J=8.4 Hz), 4.20 (bs, 1H), 3.67 (t, 2H, J=8.6 Hz), 3.06 (t, 2H, J=8.6 Hz)
33%
Reference Example 7 2,3-dihydro-1H-indole-5-carbonitrile; To a solution (5 mL) cooled to 0C in an ice bath of 1H-indole-5-carbonitrile (640 mg, 4.50 mmol) in acetic acid was added sodium cyanotrihydroborate (848 mg, 13.5 mmol). The reaction mixture was stirred at room temperature for 5 hr and diluted with water. The mixture was basified with 8N aqueous sodium hydroxide solution and extracted with ethyl acetate. The organic layer was extracted with 1N hydrochloric acid and the aqueous layer was again basified with 8N aqueous sodium hydroxide solution and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate and filtered. The filtrate was concentrated. The residue was crystallization from ethyl acetate to give the title compound as pale-yellow crystals (213 mg, yield 33%). 1H-NMR (300 MHz, CDCl3)delta:3.06 (t, J = 8.5 Hz, 2 H), 3.67 (t, J = 8.7 Hz, 2 H), 3.83 (br s, 1 H), 6.45 - 6.59 (m, 1 H), 7.24 - 7.33 (m, 2 H).
With triethylsilane; In trifluoroacetic acid; at 0℃; for 4h;
To a stirred solution of 5-cyanoindole (20.0 g, 140 mmol) in trifluoroacetic acid (150 mL) at 0 C. was added triethylsilane (45.2 mL, 281 mmol). The reaction mixture was stirred for 4 h at 0 C. The reaction mixture was diluted with EtOAc (200 mL) and the organic layer was extracted with 1N HCl (200 mL). The aqueous layer was basified with 50% NaOH solution to pH 10-11 and extracted with EtOAc (2*200 mL). The combined organic layers were dried over anhydrous Na2SO4 and evaporated to dryness to give 2,3-dihydro-1H-indole-5-carbonitrile (6.2 g, 31%) as a pale yellow solid. The material was used without further purification.
With iodine; potassium hydroxide; In N,N-dimethyl-formamide; at 10℃; for 0.5h;Inert atmosphere; Darkness;
Example 1 Synthesis of 3-iodo-1H-indole-5-carbonitrile (V) 1H-indole-5-carbonitrile (5 g, 35.2 mmol), KOH (7.90 g, 141 mmol) and I2 (8.90 g, 35.2 mmol) are suspended in 25 mL of DMF under inert atmosphere. The reaction is maintained under stiffing in the dark for 30 min at 10 C., and then treated with an 0.1M solution of Na2S2O3 (150 mL) The resulting suspension is maintained under stirring for 30 min, then filtered, and the resulting solid is washed with water and dried at 50 C. under vacuum to constant weight. Product (V) (9.0 g) is obtained as a white solid with a yield of 95%. 1H-NMR (400 MHz, CDCl3), delta: 8.78 (1H, bs); 7.80 (1H, s); 7.46-7.40 (3H, m).
94.5%
With iodine; potassium hydroxide; In N,N-dimethyl-formamide; at 20℃;
Preparation of compound 69a: 3-iodo-l/7-indole-5-carbonitrileTo a solution of l//-indole-5-carbonitrile (lg, 7.048 mmol) in DMF (10 mL) was added KOH pellets (1.15 g, 21.14 mmol), followed by I2 (3.57 g, 14.08 mmol) portionwise. The reaction was stirred for 1 h at RT. 10% aq sodium bisulphite solution (10 mL) was added to the mixture and an off-white solid precipitate was formed. The suspension was filtered and the solid was washed with H20 (10 mL) and dried under vacuum to give 3-iodo-lH- indole-5-carbonitrile (1.7 g, 94.5%) as an off white solid. .H NMR (400MHz, DMSO-d6): delta 12.18 (brs, 1H), 7.78 (s, 1H), 7.75 (d, J=1.2Hz, 1H), 7.59 (dd, J=8.4, 0.4Hz, 1H), 7.52 (dd, J=8.4, 1.6Hz, 1H).
93%
With N-iodo-succinimide; In dimethyl sulfoxide; at 30℃; for 1h;
General procedure: To a 5 mL round-bottom flask containing 0.1 g of indole dissolved in 2.0 mL of DMSO, NIS (1.05 equiv) was added, and the reaction mixture was stirred for 0.5-24 h (cf. Table 3). After completion of the reaction as monitored by TLC, the reaction mixture was diluted with 20 mL of ethyl acetate, and it was washed with saturated solution of Na2S2O3. The organic phase was subsequently dried over anhyd. sodium sulfate and the solvent was removed in vacuo. The resultant crude product compound was subjected silica gel column chromatography to isolate the product; in the case of 5-methoxy-3-iodoindole, neutral alumina was employed.
88%
With iodine; potassium iodide; sodium hydroxide; In methanol; water; at 25℃; for 6h;
Compound 13 (15 g) was taken in methanol (300 ml) and water (30 ml) mixture and NaOH (4.6 g) and KI (20.89 g) were added. Iodine (30.51 g) was added portion wise at RT. Reaction mix- ture was stirred at RT for 6 hr. Reaction was monitored using LCMS & TLC. Reaction mixture was diluted with water (1000 ml) and the precipitated solid was filtered-off and dried under vac- uum. Yellow solid obtained was dissolved in DCM and dried over sodium sulphate and concen- trated under vacuum.25 g of pure solid compound 14 was obtained. 1H NMR (300 MHz, DMSO-d6) delta 12.09 (s, 1H), 7.82- 7.73 (m, 2H), 7.64- 7.47 (m, 2H).
88%
With iodine; potassium iodide; sodium hydroxide; In methanol; water; for 6h;
Compound 14 (15 g) was taken in methanol (300 ml) and water (30 ml) mixture and NaOH (4.6 g) and Kl (20.89 g) were added. Iodine (30.51 g) was added portion wise at RT. Reaction mixture was stirred at RT for 6 hr. Reaction was monitored using LCMS & TLC. Reaction mixture was diluted with water (1000 ml) and the precipitated solid was filtered-off and dried under vac- uum. Yellow solid obtained was dissolved in DCM and dried over sodium sulphate and concentrated under vacuum. 25 g of pure solid compound 15 was obtained. (0552) 1 H NMR 300 MHz, DMSO-d6: delta 12.09 (s, 1 H), 7.82 - 7.73 (m, 2H), 7.64 - 7.47 (m, 2H).
75%
With iodine; sodium hydroxide; In N,N-dimethyl-formamide; at 20℃; for 0.416667h;
5-Cyanoindole (0.500 g, 3.52 mmol) was dissolved in DMF (25 mL), and sodium hydroxide (0.493 g, 8.79 mmol) and iodine (0.902 g, 3.55 mmol) were added thereto, then the mixture was stirred at room temperature for 25 minutes. A saturated aqueous sodium thiosulfate solution was added to the mixture, and the precipitated solid was filtered off to give 5-cyano-3-iodoindole(0.707 g, 75%). ESI-MS: m/z 269 [M + H]+.
With potassium hydroxide; iodine; In DMF (N,N-dimethyl-formamide); at 10℃; for 0.5h;
5-cyanoindole (4.0 g, 28.1 mMol) was dissolved in DMF (20 mL) and potassium hydroxide (4.74 g, 84.4 mMol) was added. The reaction was cooled in a water bath at 10 C. and iodine (7.12 g, 28.1 mMol) was added. After stirring for 30 min the reaction was poured into water (100 mL) with sodium thiosulfate (2 g). The resulting solid 5-cyano-3-iodo-lindole was collected by filtration and recrystallized from ethyl acetate and hexanes.The crystals were dissolved in acetonitrile (60 mL) and N,N-diisopropylethylamine (5.64 mL, 32.3 mMol) and solid p-toluenesulfonyl chloride (6.17 g, 32.3 mMol) was added. After stirring for 1 h, the reaction was poured into water (100 mL) and the resulting solids were collected. The material was recrystallized from hot ethyl acetate/hexanes to provide the product as white needles (7.92 g, 67%): 1H NMR (400 MHz, CDCl3) delta 8.06 (1H, d, J=9.2 Hz), 7.79 (3H, m), 7.73 (1H, d, J=1.5 Hz), 7.61 (1H, dd, J=8.6, 1.5 Hz), 7.29 (2H, d, J=8.5 Hz), 2.38 (3H, s); MS m/e 454.9 (M+Na).
With trichlorophosphate; In N-methyl-acetamide; water;
Preparation 35 3-Formyl-1 H -indole-5-carbonitrile Phosphoryl chloride (4.24ml, 45.48mmol) was added dropwise to dimethylformamide (3.52ml, 45.48mmol) and stirred for 30mins at room temperature. A solution of 1H-indole-5-carbonitrile (5.39g, 37.9mmol) in dimethylformamide (10ml) was added dropwise. A solid precipitated, further dimethylformamide (10ml) was added to aid stirring and the reaction mixture was then stirred at room temperature for 3hr. Water was added to quench the reaction mixture which was then stirred for 18hr. The stirring was stopped and the reaction mixture was left to stand, after 24hr a pink solid had precipitated in the organic layer. The layers were separated and organic layer filtered, washed with water and dried to give the desired product (5.44g, 84%). 1H NMR (DMSO)delta 7.60-7.80 (m, 2H), 8.20-8.30 (m, 2H), 10.00 (s, 1H), 12.20-12.35 (s, br, 1H).
84%
In N-methyl-acetamide; water;
Preparation 35 3-Formyl-1H-indole-5-carbonitrile Phophoyl chloride (4.24 ml, 45.48 mmol) was added dropwise to dimethylformamide (3.52 ml, 45.48 mmol) and stirred for 30 mins at room temperature. A solution of 1H-indole-5-carbonitrile (5.39 g, 37.9 mmol) in dimethylformamide (10 ml) was added dropwise. A solid precipitated, further dimethylformamide (10 ml) was added to aid stirring and the reaction mixture was then stirred at room temperature for 3 hr. Water was s added to quench the reaction mixture which was then stirred for 18 hr. The stirring was stopped and the reaction mixture was left to stand, after 24 hr a pink solid had precipitated in the organic layer. The layers were separated and organic layer filtered, washed with water and dried to give the desired product (5.44 g, 84%). 1H NMR (DMSO) delta 7.60-7.80 (m, 2H), 8.20-8.30 (m, 2H), 10.00 (s, 1H), 12.20-12.35 (s, br, 1H).
With tetrabutylammomium bromide; sodium hydroxide In toluene at 20℃; for 1h;
5 Preparation of 1-tosyl-5-cyano-indole: Comparative Test 1
The 1L 4-neck flask was added 5-cyano-indole (10g, 0.07mol), in toluene (300ml) and 300ml 30% sodium hydroxide and tetrabutylammonium bromide (2.3g, 7mmol), at room temperature was added p-toluenesulfonic chloride (14.8g, 0.074mol). was stirred for 1 hour, extracted three times with dichloromethane, the organic layers combined, washed with brine, dried over anhydrous magnesium sulfate, filtered, and evaporated to dryness under reduced pressure to give 20.2 g of a white solid, yield 98 %, melting point 116 ~ 118 .
98%
With tetrabutylammomium bromide; sodium hydroxide In water; toluene at 20℃; for 1h;
1 1-p-toluenesulfonyl-5-cyanoindole (VI)
Add 5-cyanoindole (V) (10.0 g, 0.07 mol) to toluene (300 mL), and heat until the sample is dissolved. The reaction solution was cooled to room temperature, 15% NaOH aqueous solution (300mL) and tetrabutylammonium bromide (2.3g) were added, p-toluenesulfonyl chloride (14.8g, 0.074mol) in toluene (100mL) was added dropwise at room temperature, and stirred React for 1 hour. It was extracted with ethyl acetate (3×125 mL), the organic layers were combined, washed with saturated brine, and dried over anhydrous sodium sulfate. Suction filtration, and the filtrate was evaporated to dryness under reduced pressure to obtain 20.5 g of light red crude product. Recrystallize with absolute ethanol (120mL) to obtain 20.4g of white crystals, yield 98.0%
98.1%
With tetrabutylammomium bromide; sodium hydroxide In toluene at 20℃; for 1h;
1 1-p-toluenesulfonyl-5-cyanoindole (VI-1)
Add 5-cyanoindole (V-1) (10.0g, 0.07mol) to toluene (300mL), stir to dissolve, add 15% NaOH (300mL) and tetrabutylammonium bromide (2.3g), at room temperature A toluene (100 mL) solution of p-toluenesulfonyl chloride (14.8 g, 0.074 mol) was added dropwise, and the reaction was completed in about 1 hour.Extracted with ethyl acetate (3×125mL), the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, the filtrate was evaporated to dryness under reduced pressure to obtain a pale red crude product, and then recrystallized with absolute ethanol (120mL) to obtain pure white crystals 20.4 g, yield 98.1%,
98.01%
With tetrabutylammomium bromide; sodium hydroxide In water; toluene at 20℃; for 1h;
1 Synthesis of 1-(p-toluenesulfonyl)-5-cyanoindole (VII-1)
5-Cyanoindole (VI-1, 10.00 g, 70.34 mmol) was added to toluene (300 ml) and heated to about 45° C until it was completely dissolved. The solution was cooled to room temperature, 15% NaOH aqueous solution (300 ml) and tetrabutylammonium bromide (2.30 g) were added, and a solution of p-toluenesulfonyl chloride (14.80 g, 78.63 mmol) dissolved in toluene (100 ml) was added dropwise at room temperature and stirred the reaction was stopped after 1 h. Extract with ethyl acetate (3×125 ml), combine the organic layers, wash with saturated brine, and dry over anhydrous sodium sulfate overnight. Suction filtration and spin drying to obtain 20.5 g of pale red crude product. Recrystallization from absolute ethanol (120ml) gave 20.40g of pure white crystals (VII-1) with a yield of 98.01%.
89%
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In tetrahydrofuran; mineral oil at 0℃; for 2h; Inert atmosphere;
Stage #2: p-toluenesulfonyl chloride In tetrahydrofuran; mineral oil at 0 - 20℃; for 15h; Inert atmosphere;
71%
With N-benzyl-N,N,N-triethylammonium chloride; sodium hydroxide In dichloromethane at 20℃; for 4h;
2 Synthesis of 1-tosyl-1H-indole-5-carbonitrile (110-1)
To a mixture of 110-s (5.0 g, 35.2 mmol), TEBA (800 mg, 3.52 mmol) and NaOH (2.54 g, 63.4 mmol) in CH2Cl2 (100.0 mL) was added TsCl (8.0 g, 42.3 mmol). The reaction was stirred at room temperature for 4 h. When the reaction was completed, it was concentrated and purified by silica gel column chromatography (petrol ether/ethyl acetate = 80/1) to afford 110-1 (2.50 g, 71% yield) as a white solid.
58%
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In tetrahydrofuran; mineral oil at 0 - 23℃; Inert atmosphere;
Stage #2: p-toluenesulfonyl chloride In tetrahydrofuran; mineral oil at 0 - 23℃; Inert atmosphere;
In tetrahydrofuran; water
1 5-Cyano-1-(p-toluenesulfonyl)indole
PREPARATION 1 5-Cyano-1-(p-toluenesulfonyl)indole Sodium hydride (8.4 g, 210 mmol) was washed twice with hexane and then suspended in tetrahydrofuran (500 ml). 5-Cyanoindole (20 g, 140 mmol) in tetrahydrofuran (200 ml) was added dropwise. The resulting mixture was stirred at ambient temperature for 1 hour and then p-toluenesulfonyl chloride (26.7 g, 140 mmol) in tetrahydrofuran (200 ml) was added. The reaction was stirred 3 hours more followed by addition of water. The phases were separated and the aqueous phase was extracted twice with ethyl acetate. The combined organic phase was washed with brine, dried over calcium sulfate and concentrated. The residue was recrystallized from ether to afford 29.97 g, 72% of title product; m.p. 129°-131° C.; NMR 8.04 (d, J=8.5 Hz, 1H), 7.85 (d, J=1 Hz, 1H), 7.75 (d, J=9 Hz, 2H), 7.67 (d, J=3.5 Hz, 1H), 7.53 (m, 1H), 7.23 (m, 2H), 6.68 (d, J=3.5 Hz, 1H), 2.34 (s, 3H). IR(CHCl3 solution) 2225, 1597, 1453, 1380, 1289, 1266, 1169, 1138, 1123, 1089 (shoulder), 990. FAB HRMS calcd for C16 H13 N2 O2 S(MH+): 297.0669. Observed m/e: 297.0685.
With N-benzyl-N,N,N-triethylammonium chloride; sodium hydroxide In dichloromethane at 20℃;
With N-benzyl-N,N,N-triethylammonium chloride; sodium hydroxide In dichloromethane at 20℃; for 1h;
With N-benzyl-N,N,N-triethylammonium chloride; sodium hydroxide In dichloromethane at 20℃;
With N-benzyl-N,N,N-triethylammonium chloride; sodium hydroxide In dichloromethane at 20℃; for 2h;
2 Compound 17
The preparation method is as follows: Weigh 5-cyano-indole (1mmol), p-methylphenylsulfonyl chloride (1.2mmol), Sodium hydroxide (1.8 mmol) and TEBA (0.1 mmol) in a 50 mL flask. Add dichloromethane (5 mL), react at room temperature for 2 h, and trace the reaction to complete reaction by TLC. The reaction was quenched with water (10 mL). The organic phases were combined and the organic phase was washed with saturated sodium chloride (10 mL) Drying over anhydrous sodium sulfate and concentrating under reduced pressure afforded compound 17 which was used to afford compound 2 without isolation
In tetrahydrofuran; water
1 5-Cyano-1-(p-toluenesulfonyl)indole
PREPARATION 1 5-Cyano-1-(p-toluenesulfonyl)indole Sodium hydride (8.4 g, 210 mmol) was washed twice with hexane and then suspended in tetrahydrofuran (500ml). 5-Cyanoindole (20g, 140 mmol) in tetrahydrofuran (200 ml) was added dropwise. The resulting mixture was stirred at ambient temperature for 1 hour and then p-toluenesulfonyl chloride (26.7 g, 140 mmol) in tetrahydrofuran (200 ml) was added. The reaction was stirred 3 hours more followed by addition of water. The phases were separated and the aqueous phase was extracted twice with ethyl acetate. The combined organic phase was washed with brine, dried over calcium sulfate and concentrated. The residue was recrystallized from ether to afford 29.97 g, 72% of title product; m.p. 129°-131° C.; NMR 8.04 (d, J=8.5 Hz, 1H), 7.85 (d, J=1 Hz, 1H), 7.75 (d, J=9 Hz, 2H), 7.67 (d, J=3.5 Hz, 1H), 7.23 (m, 2H), 6.68 (d, J=3.5 Hz, 1H), 2.34 (s, 3H). IR(CHCl3 solution) 2225, 1597, 1453, 1380, 1289, 1266, 1169, 1138, 1123, 1089 (shoulder), 990. FAB HRMS calcd for C16 H13 N2 O2 S(MH+): 297.0669. Observed m/e: 297.0685.
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In tetrahydrofuran at 0℃; for 0.166667h;
Stage #2: p-toluenesulfonyl chloride In tetrahydrofuran at 20℃; for 14h;
With hydrogenchloride; potassium hydroxide; ammonium hydroxide; sodium hydroxide; In methanol; dichloromethane;
EXAMPLE 15 5-cyano-3-(1,2,5,6-tetrahydropyridin-4-yl)-1H-indole To a solution of 8.8 gm (157 mMol) potassium hydroxide in 85 mL methanol were added 8.15 gm (57.33 mMol) 5-cyano-1H-indole and 7.86 gm (51.17 mMol) <strong>[40064-34-4]4-piperidone hydrochloride monohydrate</strong>. The resulting mixture was heated to reflux for 48 hours and was then allowed to cool to room temperature. The reaction mixture was concentrated under reduced pressure to about half volume and was then treated with 1M HCl until the pH of the solution was between 1 and 2. The resulting solution was extracted twice with 100 mL of diethyl ether and the remaining aqueous phase was treated with 5N sodium hydroxide until the pH of the solution was between 12 and 14. This aqueous phase was extracted 5 times with 10% methanol in dichloromethane. These organic phases were combined, dried over sodium sulfate and concentrated under reduced pressure to a residue. This residue was subjected to flash silica gel chromatography, eluding with dichloromethane which contained 20% methanol and 2% ammonium hydroxide. Fractions shown to contain product were combined and concentrated under reduced pressure to give 6.86 gm (60%) of the title compound as a solid. m.p.=185-187 C. MS(FD): m/e=223 (M+)
6.86 gm (60%)
With hydrogenchloride; potassium hydroxide; ammonium hydroxide; sodium hydroxide; In methanol; dichloromethane;
EXAMPLE 15 5-cyano-3-(1,2,5,6-tetrahydropyridin-4-yl)-1H-indole To a solution of 8.8 gm (157 mMol) potassium hydroxide in 85 mL methanol were added 8.15 gm (57.33 mMol) 5-cyano-1H-indole and 7.86 gm (51.17 mMol) <strong>[40064-34-4]4-piperidone hydrochloride monohydrate</strong>. The resulting mixture was heated to reflux for 48 hours and was then allowed to cool to room temperature. The reaction mixture was concentrated under reduced pressure to about half volume and was then treated with 1M HCl until the pH of the solution was between 1 and 2. The resulting solution was extracted twice with 100 mL of diethyl ether and the remaining aqueous phase was treated with 5N sodium hydroxide until the pH of the solution was between 12 and 14. This aqueous phase was extracted 5 times with 10% methanol in dichloromethane. These organic phases were combined, dried over sodium sulfate and concentrated under reduced pressure to a residue. This residue was subjected to flash silica gel chromatography, eluding with dichloromethane which contained 20% methanol and 2% ammonium hydroxide. Fractions shown to contain product were combined and concentrated under reduced pressure to give 6.86 gm (60%) of the title compound as a solid. m.p.= 185-187C MS(FD): m/e=223 (M+)
a. Methyl 4-(5-cyanoindol-3-ylmethyl)-3-methoxybenzoate A mixture of 5-cyanoindole (10 g) and freshly prepared silver carbonate on diatomaceous earth (40.66 g) was stirred and heated under reflux in toluene (100 ml) for 18 hr, under an atmosphere of nitrogen. The mixture was cooled to room temperature, <strong>[70264-94-7]methyl 4-bromomethyl-3-methoxybenzoate</strong> (22.7 g) added, and stirring continued for 4 hr. Ethyl acetate (200 ml) was added, the mixture filtered through diatomaceous earth, the filter pad washed with ethyl acetate and the filtrate evaporated. The dark oil obtained was purified by flash chromatography, eluding with 45:45:10 hexane:methylene chloride:ethyl acetate, to give a foam which was crystallized from toluene to give methyl 4-(5-cyanoindol-3-ylmethyl)-3-methoxybenzoate (11.8 g, 53percent) as white crystals; mp 148°-149°; partial NMR (250 MHz, DMSO-d6): 3.83(s, 3H, OCH3), 3.91(s, 3H, OCH3), 4.08(s, 2H, ArCH2 Ar'), 8.00(s, 1H, H4 -indole), 11.49(br s, 1H, H1 -indole).
53%
In ethyl acetate; toluene;
a. Methyl 4-(5-cyanoindol-3-ylmethyl)-3-methoxybenzoate A mixture of 5-cyanoindole (10 g) and freshly prepared silver carbonate on diatomaceous earth (40.66 g) was stirred and heated under reflux in toluene (100 ml) for 18 h, under an atmosphere of nitrogen. The mixture was cooled to room temperature, <strong>[70264-94-7]methyl 4-bromomethyl-3-methoxybenzoate</strong> (22.7 g) added, and stirring continued for 4 h. Ethyl acetate (200 ml) was added, the mixture filtered through diatomaceous earth, the filter pad washed with ethyl acetate and the filtrate evaporated. The dark oil obtained was purified by flash chromatography, eluding with 45:45:10 hexane:methylene chloride:ethyl acetate, to give a foam which was crystallized from toluene to give methyl 4-(5-cyanoindol-3-ylmethyl)-3-methoxybenzoate (11.8 g, 53percent) as white crystals; mp 148°-149°; partial NMR (250 MHz, DMSO-d6): 3.83(s, 3H, OCH3), 3.91(s, 3H, OCH3), 4.08(s, 2H, ArCH2 Ar'), 8.00(s, 1H, H4 -indole), 11.49(br s, 1H, H1 -indole).
53%
In ethyl acetate; toluene;
a. Methyl 4-(5-cyanoindol-3-ylmethyl)-3-methoxybenzoate A mixture of 5-cyanoindole (10 g) and freshly prepared silver carbonate on diatomaceous earth (40.66 g) was stirred and heated under reflux in toluene (100 ml) for 18 h, under an atmosphere of nitrogen. The mixture was cooled to room temperature, <strong>[70264-94-7]methyl 4-bromomethyl-3-methoxybenzoate</strong> (22.7 g) added, and stirring continued for 4 h. Ethyl acetate (200 ml) was added, the mixture filtered through diatomaceous earth, the filter pad washed with ethyl acetate and the filtrate evaporated. The dark oil obtained was purified by flash chromatography, eluding with 45:45:10 hexane:methylene chloride:ethyl acetate, to give a foam which was crystallized from toluene to give methyl 4-(5-cyanoindol-3-ylmethyl)-3-methoxybenzoate (11.8 g, 53percent) as white crystals; mp 148-149°; partial NMR (250 MHz, DMSO-d6): 3.83(s, 3H, OCH3), 3.91(s, 3H, OCH3), 4.08(s, 2H, ArCH2 Ar'), 8.00(s, 1H, H4 -indole), 11.49(br s, 1H, H1 -indole).
With ammonium cerium (IV) nitrate In methanol at 25℃; for 0.25h;
4.1.2. General procedure for the preparation of compounds 15a-f
General procedure: The substituted indoles 14a-f (1 mmol) and ammonium thiocyanate (1.2 mmol) were dissolved in 4 mL of methanol and treated with cerium (IV) ammonium nitrate (CAN; 2.3 mmol) in methanol (25 mL) at room temperature. The reaction mixture was stirred for 15 min. It was then diluted with water (100 mL) and extracted with ethyl acetate (20 mL x 4). The combined organic layer was washed with water and brine, dried over anhydrous MgSO4, filtered and concentrated to give the crude product, which was purified by column chromatography on silica gel with light petroleum/AcOEt (v/v = 3:1).
95%
With Oxone In methanol at 20℃;
92%
With Selectfluor In acetonitrile at 20℃; for 0.166667h;
60%
With Selectfluor In acetonitrile at 20℃; for 2h; Inert atmosphere;
With 1,1,1,3',3',3'-hexafluoro-propanol; at 150℃; for 0.0833333h;Microwave irradiation;Product distribution / selectivity;
EXAMPLE 2; Following the procedure set forth in Example 1 (A) above, indole derivatives were deprotected using TFE or HFIP in a microwave reactor at 150 C. as set forth in Table 1 below.
98%
With 2,2,2-trifluoroethanol; at 150℃; for 1h;Microwave irradiation;Product distribution / selectivity;
Example 1; (A) A solution of the N-Boc protected amine (1 mmol) in TFE (2,2,2-trifluoroethanol) or HFIP (hexafluoroisopropanol) (5 mL) was placed in a sealed microwave vial. The reaction mixture was heated (100C or 150C) in a Biotage - Initiator Sixty microwave reactor with stirring until the disappearance of the starting material was observed. After cooling to room temperature, the mixture was evaporated to dryness under reduced pressure. The crude product was purified by flash-column chromatography. 1H NMR and 13C NMR were measured on Bruker Avance DPX-300 NMR or Bruker Avance-300 NMR spectrometers, operating at a proton (1H) frequency of 300.13 MHz and carbon (13C) frequency of 75.43 MHz.Example 2 Following the procedure set forth in Example 1(A) above, indole derivatives were deprotected using TFE or HFIP in a microwave reactor at 150C as set forth in Table 1 below.
98%
With 1,1,1,3',3',3'-hexafluoro-propanol; at 150℃; for 0.0833333h;Microwave irradiation;Product distribution / selectivity;
Example 1; (A) A solution of the N-Boc protected amine (1 mmol) in TFE (2,2,2-trifluoroethanol) or HFIP (hexafluoroisopropanol) (5 mL) was placed in a sealed microwave vial. The reaction mixture was heated (100C or 150C) in a Biotage - Initiator Sixty microwave reactor with stirring until the disappearance of the starting material was observed. After cooling to room temperature, the mixture was evaporated to dryness under reduced pressure. The crude product was purified by flash-column chromatography. 1H NMR and 13C NMR were measured on Bruker Avance DPX-300 NMR or Bruker Avance-300 NMR spectrometers, operating at a proton (1H) frequency of 300.13 MHz and carbon (13C) frequency of 75.43 MHz.Example 2 Following the procedure set forth in Example 1(A) above, indole derivatives were deprotected using TFE or HFIP in a microwave reactor at 150C as set forth in Table 1 below.
With ferric hydrogen sulphate In dichloromethane at 20℃; for 0.75h;
94%
With titanium(IV) oxide at 80℃; for 0.0833333h; neat (no solvent);
94%
With 1,3-bis(mesityl)imidazolium chloride In dichloromethane at 20℃; for 1h; Schlenk technique;
4.2. General procedure for the synthesis of bis(indolyl)methanes and bis(naphthyl)methane
General procedure: An oven-dried 20 mL Schlenk flask was charged with triazolium salt (0.1 mmol, 0.2 equiv),indole derivatives or naphthol (1.0 mmol, 2.0 equiv). A solution of the aldehyde (0.5 mmol, 1.0equiv, 5.0 mL, 0.1Min CH2Cl2) was then added via syringe. The reaction mixture was stirredat room temperature for 1 h. Next, the reaction was quenched with water and the aqueous mixturewas extracted with EtOAc (10 mL × 3). The extracts were combined, dried with MgSO4,and concentrated to afford the residue, which was chromatographed on silica gel (15 g, 20:1-3:1 PE/EA) to give bis(indolyl)methanes or bis(naphthyl)methane.
80%
With Graphene oxide (GO) In water at 40℃; for 7h; Green chemistry;
2.2 General experiment for the synthesis of bis(indolyl)methanes (3a as an example)
General procedure: A mixture of benzaldehyde 1a (0.5mmol), indole 2a (1.5mmol) and GO 150mg were added in 10ml water with a condenser and then stirred under air at 40°C for 3h. After the completion of the reaction (monitored by TLC), the reaction mixture was cooled to room temperature, extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous sodium sulfate and the solvent was evaporated to dryness. The crude residue was purified by flash chromatography on silica to afford pure 3,3′-(phenylmethylene)bis(1H-indole) 3a as a pink solid (92% yield).
General procecdure for synthesis of bis(indolyl)indolin-2-one derivatives
General procedure: Isatin (1 mmol), substituted indole (2 mmol) and ZnO (0.25 mmol) were taken in the round bottom flask and heated to 100 °C for 2 h. The progress of the reaction was monitored by thin layer chromatography (TLC). After completion, the reaction mixture was suspended in 6 mL (3:3 v/v) chloroform and methanol mixture and the catalyst was filtered through Whatman filter paper. The filtrate was concentrated under reduced pressure and the residue was purified by recrystallization in ethanol solvent.
85%
With iodine In dichloromethane at 20℃; for 18h;
General procedure:
General procedure: To a stirred solution of isatin (1 mmol) and indole (2 mmol) in dry dichloromethane (4 mL) was added molecular iodine (26 mg, 10 mol %) at room temperature. The resulting mixture was stirred at room temperature for the appropriate time (Table 1). After complete conversion, as indicated by TLC, the reaction mixture was quenched by saturated sodium thiosulphate solution (4 mL) and extracted with dichloromethane (3 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4, concentrated in vacuo and purified by column chromatography on silica gel (Merck, 60-120 mesh, ethyl acetate-hexane, 3:7) to afford the pure di(indolyl)indolin-2-one.
84%
With proline, oxalic acid at 20℃; for 0.283333h; Green chemistry;
5 2.3. Typical procedure for the synthesis of 3,3-di(indolyl)indolin-2-one (3a)
General procedure: Indole (0.234 g, 2 mmol) and isatin (0.147 g, 1 mmol) were mixed in oxalic acid: proline (5 ml) as solvent and the resultant mixture was stirred at room temperature for appropriate time. After the completion of the reaction indicated by TLC, 5 ml distilled water was added. The insoluble crude product was filtered, washed with distilled water and recrystallized from ethanol.
78%
With rhodium(III) chloride hydrate In methanol at 50℃; for 0.5h;
With N-benzyl-N,N,N-triethylammonium chloride; sodium hydroxide In dichloromethane at 20℃;
With N-benzyl-N,N,N-triethylammonium chloride; sodium hydroxide In dichloromethane at 20℃; for 2h;
4 Compound 19
The preparation method is as follows: Weigh 5-cyanoindole (1 mmol), p-ethylbenzenesulfonyl chloride (1.2 mmol), sodium hydroxide (1.8 mmol) and TEBA (0.1 mmol) in a 50 mL flask, add dichloromethane (5 mL), react at room temperature for 2 h, and trace the reaction to complete reaction by TLC. The reaction was quenched with water (10 mL). The organic phases were combined and the organic phase was washed with saturated sodium chloride (10 mL) drying over anhydrous sodium sulfate and concentrating under reduced pressure afforded compound 19, which was used directly to afford compound 4 without isolation
With hydroxylamine hydrochloride; triethylamine In ethanol at 80℃;
General Procedure A: Amidoxime Formation.
General procedure: Cyanoindole (1equiv) and HONH2HCl (3 equiv), and TEA (3 equiv) were added to around bottom flask containing EtOH (10 mL EtOH/1 mmol cyanoindole).The reaction mixture was heated to 80 C for 12 h oruntil the reaction appeared to have gone to completion via monitoringby TLC. The solutionwas cooled to room temperature and the solvent was removed under reduced pressure. The resulting solidwas loaded onto Celite and purified on a silica gel column with0e10% MeOH in EtOAc to afford the corresponding amidoximeproduct.
90%
With hydroxylamine hydrochloride; sodium carbonate In ethanol; water for 3h; Reflux;
General Procedure A:
General procedure: A solution of nitrile (2.5 mmol), hydroxylamine hydrochloride (695 mg, 10.0 mol), sodium carbonate (530 mg, 5.0 mol), water (6 mL) and ethanol (9 mL) was refluxed for 1 h (for 2c,d,f-i,l) or 3 h (2e,j,k). The reaction was allowed to cool and the ethanol was removed under reduced pressure. The aqueous layer was extracted with ethyl acetate (3 × 10 mL), the combined organic fractions were dried over anhydrous Na2SO4 and the solvent removed under reduced pressure to afford the desired amidoximes 2, which were sufficiently pure for use without further purification.
82.3%
With ethanol; hydroxylamine hydrochloride; triethylamine at 0 - 80℃;
1 Preparation of the compound of formula 2
General procedure: The powdery pairCyano indole (Formula 1) was dissolved in 50 ml of absolute ethanol,Ice bath to 0 ° C,4.04 g of triethylamine and 1.67 g of hydroxylamine hydrochloride were added and stirred for 0.5 h,Heated to reflux (80 ° C) overnight. TLC detection reaction is complete,The reaction solution was concentrated. Ethyl acetate (50 ml * 3), the combined organic layers were washed three times with saturated sodium chloride solution,Dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel chromatography to give a yellow viscous materialN'-hydroxy-1H-indole-5-carboxamidine(Equation 2) 1.44 g, yield 82.3%
40%
With hydroxylamine hydrochloride; triethylamine In ethanol at 20℃; for 54h; Inert atmosphere; Reflux;
With hydroxylamine hydrochloride; potassium hydrogencarbonate In ethanol for 16h; Reflux;
3-(lH-indol-5-yl)-5-(4-phenyl-5-(trifluoromethyl)thiophen-2-yl)-l,2,4- oxadiazoleTo a solution of lH-indole-5-carbonitrile (2g, 14.06mmol) in ethanol under reflux were added, in three equal portions, hydroxylamine hydrochloride (4.88g, 70.3 mmol) and potassium bicarbonate (7.04g, 70.3 mmol), the reaction was reflux for 16h. The mixture was cooled to room temperature and the solid was filtered. The organic solvent was concentrated under reduced pressure and the ./V-hydroxyimidamide was used in the next step without further purification.To a solution of 4-phenyl-5-(trifluoromethyl)-2-thiophenecarboxylic acid in 1,4-dioxane were added under nitrogen atmosphere EDCI (125mg, 0.65mmol) and ηOBt (88mg, 0.65mmol). The reaction was stirred at room temperature for 30 min followed by addition of ./V-hydroxyimidamide (114 mg, 0.65mmol), the reaction was stirred for 30 additional minutes at room temperature followed by 16 h at 95 0C. The reaction was concentrated under reduced pressure. The crude was diluted with EtOAc (8OmL) and washed with a saturated solution of NaHCO3 (2X50ml). The organic phase was dried over Na2SO4 anhydrous and concentrated under reduced pressure. The product was purified by column chromatography using CH2Cl2:Me0H (9:1) to yield 91mg (46.7%) of the product. MS (EI) m/z: 412 (M+).
With hydroxylamine In ethanol; water for 3h; Reflux;
With hydroxylamine In acetonitrile for 3h; Reflux;
potassiumhexacyanoferrate(II) trihydrate[ No CAS ]
[ 17422-32-1 ]
[ 15861-24-2 ]
Yield
Reaction Conditions
Operation in experiment
91%
With 2-[2-(dicyclohexylphosphino)-phenyl]-1-methyl-1H-indole; palladium diacetate; sodium carbonate; triethylamine In water; acetonitrile at 70℃; for 18h;
64%
Stage #1: potassiumhexacyanoferrate(II) trihydrate; 5-chloro-1H-indole With sodium 2'‐(dicyclohexylphosphaneyl)‐2,6‐diisopropyl‐[1,1'‐biphenyl]‐4‐sulfonate; palladium diacetate; potassium carbonate In water at 120℃; for 12h; Inert atmosphere; Sealed tube;
Stage #2: potassiumhexacyanoferrate(II) trihydrate; 5-chloro-1H-indole With potassium carbonate In water at 120℃; for 12h; Inert atmosphere; Sealed tube;
2.3 General Procedure forPalladium-CatalyzedCyanation ofAryl ChlorideswithK4[Fe(CN)6]·3H2O inPEG-400/H2O
General procedure: A pressure tube equipped with a magnetic stir bar wascharged with Pd(OAc)2 (4.5mg, 0.02mmol), XPhos-SO3Na(20.9 mg, 0.04 mmol), K4[Fe(CN)6]·3H2O (105.6 mg,0.25mmol), K2CO3(35mg, 0.25mmol) and PEG-400(1.0mL). The reaction tube was evacuated and backfilledwith argon (this sequence was carried out three times) andthen aryl chloride (1.0mmol, if liquid) and water (1.0mL)were added by syringe (aryl chlorides that were solids atroom temperature were added with the palladium catalystand ligand). The reaction tube was sealed and the reactionmixture was stirred for 12h at the indicated temperature.After being cooled to room temperature, the mixture wasextracted with cyclohexane (3 × 10mL). The combinedcyclohexane phase was concentrated under reduced pressure,and the residue was purified by flash column chromatographyon silica gel (light petroleum ether-ethyl acetate)to afford the desired aryl nitrile 2.The residue of the extraction was heated to 50C invacuo for 30min to remove the residual cyclohexane, andthen subjected to a second cycle of the cyanation reactionby charging with the same substrates (aryl chloride,K4[Fe(CN)6]·3H2O and K2CO3)under the same conditionswithout further addition of Pd(OAc)2 and Xphos-SO3Na.
potassiumhexacyanoferrate(II) trihydrate[ No CAS ]
[ 15861-24-2 ]
Yield
Reaction Conditions
Operation in experiment
92%
With tris-(dibenzylideneacetone)dipalladium(0); tris(2-morpholinophenyl)phosphine; potassium carbonate In water; <i>tert</i>-butyl alcohol at 85℃; for 10h; Schlenk technique; Inert atmosphere;
85%
With tetrakis(triphenylphosphine) palladium(0); 1,8-diazabicyclo[5.4.0]undec-7-ene In water; <i>tert</i>-butyl alcohol at 85℃; for 8h; Inert atmosphere;
76%
With sodium carbonate In water; N,N-dimethyl-formamide at 120℃; for 24h;
3.29. 1H-indole-5-carbonitrile (2ad)
A round-bottomed flask (10 mL) was charged with 5-bromo-1H-indole(1.5 mmol, 0.294 g), Na2CO3 (1.8 mmol, 0.190 g), the catalyst (0.0075 g, 1.7 mol%) and 1:1 (V/V) DMF/H2O (5 mL). The reaction mixture was heated to 120 °C and K4[Fe(CN)6].3H2O (0.60mmol, 0.253 g) was then added. The final mixture was stirred for 24 h. at 120 °C. After allowing the mixture to cool to room temperature, the catalyst was filtrated.Then H2O (15 mL) was added to the mixture and the product was extracted with ethyl acetate (3 10 mL). The organic extracts were collective and dried over anhydrous Na2SO4. Evaporation of the solvent afforded the crude desired product, which was purified by column chromatography using hexane/EtOAc 20:2 (v/v) as the eluents, to afford the title compound. The spectral data for this compound corresponds to those reported in the literature. Yield: 76%. Yellow solid.1H-NMR (300 MHz, CDCl3/TMS) δ (ppm): 8.59 (s, 1H), 7.97-7.88 (m, 1H), 7.44-7.30 (m, 2H), 7.27 (dd, J = 3.2, 2.5 Hz, 1H), 6.56 (m, 1H). Anal.Calcd.forC9H6N2 (142.16): C, 76.04; H, 4.25; N, 19.71. Found: C, 75.97; H, 4.17; N, 19.65.
With potassium penicillin V; sodium thiosulfate; In N,N-dimethyl-formamide;
Example 1 Synthesis of 3-iodo-1H-indole-5-carbonitrile (V) 1H-indole-5-carbonitrile (5 g, 35.2 mmol), KOH (7.90 g, 141 mmol) and I2 (8.90 g, 35.2 mmol) are suspended in 25 mL of DMF under inert atmosphere. The reaction is maintained under stirring in the dark for 30 min. at 10C, and then treated with an 0.1M solution of Na2S2O3 (150 mL). The resulting suspension is maintained under stirring for 30 min, then filtered, and the resulting solid is washed with water and dried at 50C under vacuum to constant weight. Product (V) (9.0 g) is obtained as a white solid with a yield of 95%. 1H-NMR (400 MHz, CDCl3), delta: 8.78 (1H, bs); 7.80 (1H, s); 7.46-7.40 (3H, m).
With lithium tert-butoxide In N,N-dimethyl-formamide at 100℃; for 24h;
Representative procedure for the direct C-H carboxylation of indole derivatives 1a-p
General procedure: In a dried two-necked test tube was charged with LiOtBu (160 mg, 2.00 mmol) and indole 1a (23.4 mg, 0.4 mmol). The reaction vessel was evacuated under high vacuum and the atmosphere was replace with a balloon of CO2. Then DMF (2 mL) was added and the mixture was stirred for 24 h at 100°C. Then the result mixture was cooled and carefully quenched with a solution of HCl (2 N) and extracted with EtOAc (5x). The combined organic layers were washed with water (2x), brine (1x) and dry over MgSO4. The dried organics were concentrated under reduce pressure and the residue was purified by preparative TLC (hexane:acetone = 1:1) to afford the desired product 2a (153.0 mg, 95%) as a white solid.
90%
Stage #1: 1H-indole-5-carbonitrile; carbon dioxide With lithium tert-butoxide In N,N-dimethyl-formamide at 100℃; for 24h;
Stage #2: With hydrogenchloride In water; N,N-dimethyl-formamide
With pyridine; bis-[(trifluoroacetoxy)iodo]benzene; bis(pinacol)diborane In toluene at 60℃; for 11h;
2
To a solution of the above formula (I), 12 mmol of a two-component catalyst (in the presence of a mixture of pyridine and toluene in a volume ratio of 1: 4) in an air atmosphere, for 3 mmol of triphenylphosphine chloride9 mmol of a mixture of silver tetrachlorophthalate), 35 mmol of an oxidizing agent Phi (TFA) 2 and 20 mmol of a accelerater (i.e., the above-mentioned cyclic borate) were added and then the temperature was raised to 60 ° C and the reaction was stirred at that temperature for 11 hours. After the reaction was completed, the deionized water was added to the filtrate and the ρ H value of the system was adjusted to 6-7. Then, the mixture was extracted with chloroform to give 2-3, and the organic phase was combined and dried over anhydrous magnesium sulfate. And the residue was separated by 200-300 mesh silica gel column chromatography, and the mixture of petroleum ether and acetone in a volume ratio of 1: 2 was used as the eluent to obtain the compound of the above formula (II) in a yield of 97.9%
56%
With morpholine; copper(I) oxide; tert.-butylhydroperoxide; oxygen In chloroform at 20℃; for 18h; regioselective reaction;
37%
With pyridine; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; water; oxygen; silver nitrate; benzoic acid at 65℃; for 48h;
15%
With 2,2,6,6-tetramethyl-piperidine-N-oxyl; benzoic acid In acetonitrile at 65℃; for 72h;
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In tetrahydrofuran for 0.25h; Inert atmosphere; Sonication;
Stage #2: berberine chloride In tetrahydrofuran at 20℃; Inert atmosphere;
1 4.3.1 General procedure for nucleophilic addition of indoles, and 7-azaindole to a quaternary protoberberine skeleton
General procedure: NaH (0.0096 g, 0.24 mmol; 1.2 eq) was added into a 25 ml pear-shaped dried flask containing freshly distilled THF (15 ml) and 5-methoxyindole (0.03237 g, 0.22 mmol; 1.1 eq) under argon atmosphere. The reaction mixture was sonicated for 15 min followed by addition of previously dried (90 °C for 2-3 h) berberine chloride (0.07436 g, 0.20 mmol). The mixture was magnetically stirred at room temperature overnight. When the suspension settled down, the organic liquid phase was taken carefully through a syringe and filtered through HPLC filter. Filtrate was collected and the solvent was evaporated under reduced pressure.
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0 - 20℃; for 1h;
Stage #2: 2-chloropyrimidine In N,N-dimethyl-formamide; mineral oil at 130℃; for 5h;
43 Synthesis of compound (III-7):
0.142g (1mmol) of 5-cyanoindole (purchased from Beijing Bailingwei Technology Co., Ltd.) was dissolved in 2ml of anhydrous DMF, and 0.044g (1.1mmol) was added at 0°C for hydrogenation Sodium (60%, dispersed in mineral oil). The reaction was warmed to room temperature and stirred for 30 minutes. After 30 minutes, 0.137g (1.2mmol) of 2-chloropyrimidine was added, and the reaction was heated to 130°C for 5.0 hours. The reaction was followed by TLC. The indole spot disappeared and the reaction was complete. 10ml of saturated aqueous sodium chloride solution was added to precipitate a solid, and the solid was collected by suction filtration and dried to obtain 0.202 g of compound (III-7) with a yield of 91.8%.
85%
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide at 25℃; for 0.5h; Inert atmosphere; Cooling with ice;
Stage #2: 2-chloropyrimidine In N,N-dimethyl-formamide at 130℃; Cooling with ice;
8.a Step a.
Under N2 atmosphere, two-necked 50mL flask was added indole A, i.e. 5-cyanoindole (5.0mmol, 710.8mg), was dissolved in DMF (15mL), and stirred with ice bath cooling. To the solution was added NaH (6.0mmol, 144mg), naturally warmed to room temperature (room temperature in this embodiment 25 deg. C) stirring was continued for 0.5h. Again ice-cooled, 2-chloropyrimidine (6.0mmol, 687.2mg) in DMF (5mL) was slowly added dropwise, the reaction in an oil bath at 130 deg. C, TLC (thin layer chromatography) to detect the reaction was complete. Purification treatment: Thereto was added H2O (15mL) to quench the reaction then transferred to a separatory funnel, and extracted with EtOAc (15mL × 2), the combined organic phases, and washed with saturated NaCl (20mL × 1). After liquid separation, dried over anhydrous magnesium sulfate, the organic solvent was removed under reduced pressure, and purified by silica gel column chromatography [the V (petroleum ether): the V (ethyl acetate) = 3: 1], to obtain pure product I- 2 (R1 = cyano), yield: 85%.
63%
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0℃; for 0.5h; Inert atmosphere; Sealed tube;
Stage #2: 2-chloropyrimidine In N,N-dimethyl-formamide; mineral oil at 0 - 130℃; for 24h; Inert atmosphere; Sealed tube;
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0℃; for 0.5h;
Stage #2: 2-chloropyrimidine In N,N-dimethyl-formamide; mineral oil at 130℃; for 24h;
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0℃; for 0.5h;
Stage #2: 2-chloropyrimidine In N,N-dimethyl-formamide; mineral oil at 130℃; for 24h;
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0℃; for 0.5h;
Stage #2: 2-chloropyrimidine In N,N-dimethyl-formamide; mineral oil at 100℃; for 24h;
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0℃; for 0.5h;
Stage #2: 2-chloropyrimidine In N,N-dimethyl-formamide; mineral oil at 130℃; Schlenk technique;
General procedure for synthesis of N-pyrimidyl indoles1
General procedure: NaH(60% dispersion in mineral oil, 3.6 mmol) was added in portions at 0 °C to a stirred solution of indole (3.0 mmol) in DMF (10 mL). After stirring for 30 min at 0°C, chloropyrimidine(3.6 mmol) was added and the mixture was stirred at 130 °C for 4-24 h. Then,the reaction mixture was cooled to ambient temperature, poured into H2O(20 mL) and extracted with EtOAc (25 mL×2). The combined organic phase was dried over Na2SO4. After filtration and evaporation ofthe solvents under reduced pressure, the crude product was purified by column chromatography on silica gel to afford the corresponding product.
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0℃; for 0.5h;
Stage #2: 2-chloropyrimidine In N,N-dimethyl-formamide; mineral oil at 130℃; for 24h;
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0℃; for 0.5h;
Stage #2: 2-chloropyrimidine In N,N-dimethyl-formamide; mineral oil at 130℃;
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0℃; for 0.5h;
Stage #2: 2-chloropyrimidine In N,N-dimethyl-formamide; mineral oil at 20℃; for 24h;
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide; mineral oil at 20℃;
Stage #2: 2-chloropyrimidine In N,N-dimethyl-formamide; mineral oil at 120 - 140℃; for 12h;
Stage #1: 1H-indole-5-carbonitrile With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0℃; for 0.5h;
Stage #2: 2-chloropyrimidine In N,N-dimethyl-formamide; mineral oil at 130℃; for 24h;
With caesium carbonate In dimethyl sulfoxide at 110℃; for 1h;
General procedure for N-arylation of N-heterocycles
General procedure: A mixture of CuatCu2O NPs nanocomposite (5 mol% ofCu), Cs2CO3(1.5 mmol), N-heterocycle (1.0 mmol), aryl halide(1.0 mmol), and DMSO (2 mL) under air was stirred for 1 h at 110 °C.After completion of the reaction as indicated by TLC, the heterogeneous mixture was cooled to room temperature and diluted with ethyl acetate (10 mL). The mixture was filtered through a pad of celite. The filtrate was concentrated and then residue was purified by column chromatography (SiO2, ethyl acetate and n-hexane) to yield pure product. The catalysts were recovered by simple filtration and washed extensively with acetone and deionized water and then drying in the air.
3-(p-tolylthio)-1H-indole-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85%
With sodium hydroxide In dimethyl sulfoxide at 70℃; for 6h;
78%
With Graphene oxide In 1,2-dichloro-ethane at 20℃; for 12h; Irradiation; Schlenk technique; Green chemistry;
3.2. General Procedure of the Products 6
General procedure: In a 10 mL Schlenk tube, indole (0.3 mmol), GO (17.6 mg), and thiol (0.36 mmol) werestirred in DCE (1 mL) for 12 h at room temperature under an air atmosphere irradiated byblue LEDs. The reaction mixture was concentrated under reduced pressure. The residuewas purified by flash chromatography on silica gel (eluent: EtOAc/PE = 1:10) to yield thecorresponding product.
47%
With manganese(IV) oxide; iodine; oxygen at 80℃; for 16h; Schlenk technique;
With water; iodine; oxygen; sodium carbonate; In 1,4-dioxane; at 100℃; under 760.051 Torr; for 36h;Schlenk technique; Sealed tube;
General procedure: Under air, a 20 mL of Schlenk tube equipped with a stir bar was charged with indole 1 (0.2 mmol, 1 equiv),TMEDA (75 muL, 0.5 mmol, 2.5 equiv), Na2CO3 (42.4 mg, 0.4mmol, 2.0 equiv), 1,4-dioxane (0.5 mL) and H2O (100 muL). Then I2 (101.5 mg, 0.4 mmol, 2.0 equiv) was added and the tube was sealed with a rubber plug and charged with O2. The reaction mixture was stirred at 100 C for 36 h in oil bath. After cooling to room temperature, the resultant mixture was evaporated with EtOAc (20 mL) under reduced pressure and the residue was purified by flash column chromatography on a silica gel to give the products.
Stage #1: 4-bromobutyroyl chloride With aluminum (III) chloride In 1,2-dichloro-ethane at 0℃; for 0.833333h;
Stage #2: 1H-indole-5-carbonitrile In 1,2-dichloro-ethane at 0 - 20℃; for 2.5h;
2 Example 2: Preparation of 3- (4-bromo-butyryl) indole-5-carbonitrile
At 0 , equipped with mechanical stirring three 1L flask was added 1,2-dichloroethane (200 mL) and anhydrous aluminum chloride (15.4g, 0.12moL). At the same temperature was added dropwise 4-bromo-butyryl chloride (25.0g, 0.13moL), about 20min addition was complete, stirring was continued for 30 min. Was then added 5-cyano-indole (14.0g, 0.096moL) in 1,2-dichloroethane (200mL), keeping the temperature at 0 ~ 5 . The addition was complete within 30min. The ice bath was removed, brought to room temperature and then stirring was continued for 2h. document.write(""); Subsequently, the reaction mixture was added to 100g of ice and 100mL of concentrated hydrochloric acid. The reaction was stirred at room temperature overnight. Filtered and dried under vacuum to give a brown solid 3- (4-bromo-butyryl) indole-5-carbonitrile 21.8g, 78% yield.
1-(4-(2-methoxyethoxy)phenyl)-1H-indole-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
42%
With copper(I) oxide; 1,10-Phenanthroline; tetrabutyl ammonium fluoride; at 150℃; for 2h;Inert atmosphere;
[00214] A mixture of 1 ,10-phenanthroline (0.127 g, 0.703 mmol), 1 mol/L TBAF (10.55 ml, 10.55 mmol), Cu20 (0.05 g, 0.352 mmol), 1 -bromo-4-(2-methoxyethoxy)benzene (0.975 g, 4.22 mmol) and compound 1 (0.5 g, 3.52 mmol) was concentrated in high vacuum. The solvent free residue was warmed to 150 C under nitrogen protection for 2 hr. The mixture was dissolved in DCM (30 ml_), filtered and the filtrate was purified by silica column to afford the title compound (450 mg, 42%) as a white solid. LC-MS: [M+H]+ = 293.1.
methyl 2-(5-cyano-1H-indol-1-yl)-5-methoxybenzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
21.7%
With copper(I) oxide; 1,10-Phenanthroline; tetrabutyl ammonium fluoride at 150℃; for 5h;
56 Intermediate 56.1: methyl 2-(5-cyano-1H-indol-1-yl)-5-methoxybenzoate
[00224] To a mixture of 1 (1 g, 7.03 mmol), methyl 2-bromo-5-methoxybenzoate (2.6 g, 10.55 mmol), 1 ,10-phenanthroline (506 mg, 2.8 mmol), Cu20 (200 mg, 1 .4 mmol) in THF (20 mL) was added TBAF 3H20 (5.5 g, 21 .1 mmol). The solvent was removed after TBAF 3H20 was dissolved completely. The reaction was stirred at 150 °C for 5 h in N2 atmosphere. The reaction mixture was extracted with EA, washed with water and brine. The combined organic layers was dried over anhydrous Na2S04 and filtered. The filtrate was concentrated to give the crude which was purified by column chromatography on silica gel (PE: EA=40:1 ~15:1) to give the title compound (560 mg, 21 .7%). 1H NMR (300 MHz, DMSO-c/6) δ ppm 8.24 (s, 1 H), 7.67 (d, 1 H), 7.61 - 7.49 (m, 3H), 7.43 (dd, 1 H), 7.17 (d, 1 H), 6.84 (d, 1 H), 3.96 (s, 3H), 3.48 (s, 3H). LC-MS: [M+H]+ = 307.4
1-(3-chloro-4-methoxyphenyl)-1H-indole-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
47%
With copper(I) oxide; 1,10-Phenanthroline; tetrabutyl ammonium fluoride; at 150℃; for 5h;
[00288] The mixture of compound 1 (300 mg, 2.1 1 mmol), 4-bromo-2-chloro-1 - methoxybenzene (701 mg, 3.165 mmol), 1 ,10-phenanthroline (152 g, 0.844 mmol), Cu20 (60 mg, 0.419 mmol) and the solution of TBAF in THF (1 N, 6.4 ml_) was concentrated under vacuo to remove solvent. The residue was then heated to 150 C for 5 h. After cooling down, the residue was diluted with water and extracted with EA for 3 times. The organic layer was dried over anhydrous Na2S04, filtered and concentrated to give a crude product which was purified by column chromatography on silica gel (PE/EA=4:1) to give the title compound (280 mg, 47%). 1H NMR (300 MHz, DMSO-cf6) delta ppm 8.26 (s, 1 H), 7.87 (d, 1 H), 7.77 (d, 1 H), 7.68 - 7.54 (m, 3H), 7.41 (d, = 1 H), 6.89 (d, 1 H), 4.00 (s, 3H). LC-MS: [M+H]+ = 283.3.
With palladium on activated charcoal; deuterium In hexane; water-d2; ethyl acetate at 100℃; for 24h;
19 Example 19In this Example, an aromatic deuterated methyl compound was prepared by the following preparation method,The reaction is as follows:
5-Cyanoindole (1 g) was mixed with palladium on carbon (0.1 g) in n-hexane: ethyl acetate:Deuterium water (20 mL: 2 mL: 2 mL), 2 MPa deuterium gas pressure,100 degrees stirring for 24 hours, after the reaction, filtered to give deuterated product,Yield 90%, deuterium rate 98%.
With 1,4-diaza-bicyclo[2.2.2]octane; copper(I) oxide; phenylsilane; ammonia In 1-methyl-pyrrolidin-2-one at 180℃; for 24h; Sealed tube; chemoselective reaction;
General procedure for the cyanation of aryl iodide with CO2 and NH3
General procedure: Under nitrogen atmosphere, Cu2O (10 mol %), DABCO (25 mol %), and a stirring bar were added into a 10 mL oven-dried sealed glass tube (as shown in Figure S1). Then NMP (0.5 mL), aryl iodides (0.125 mmol, 1.0 equiv.) and PhSiH3 (0.75 mmol, 6 equiv.) were injected by syringe. The tube was then sealed and CO2 (0.67 mmol, 5.4 equiv., 15 mL) as well as NH3 (0.67 mmol, 5.4 equiv., 15 mL) were injected by syringe after N2 was removed under vacuum. Finally, the mixture was stirred for 24 hr in a pre-heated-to-130 °C alloyed block. After the reaction was finished, the tube was cooled to room temperature and the pressure was carefully released. The yield of were measured by GC analysis using dodecane as the internal standard or by flash chromatography on silica gel (petroleumether/ethyl acetate).
92 %Chromat.
With ammonia; hydrogen; nickel dibromide In N,N-dimethyl-formamide at 160℃; for 10h; Sealed tube;
23 Example 23: Preparation of 5-cyanoguanidine from 5-Iodo iodine (23)
Under a nitrogen atmosphere, nickel bromide (20 mol%, 5.5 mg) and a magnetite were charged into a previously baked 10 mL glass pressure tube. Then N,N-dimethylformamide (0.5 mL) and 5-iodoguanidine (0.125 mmol, 1.0 equiv., 30.4 mg) were added. The pressure tube was sealed, the tube air was removed and charged with carbon dioxide (5.0 equiv., 15 mL), ammonia (5.0 equiv., 15 mL) and hydrogen (5.0 equiv., 15 mL). After the addition was completed, the glass pressure tube was placed in a metal module that had been preheated to 160° C. and stirred for 10 hours. After the reaction was completed, the reaction system was cooled to room temperature and pressure was slowly released. use Dodecane was used as an internal standard and its yield was determined to be 92% by gas chromatographic working curve
3-(3-chloropropanoyl)-1H-indole-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
89%
With Aluminum Chloride In dichloromethane at 0 - 20℃;
General procedure for the synthesis of intermediates 6(d-k)-1.
General procedure: 4-chlorobutyryl chloride (45 mmol, 5.04 mL) was added to the suspension of AlCl3 (60 mmol, 8.000 g) in CH2Cl2 (150 mL) at 0 °C, the mixture solution was stirred for a moment and a solution of 5-substituted indole (30 mmol) in CH2Cl2 was added dropwise. Then return to room temperature and continue to react for 2 h. Detect the reaction process by TLC, pour the mixture solution to ice water when the reaction finished and a mass of solid was separated out, filter and collect the precipitate, then drying it to afford intermediate 6(d-k)-1.
89%
Stage #1: 2-chloropropionyl chloride With Aluminum Chloride In dichloromethane at 0℃; for 0.166667h;
Stage #2: 1H-indole-5-carbonitrile In dichloromethane at 20℃; for 2h;
3-(3-Chloropropanoyl)-1H-indole-5-carbonitrile (M73)
To the suspension of AlCl3 (60 mmol, 8.00 g) in CH2Cl2 (150 mL), 3-chloropropionyl chloride (45 mmol) was added under stirring at 0 °C. After 10 minutes, a solution of 1H-indole-5-carbonitrile (45 mmol) in CH2Cl2 (100 mL) was added dropwise. Then the reaction mixture was returned to room temperature and continued stirring for 2 h. Monitored by TLC, the mixture was poured into ice water when it reached end point, and a mass of solid was separated out. After filtration, the residue was collected and dried to obtain intermediate M73 as yellow solid in 89% yield. 1H NMR (400 MHz, DMSO-d6) δ 12.54 (s, 1H), 8.65 (d, J = 3.0 Hz, 1H), 8.56 (s, 1H), 7.68 (d, J = 8.4 Hz, 1H), 7.62 (dd, J = 8.5, 1.4 Hz, 1H), 3.98 (t, J = 6.3 Hz, 2H), 3.43 (t, J = 6.3 Hz, 2H).
89%
With Aluminum Chloride In dichloromethane at 0 - 20℃; for 2h;
3-(3-Chloropropanoyl)-1H-indole-5-carbonitrile (m50-a).
4-chlorobutyryl chloride (1.34 g, 10.55 mmol) was added to the suspension of AlCl3 (1.88 g, 14.07 mmol) in 50 mL DCM at 0 °C, the mixture solution was stirred for a moment and a solution of 5-carbonitrile indole (1.00 g, 7.03 mmol) in DCM was added dropwise. Then return to room temperature and continue to react for 2 h. Detect the reaction process by TLC, pour the mixture solution to ice water when the reaction finished and a mass of solid was separated out, filter and collect the precipitate, then drying it to afford intermediate m50-a. Yellow solid, 89% yield. 1H NMR (400 MHz, DMSO-d6) δ 12.54 (s, 1H), 8.65 (d, J = 3.0 Hz, 1H), 8.56 (s, 1H), 7.68 (d, J = 8.4 Hz, 1H), 7.62 (dd, J = 8.5, 1.4 Hz, 1H), 3.98 (t, J = 6.3 Hz, 2H), 3.43 (t, J = 6.3 Hz, 2H).
89%
With Aluminum Chloride In dichloromethane at 0 - 20℃; for 2h;
3-(3-Chloropropanoyl)-1H-indole-5-carbonitrile (m50-a).
4-chlorobutyryl chloride (1.34 g, 10.55 mmol) was added to the suspension of AlCl3 (1.88 g, 14.07 mmol) in 50 mL DCM at 0 °C, the mixture solution was stirred for a moment and a solution of 5-carbonitrile indole (1.00 g, 7.03 mmol) in DCM was added dropwise. Then return to room temperature and continue to react for 2 h. Detect the reaction process by TLC, pour the mixture solution to ice water when the reaction finished and a mass of solid was separated out, filter and collect the precipitate, then drying it to afford intermediate m50-a. Yellow solid, 89% yield. 1H NMR (400 MHz, DMSO-d6) δ 12.54 (s, 1H), 8.65 (d, J = 3.0 Hz, 1H), 8.56 (s, 1H), 7.68 (d, J = 8.4 Hz, 1H), 7.62 (dd, J = 8.5, 1.4 Hz, 1H), 3.98 (t, J = 6.3 Hz, 2H), 3.43 (t, J = 6.3 Hz, 2H).
65%
With Aluminum Chloride In dichloromethane at 0 - 20℃; for 3h;
7
5-cyano-1H-indole (I) (0.1 mol) and AlCl3 (0.1 mol)Soluble in dichloromethane (200 mL),Cool down to 0 ° C and slowly add 3-chloropropionyl chloride (II) (0.1 mol) with stirring.A solution of dichloromethane (50 mL). In accordance with the general law,Intermediate 14. (colorless liquid) 15.12 g was obtained in a yield of 65%.
With sodium carbonate In acetonitrile at 120℃; for 24h; Schlenk technique; regioselective reaction;
Na2CO3-Catalyzed N-Acylation of Indoles; General Procedure
General procedure: A mixture of indole 1 (0.50 mmol), Na2CO3 (10.6 mg, 0.10 mmol, 20 mol%), and alkenyl carboxylate 2 (2.0 mmol, 4.0 equiv) in MeCN (3 mL) was added into a Schlenk flask (25 mL) and stirred at 120 °C until completion of the reaction. Then the solvent was evaporated under reduced pressure and the residue was purified by column chromatography (petroleum ether/EtOAc 20:1 to 5:1) to afford the desired acylindoles 3 (Tables 2 and 3).
88%
With sodium hydroxide In ethanol at 80℃; for 30h; Schlenk technique;
3 Example 3: Synthesis of 1-acetyl-1H-indole-5-carbonitrile (3c)
Accurately weigh 5-cyanoindole (71.1 mg, 0.5 mmol), vinyl acetate (215.2 mg, 2.5 mmol) and sodium hydroxide (10.0 mg, 0.25 mmol), and successovely add them to a 50 mL Schlenk flask. Add ethanol (10.0 mL). Place in an oil bath at 80 ° C for 30 h. After the reaction was completed, the solvent was removed under reduced pressure. Separation of silica gel column using petroleum ether/ethyl acetate as eluent. The yield of 1-acetyl-5-cyanoindole was 88%.
With formic acid; (4,4'-di-tert-butyl-2,2'-dipyridyl)-bis-(2-phenylpyridine(-1H))-iridium(III) hexafluorophosphate; N-ethyl-N,N-diisopropylamine In acetonitrile at 20℃; for 24h; Inert atmosphere; Sealed tube; Irradiation;
87%
With sodium hydride In N,N-dimethyl acetamide at 60℃; for 2h; Inert atmosphere;
General procedure for the detoyslation reaction.
General procedure: To a suspension solution of NaH (0.4 mmol, 2.0 equiv) in dry DMA (1.0 mL) under N2 was added dropwise the solution of 4 or 5 (0.2 mmol, 1 equiv) in dry DMA (0.5 mL) by syringe. Then the mixture was heated at 60 °C until TLC showed the completion of the reaction. A saturated solution of NH4Cl was added to quench the reaction and extracted with EtOAc for one time. The organic layer was washed with water for three times, and the combined aqueous layers were extracted with EtOAc for one time. The combined organic layers were washed with brine and dried over sodium sulfate and concentrated in vacuo. The residue was purified by column chromatography to give corresponding products 3 or 6.
84%
With water; sodium hydroxide In methanol at 60℃; for 2h;
8 Example 8Synthesis of 5-cyanoindole:
Add 5 g of 5-cyano-1-p-toluenesulfonyl-indole to a 100 mL reaction flask,2g sodium hydroxide, 30mL methanol, 10mL water; heated to 60 ° C for 2h;After that, the methanol was concentrated under reduced pressure; the residue was added to a mixed solution of 30 mL of water and 30 mL of ethyl acetate; the liquid was extracted and separated, and the organic phase was dried and concentrated to obtain 3.0 g of a solid;It was washed with 3 mL of isopropyl alcohol at 5 ° C, and crystals were dried to obtain 2.0 g of 5-cyanoindole solid.Yield: 84%, purity: 97%;
With hydroxylamine hydrochloride; sodium carbonate In dimethyl sulfoxide at 40 - 80℃;
4.1.2 General procedure for the preparation of N-hydroxy-1-alkyl-1H-indole-5/6-carboximidamides (7a-7u, 8c, 8e, 8h and 8n)
1-Alkyl-1H-indole-5/6-carbonitrile (3, 5b-5u, 6c, 6e, 6h and 6n, 60mmol), sodium carbonate (120mmol) and hydroxylamine hydrochloride (72mmol) were suspended in 60mL of DMSO. The reaction mixture was stirred at 40°C for 30min and then stirred at 80°C for 2-3h. After completion, monitored by thin-layer chromatography, the solvent was cooled to room temperature and poured into cold water with vigorous stirring. The resulting precipitate was filtered, washed with water and dried to obtain the corresponding title compound, which was analytically pure and used without further purification. 4.1.2.1 N-Hydroxy-1H-indole-5-carboximidamide (7a) (0044) A white crystalline powder; yield 85.9%; M.p 143.6°C-145.3°C; 1H NMR (400MHz, DMSO-d6) δ 11.14 (s, 1H), 9.37 (s, 1H), 7.87 (d, J=1.6Hz, 1H), 7.47 (dd, J=8.6, 1.7Hz, 1H), 7.38-7.33 (m, 2H), 6.46 (t, J=2.7Hz, 1H), 5.69 (s, 2H); ESI-MS m/z 176.06 [M+H]+.
85.9%
With hydroxylamine hydrochloride; sodium hydrogencarbonate In dimethyl sulfoxide at 80℃; for 1.5h;
2.2.1 2.1 Preparation of N'-hydroxy-1H-indole-5-carboximidamide (501B, R1 is hydrogen)
Add 80mL DMSO, hydroxylamine hydrochloride (8.90g, 0.128mol) in a 250mL single-mouth bottle,Sodium bicarbonate (16.13g, 0.192mol), heated to 50 dissociation for 1 hour. 5-cyanoindole (9g, 0.064mol) was added and reacted at 80 ° C for 1.5 hours.After the reaction was completed, the reaction solution was cooled to 15 ° C, 100 mL of cold water was added, and stirred for 20 min.Filter with suction, wash the filter cake twice with water, and dry to obtain 9.5 g of white solid powder, yield: 85.9%,
With hydroxylamine hydrochloride; potassium carbonate In ethanol at 85℃; for 12h;
17; 18 Preparation of N-hydroxy-1H-indole-5-carboxamidine (W-1):
30.0g (0.211mol) of 5-cyanoindole,Potassium carbonate 58g (0.422mol), hydroxylamine hydrochloride 21.7g (0.316mol),Add to 500mL75% ethanol, react at 85 for 12h,TLC monitoring, after the completion of the reaction, suction filtration, the filtrate was evaporated to dryness under reduced pressure,A red paste-like product was obtained, which was directly used in the next reaction without purification.
With hydroxylamine hydrochloride; potassium carbonate In ethanol at 85℃; for 12h;
4.1.9. Synthesis of N-hydroxy-1H-indole-5-carboximidamide (17)
A mixture of 30.0 g 1H-indole-5-carbonitrile (16) (0.211 mol), 58g K2CO3 (0.422 mol), and 21.7g hydroxylamine hydrochloride (0.316mol) was added to 75% ethanol (500 mL). The mixturewas refluxedat 85 for 12 h. After completion of the reaction, the precipitatewas filtered, and the reaction mixture was concentrated underreduced pressure to give compound 17 (red solid), which was usedfor the next reaction without further purification.
1-(3-bromobenzyl)-1H-indole-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90.7%
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
General procedure: A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound.
1-(3-chlorobenzyl)-1H-indole-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
91.5%
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
General procedure: A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound.
1-(3-fluorobenzyl)-1H-indole-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
91.6%
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
General procedure: A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound.
1-(3-methoxybenzyl)-1H-indole-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
93.4%
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
General procedure: A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound.
1-(3-methylbenzyl)-1H-indole-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
87.1%
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
General procedure: A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound.
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
General procedure: A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound.
1-(4-bromobenzyl)-1H-indole-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
94.4%
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
General procedure: A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound.
1-(4-chlorobenzyl)-1H-indole-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
95.2%
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
General procedure: A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound.
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
General procedure: A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound.
1-(4-methoxybenzyl)-1H-indole-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
96.1%
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
General procedure: A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound.
1-(4-methylbenzyl)-1H-indole-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
92.6%
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
General procedure: A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound.
1-(4-tert-butylbenzyl)-1H-indole-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85.2%
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
General procedure: A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound.
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
General procedure: A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound.
1-(cyclohexylmethyl)-1H-indole-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
78.2%
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
General procedure: A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound.
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
General procedure: A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound.
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
General procedure: A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound.
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
General procedure: A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound.
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
General procedure: A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound.
With sodium hydride; potassium iodide In mineral oil at 0 - 80℃;
4.1.1 General procedure for the preparation of 1-alkyl-1H-indole-5/6-carbonitriles (5b-5u, 6c, 6e, 6h and 6n)
A solution of 3 (or 4, 80mmol) in DMF (80mL) at 0°C was treat with sodium hydride (60% dispersion in mineral oil, 90mmol) portionwise, and the reaction was stirred until the hydrogen was completely evolved. The appropriate alkyl halide (100mmol) and potassium iodide (8mmol) were then added to the reaction mixture and the reaction was stirred at 25-80°C. After completion, the reaction was quenched with caution by the dropwise addition of water, treated with saturated sodium chloride, and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the title compound. 4.1.1.1. 1-Methyl-1H-indole-5-carbonitrile (5b). A white crystallinepowder; yield 93.6%; M.p 77.1Ce77.8 C; 1H NMR (400 MHz, DMSO-d6) δ 8.07 (d, J 1.6 Hz, 1H), 7.62 (d, J 8.5 Hz, 1H), 7.53 (d,J 3.2 Hz, 1H), 7.48 (dd, J 8.5, 1.6 Hz, 1H), 6.59 (d, J 3.2 Hz, 1H),3.84 (s, 3H); ESI-MS m/z 157.05 [MH].
With zinc(II) chloride; In dichloromethane; at -10 - 5℃; for 2h;
Compound 1 (142.2g, 1.0mol) was dissolved in dichloromethane (1.5L), the internal temperature was controlled at 0-5 C, and anhydrous zinc chloride (150g, 1.1mol) was added.After the addition was completed, the temperature was lowered to about -10 C, and 1-bromo-4-chlorobutane (Compound 3) (188.6g, 1.1mol) was added dropwise.After the dropwise addition was completed, the temperature was raised to a reaction temperature of 0 C and the reaction was conducted for 2 hours.After the reaction was completed, dichloromethane (0.5 L) was diluted, and ice water (1 L) was destroyed. Subsequently, 1 N sodium hydroxide solution was added to neutralize to adjust pH = 7, and extraction and liquid separation were performed.The organic phase was washed with saturated brine (0.5 L) and separated.After the organic phase was concentrated to dryness, the residue was recrystallized from isopropyl acetate (0.5 L),205 g of the product (I) was obtained, the yield was 88%, and the HPLC purity was 99.62%.
1-(2-phenylacryloyl)-1H-indole-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
57%
Stage #1: 2-phenylacrylic acid With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 20℃; for 3h;
Stage #2: 1H-indole-5-carbonitrile With tetrabutyl ammonium fluoride; potassium hydroxide In dichloromethane; toluene at 20℃;
With water; iodine In N,N-dimethyl-formamide at 150℃; for 0.133333h; Flow reactor;
3. C3-formylation of indoles in the continuous flow reactor
General procedure: The continuous flow reactor system consisted of the dual pump (KP-22-13, SS), PTFE tubing (length: 16 m, ID: 1 mm), T-junction (1.25 mm thru-hole), back pressure regulator (40, 75, 100, 250 psi), oil bath and hot plate. In this experiment, we used BOLA PTFE tubing to experiment at high temperature. In order to react stably at the temperature above the boiling point of water, looked for the maximum temperature conditions according to the pressure of the back pressure regulator in which the bubble does not occur. The reaction started in the concentration of indole (1.0 mmol), HMTA (2.0 mmol) and I 2 (1.0 mmol) in solvent (10 mL). Since this reaction proceeds in a single phase of liquid, the reaction time was set by the inner volume of tubing and the flow rate in the continuous flow reactor. After the reaction is over, the product was quantified using 1 H NMR and the NMR yield was obtained.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






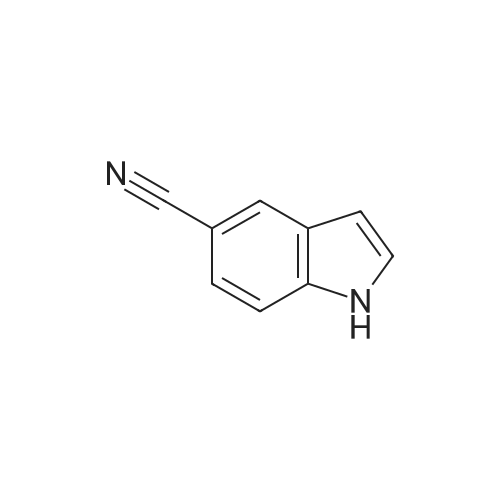

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping