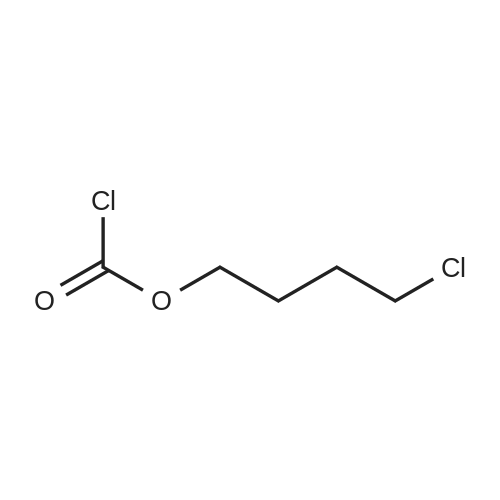
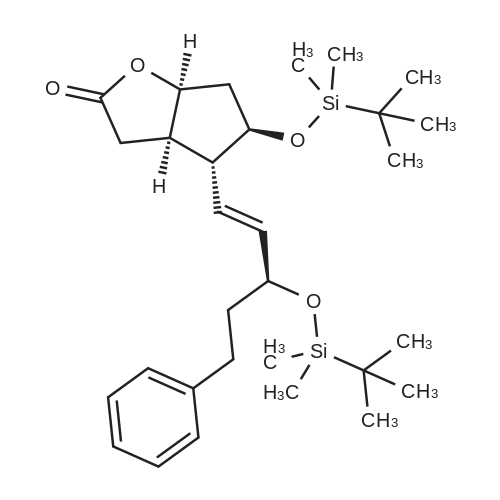
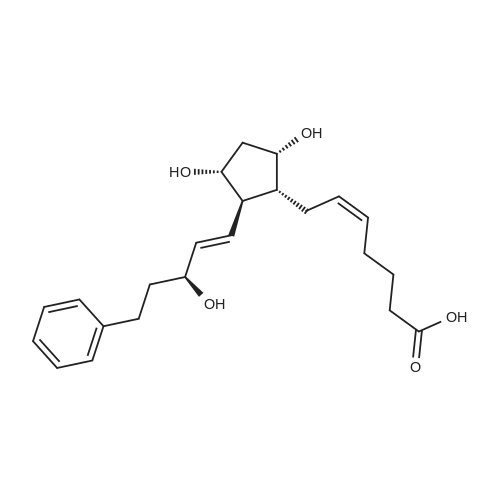
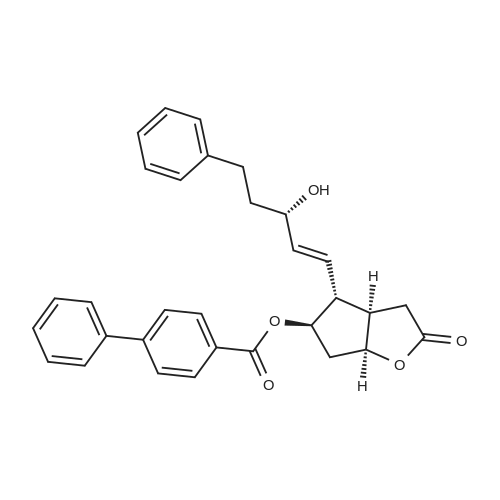
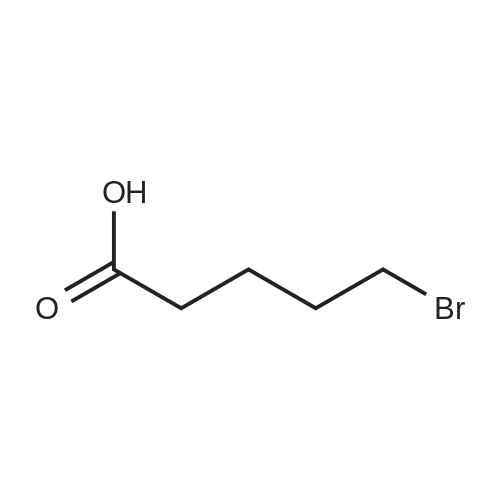
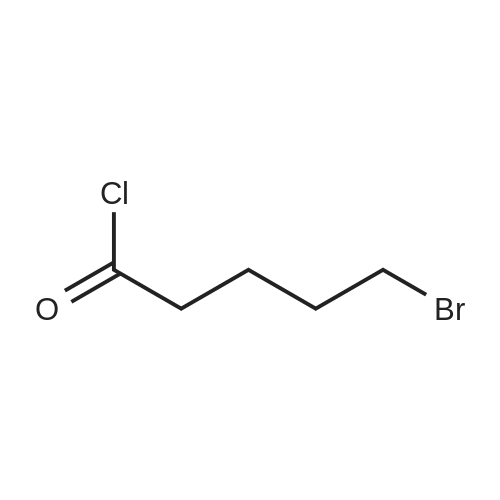
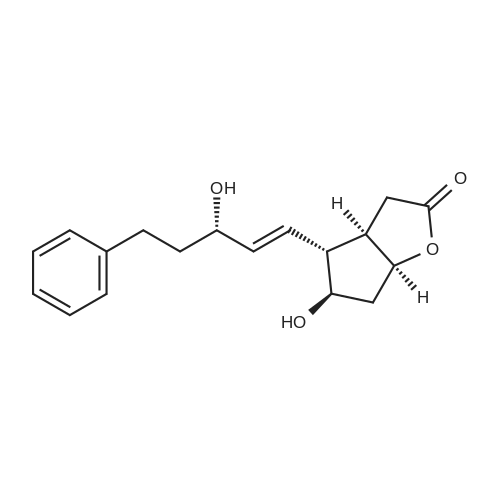
| 12.4%; 1.7% |
With DMAP resin (PS-DMAP); In dichloromethane; at 0 - 20℃; for 18.3333h;Inert atmosphere; |
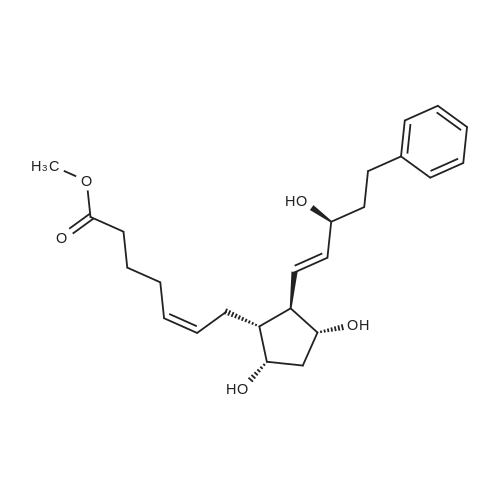
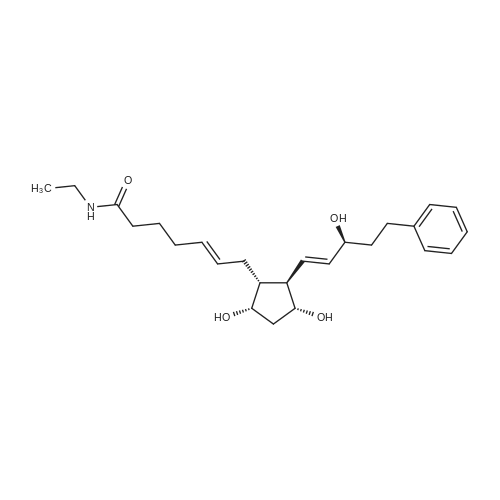
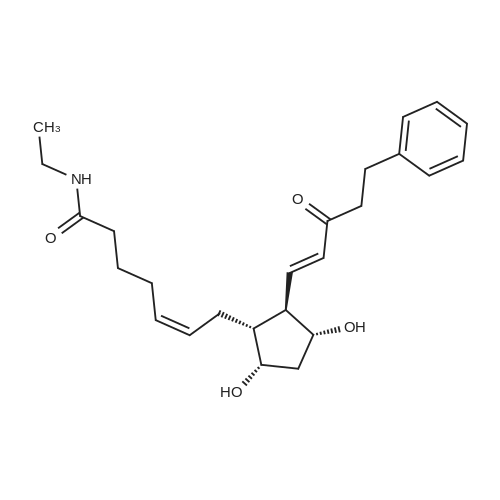
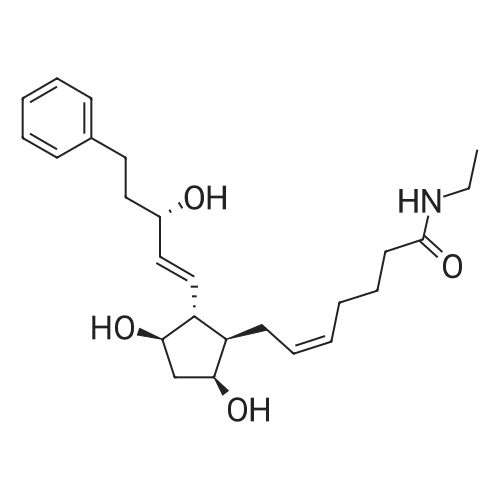
6-(Nitrooxy)hexanoic acid (1.77 g, 10.0 mmol) was dissolved in anhydrous dichloromethane (80 ml_) under nitrogen at ambient temperature. The solution was cooled to 0C, successively added 4 drops of anhydrous DMF and oxalyl chloride (870 uL, 10 mmol), and allowed to warm to ambient temperature. After 12h, the mixture was filtered through a silica plug, which was washed with anhydrous dichloromethane (50 mL). The solvent was evaporated to give 1.71g (88%) of crude presumed acid chloride (6-nitroxy-hexanoyl chloride) as a yellow oil, which was immediately used without further purification. To a suspension of bimataprost (Cayman Chemicals; 831 mg, 2.00 mmol) in dichloromethane (10 mL) at 0C were added DMAP resin (Argonaut (Biotage "PS- DMAP"); 2.75 g of 1.60 mmol/g, 4.20 mmol) and the above crude 6-nitroxy-hexanoyl chloride (822 mg, 2.1 mmol) under nitrogen. After 20 min at 0C, the cooling bath was removed and stirred at ambient temperature 18 hours. The resin was filtered off, and the filtrate concentrated, then the crude mixture was separated by preparative reverse phase HPLC to provide 15-ester F-1 (142 mg, 12.4%) as colorless oil, 9-ester F-2 (20 mg, 1.7%) as a white solid, 9,11-diacyl nitrate F-3 as colorless oil, and 11.15-diacyl ester F-4 as colorless oil, respectively.; Example F-11H NMR (700 MHz, DMSO-d6) delta 0.98 (t, J=7.30 Hz, 3 H), 1.31 (dq, J=7.74, 7.59 Hz, 2 H), 1.42 (dd, J=15.48, 3.98 Hz, 1 H), 1.45 - 1.50 (m, 2H), 1.50 - 1.54 (m, 2 H), 1.57 - 1.63 (m, J=7.35, 7.35, 7.19, 6.86 Hz, 2 H), 1.63 - 1.70 (m, 2 H), 1.95 (d, J=7.08 Hz, 2 H), 1.98 (t, J=7.52 Hz, 3 H), 2.11 (t, J=15.70 Hz, 1 H), 2.25 (t, J=7.30 Hz, 2 H), 2.33 (ddd, J=14.82, 9.29, 5.53 Hz, 1 H), 2.45 (t, J=8.18 Hz, 1 H), 2.55 - 2.64 (m, 2 H), 2.98 - 3.06 (m, 2H), 3.91 (qd, J=5.68, 5.53 Hz, 1 H), 3.95 (d, J=3.98 Hz, 1 H), 4.46 (t, J=6.63 Hz, 2 H), 4.61 (d, J=3.98 Hz1 1 H), 4.75 (d, J=4.87 Hz, 1 H), 4.77 (dd,J=7.74, 4.64 Hz, 1 H), 5.24 - 5.32 (m, 1 H), 5.38 - 5.49 (m, 3 H), 7.16 (d, J=7.52 Hz, 3 H), 7.25 (t, J=7.74 Hz, 2 H), 7.70 (t, J=4.64 Hz, 1 H). 13C NMR (176 MHz, DMSO-cfe): delta 14.78, 24.04, 24.34, 24.49, 25.29, 25.67,26.31, 31.14, 33.20, 33.40, 34.90, 40.00, 41.30, 48.67, 50.77, 69.27, 69.92, 73.88, 78.27, 125.54, 128.17, 128.22, 128.94, 129.24, 129.84, 136.07, 142.23, 171.57, 172.62.Confirmed the structure as depicted, based upon detailed inspection of proton chemical shifts, integration, couplings, as well as key homo and hetero-nuclear correlation observed in 2D spectra. The absence of carbon correlations in the HSQC spectrum allowed for the identification of hydroxyl protons. The observation of correlations from the hydroxyl protons to neighboring methine and methylene protons in the COSY spectrum enabled the determination of substitution of the cyclopentyl ring.Key COSY correlations observed: <n="45"/> Key HMBC correlations observed : A key correlation for the site of attachment was found in the HMBC spectrum for proton at C-11 (numbered 1 above) assigned to resonance at delta 4.77 (dd, J=7.74, 4.64 Hz1 1 H) to the carbonyl of the ester (numbered 7 above) at delta 172.62.LCMS ESI: m/z 597.2 (M+Na+). HRMS. Calcd for C29H42BrNO5Na [M+Na]+: 597.3146. Found: 597.3134 <n="46"/>Example F-2 1H NMR (700 MHz, DMSO-d6) delta 0.98 (t, J=7.30 Hz, 3 H), 1.17 - 1.30 (m, 2 H), 1.33 - 1.41 (m, 4 H), 1.47 (dt, J=15.48, 7.74 Hz, 2 H), 1.56 (qd, J=7.67, 7.52 Hz, 2 H), 1.59 - 1.74 (m, 5 H), 1.86 (dd, J=13.71 , 7.96 Hz, 1 H), 1.90 - 1.96 (m, 1 H), 1.98 (t, J=7.52 Hz, 2 H)1 2.02 (t, J=5.09 Hz, 2 H), 2.17 (dt, J=11.94, 7.96 Hz, 1 H), 2.29 (q,J=7.08 Hz, 2 H), 2.36 (ddd, J=14.82, 8.62, 6.19 Hz, 1 H), 2.56 - 2.66 (m, 2 H), 3.03 (dd, J=7.30, 5.53 Hz, 2 H), 3.76 (dd, J=13.71 , 2.65 Hz, 1 H), 3.88 - 3.95 (m, 1 H), 4.50 (t, J=6.63 Hz, 2 H), 4.73 (d, J=4.42 Hz, 1 H), 4.75 (d, J=5.75 Hz, 1 H), 4.91 (t, J=4.64 Hz, 1 H), 5.29 (d, J=4.42 Hz, 1 H), 5.36 - 5.44 (m, 1 H), 5.46 - 5.53 (m, 1 H), 7.10 - 7.20 (m, 3 H), 7.26 (t, J=7.52 Hz, 2 H), 7.70 (qd, J=5.68, 5.53 Hz, 1 H).13C NMR (176 MHz, DMSO-d6) delta 14.78, 24.00, 24.58, 24.64, 25.23, 25.73, 26.33, 31.32, 33.19, 33.57, 34.85, 40.01, 41.45, 46.55, 54.70, 70.30, 73.67, 73.76, 75.04, 125.57, 128.05, 128.23, 128.26, 129.73, 130.77, 136.00, 142.27, 171.43, 172.27.Confirmed the structure as depicted based upon detailed inspection of proton chemical shifts, integration, couplings, as well as key homo and hetero-?uclear correlation observed in 2D spectra. The absence of carbon correlations in the HSQC spectrum allowed for the identification of hydroxyl protons. The observation of correlations from the hydroxyl protons to neighboring methine and methylene protons in the COSY spectrum enabled the determination of substitution of the cyclopentyl ring. Key COSY correlations observed: <n="47"/> The COSY and HSQC helped assign the hydroxyl protons to resonances at delta 4.73 (d, J=4.42 Hz, 1 H), 4.75 (d, J=5.75 Hz, 1 H) and methine protons; C-9 (numbered 4 above) to delta 4.91 (t, J=4.64 Hz, 1 H), C-11 (numbered 1 above) to ... |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping