| 98.8% |
With pyrographite; sodium sulfate; In tetrahydrofuran; at 8 - 15℃; for 2h; |
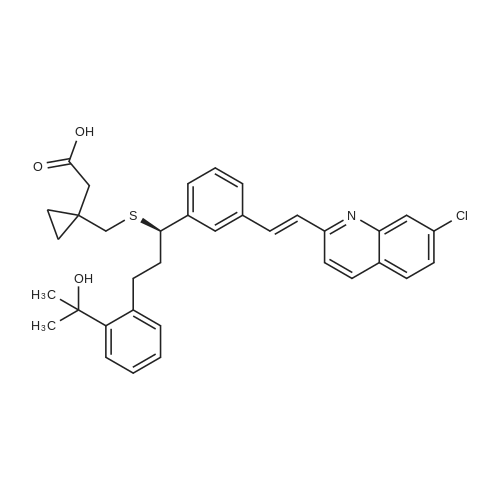
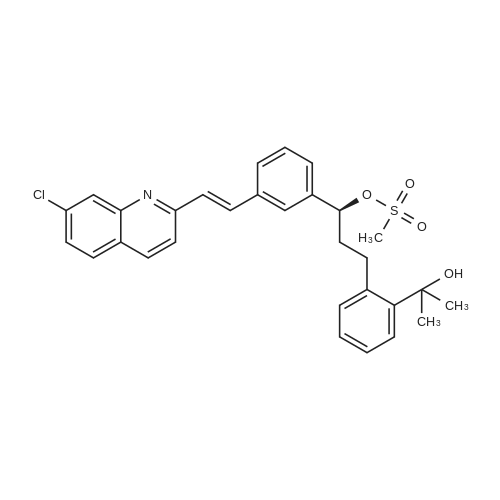
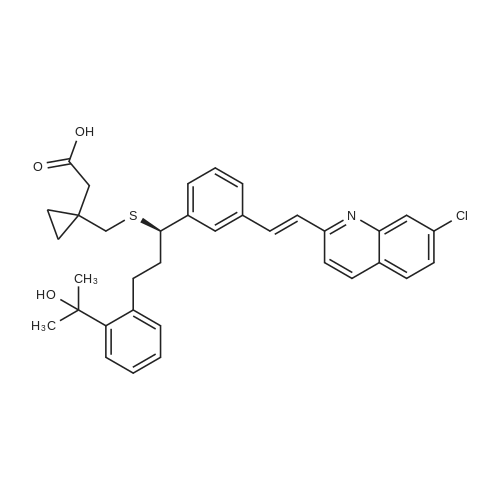
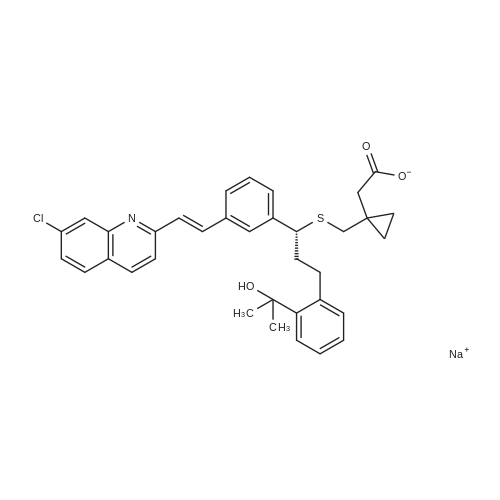
4) To a 1000 mL three-necked flask, 30 g of montelukast acid, 300 mL of tetrahydrofuran were added successively, and 2.05 g1.5g of activated carbon, 4.5g of anhydrous sodium sulphate were added in succession, the temperature was controlled at 8-15 C for 2 hours, filtered, 150 mL of cyclohexane was added and the temperature was controlled at 2 to 10 C for 1 hour , Filtering and vacuum drying for 24 hours at 70 DEG C to obtain montelukast sodium, the yield is 98.8%, the purity is 99.9%, and the impurity content of styrene and sulfoxide is less than 0.02%. |
| 98% |
With sodium hydroxide; In methanol; at 20℃; for 0.5h;Darkness; |
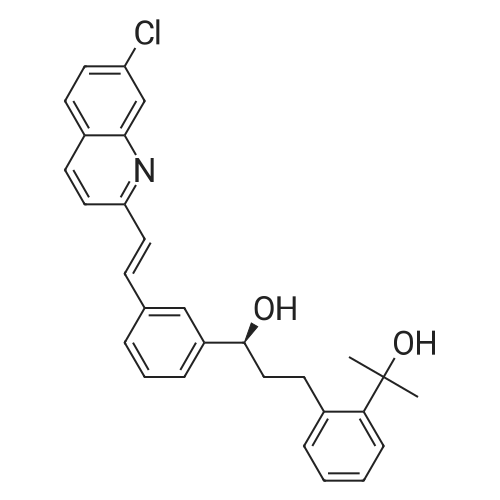
Room temperature, dark room,0.70 g of sodium hydroxide and 50 mL of methanol were placed in a round bottom flask,Stirred for 15 minutes, and then added to the lye dissolved l0gMontelukast acid in 50ml methanol,The reaction was stirred for 30 minutes,Then add activated carbon lg, stirring 30 minutes, and then filtered, the filtrate was concentrated to dry, vacuum drying the product 10. 2g, the yield was 98.0%, HPLC> 99.6%, isomer content <0 ? 1% . |
| 95% |
With sodium hydroxide; In methanol; at 20℃; for 0.5h;pH 9-10; |
A solution of the compound of formula I '(9.8 g, 16.7 mmol)Dissolved in 60 mL of methanol,Methanol solution of sodium hydroxide was slowly added dropwise at room temperature,When the pH of the reaction solution is 9 to 10,Stop dropping, stirring 30min,The solvent was evaporated under reduced pressure and the residue was dissolved in 30 mL of toluene,Then 100 mL of n-heptane was slowly added dropwise,After the addition of 4h,Filtration, the solid at 50 ~ 60 vacuum drying 12h,That is, montelukast sodium 9.6g, the yield was 95%. |
| 94% |
With sodium hydroxide; In methanol;Inert atmosphere; |
In a nitrogen atmosphere, 25 ml of methanol was added to 5.0 g of montelukast free acid and stirred A solution obtained by dissolving 0.35 g of sodium hydroxide in 10 ml of methanol Was added. After confirming that all of the solid was dissolved, the solvent was distilled off under reduced pressure to obtain an oily matter Crude montelukast sodium was obtained. 25 ml of isopropyl acetate was added to the obtained crude montelukast sodium, and the mixture was stirred and mixed , The obtained solution was added dropwise to 150 ml of heptane cooled to 0 C. and kept at the same temperature for 30 minutes . The precipitated solid was collected by filtration by pressure filtration, washed twice with 5 ml of heptane, and the obtained wet body Was dried at 80 C. under reduced pressure for 24 hours to obtain 4.9 g of amorphous montelukast sodium ( Purity 99.8%, isopropyl acetate residual amount 401 ppm, heptane residual amount 991 ppm) (Yield 94%). Amorphous of montelukast sodium obtained is white It was. |
| 92.8 - 97.73% |
With sodium hydroxide; In water; at 20℃;Product distribution / selectivity; |
Step d) Preparation of sodium 1- [ [ [ (IR) -1- [3- [ (IE) -2- (7- chloroquinoline-2-yl) ethenyl]phenyl] -3- [2- (1-hydroxy-l- methylethyl)phenyl]propyl] thio]methyl] cyclopropaneacetate (Ib); Montelukast (Ia) obtained in the previous step was added on an aqueous solution containing 1 equivalent of NaOH. The resulting mixture was stirred at room temperature till a solution was obtained. The resulting solution was filtered and dried at the rotatory evaporator at 50 0C, resulting in the isolation of montelukast sodium (Ib) (92.8% yield, 98.78% purity by HPLC, Titration = 100.29%, Water content (Karl Fischer (K.F.)) = 2.90%); Example 5. Preparation of montelukast sodium (Ib) from a basic aqueous solution of the montelukast (Ia); 50.05 g (titration = 99.91%, 0.085 mols) of montelukast (Ia) (obtained following the same protocol as described in example 4) were added to a basic aqueous solution prepared by mixing 3.927 g (0.097 mols) of sodium hydroxide in 865.00 ml of water. The resulting suspension was stirred until a yellow solution was obtained, which was titrated with tetra n-butyl ammonium hydroxide to check that the salification was complete.The yellow solution was filtered to remove any particulates impurities, resulting in a clear solution, that was lyophilized in a LYOBETA 25 (cycle: 3.3Oh freezing at -45C and 17 h primary drying at -1O0C, 0.200 mbar)The process took one day, after which a yellow porous solid (Ib) was obtained (53.70 g, 97.73% yield, 97.38% purity by HPLC, assay 99.85%, water content (KF) = 5.76%, lod (loss on drying) = 3.47% (8O0C, 3 hours), [alpha] = +91.17, (c = 1%; D = Methanol; Ta = 200C) EPO <DP n="19"/> |
| 87% |
In methanol; ethanol; at 25 - 50℃; for 0.5h;Product distribution / selectivity; |
Dissolved Montelukast free acid (100 g) in methanol (800 ml) by raised the temperature to 50 C., cooled the clear solution to 25 C.-30 C. and added 0.486 molar solution of 1% aqueous ethanol solution (352 ml) over 30 min. Maintained the mass at 25 C.-30 C. for 30 min. and treated the solution with activated carbon, Filtered off the carbon, distilled the solvents from filtrate at temperature below 40 C. under reduced pressure to get residue. Added toluene (100 ml) and again distilled off under reduced pressure to remove traces of methanol, Ethanol. Dissolved the residue in toluene (1000 ml), raised the temperature and maintained at 45 C.-50 C. Cooled the solution to 30 C.-35 C., added carbon, mixed for 15 min and filtered off the carbon. Added n-Heptane (3000 ml) slowly to the clear filtrate over 1 hr at temperature 25 C.-30 C. and maintained for 3 hrs. Filtered the product, washed with n-Heptane (50 ml) and dried at 80 C.-90 C. under vacuum till constant weight.Dry weight of Montelukast sodium is 90 g (87%) |
| 87% |
With sodium hydroxide; In methanol; ethanol; water; at 25 - 50℃;Product distribution / selectivity; |
Example- 10: Preparation of Montelukast sodium from Montelukast free acid Dissolved Montelukast free acid (100 g) in methanol (800 ml) by raised the temperature to 50C, cooled the clear solution to 25C - 30C and added 0.486 molar solution of 1% aqueous ethanol solution (352 ml) over 30 min. Maintained the mass at 25C - 30C for 30 min. and treated the solution with activated carbon, Filtered off the carbon, distilled the solvents from filtrate at temperature below 40C under reduced pressure to get residue. Added toluene (100 ml) and again distilled off under reduced pressure to remove traces of methanol, Ethanol. Dissolved the residue in toluene (1000 ml), raised the temperature and maintained at 45C - 50C. Cooled the solution to 30C - 35C, added carbon, mixed for 15 min and filtered off the carbon. Added n-Heptane (3000 ml) slowly to the clear filtrate over 1 hr at temperature 25C - 30C and maintained for 3 hrs. Filtered the product, washed with n-Heptane (50 ml) and dried at 80C - 90C under vacuum till constant weight. Dry weight of Montelukast sodium is 90 g (87%) The product is identical with the Montelukast sodium obtained in the prior art methods. |
| 80 - 90% |
With sodium methylate; In methanol;pH 8.43;Product distribution / selectivity; |
Example 6; Purified montelukast acid (85 mmol) was treated with 248 mL 0.31 N (77 mmol) sodium methoxide methanol solution (previously degassed), and additional 16.5 mL sodium methoxide was added until a pH of 8.43 was obtained. Most of the methanol was then removed under reduced pressure at less than 50 C. Water was added to the resulting crude oil, and the residual methanol was distilled off. Additional water was added to a final volume of 1,060 mL. The aqueous solution was then filtered, the residual methanol being 3.0% as determined by GC and the purity being 99.8% as determined by HPLC. The aqueous montelukast sodium was subjected to spray drying, and the results were listed in Table 6. |
| 52.8% |
|
Example - 1: Preparation of amorphous montelukast sodium; Montelukast free acid (70 g) is dissolved in methanol (210 ml), 0.486 molar solution of sodium hydroxide in ethanol (230 ml) is added, mixed for about 10-15 min, filtered the resultant reaction mass through hyflow bed, washed the bed with 35 ml methanol. The clear solution is spray dried with spray drier at inlet temperature of HO0C, outlet temperature of 730C to 750C, nitrogen flow rate of 40-50 mm, with a solution feed rate of 30%.Dry wt of the amorphous sodium is 62.0 g, (yield of 52.8%). |
|
With sodium hydroxide; In acetone; at 20℃; for 5h;Product distribution / selectivity; |
EXAMPLE 5 Preparation of Montelukast Sodium in Acetone and Instant Drying Montelukast acid (3 g), NaOH powder (0.24 g, 1.2 eq), and acetone (9 mL) were added to a 100 mL flask equipped with a mechanical stirrer. The reaction was stirred at ambient temperature for 5 hours. The reaction mixture was slowly added dropwise into a 500 mL reactor under vacuum of 1 mm Hg and heated at 35 C. Every drop added was immediately evaporated before reaching the sides or bottom of the reactor. After the end of the addition, the mechanical stirrer was switched on to break the foam formed. The reactor was inverted to collect the powder produced. |
|
With sodium hydroxide; In methanol; at 20℃; for 2h;Product distribution / selectivity; |
EXAMPLE 7 Preparation of Montelukast Sodium in MeOH and Instant Drying Montelukast acid (3 g), NaOH powder (0.24 g, 1.2 eq), and MeOH (15 mL) were added to a 100 mL flask equipped with a mechanical stirrer. The reaction was stirred at ambient temperature for 2 hours until a clear solution was obtained. The reaction mixture was slowly added dropwise into a 500 mL reactor under vacuum of 1 mm Hg and heated at 25 C. Every drop added immediately evaporated before reaching the sides or bottom of the reactor. After the end of the addition, the mechanical stirrer was switched on to break the foam formed. Then the reactor was inverted to collect the powder produced. The characterization of mupirocin calcium is carried out according to WO03/06595 (US 2004/024052). |
|
With sodium hydroxide; In ethanol;Product distribution / selectivity; |
Step d) Preparation of sodium 1- [ [ [ (IR) -1- [3- [ (IE) -2- (7- chloroquinoline-2-yl) ethenyl]phenyl] -3- [2- (1-hydroxy-l- methylethyl)phenyl]propyl] thio]methyl] cyclopropaneacetate (Ib); One equivalent of IN NaOH is added to an ethanolic solution of montelukast (Ia) , obtained in the previous step c) . The solvent is evaporated and water is added to the resulting residue till a solution is obtained. The resulting solution is concentrated to dryness with a rotatory evaporator at 50 0C, resulting in the isolation of compound (Ib) . |
|
With sodium hydroxide; In tetrahydrofuran; water; at 20 - 25℃; for 0.5h;Product distribution / selectivity; |
EXAMPLE 5 1-[1 (R)-(3-(2-(7-chloro-2-quinolinyl)ethenyl)-phenyl)-3-(2-(1-hydroxy-1-methylethyl)phenyl)propyl)thiomethyl)-cyclopropaneacetic acid sodium salt (I) The solution of the acid from Example 4 was transferred into a 250 ml flask fitted with a mechanical stirrer and a thermometer. 1 equivalent of 1N NaOH was added and stirring of the mixture was continued at 20-25 C. for 30 minutes. The phases were separated and the aqueous phase was freeze-dried. 45 g of a light yellow solid were obtained in this manner (HPLC A%=99.5%). 1H-NMR(CDCl3): ppm 0.15-0.20 (d, 2H); 0.40 (d, 2H); 1.50 (s, 3H); 1.55 (s, 3H); 2.1-2.25 (m, 4H); 2.3-2.5 (dd, 2H); 2.70 (t, 1H); 3.25 (t, 1H); 4.5 (s, 1H) 6.95-8.0 (m, 15H). |
|
|
Example - 1: Preparation of amorphous montelukast sodiumMontelukast free acid (70 g) is dissolved in methanol (210 ml), 0.486 molar solution of sodium hydroxide in ethanol (230 ml) is added, mixed for about 10-15 min, filtered the resultant reaction mass through hyflow bed, washed the bed with 35 ml methanol. The clear solution is spray dried with spray drier at inlet temperature of HO0C, outlet temperature of 730C to 750C, nitrogen flow rate of 40-50 mm, with a solution feed rate of 30%.Dry wt of the amorphous sodium is 62.0 g, (yield of 52.8%). EPO <DP n="7"/>XRD is as shown in Fig.1.The various solvents used for dissolution of montelukast free acid and the parameters of Spray drier to obtain amorphous montelukast sodium is given in below (Table - 1) |
|
|
Example - 1: Preparation of amorphous montelukast sodiumMontelukast free acid (70 g) is dissolved in methanol (210 ml), 0.486 molar solution of sodium hydroxide in ethanol (230 ml) is added, mixed for about 10-15 min, filtered the resultant reaction mass through hyflow bed, washed the bed with 35 ml methanol. The clear solution is spray dried with spray drier at inlet temperature of HO0C, outlet temperature of 730C to 750C, nitrogen flow rate of 40-50 mm, with a solution feed rate of 30%.Dry wt of the amorphous sodium is 62.0 g, (yield of 52.8%). EPO <DP n="7"/>XRD is as shown in Fig.1.The various solvents used for dissolution of montelukast free acid and the parameters of Spray drier to obtain amorphous montelukast sodium is given in below (Table - 1) |
|
|
Example - 1: Preparation of amorphous montelukast sodiumMontelukast free acid (70 g) is dissolved in methanol (210 ml), 0.486 molar solution of sodium hydroxide in ethanol (230 ml) is added, mixed for about 10-15 min, filtered the resultant reaction mass through hyflow bed, washed the bed with 35 ml methanol. The clear solution is spray dried with spray drier at inlet temperature of HO0C, outlet temperature of 730C to 750C, nitrogen flow rate of 40-50 mm, with a solution feed rate of 30%.Dry wt of the amorphous sodium is 62.0 g, (yield of 52.8%). EPO <DP n="7"/>XRD is as shown in Fig.1.The various solvents used for dissolution of montelukast free acid and the parameters of Spray drier to obtain amorphous montelukast sodium is given in below (Table - 1) |
|
|
Example - 1: Preparation of amorphous montelukast sodiumMontelukast free acid (70 g) is dissolved in methanol (210 ml), 0.486 molar solution of sodium hydroxide in ethanol (230 ml) is added, mixed for about 10-15 min, filtered the resultant reaction mass through hyflow bed, washed the bed with 35 ml methanol. The clear solution is spray dried with spray drier at inlet temperature of HO0C, outlet temperature of 730C to 750C, nitrogen flow rate of 40-50 mm, with a solution feed rate of 30%.Dry wt of the amorphous sodium is 62.0 g, (yield of 52.8%). EPO <DP n="7"/>XRD is as shown in Fig.1.The various solvents used for dissolution of montelukast free acid and the parameters of Spray drier to obtain amorphous montelukast sodium is given in below (Table - 1) |
|
|
Example - 1: Preparation of amorphous montelukast sodiumMontelukast free acid (70 g) is dissolved in methanol (210 ml), 0.486 molar solution of sodium hydroxide in ethanol (230 ml) is added, mixed for about 10-15 min, filtered the resultant reaction mass through hyflow bed, washed the bed with 35 ml methanol. The clear solution is spray dried with spray drier at inlet temperature of HO0C, outlet temperature of 730C to 750C, nitrogen flow rate of 40-50 mm, with a solution feed rate of 30%.Dry wt of the amorphous sodium is 62.0 g, (yield of 52.8%). EPO <DP n="7"/>XRD is as shown in Fig.1.The various solvents used for dissolution of montelukast free acid and the parameters of Spray drier to obtain amorphous montelukast sodium is given in below (Table - 1) |
|
|
Example - 1: Preparation of amorphous montelukast sodiumMontelukast free acid (70 g) is dissolved in methanol (210 ml), 0.486 molar solution of sodium hydroxide in ethanol (230 ml) is added, mixed for about 10-15 min, filtered the resultant reaction mass through hyflow bed, washed the bed with 35 ml methanol. The clear solution is spray dried with spray drier at inlet temperature of HO0C, outlet temperature of 730C to 750C, nitrogen flow rate of 40-50 mm, with a solution feed rate of 30%.Dry wt of the amorphous sodium is 62.0 g, (yield of 52.8%). EPO <DP n="7"/>XRD is as shown in Fig.1.The various solvents used for dissolution of montelukast free acid and the parameters of Spray drier to obtain amorphous montelukast sodium is given in below (Table - 1) |
|
|
Example - 1: Preparation of amorphous montelukast sodiumMontelukast free acid (70 g) is dissolved in methanol (210 ml), 0.486 molar solution of sodium hydroxide in ethanol (230 ml) is added, mixed for about 10-15 min, filtered the resultant reaction mass through hyflow bed, washed the bed with 35 ml methanol. The clear solution is spray dried with spray drier at inlet temperature of HO0C, outlet temperature of 730C to 750C, nitrogen flow rate of 40-50 mm, with a solution feed rate of 30%.Dry wt of the amorphous sodium is 62.0 g, (yield of 52.8%). EPO <DP n="7"/>XRD is as shown in Fig.1.The various solvents used for dissolution of montelukast free acid and the parameters of Spray drier to obtain amorphous montelukast sodium is given in below (Table - 1) |
|
With sodium hydroxide; In methanol; water; toluene; at 25 - 50℃; for 0.833333h; |
Example 16 - Montelukast sodium amorphous[0105] 2.0 g of Montelukast was dissolved in 5 ml of a toluene-methanol mixture (4: 1 v/v) at 500C. The turbid solution was cooled under stirring to 25C. Atthis temperature, 0.33 ml of aqueous NaOH solution (0.15 g, 3.75 mmol) was addeddropwise within 20 minutes. The solution was maintained at this temperature for 30minutes, 0.05 g of activated charcoal was added and the suspension was filteredthrough Celite filter. The Celite was washed with 2x2 ml of toluene. The combined solution was evaporated almost to dryness under reduced pressure at bath temperature38C . Yellow viscous oil was obtained.[0106] The oil was added dropwise into 10 ml of stirred n-heptane and stirredfor 15 minutes at 25C. White precipitate was formed. Then the mixture was stirred at250C for next 17 hours. The precipitate was filtered by suction, washed with 2x10 ml of n-heptane and dried at room temperature protected from light. Yield 1.85 g of whiteamorphous solid. |
|
With sodium hydroxide; In dichlorometnane; water; at 20℃;pH 10.5; |
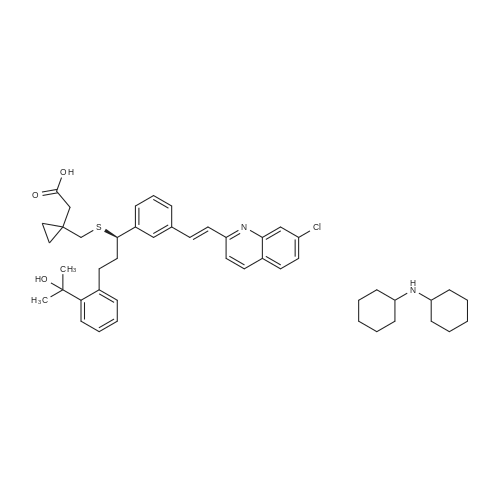
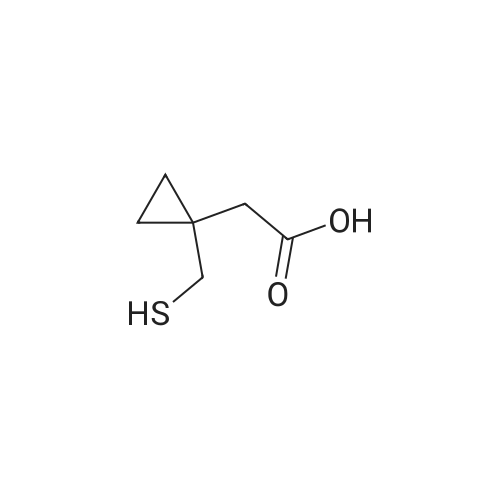
Another three-necked flask equipped with a thermometer, a nitrogen inlet and a magnetic stirrer was charged at room temperature with 6.7 g (0.0459 moles) of 1-(mercaptomethyl)cyclopropaneacetic acid and 48 ml of NMP under stirring and under nitrogen atmosphere to obtain a solution. 4.5 g of NaOH flakes (0.1125 moles) was added in one portion at room temperature followed by addition of 2.4 ml of water, and stirring was maintained for 1 hour to afford a suspension. The solution of compound (III) in about 50 ml THF, which was kept at 5 C., was added in portions at ambient temperature. After completing the addition, the mixture was stirred for 2 hours and reaction completion was checked by HPLC. 130 ml of toluene was added to the reaction mixture along with 130 ml of 5% sodium chloride solution, and the mixture was stirred for 20 minutes. Then, the layers were separated and the upper organic layer was washed with 130 ml of 5% sodium chloride solution, and the layers were separated. 84 ml of 0.5 M tartaric acid solution was added to the upper layer and the layers were separated. The upper layer was washed with 40 ml of water and again separated. The organic layer was distilled to dryness to afford an oily residue. 90 ml of toluene was added to the residue under stirring to obtain a solution. 3.1 ml of cyclooctylamine (0.0226 moles) was added and stirring was maintained for few minutes and the solution was seeded with crystalline montelukast acid cyclooctyl ammonium salt. Stirring was maintained at room temperature to afford a suspension, which was filtered off to obtain a cake. The cake was washed with toluene and dried at 40 C. in vacuum to afford 9.88 g of dry crude montelukast acid cyclooctyl ammonium salt in 70% yield, having 98% purity (according to HPLC). The crude montelukast cyclooctyl ammonium salt was crystallized from toluene containing about 2% of methanol to obtain a product having 99% purity (according to HPLC). 30 ml of dichloromethane was added followed by addition of 17 ml of 0.5M citric acid solution. The mixture was stirred at room temperature for half an hour to afford a two phase system. The layers were separated and the organic layer (containing the montelukast acid) was washed with 3×15 ml water, the layers were separated and the aqueous layer was removed. 20 ml of water was added under stirring followed by addition of 4 ml of 1M NaOH solution and stirring was maintained for about 5 minutes. The dichloromethane was distilled off at a temperature lower than 35 C. The pH was checked and 1 M NaOH solution was added drop-wise until the pH value was about 10.5. The aqueous layer (containing the desired end product) was freeze-dried to obtain 8.3 g of montelukast sodium in 99% yield having a purity of 99.8% (according to HPLC). |
|
With sodium methylate; In methanol; toluene; for 0.5h; |
Example 2 - Preparation of (RVl-ri-(3-('2-(7-chloro-2-quinolinylVethenyl)-phenylV3-fr2-ri- hvdroxy-l-methylethvD-phenvD-propylVthioi-cyclopropaneacetic acid sodium salt, Montelukast Sodium (Compound 10, Scheme 2); To a stirred solution of the title compound of Example 1, montelukast free acid, (5.7 g), in dry toluene (4OmL), sodium methoxide (0.55g) in methanol was added. The resulting mixture was stirred for 30 minutes and diisopropyl ether (25OmL) was added dropwise over a period of 60 minutes. White solid started to precipitate. The white solid was then filtered off, washed with diisopropyl ether and dried under vacuum. The title compound, montelukast sodium, was obtained as an amorphous white solid (5.2g, 98.2 area%). |
|
With sodium hydroxide; In methanol; at 25 - 30℃; for 1h; |
Example 16 : Preparation of Montelukast sodiumMontelukast -Isopropylamine salt (10 gm), methylene chloride (100 ml) and water (100 ml) was charged under nitrogen atmosphere and stirred for 10 minutes. The pH of the reaction mass was adjusted to 4.0-4.5 with 10% acetic acid solution, separated methylene chloride layer and extracted the aqueous layer with methylene chloride (50 ml), the combined methylene chloride layer was dried over sodium sulphate (5 gm) and distilled off methylene chloride completely under vacuum at 25-3O0C to residue. The residue was dissolved in methanol (60 ml), a solution of sodium hydroxide in methanol (0.68 gm in 30 ml of methanol) was added and stirred for 1 hour at 25-3O0C, clarified, distilled off methanol completely under vacuum at less than 350C, stripped off methanol with ethyl acetate (50 ml x 3) to residue, the residue was dissolved in toluene (30 ml), n-heptane (100 ml) was added dropwise under nitrogen atmosphere at 25-300C and stirred for 30 minutes, filtered the material so obtained was dried under vacuum at 70-800C for 24 hours to give Montelukast sodium (8.5 gm, 90% yield, 99% HPLC purity). |
|
With sodium hydroxide; In methanol; toluene; at 20℃; for 1h;Product distribution / selectivity; |
Montelukast arginine salt (4.14 g) was dissolved in toluene (100 mL) . Acetic acid (1 mL) in water {100 mL) was added. The mixture was stirred at rt for 30 min, and layers separated. To organic layer, solution of sodium hydroxide (218 mg) in methanol (12 mL) was added, and stirred at rt for 1 hr . The solvent is completely disstiled off under vacuum at below 500C to afford the residue. The residue is dissolved in toluene (40 mL} and charcoal was added and stirred at 400C for 2 hrs . The reaction mixture was filtered, and concentrated under vacuum to -20 mL. The mixture was dropwise added to hexane (60 mL) and stirred under nitrogen 18 hrs. The product is filtered under nitrogen and washed with hexane (20 mL) , and dried under vacuum at 4O0C for 12 hrs to yield amorphous form of montelukast sodium (2.07 g) .; Example 171 Montelukast sodiumTo the solution of montelukast acid (2.5 g) prepared by the processes as disclosed in the previous examples or by any processes known from the prior art in toluene (50 mL) solution of sodium hydroxide (218 mg) in methanol (12 mL) was added, and stirred at rt for 1 hr. The solvent is completely disstiled off under vacuum at below 500C to afford the residue. The residue is dissolved in toluene (40 mL) and charcoal was added and stirred at 400C for 2 hrs. The reaction mixture was filtered, and concentrated under vacuum to -20 mL. The mixture was dropwise added to hexane (60 mL) and stirred under nitrogen 18 hrs. The product is filtered under nitrogen and washed with hexane (20 mL) , and dried under vacuum at 400C for 12 hrs to yield amorphous form of montelukast sodium (1.95 g) . |
|
With sodium hydroxide; In tetrahydrofuran; water;Product distribution / selectivity; |
Example 5; Montelukast cyclohexylamine salt suspended in toluene (5 parts) was treated with aqueous acetic acid (5 parts) until a pH of 5.05.5 was reached. The organic phase was separated and washed with water (5 parts) twice, and the resulting organic layer was concentrated to provide a crude oil of montelukast acid. The montelukast acid (100.0 g, 170.2 mmol) thus obtained was dissolved in 500 mL THF, and then 78 mL of ca. 2N NaOH aqueous solution was added under nitrogen. After stirring for several minutes, the solvent was removed under reduced pressure at 30 C. to afford montelukast sodium as a crude oil. The crude oil was taken up in 2.5 L water and concentrated to remove the remaining THF until the total volume was about 2.7 L (THF remained as analyzed by GC was 1.7%). The aqueous solution of montelukast sodium was filtered, and its purity was determined by HPLC (99.7%). The filtered solution was subjected to spray drying at various conditions, and the results were listed in Table 5. |
|
With sodium hydroxide; In methanol; at 30℃; for 0.5h; |
EXAMPLE 10: PREPARATION OF MONTELUKAST SODIUM (FORMULA I).; Sodium hydroxide pellets (0.34 g) were dissolved in methanol (25 mL) and stirred for about 30 minutes. Montelukast acid (5 g) obtained by the procedure of Example 2 was charged into a round bottom flask followed by addition of methanol (25 mL) and stirring for about 15 minutes. The sodium hydroxide and montelukast acid solutions were combined and stirred for about 30 minutes at 30+/-5C. Carbon (0.5 g) was charged and the mixture was filtered through a Hyflow bed and washed with methanol (10 mL). The filtrate was distilled completely under reduced pressure at a temperature of about 500C and the residue was dried at about 700C for 4 hours to yield the title compound. Yield 4.6 g, chemical purity by HPLC 99.7%, sulphoxide impurity 0.03%, stryrene impurity 0.057%, keto impurity 0.07%, chiral purity by HPLC 99.9%. |
|
With sodium hydroxide; In methanol; at 20 - 25℃; for 5h; |
Example 6: Preparation of [R- (E)]-l-[[[l-[3-[2-(7-chloro-2-quinolinyl) ethenyl] phenyl]-3-[2-(l-hydroxy-l-methylethyl) phenyl] propyl] thio] methyl] cyclopropaneacetic acid sodium salt <n="17"/>; Sodium hydroxide (1.2 gm) was added to a solutin of [R- (E)]-l-[[[l-[3-[2-(7-chloro- 2-quinolinyl) ethenyl] phenyl]-3-[2-(l-hydroxy-l-methylethyl) phenyl] propyl] thio] methyl] cyclopropaneacetic acid (7 gm) in methanol ( 42 ml) at 25C. The resulting reaction mixture was stirred 5 hours at 20C to 25C and then concentrated under reduced pressure to get slurry. The slurry was treated with acetonitrile (40 ml) to get [R- (E)]-l-[[[l-[3-[2-(7-chloro-2-quinolinyl) ethenyl] phenyl] -3 -[2-(I -hydroxy- 1- methylethyl) phenyl] propyl] thio] methyl] cyclopropaneacetic acid sodium salt. Yield: 6.2 gm |
|
With sodium hydroxide; In 4-methyl-2-pentanone; at 0 - 20℃; for 0.5h;Inert atmosphere;Product distribution / selectivity; |
In a 5 ltr 4-necked flask equipped with a thermometer and mechanical stirrer, 100 g (0.1706 moles) montelukast acid was suspended in methyl isobutyl ketone (800 ml) under nitrogen atmosphere at room temperature, stir for 30 min and cool to 0-10 0C. Add 0.5 M NaOH solution in methanol (360 ml) at 0-10 0C and stir for 30 min. Charcoalise the product for 1 5 min. and filter through hyflo. Solvent was recovered under vacuum at 40-45 0C. Add methyl isobutyl ketone (100 ml) and recovery continued under vacuum. Remove traces of methyl isobutyl ketone under vacuum at 40-45 0C. Add n-Heptane ( 1 ltr.) to the product and stir for 4-6 hrs. Filter the product under nitrogen atmosphere and dry the material at 45-50 0C to get montelukast sodium (95 g, HPLC purity 99.4%) |
|
With sodium hydroxide; In Isopropyl acetate; water; at 20 - 25℃; for 0.5h;Product distribution / selectivity; |
EXAMPLE 5 1-[1(R)-(3-(2-(7-chloro-2-quinolinyl)ethenyl)-phenyl)-3-(2-(1-hydroxy-1-methylethyl)phenyl)propyl)thiomethyl)-cyclopropaneacetic acid sodium salt (I) The solution of the acid from Example 4 was transferred into a 250 ml flask fitted with a mechanical stirrer and a thermometer. 1 equivalent of IN NaOH was added and stirring of the mixture was continued at 20-25C for 30 minutes. The phases were separated and the aqueous phase was freeze-dried. 45 g of a light yellow solid were obtained in this manner (HPLC A% = 99.5%). 1H-NMR(CDCl3): ppm 0.15-0.20 (d, 2H); 0.40 (d, 2H); 1.50 (s, 3H); 1.55 (s, 3H); 2.1-2.25 (m, 4H); 2.3-2.5 (dd, 2H); 2.70 (t, 1H); 3.25 (t, 1H); 4.5 (s, 1H) 6.95-8.0 (m, 15H). EXAMPLE 8 1-[1 (R)-(3-(2-(7-chloro-2-quinolinyl)ethenyl)-phenyl)-3-(2-(1-hydroxy-1-methylethyl)phenyl)propyl)thiomethyl)-cyclopropaneacetic acid sodium salt (I) The solution of the acid from Example 7 was transferred into a 250 ml flask fitted with a mechanical stirrer and a thermometer. 1 equivalent of IN NaOH was added and stirring of the mixture was continued at 20-25C for 30 minutes. The phases were separated and the aqueous phase was freeze-dried. 45 g of a light yellow solid were obtained in this manner (A% 99.5%). |
|
With sodium hydroxide; In ethanol; di-isopropyl ether; at 8 - 12℃; for 1.5h; |
Montelukast free acid (100 gms.) is suspended in Di-isopropyl ether (700 ml) under Nitrogen at 25-300C. Stirred for 5 minutes and cooled at 8C under Nitrogen. 0.486- M Ethanolic NaOH solution (350ml) is slowly added at 8-12C under Nitrogen over 60 minutes. Stirred at 8-12C under Nitrogen for 30 minutes and treated with activated carbon at 8-120C. Filtered the carbon over hyflow bed and washed the bed with Diisopropyl ether (200 ml) at 10-150C. Filtrate is distilled off completely under vacuum at temperature below 400C and apply high vacuum for removing the traces to get white sticky material. n-Heptane (100ml) is charged to the sticky mass and distilled off solvent traces under vacuum at temperature below 400C to get sticky material. Again n-Heptane (2000 ml) is charged at 25-35C under Nitrogen. Stirred at 25-35C under Nitrogen for 8 hours. Precipitated product is filtered and washed with n-Heptane (200ml). Dried the product at 50-550C under high vacuum for 2 hours 700C / 6 hrs and at 85-95C for 3 hours. Out put: 90gms |
|
|
The reactor is charged with toluene (8.8 kg) and montelukast acid (0.6 kg, 1.024 mol) and the reactor is cooled to 0-5C. The ethanolic solution of sodium hydroxide (47 g of NaOH in 2.2 L of ethanol) is added dropwise to the solution of montelukast acid in toluene during 30 min, maintaining the temperature of solution below 5C. Thereafter, the reaction mixture is stirred at 20-30C for 2 hrs, than activated carbon is added and solution stirred at the same temperature for 2 hr. The reaction mixture is filtrated and activated charcoal is washed with the mixture of ethanol and toluene. Filtrate is concentrated to about 1/4 of the volume. To the crystallization reactor heptane (10 kg) is added and the solution of Montelukast sodium in toluene is added dropwise for 2 hrs (temperature is 15-25C), and after the addition, the reaction mixture is stirred for 2 hrs at the same temperature. The solution is transferred to the centrifuge, filtrated and crystalline product is washed with heptane. The material is transferred to a drier, and dried. The final product is amorphous montelukast sodium. |
|
With sodium hydroxide; In ethanol; toluene; at 0 - 30℃; for 2.5h;Industry scale; |
The reactor is charged with toluene (8.8 kg) and monteiukast acid (0.6 kg, 1.024 mol) and the reactor is coo.ed to 0-5C. The ethanolic solution of sodium hydroxide (47 g of NaOH in 2.2 L of ethanol) is added dropwise to the solution of montelukast acid in toluene during 30 min, maintaining the temperature of solution below 5C. Thereafter, the reaction mixture is stirred at 20-30C for 2 hrs, than activated carbon is added and solution stirred at the same temperature for 2 hr. The reaction mixture is filtrated and activated charcoal is washed with the mixture of ethanol and toluene. Filtrate is concentrated to about ¼ of the volume. To the crystallization reactor heptane (10 kg) is added and the solution of Montelukast sodium in toluene is added dropwise for 2 hrs (temperature is 15-25C), and after the addition, the reaction mixture is stirred for 2 hrs at the same temperature. The solution is transferred to the centrifuge, filtrated and crystalline product is washed with heptane. The material is transferred to a drier, and dried.The final product is amorphous montelukast sodium. |
|
With methanol; sodium hydroxide; at -5 - 30℃; for 1h; |
Step 2: preparation of montelukast sodium: Sodium hydroxide (3.6 g, 0.09 mol) was dissolved in methanol (250 ml) under an atmosphere of nitrogen gas at 25-30 C. After complete dissolution, the reaction mixture was cooled slowly to 0 to -5 C. Organic layer (as obtained in step I) was added to the resulting reaction mixture and at 0 to -5 C for 30 minutes. Reaction mixture was heated to 25-30 C and stirred for 30 minutes. Activated charcoal was added to the resulting solution and stirred for 1 hour at 25-30 C. Reaction mixture was filtered through a hyflo-bed and washed with methanol (50 ml). Solvent was evaporated under vacuum and n-heptane (500 ml) was added to the resulting residue. Mixture was stirred for 5 hours at 25-30 C. Solid thus precipitated was filtered, washed with n-heptane (100 ml) and dried at 35-40 C to give 50 g of the title compound as a very hygroscopic white powder having purity 99.70 % by HPLC. |
|
With sodium hydroxide; In methanol; at 0 - 30℃;pH 10.3 - 10.6; |
Conversion of Montelukast (crude) to crude Montelukast sodium mixture: Methanolic sodium hydroxide 6.8 grams (0.17 mole in 100 ml methanol) was added slowly to (Crude) Montelukast 100 grams (0.17 mole) dissolved in 400 ml of methanol at 25-30 C., under stirring keeping the temperature at 0-5 C. to adjust the pH of the contents to 10.3-10.6. Stirring was continued for another 30 minutes at the temperature of 0-5 C. Temperature was then gradually increased to 25-30 C. to obtain a clear reaction solution. The clear reaction solution was charcolised by adding 10 grams activated charcoal and reaction solution was filtered through a 0.45 micron filter. The filter bed was washed with 100 ml methanol and filtrate was collected. Methanol was removed from the filtrate under reduced pressure at the temperature not exceeding 50-55 C. The resulting solids were further stripped with heptane (2×50 ml) to remove any remaining methanol. As a final step, 500 ml heptane was added under stirring and the stirring was continued for an hour at 25-30 C. The resulting precipitated crude Montelukast sodium mixture was further cooled to 10-15 C. and product was filtered off and dried under vacuum at 50-55 C. The yield of solid crude Montelukast sodium mixture was 101 grams. |
|
With sodium methylate; In methanol; ethyl acetate; toluene; at 20 - 25℃;Product distribution / selectivity; |
Example 5Preparation of montelukast sodium (11)(19, amine = diisobutylamine)NaOMe(11 )40 g of montelukast diisobutylamine, 400 mL of AcOEt and a solution of 11.9 g of citric acid and 200 mL of H20 were mixed in a round bottom flask under nitrogen. After stirring during 15 minutes, the formed phases were separated and partially distilled under vacuum at 30-35C until the volume was reduced to 100 mL of AcOEt. 120 mL of toluene and 10.6 mL of NaOMe (30% in MeOH) were added to the distilled organic phase and the solution was stirred at 20-25C. Afterwards, 400 mg of activated charcoal was added to the solution and after stirring for 30 minutes at 50-55C, the mixture was filtered through celite and washed with 80 mL of toluene. The obtained filtrate was partially distilled off at atmospheric pressure under nitrogen until the volume was reduced to 120 mL. The distilled solution was added dropwise into 360 mL of cyclohexane. The product mass was stirred for 1 hour and 30 min. at 20-25C and filtered. The product was dried at 80C under vacuum to get 33.5 g of montelukast sodium (11) in amorphous form. Yield: 98%. Purity (HPLC): 99.55%. |
|
With sodium hydroxide; In methanol; for 1h; |
30 ml of toluene and 15 ml of water were added to 3.0 g of montelukast dipropylamine salt crystals, stirred- mixed, and 0.30 g of acetic acid was added to the resulting solution, stirred for 30 minutes, and then the aqueous layer was removed and the obtained organic layer was cooled to 4 C, and the precipitated crystals were collected by filtration by pressure filtration and dried under reduced pressure to obtain crystals of montelukast free acid. To the solution of the montelukast free acid thus obtained was added 30 ml of methanol and stirred and mixed, and 0.15 g of sodium hydroxide was added and the mixture was stirred for 1 hour, then the solvent was distilled off and the precipitated crystals were collected by filtration by pressure filtration and dried under reduced pressure to obtain 2.9 g of montelukast sodium (montelukast sodium purity 99.9%, methylstyrene content 0.02%) (yield 93.2%). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping