* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Organic Letters, 2013, vol. 15, # 22, p. 5890 - 5893
[2] Chemistry - A European Journal, 2015, vol. 21, # 14, p. 5561 - 5583
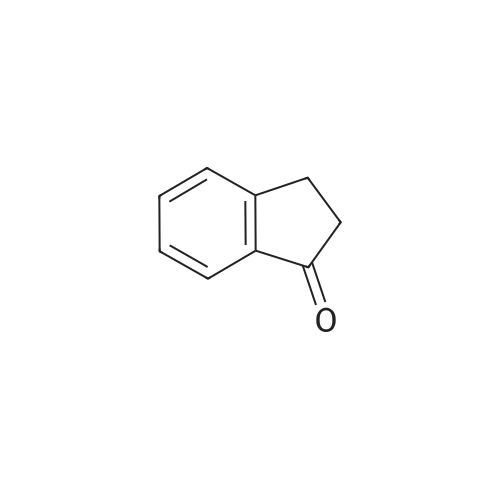
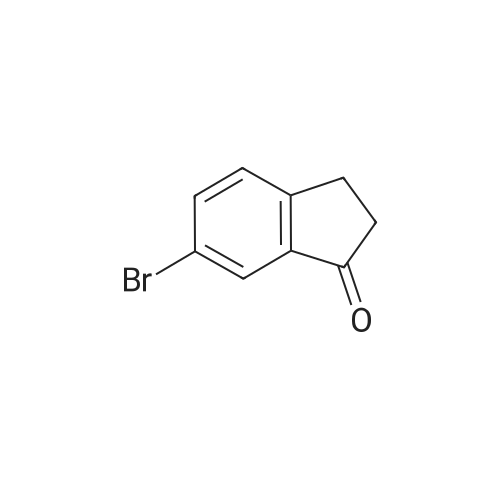
2
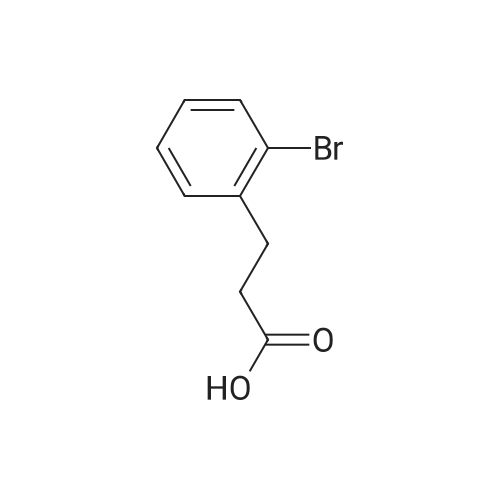
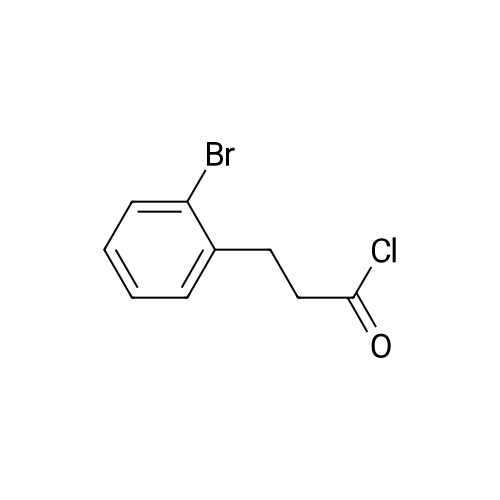
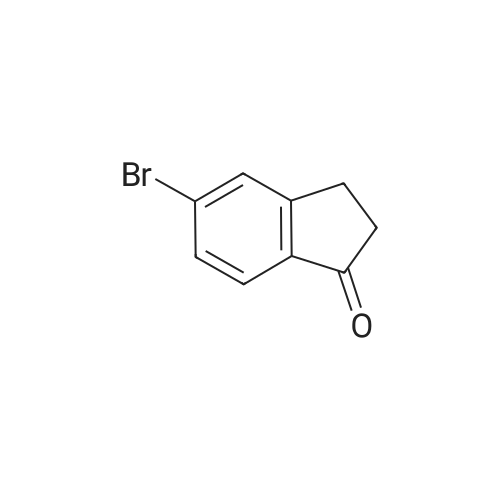
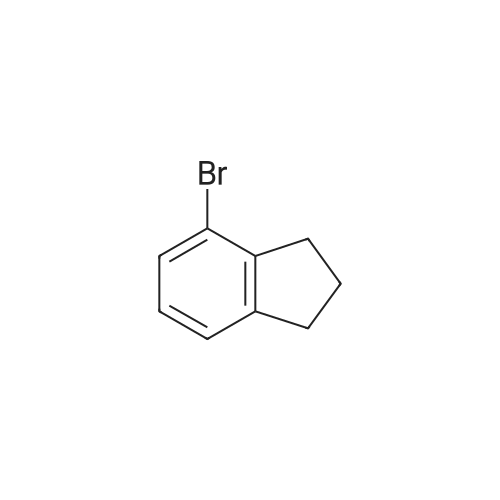
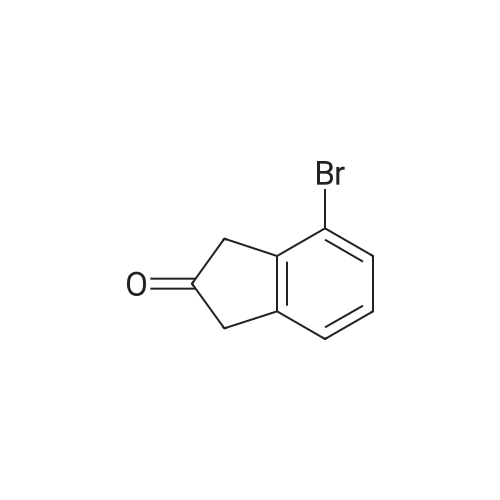
[ 15115-58-9 ]
[ 15115-60-3 ]
Yield
Reaction Conditions
Operation in experiment
100%
With trifluorormethanesulfonic acid In dichloromethane at 80℃; for 1 h; High pressure; Inert atmosphere; Green chemistry
General procedure: Trifluoromethane sulfonic acid (3 eq.) was gently added to a cooled (0 °C) solution of a 3-Phenylpropionic acid (0.5 mmol) in dry CH2Cl2 (1.0 mL) in a 12 mL Q-tube™ pressure tube, furnished by QLabtech. The temperature was raised to room temperature. A Teflon septum was placed on the top of the tube and the appropriate cap and pressure adapter were used. The mixture was heated in an oil bath at 80 °C. The reaction was monitored by TLC and GC/MS until the reactant disappeared. The mixture was poured into ice and extracted three times with CH2Cl2. The organic phase collected was dried on Na2SO4, filtered and concentrated under vacuum. The desired pure product was separated from the crude by flash chromatography.
95%
Stage #1: With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 20℃; for 1 h; Stage #2: With aluminum (III) chloride In dichloromethane at 0℃; for 1 h;
To a solution of 3-(2-bromophenyl)propanoic acid (1.20 g, 5.24 mmol, CAS 15115-58-9) in a mixture of solvent dichloromethane (20 mL) and dimethylformamide (3.83 mg, 52.4 umol) was added oxalyl chloride (1.33 g, 10.4 mmol) and the reaction mixture was stirred at 20 °C for 1 hr. On completion, the mixture was concentrated in vacuo and the residue was dissolved in dichloromethane (20 mL) and cooled to 0 °C. Then aluminum trichloride (838 mg, 6.29 mmol) was added at 0 °C and the reaction mixture was stirred at 0 °C for 1 hr. On completion, the reaction mixture was poured into 100 mL cool water and extracted with DCM (3 X 50 mL). The organic layer was dried over Na2SO4, filtered and concentrated in vacuo to give the title compound (1.10 g, 95percent yield) as a light yellow solid.1H NMR (400MHz, CDCl3) δ = 7.77 (d, J = 7.2 Hz, 1H), 7.73 (d, J = 7.2 Hz, 1H), 7.30 (dd, J = 7.2, 7.2 Hz, 1H), 3.12 (t, J = 7.2 Hz, 2H), 2.75 (t, J = 7.2 Hz, 2H).
86%
With aluminium trichloride In dichloromethane; 1,2-dichloro-ethane
4-Bromo-2,3-dihydro-1H-inden-1-one (2) 3-(2-Bromophenyl)propanoic acid (1) (550 g, 2.4 mol, 1 equiv) was dissolved in 1,2-dichloroethane (5.5 L). Thionyl chloride (437.8 mL, 6 mol, 2.5 equiv) was added to the solution and the mixture was refluxed for 24 hours. The reaction was cooled to room temperature and concentrated under reduced pressure. The residue was dissolved in methylene chloride (1 L) and added dropwise to a mechanically stirred suspension of anhydrous aluminum chloride (526.9 g, 3.96 mol, 1.65 equiv) in dichloromethane (1 L) while keeping the reaction temperature below 27° C. The reaction was stirred at room temperature for three hours before being quenched into a five gallon bucket which was half-full of ice. The resulting mixture was extracted with dichloromethane (3*3 L). The combined organic layers were washed sequentially with saturated brine (2 L) and saturated sodium bicarbonate (2 L). The organic layer was dried over sodium sulfate, and concentrated under reduced pressure. The resulting solid was dried overnight in a vacuum oven at 30° C. to give compound 2 (435 g, 86percent yield) as an off-white solid.
86%
Stage #1: With thionyl chloride In 1,2-dichloro-ethane for 24 h; Reflux Stage #2: With aluminum (III) chloride In dichloromethane at 20 - 27℃;
[00161] 4-Bromo-2,3-dihydro-lH-inden-l-one (b): 3-(2-Bromophenyl)propanoic acid (a) (550 g, 2.4 mol, 1 equiv) was dissolved in 1 ,2-dichloroethane (5.5 L). Thionyl chloride (437.8 niL, 6 mol, 2.5 equiv) was added to the solution and the mixture was refluxed for 24 hours. The reaction was cooled to room temperature and concentrated under reduced pressure. The residue was dissolved in methylene chloride (1 L) and added dropwise to a mechanically stirred suspension of anhydrous aluminum chloride (526.9 g, 3.96 mol, 1.65 equiv) in dichloromethane (1 L) while keeping the reaction temperature below 27°C. The reaction was stirred at room temperature for three hours before being quenched into a five gallon bucket which was half-full of ice. The resulting mixture was extracted with dichloromethane (3 x 3 L). The combined organic layers were washed sequentially with saturated brine (2 L) and saturated sodium bicarbonate (2 L). The organic layer was dried over sodium sulfate, and concentrated under reduced pressure. The resulting solid was dried overnight in a vacuum oven at 30°C to give compound b (435 g, 86percent yield) as an off-white solid.
76%
Stage #1: With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 0℃; Stage #2: at 0℃; for 1 h;
Example NINETEEN-1 (Compound 149) [4-BROMO-INDAN-1-ONE] was obtained by the following procedure: A solution of [3- (2-BROMO-PHENYL)-PROPIONIC] acid (commercially available from Oakwood Products) (15.0 g, 65.5 mmol) in [CHUCK] at [0 °C] was reacted with oxalyl chloride (7.2 mL, 1.5 eq) followed by 2-3 drops of DMF. The mixture was stirred until no more gas evolution was observed. As the mixture was concentrated and the residue was dissolved in [CH2C12,] cooled to [0 °C,] and treated with [AIDS] (9.6 g, 1.1 eq). After 1 h the mixture was quenched with water and the layers were separated. The aqueous layer was extracted with Et2O (3 x 150 [ML)] and the combined organic extracts were washed with H20 (3 x 100 mL), saturated NaHCO3 (3 x 100 mL), brine (1 x 100 [ML),] dried over MgS04 and concentrated. [4-BROMOINDAN-L-ONE,] 10.5 g (76percent) was obtained by chromatography using 10 percent EtOAc: hexane as eluant. Use of 4-bromo- [INDAN-1-ONE] in Method NINETEEN produced [4- (4-BROMO-INDAN-2-YL)-1,] 3-dihydro- imidazole-2-thione (Compound 149). 1H NMR (300 MHz, DMSO-d6) 8 12.0 (s, 1H), 11.7 (s, 1H), 7.34 (d, J= 7.8 Hz, 1H), 7.22 (d, [J=] 7.5 Hz, 1H), 7.09 (t, J= 7.5 Hz, [1H),] 6.63 (s, [1H),] 3.50-3. 40 (m, [1H),] 3.30-3. 12 (m, 2H), 3.06-2. 85 (m, 2H).
Reference:
[1] Molecules, 2014, vol. 19, # 5, p. 5599 - 5610
[2] Patent: WO2018/106636, 2018, A1, . Location in patent: Paragraph 100231
[3] Patent: US2015/25205, 2015, A1, . Location in patent: Page/Page column
[4] Patent: WO2015/95188, 2015, A1, . Location in patent: Paragraph 00161
[5] Patent: WO2003/99795, 2003, A1, . Location in patent: Page 111
[6] Bulletin de la Societe Chimique de France, 1966, p. 3618 - 3625
[7] Journal of Organic Chemistry, 1984, vol. 49, # 22, p. 4226 - 4237
[8] Tetrahedron, 1976, vol. 32, p. 257 - 260
[9] Bulletin of the Chemical Society of Japan, 1961, vol. 34, p. 1189 - 1194
[10] Patent: US9249239, 2016, B2,
[11] Patent: US2008/255230, 2008, A1,
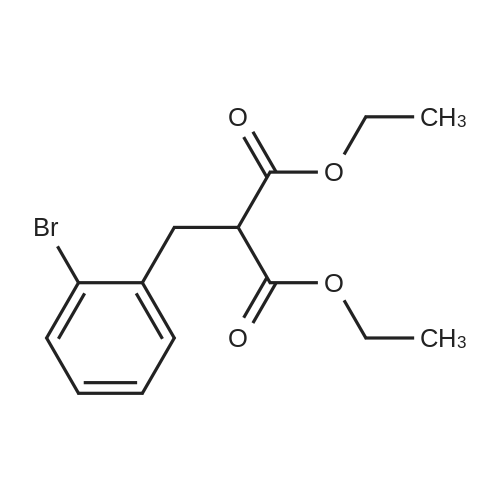
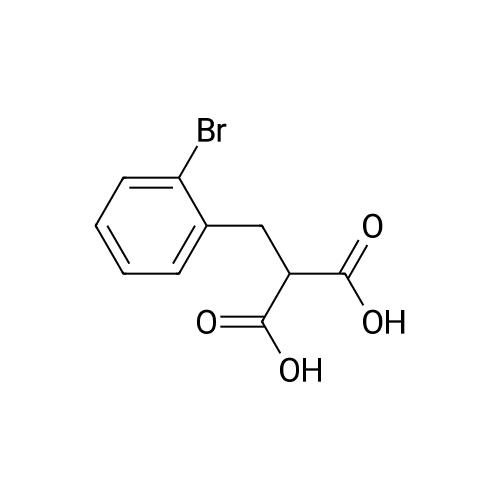
3
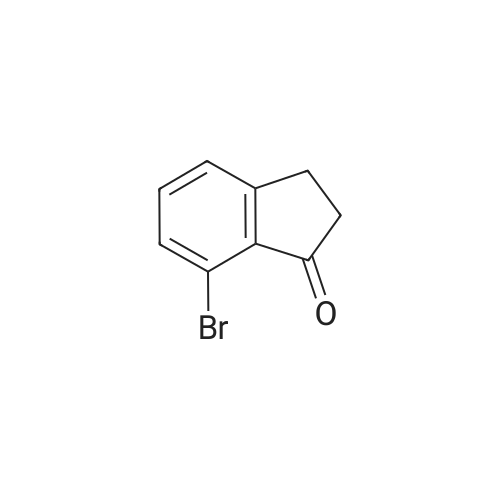
[ 90725-40-9 ]
[ 15115-60-3 ]
Yield
Reaction Conditions
Operation in experiment
435 g
With aluminum (III) chloride In dichloromethane at 23 - 27℃; for 3 h;
3-(2-Bromophenyl)propanoic acid (1) (550 g, 2.4 mol, 1 equiv) was dissolved in 1,2-dichloroethane (5.5 L). Thionyl chloride (437.8 mL, 6 mol, 2.5 equiv) was added to the solution and the mixture was refluxed for 24 hours. The reaction was cooled to room temperature and concentrated under reduced pressure. The residue was dissolved in methylene chloride (1 L) and added dropwise to a mechanically stirred suspension of anhydrous aluminum chloride (526.9 g, 3.96 mol, 1.65 equiv) in dichloromethane (1 L) while keeping the reaction temperature below 27° C. The reaction was stirred at room temperature for three hours before being quenched into a five gallon bucket which was half-full of ice. The resulting mixture was extracted with dichloromethane (3×3 L). The combined organic layers were washed sequentially with saturated brine (2 L) and saturated sodium bicarbonate (2 L). The organic layer was dried over sodium sulfate, and concentrated under reduced pressure. The resulting solid was dried overnight in a vacuum oven at 30° C. to give compound 2 (435 g, 86percent yield) as an off-white solid.
Reference:
[1] Journal of Organic Chemistry, 1984, vol. 49, # 22, p. 4226 - 4237
[2] Journal fuer Praktische Chemie (Leipzig), 1934, vol. <2> 139, p. 242
[3] Journal of the Chemical Society, 1939, p. 794,797
[4] Anales de la Real Sociedad Espanola de Fisica y Quimica, 1944, vol. 40, p. 1182,1188
[5] Tetrahedron, 1976, vol. 32, p. 257 - 260
[6] Bulletin of the Chemical Society of Japan, 1961, vol. 34, p. 1189 - 1194
[7] Patent: US9249239, 2016, B2, . Location in patent: Page/Page column 51
4
[ 335159-82-5 ]
[ 15115-60-3 ]
Reference:
[1] Advanced Synthesis and Catalysis, 2012, vol. 354, # 4, p. 651 - 662
[2] Patent: US2008/255230, 2008, A1, . Location in patent: Page/Page column 13
5
[ 83-33-0 ]
[ 15115-60-3 ]
[ 14548-39-1 ]
Reference:
[1] Synthetic Communications, 1994, vol. 24, # 19, p. 2777 - 2788
[2] Patent: US5753655, 1998, A,
6
[ 79-37-8 ]
[ 15115-58-9 ]
[ 15115-60-3 ]
Reference:
[1] Patent: US2005/75366, 2005, A1,
7
[ 66192-11-8 ]
[ 15115-60-3 ]
Reference:
[1] Journal of Organic Chemistry, 1984, vol. 49, # 22, p. 4226 - 4237
[2] Bulletin of the Chemical Society of Japan, 1961, vol. 34, p. 1189 - 1194
8
[ 58380-12-4 ]
[ 15115-60-3 ]
Reference:
[1] Journal of Organic Chemistry, 1984, vol. 49, # 22, p. 4226 - 4237
[2] Tetrahedron, 1976, vol. 32, p. 257 - 260
9
[ 3433-80-5 ]
[ 15115-60-3 ]
Reference:
[1] Journal of Organic Chemistry, 1984, vol. 49, # 22, p. 4226 - 4237
[2] Bulletin of the Chemical Society of Japan, 1961, vol. 34, p. 1189 - 1194
10
[ 32846-64-3 ]
[ 15115-60-3 ]
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1980, p. 444 - 447
[2] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1980, p. 444 - 447
11
[ 70234-46-7 ]
[ 15115-60-3 ]
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1980, p. 444 - 447
12
[ 83-33-0 ]
[ 15115-60-3 ]
[ 14548-39-1 ]
[ 34598-49-7 ]
[ 103515-98-6 ]
Reference:
[1] European Journal of Organic Chemistry, 2001, # 11, p. 2107 - 2114
13
[ 83-33-0 ]
[ 15115-60-3 ]
[ 14548-39-1 ]
[ 34598-49-7 ]
Reference:
[1] European Journal of Organic Chemistry, 2001, # 11, p. 2107 - 2114
14
[ 95-46-5 ]
[ 15115-60-3 ]
Reference:
[1] Bulletin of the Chemical Society of Japan, 1961, vol. 34, p. 1189 - 1194
15
[ 18982-54-2 ]
[ 15115-60-3 ]
Reference:
[1] Tetrahedron, 1976, vol. 32, p. 257 - 260
16
[ 13463-40-6 ]
[ 70234-46-7 ]
[ 15115-60-3 ]
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1980, p. 444 - 447
17
[ 7664-93-9 ]
[ 15115-58-9 ]
[ 15115-60-3 ]
Reference:
[1] Chemische Berichte, 1892, vol. 25, p. 2115
18
[ 75-15-0 ]
[ 7446-70-0 ]
[ 90725-40-9 ]
[ 15115-60-3 ]
Reference:
[1] Chemische Berichte, 1928, vol. 61, p. 1974
19
[ 15115-60-3 ]
[ 6134-53-8 ]
Yield
Reaction Conditions
Operation in experiment
85%
With triethylsilane; trifluoroacetic acid In tetrahydrofuran; dichloromethane at -20 - 20℃; for 2 h;
To a solution of 4-bromo-2,3-dihydro-lH-inden-l-one (2 g, 10 mmol) in DCM (lOmL) was added TFA, then cooled to - 20°C. Et3SiH (15 ml_, IN in THF, 15 mmol) was added drop wise. After stirring for 2h at RT the reaction mixture was poured into NH4CI aqueous and extracted with DCM (10 ml_*2), washed with brine (10 ml_*2), dried by MgS04, concentrated, purified by FC to afford compound (1.7 g, yield : 85percent); m/z (ES+) : 197 [M + H] +
80%
With triethylsilane; trifluorormethanesulfonic acid In dichloromethane at 0 - 20℃; for 16 h;
To a solution of 4-bromoindan-1-one (200 mg, 947 umol) in dichloromethane (10 mL) was added a solution of trifluoromethanesulfonic acid (426 mg, 2.84 mmol) in dichloromethane (500 uL) and the reaction mixture was cooled to 0 °C. Then triethylsilane (220 mg, 1.90 mmol) in dichloromethane (500 uL) was added dropwise to the reaction mixture and the mixture was stirred at 0 °C for 0.5 hr. TLC detected most starting material remained. Then another batch of trifluoromethanesulfonic acid (426 mg, 2.84 mmol) and triethylsilane (220 mg, 1.90 mmol) was added in turn and the reaction mixture was stirred at 20 °C for 15.5 hrs. On completion, the reaction mixture was diluted with 15 mL DCM and washed with saturated sodium bicarbonate until pH = 7. The organic layer was dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel chromatography (petroleum ether) to give the title compound (1.00 g, 80percent yield) as colorless oil.1H NMR (400MHz, CDCl3) δ = 7.31 (d, J = 7.6 Hz, 1H), 7.16 (d, J = 7.6 Hz, 1H), 7.02 (dd, J = 7.6, 7.6 Hz, 1H), 3.04 (t, J = 7.2 Hz, 2H), 2.98 (t, J = 7.2 Hz, 2H), 2.11 (m, 2H).
Reference:
[1] Patent: WO2015/24878, 2015, A1, . Location in patent: Page/Page column 110
[2] Patent: WO2018/106636, 2018, A1, . Location in patent: Paragraph 00232
[3] Journal of the Chemical Society, 1939, p. 794,797
[4] Journal of the American Chemical Society, 1935, vol. 57, p. 2174
[5] Bulletin of the Chemical Society of Japan, 1961, vol. 34, p. 1189 - 1194
[6] Bulletin de la Societe Chimique de France, 1966, p. 3618 - 3625
20
[ 83-33-0 ]
[ 15115-60-3 ]
[ 14548-39-1 ]
[ 34598-49-7 ]
[ 103515-98-6 ]
Reference:
[1] European Journal of Organic Chemistry, 2001, # 11, p. 2107 - 2114
21
[ 83-33-0 ]
[ 15115-60-3 ]
[ 14548-39-1 ]
[ 34598-49-7 ]
Reference:
[1] European Journal of Organic Chemistry, 2001, # 11, p. 2107 - 2114
22
[ 15115-60-3 ]
[ 56461-20-2 ]
Reference:
[1] Patent: WO2014/155301, 2014, A1,
23
[ 67-56-1 ]
[ 201230-82-2 ]
[ 15115-60-3 ]
[ 55934-10-6 ]
Yield
Reaction Conditions
Operation in experiment
64%
for 48 h;
Preparative Example - Methyl l-oxo-2,3-dihydro-lH-indene-4-carboxylate (PrepEx-11)PrepEx-11[00203] To a solution of 4-bromo-2,3-dihydro-lH-inden-l -one (5 g, 24 mmol) in MeOH (120 mL) and Et3N (40 mL) was added Pd(PPh3)2Cl2 (200 mg) and the mixture was stirred under 2.5 MPa of CO for 48 hours. The reaction solution was concentrated under reduced pressure and the residue was purified by silica gel chromatography using 2:1 petroleum ether:EtOAc to afford methyl l-oxo-2,3-dihydro-lH-indene-4-carboxylate (2.9 g, 64 percent). MS ESI calcd for CnHi0O3 [M + H]+ 191, found 191.
63%
at 100℃; for 24 h;
Into a 1000-mL pressure tank reactor was placed 4-bromo-2,3-dihydro-lH-inden-1-one (30 g, 142 mmol, 1 equiv), MeOH (450 mL), Pd(dppf)Cl2'CH2Cl2 (25 g, 0.1 equiv), and Et3N (150 mL). The resulting mixture was stirred for 24 h at 100 °C under an atmosphere of 50 MPa CO (g). After cooling to room temperature, the mixture was filtered through a pad of celite and then concentrated under vacuum. The residue was purified by normal phase chromatography on silica gel using EtO Ac/petroleum ether (1 : 10). The collected fractions were concentrated under vacuum to afford 17 g (63percent yield) of the title compound as a white solid. MS: (ES, m/z): 191 [M+H]+.
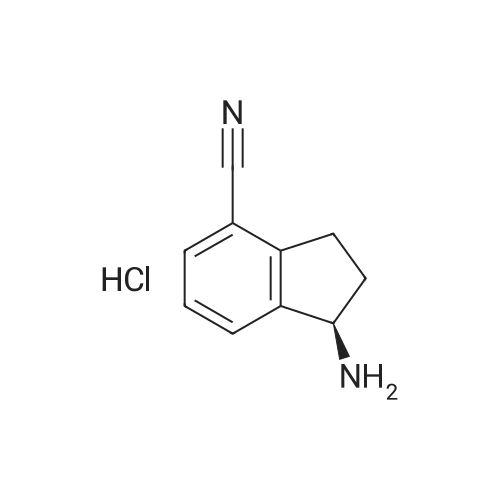
[0435] l-oxo-2,3-dihydro-lH-indene-4-carbonitrile (INT-45)[0436] To a stirring solution of 4-bromo-2,3-dihydro-lH-inden-l-one (100.0 g, 0.48 mol) in150 mL of l-methy-2-pyrrolidine (NMP) was added zinc cyanide (111.8 g, 0.95 mol) and tetrakis(triphenylphosphine)palladium [Pd(PPh3)4] (2.75 g, 0.024 mol). The solution was degassed with N2 and the reaction mixture heated at 95°C for 7 h. Upon cooling, the reaction mixture was poured onto ice water (3.5 L). The compound and inorganic Zn salts precipitated. The solid was collected and partitioned between DCM and water. The organic layers were filtered to remove the Zn salts, and the filtrate was concentrated and crystallized from a 4:1 mixture of EtOH and MeOH (400 mL) to give 45.5 g (60 percent) of l-oxo-2,3- dihydro-lH-indene-4-carbonitrile INT-45 as a light yellow solid. LCMS-ESI (m/z) calculated for C10H7NO: 157.2; found 158.1 [M+H]+, tR = 2.67 min. 1H NMR (400 MHz, CDC13) 6 8.00 - 7.90 (m, 1H), 7.86 (dd, J = 7.5, 1.1, 1H), 7.50 (t, J = 7.6, 1H), 3.40 - 3.19 (m, 2H), 2.90 - 2.61 (m, 2H). 13C NMR (101 MHz, CDC13) δ 204.70, 157.90, 138.38, 137.88, 128.44, 128.28, 116.31, 111.70, 36.01, 25.49.
60%
With tetrakis(triphenylphosphine) palladium(0) In 1-methyl-pyrrolidin-2-one at 95℃; for 7 h; Inert atmosphere
To a stirred solution of 4-bromo-2,3-dihydro-lH-inden-l-one (100.0 g, 0.48 mol) in 150 mL of l-methy-2-pyrrolidine (NMP) was added zinc cyanide (111.8 g, 0.95 mol) and tetrakis(triphenylphosphine)palladium [Pd(PPh3)4] (2.75 g, 0.024 mol). The solution was degassed with N2 and the reaction mixture heated at 95°C for 7 h. Upon cooling, the reaction mixture was poured onto ice water (3.5 L). The compound and inorganic Zn salts precipitated. The solid was collected and partitioned between DCM (3 X 100 mL) and water. The organic layers were filtered to remove the Zn salts, and the filtrate was concentrated and crystallized from a 4: 1 mixture of EtOH and MeOH (400 mL) to give 45.5 g (60 percent) of l-oxo-2,3-dihydro-7H-indene-4-carbonitrile INT-1 as a light yellow solid. LCMS-ESI (m/z) calculated for Ci0H7NO: 157.2; found 158.1 [M+H]+, tR = 2.67 min. 1H NMR (400 MHz, CDC13) δ 8.00 - 7.90 (m, 1H), 7.86 (dd, J = 7.5, 1.1, 1H), 7.50 (t, J = 7.6, 1H), 3.40 - 3.19 (m, 2H), 2.90 - 2.61 (m, 2H). 13C NMPv (101 MHz, CDC13) δ 204.70, 157.90, 138.38, 137.88, 128.44, 128.28, 116.31, 111.70, 36.01, 25.49.
60%
With tetrakis(triphenylphosphine) palladium(0) In N,N-dimethyl-formamide at 165℃; for 1 h; Microwave irradiation
To a solution of 4-bromo-2,3-dihydroinden-1-one SM1 (1 g, 4.74 mol) in DMF (5 mL) were added Zn(CN)2 (0.55 g, 4.74 mmol) and Pd(PPh3)4 (0.14 g, 0.12 mmol), the reaction mixture was under microwave irradiation for 1 hour at 165 °C . The solvent was removed under reduced pressure. The cmde was purified by flash chromatography on silica gel using a mixture of (PE/EtOAc 5:1) as eluent to afford compound 1 (0.45 g, 60percent) as an solid. LC-MS: m/z =158[M+H]
60%
With tetrakis(triphenylphosphine) palladium(0) In 1-methyl-pyrrolidin-2-one at 95℃; for 7 h; Inert atmosphere
To a stirred solution of 4-bromo-2,3-dihydro-1H-inden-1-one (100.0 g, 0.48 mol) in 150 mL of 1-methy-2-pyrrolidine (NMP)was added zinc cyanide (111.8 g,0.95 mol) and tetrakis(triphenylphosphine)palladium [Pd(PPh3)4] (2.75 g, 0.024 mol).The solution was degassed with N2 and the reaction mixture heated at 95°C for 7 h. Upon cooling, the reaction mixture was poured onto ice water (3.5 L). The compound and inorganic Zn salts precipitated. The solid was collected and partitioned between DCM (3 X 100 mL) and water. The organic layers were filtered to remove the Zn salts, and the filtrate was concentrated and crystallized from a 4:1 mixture of EtOH andMeOH (400 mL) to give 45.5 g (60 percent) of 1-oxo-2,3-dihydro-JH-indene-4-carbonitrile INT-1 as a light yellow solid. LCMS-ESI (m/z) calculated for C,0H7N0: 157.2; found 158.1 [M+H], tR = 2.67 mm. ‘H NIVIR (400 MHz, CDC13) 8.00 — 7.90 (m, 1H), 7.86 (dd, J 7.5, 1.1, 1H), 7.50 (t, J= 7.6, 1H), 3.40—3.19 (m, 2H), 2.90—2.61 (m, 2H).‘3CNIVIR(1o1 MHz, CDCl3)204.70, 157.90, 138.38, 137.88, 128.44, 128.28, 116.31,111.70, 36.01, 25.49.
0.18 g
at 150℃; for 1 h; Microwave irradiation
To a solution of 4-bromo-2,3-dihydro-1H-inden-1-one (300 mg, 1.4214 mmol) in dimethylamine (3 mL) was added dicyanozinc (0.1669 g, 1.4214 mmol) and Pd(PPh3)4 (0.0822 g, 0.0711 mmol). The reaction was stirred at 150 °C in microwave for 1 h. Water (20 mL) was added to the reaction vessel and the resulting biphasic mixture was transferred to a separatory funnel. The layers were separated and the aqueous phase was extracted with EtOAc (3 x 20 mL). The combined organics were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The resulting oil was purified by flash column chromatography with a gradient elution of EtOAc (5percent) and hexane (95percent) to EtOAc (10percent) and EtOAc (90percent) to provide 1-oxo-2,3-dihydro-1H-indene-4-carbonitrile (0.18 g, 1.1453 mmol) as a white solid.
With copper(l) iodide In N,N-dimethyl-formamide at 120℃; for 16 h;
l-oxo-2,3-dihydro-lH-indene-4-carbonitrile: To a solution of 4-bromo-2,3- dihydro-lH-inden-l-one (50 g, 236.91 mmol, 1.00 equiv) in N,N-dimethylformamide (250 mL) was added CuCN (63.67 g, 710.92 mmol, 3.00 equiv), Cul (4.5 g, 23.63 mmol, 0.10 equiv). The resulting solution was stirred for 16 h at 120 °C. The reaction mixture was cooled. The resulting solution was diluted with water (700 mL). The solids were filtered out. The resulting solution was extracted with ethyl acetate (4 x 300 mL) and the organic layers were combined, dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by a silica gel column eluting with ethyl acetate/petroleum ether (1:2) to afford 24 g (64percent) of l-oxo-2,3-dihydro-lH-indene-4-carbonitrile as a light brown solid.
60%
at 20℃; for 22 h; Reflux
To a solution of 4-bromo-1-indanone (10 g, 47.37 mmol) in DMF (100 mL) was added CuCN (12.72 g, 142.11 mmol) and the reaction mixture heated under reflux for 6h. The reaction mixture was cooled to RT and stirred for 16h. The reaction mixture was filtered through Celite®, and washed with EtOAc. The combined filtrate was diluted with EtOAc(500 mL) and washed with ice-water (3 x 300 mL) and the combined organic layers were washed with brine, dried over Na2504 and concentrated in vacuo. The crude material was purified by silica gel column chromatography, eluting with 10-20percent EtOAc/pet. ether to afford 1-oxo-indan-4-carbonitrile (4.5 g, 60percent) as a yellow solid.Rt: 0.6 (30percent EtOAc/pet ether).1H NMR (400MHz, CDCI3): O 7.97 (d, J= 7.6 Hz, 1H), 7.89 (dd, J= 6.0, 1.2 Hz, 1H), 7.52 (dd, J= 4.4, 3.6 Hz, 1H), 3.33 (t, J= 6.0 Hz, 2H), 2.82-2.79 (m, 2H).
Reference:
[1] Patent: US5902803, 1999, A,
[2] Patent: US5789406, 1998, A,
28
[ 15115-60-3 ]
[ 16657-07-1 ]
Reference:
[1] Journal of Organic Chemistry, 1984, vol. 49, # 22, p. 4226 - 4237
[2] Anales de la Real Sociedad Espanola de Fisica y Quimica, 1956, vol. <B> 52, p. 117,121
Stage #1: With hydroxylamine hydrochloride In methanol for 1 h; Reflux Stage #2: With diisobutylaluminium hydride In dichloromethane; toluene at 0 - 20℃;
Compound e (1.05 g, 5 mmol) was dissolved in methanol (14 ml) and hydroxylamine hydrochloride (1.4 g, 20 mmol) was added and refluxed for 1 h.Concentrate in vacuo, add dichloromethane and 50percent NaHCO3 for 20 minutes, dry the organic layer and concentrate to a white solid. Yield: 99percent.The resulting white solid (5 mmol) was dissolved in methylene chloride (30 ml), cooled to 0 °C, AlH (Bu-i) 2 (20 ml, 30 mmol, 1.5 M in toluene) was slowly added, and the mixture was stirred for 30 min.Move to room temperature and stir overnight. Quenching with water under ice bath conditions, dichloromethane extraction, saturated brine,After drying over anhydrous sodium sulfate, it was concentrated and purified by silica gel column (PE:EA=3:1) to give 600 mg of pale yellow solid f, yield: 56.6percent.
Reference:
[1] Patent: CN107814785, 2018, A, . Location in patent: Paragraph 0062; 0081; 0082; 0083
32
[ 15115-60-3 ]
[ 880094-83-7 ]
Yield
Reaction Conditions
Operation in experiment
67%
With sodium azide; sulfuric acid In chloroform at 20 - 45℃; for 24 h;
[0105] A triphasic mixture of sodium azide (18.5 g, 284 mmol), sulfuric acid (18.8 M, 4.8 mL, 90 mmol), water (36 mL) and chloroform (144 mL) was stirred at 0°C for 2.5 h. The layers were separated and the organic layer was dried over sodium sulfate and filtered. The filtrate was added to a solution of 4-bromoindan-l-one (12.0 g, 56.9 mmol) in chloroform (215 mL). To this solution was added sulfuric acid (18.8 M, 18.7 mL, 351.6 mmol) dropwise over 10 min. The reaction mixture was stirred at 45°C for 4 h, then cooled to room temperature and stirred for 20 h. The mixture was poured onto ice (200 g) and neutralized by addition of 10percent aqueous sodium hydroxide (50 mL). The layers were separated and the aqueous layer was extracted with chloroform (100 mL). The combined organic layers were dried over sodium sulfate and evaporated. The crude product was recrystallized from ethanol (55 mL) to give the title compound (8.65 g, 38.2 mmol, 67percent) as an off-white powder. LCMS: 98percent, Rt 1.290, ESMS m/z 226 (M+H)+.
Reference:
[1] Tetrahedron Letters, 2010, vol. 51, # 39, p. 5210 - 5212
[2] Bioorganic and Medicinal Chemistry Letters, 2007, vol. 17, # 12, p. 3473 - 3479
34
[ 15115-60-3 ]
[ 1109230-25-2 ]
Yield
Reaction Conditions
Operation in experiment
93%
With sodium azide; methanesulfonic acid In dichloromethane at 0 - 20℃; for 16 h;
[0107] To a mixture of 4-bromoindan-l-one (4.00g, 18.9 mmol) and methanesulfonic acid (20.2 mL, 310 mmol) in dichloromethane (180 mL) was added sodium azide (2.46 g, 37.9 mmol) at 0°C. The mixture was warmed to room temperature and stirred for 16 h. The reaction mixture was poured into 10percent aqueous sodium hydroxide (200 mL) and extracted with dichloromethane (100 mL). The combined organic layers were dried over sodium sulfate and evaporated. The crude product was recrystallized from ethyl acetate (40 mL) to give the title compound (3.98 g, 17.6 mmol, 93percent) as a white powder. LCMS: 96percent, Rt 1.225, ESMS m/z 226 (M+H)+.
93%
With sodium azide; methanesulfonic acid In dichloromethane at 0 - 20℃; for 16 h;
2H-isoquinolin-l-one. [00112] To a mixture of 4-bromoindan-l-one (4.00g, 18.9 mmol) and methanesulfonic acid (20.2 mL, 310 mmol) in dichloromethane (180 mL) was added sodium azide (2.46 g, 37.9 mmol) at 0 °C. The mixture was warmed to room temperature and stirred for 16 h. The reaction mixture was poured into 10percent aqueous sodium hydroxide (200 mL) and extracted with dichloromethane (100 mL). The combined organic layers were dried over sodium sulfate and evaporated. The crude product was recrystallized from ethyl acetate (40 mL) to give the title compound (3.98 g, 17.6 mmol, 93percent) as a white powder. LCMS: 96percent, t 1.225, ESMS m/z 226 (M+H)+.
93%
With sodium azide; methanesulfonic acid In dichloromethane at 0 - 20℃; for 16 h;
To a mixture of 4-bromoindan-1-one (4.OOg, 18.9 mmol) and methanesulfonic acid (20.2 mL, 310 mmol) in dichloromethane (180 mL) was added sodium azide (2.46 g, 37.9 mmol) at 0 °C. The mixture was warmed to room temperature and stirred for 16 h. The reaction mixture was poured into 10percent aqueous sodium hydroxide (200 mL) and extracted with dichloromethane (100 mL). The combined organic layers were dried over sodium sulfate and evaporated. The crude product was recrystallized from ethyl acetate (40 mL) to give the title compound (3.98 g, 17.6 mmol, 93percent) as a white powder. LCMS: 96percent, Rt 1.225, ESMS m/z 226 (M+H).
93%
With sodium azide; methanesulfonic acid In dichloromethane at 0 - 20℃; for 16 h;
100106] To a mixture of 4-bromoindan-1-one (4.OOg, 18.9 mmol) and methanesulfonic acid (20.2 mL, 310 mmol) in dichloromethane (180 mL) was added sodium azide (2.46 g, 37.9 mmol) at 0 °C. The mixture was warmed to room temperature and stirred for 16 h. The reaction mixture was poured into 10percent aqueous sodium hydroxide (200 mL) and extracted with dichloromethane (100 mL). The combined organic layers were dried over sodium sulfate and evaporated. The crude product was recrystallized from ethyl acetate (40 mL) to give the title compound (3.98 g, 17.6 mmol, 93percent) as a white powder. LCMS: 96percent, Rt 1.225, ESMS m/z 226 (M+H).
46%
With sodium azide In methanesulfonic acid; dichloromethane at 0 - 20℃;
General Procedure: To an ice-cooled solution of the benzocycloketone (1.4 mmol) in 1:1 CH2Cl2/methanesulfonic acid (10 mL) (except for compound 17, where concentrated HCl was used as solvent), sodium azide (0.2g, 2.8 mmol) was added in portions. The resulting mixture was allowed to stir at 0 °C for 1h, and then warmed up to room temperature while stirring overnight. Then, the reaction mixture was poured into ice-water (20 mL), basified with 5M NaOH (aq) until pH 9, and extracted with CH2Cl2 (3 .x. 10 mL). The organic layers were combined, dried over anhydrous Na2SO4, and concentrated under reduced pressure to give the crude lactam. Purification on silicagel chromathography (1:1 AcOEt/Hexane --> AcOEt) provided the desired 2H-benzolactam. The isomeric 1H benzolactam was also isolated in lower yield.
Reference:
[1] Patent: WO2014/153055, 2014, A2, . Location in patent: Paragraph 0106-0107
[2] Patent: WO2015/20553, 2015, A1, . Location in patent: Paragraph 00111-00112
[3] Patent: WO2015/50472, 2015, A1, . Location in patent: Paragraph 00109; 00110
[4] Patent: WO2015/50471, 2015, A1, . Location in patent: Paragraph 00105; 00106
[5] Bioorganic and Medicinal Chemistry Letters, 2011, vol. 21, # 9, p. 2670 - 2674
[6] Journal of Medicinal Chemistry, 2012, vol. 55, # 5, p. 2452 - 2468
With triethylsilane; trifluoroacetic acid; In tetrahydrofuran; dichloromethane; at -20 - 20℃; for 2h;
To a solution of 4-bromo-2,3-dihydro-lH-inden-l-one (2 g, 10 mmol) in DCM (lOmL) was added TFA, then cooled to - 20C. Et3SiH (15 ml_, IN in THF, 15 mmol) was added drop wise. After stirring for 2h at RT the reaction mixture was poured into NH4CI aqueous and extracted with DCM (10 ml_*2), washed with brine (10 ml_*2), dried by MgS04, concentrated, purified by FC to afford compound (1.7 g, yield : 85%); m/z (ES+) : 197 [M + H] +
80%
With triethylsilane; trifluorormethanesulfonic acid; In dichloromethane; at 0 - 20℃; for 16h;
To a solution of 4-bromoindan-1-one (200 mg, 947 umol) in dichloromethane (10 mL) was added a solution of trifluoromethanesulfonic acid (426 mg, 2.84 mmol) in dichloromethane (500 uL) and the reaction mixture was cooled to 0 C. Then triethylsilane (220 mg, 1.90 mmol) in dichloromethane (500 uL) was added dropwise to the reaction mixture and the mixture was stirred at 0 C for 0.5 hr. TLC detected most starting material remained. Then another batch of trifluoromethanesulfonic acid (426 mg, 2.84 mmol) and triethylsilane (220 mg, 1.90 mmol) was added in turn and the reaction mixture was stirred at 20 C for 15.5 hrs. On completion, the reaction mixture was diluted with 15 mL DCM and washed with saturated sodium bicarbonate until pH = 7. The organic layer was dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel chromatography (petroleum ether) to give the title compound (1.00 g, 80% yield) as colorless oil.1H NMR (400MHz, CDCl3) delta = 7.31 (d, J = 7.6 Hz, 1H), 7.16 (d, J = 7.6 Hz, 1H), 7.02 (dd, J = 7.6, 7.6 Hz, 1H), 3.04 (t, J = 7.2 Hz, 2H), 2.98 (t, J = 7.2 Hz, 2H), 2.11 (m, 2H).
With hydrogenchloride; sodium borohydrid; In ethanol; dichloromethane;
4-Bromo-2,3-dihydro-1H-inden-1-ol (3) A solution of compound 2 (435 g, 2.06 mol, 1 equiv) in ethanol (5 L) was treated with sodium borohydride (101.6 g, 2.68 mol, 1.3 equiv) and stirred overnight at room temperature. The reaction was concentrated under reduced pressure and the residue partitioned between 4 L of dichloromethane and 4 L of 10% aqueous hydrochloric acid. The layers were separated and the aqueous layer was extracted with dichloromethane (3*1 L). The combined organic layers were washed with saturated brine (2 L), dried over sodium sulfate and concentrated under reduced pressure. The resulting solid was dried overnight in a vacuum oven at 30 C. to give compound 3 (422 g, 96% yield) as an off-white solid.
96%
With sodium tetrahydroborate; In ethanol; at 20℃;
00162] 4-Bromo-2,3-dihydro-lH-inden-l-ol (c): A solution of compound b (435 g, 2.06 mol, 1 equiv) in ethanol (5 L) was treated with sodium borohydride (101.6 g, 2.68 mol, 1.3 equiv) and stirred overnight at room temperature. The reaction was concentrated under reduced pressure and the residue partitioned between 4 L of dichloromethane and 4 L of 10% aqueous hydrochloric acid. The layers were separated and the aqueous layer was extracted with dichloromethane (3 x 1 L). The combined organic layers were washed with saturated brine (2 L), dried over sodium sulfate and concentrated under reduced pressure. The resulting solid was dried overnight in a vacuum oven at 30C to give compound c (422 g, 96% yield) as an off-white solid.
96%
With sodium tetrahydroborate; In ethanol; at 23℃;
4-Bromo-2,3-dihydro-1H-inden-1-ol (3): A solution of compound 2 (435 g, 2.06 mol, 1 equiv) in ethanol (5 L) was treated with sodium borohydride (101.6 g, 2.68 mol, 1.3 equiv) and stirred overnight at room temperature. The reaction was concentrated under reduced pressure and the residue partitioned between 4 L of dichloromethane and 4 L of 10% aqueous hydrochloric acid. The layers were separated and the aqueous layer was extracted with dichloromethane (3×1 L). The combined organic layers were washed with saturated brine (2 L), dried over sodium sulfate and concentrated under reduced pressure. The resulting solid was dried overnight in a vacuum oven at 30 C. to give compound 3 (422 g, 96% yield) as an off-white solid.
86.8%
With methanol; sodium tetrahydroborate; at 5 - 20℃; for 2h;
Compound I-15A (10.5 g, 50 mmol) was dissolved in methanol (100 ml), cooled to below 5 C in an ice bath, and then a portion of sodium borohydride was added. After the addition, the reaction was allowed to stand at room temperature for two hours. The reaction was quenched with 10 mL of water. EtOAc was evaporated.Extracted with ethyl acetate (150 mL×3).The combined organic layers were washed with brine.Then dried over anhydrous sodium sulfate, filtered to remove the desiccant, and desolvated under reduced pressure.The residue was purified by silica gel column chromatography(petroleum ether/ethyl acetate = 10/1),The compound I-15B (9.2 g, pale yellow liquid) was obtained, yield: 86.8%.
85%
With sodium tetrahydroborate; ethanol; silica gel; at 0 - 20℃; for 2.33333h;
[0393] 4-bromo-2,3-dihydro-lH-inden-l-ol (INT-28)[0394] To a stirring solution of 4-bromoindanone (3 g, 14.2 mmol) in anhydrous EtOH(30mL) were added sodium borohydride (0.36 g, 9.5 mmol) and silica gel (2g) at 0C. The reaction was stirred at 0C for 20 min and was allowed to stir at room temperature for 2 h. The reaction mixture was quenched with saturated NaHC03 and concentrated to remove EtOH. The aqueous layer was extracted with EA and the organic phase was dried over MgS0 . After concentration, the crude product was purified by chromatography (EA / hexane) to yield 4-bromo-2,3-dihydro-lH-inden-l-ol INT-28 (2.56 g, 85%) as white solid. LCMS-ESI (m/z) calculated for C9H9BrO: 213.1; found 195.0 [M-H20]+, tR = 3.07 min. NMR (400 MHz, CDC13) δ 7.35 (d, J = 7.9, 1H), 7.27 (d, J = 7.4, 1H), 7.05 (t, J = 7.7, 1H), 5.23 (t, J = 6.2, 1H), 3.00 (ddd, J = 16.6, 8.8, 4.6, 1H), 2.84 - 2.66 (m, 1H), 2.45 (dddd, J = 13.2, 8.4, 7.0, 4.6, 1H), 1.96 - 1.70 (m, 2H).
85%
With sodium tetrahydroborate; ethanol; at 0 - 20℃; for 2.33333h;
(+-)-4-bromo-2,3-dihydro-1H-inden-1-ol (IND INT-5) To a stirring solution of 4-bromoindanone (3 g, 14.2 mmol) in anhydrous EtOH (30 mL) was added sodium borohydride (0.36 g, 9.5 mmol) and silica gel (2 g) at 0 C. The reaction was stirred at 0 C. for 20 min and was allowed to stir at room temperature for 2 h. The reaction mixture was quenched with saturated NaHCO3 solution (10 mL) and concentrated to remove EtOH. The aqueous layer was extracted with EA (3*20 mL) and the organic phase was dried over MgSO4. After concentration, the crude product was purified by chromatography (EA/hexane) to yield (+-)-4-bromo-2,3-dihydro-1H-inden-1-ol IND INT-5 (2.56 g, 85%) as a white solid. LCMS-ESI (m/z) calculated for C9H9BrO: 213.07. found 195.0 [M-H2O]+, tR=3.07 min. 1H NMR (400 MHz, CDCl3) δ 7.35 (d, J=7.9, 1H), 7.27 (d, J=7.4, 1H), 7.05 (t, J=7.7, 1H), 5.23 (t, J=6.2, 1H), 3.00 (ddd, J=16.6, 8.8, 4.6, 1H), 2.84-2.66 (m, 1H), 2.45 (dddd, J=13.2, 8.4, 7.0, 4.6, 1H), 1.96-1.70 (m, 2H).
With sodium tetrahydroborate; In methanol; at 20℃; for 1h;
Sodium borohydride (0.36 g, 9.52 mmol) was added to a methanol solution (15 mL) of 4-bromo-1-indanone (1.0 g, 4.74 mmol). The mixture was stirred at room temperature for 1 h. The reaction mixture was diluted with water, and extracted with ethyl acetate. The organic phase was dried over anhydrous magnesium sulfate, and the solvent was evaporated. The resulting alcohol and triphenylphosphonium bromide (1.63 g, 4.74 mmol) were suspended in benzene (10 mL) and refluxed for 12 h. The precipitate was collected by filtration and dried. The obtained salt (0.68 g, 1.26 mmol), 3-furaldehyde (0.12 g, 1.25 mmol) and 18-crown-6 (0.04 g) were dissolved in dichloromethane (3 mL), and a 50% potassium carbonate aqueous solution (1.4 mL) was added. The mixture was stirred vigorously overnight. Then, the reaction was extracted with ethyl acetate several times. The organic layers were combined and dried with anhydrous magnesium sulfate. The solvent was removed with a rotary evaporator, and the residue was purified via chromatography using hexane for elution to generate 3-(4-bromo-indan-1-ylidenemethyl)furan (0.26 g, 20% yield).
With sodium tetrahydroborate; ethanol; at 20℃; for 12h;
Compound 50-1 (2.00 g, 9.48 mmol) was dissolved in ethanol (20.0 mL), and sodium borohydride (466 mg, 12.3 mmol) was added. The reaction was stirred at 20 C. for 12 hours, then concentrated under reduced pressure. The residue was dissolved in dichloromethane (40 mL). Then, 1M hydrochloric acid (20 mL) was added to the reaction mixture, and the aqueous layer was extracted with ethyl acetate (20 mL×2). The organic layers were combined and washed with saturated brine (30 mL×2), dried over anhydrous sodium sulfate and concentrated under reduced pressure to give Compound 50-2. (0611) 1H NMR: (400 MHz, CDCl3) δ 7.43 (d, J=7.6 Hz, 1H), 7.36 (d, J=7.6 Hz, 1H), 7.13 (t, J=7.6 Hz, 1H), 5.35-5.27 (m, 1H), 3.11-3.04 (m, 1H), 2.91-2.76 (m, 1H), 2.60-2.44 (m, 1H), 2.02-1.91 (m, 1H), 1.87 (s, 1H).
With trifluorormethanesulfonic acid In dichloromethane at 80℃; for 1h; High pressure; Inert atmosphere; Green chemistry;
3.4. General Q-tube-Assisted Procedure
General procedure: Trifluoromethane sulfonic acid (3 eq.) was gently added to a cooled (0 °C) solution of a 3-Phenylpropionic acid (0.5 mmol) in dry CH2Cl2 (1.0 mL) in a 12 mL Q-tube pressure tube, furnished by QLabtech. The temperature was raised to room temperature. A Teflon septum was placed on the top of the tube and the appropriate cap and pressure adapter were used. The mixture was heated in an oil bath at 80 °C. The reaction was monitored by TLC and GC/MS until the reactant disappeared. The mixture was poured into ice and extracted three times with CH2Cl2. The organic phase collected was dried on Na2SO4, filtered and concentrated under vacuum. The desired pure product was separated from the crude by flash chromatography.
95%
Stage #1: 3-(2-bromophenyl)propionic acid With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 20℃; for 1h;
Stage #2: With aluminum (III) chloride In dichloromethane at 0℃; for 1h;
1 Step 1 - 4-Bromoindan-1-one
To a solution of 3-(2-bromophenyl)propanoic acid (1.20 g, 5.24 mmol, CAS 15115-58-9) in a mixture of solvent dichloromethane (20 mL) and dimethylformamide (3.83 mg, 52.4 umol) was added oxalyl chloride (1.33 g, 10.4 mmol) and the reaction mixture was stirred at 20 °C for 1 hr. On completion, the mixture was concentrated in vacuo and the residue was dissolved in dichloromethane (20 mL) and cooled to 0 °C. Then aluminum trichloride (838 mg, 6.29 mmol) was added at 0 °C and the reaction mixture was stirred at 0 °C for 1 hr. On completion, the reaction mixture was poured into 100 mL cool water and extracted with DCM (3 X 50 mL). The organic layer was dried over Na2SO4, filtered and concentrated in vacuo to give the title compound (1.10 g, 95% yield) as a light yellow solid.1H NMR (400MHz, CDCl3) δ = 7.77 (d, J = 7.2 Hz, 1H), 7.73 (d, J = 7.2 Hz, 1H), 7.30 (dd, J = 7.2, 7.2 Hz, 1H), 3.12 (t, J = 7.2 Hz, 2H), 2.75 (t, J = 7.2 Hz, 2H).
86%
With aluminium trichloride In dichloromethane; 1,2-dichloro-ethane
4-Bromo-2,3-dihydro-1H-inden-1-one (2)
4-Bromo-2,3-dihydro-1H-inden-1-one (2) 3-(2-Bromophenyl)propanoic acid (1) (550 g, 2.4 mol, 1 equiv) was dissolved in 1,2-dichloroethane (5.5 L). Thionyl chloride (437.8 mL, 6 mol, 2.5 equiv) was added to the solution and the mixture was refluxed for 24 hours. The reaction was cooled to room temperature and concentrated under reduced pressure. The residue was dissolved in methylene chloride (1 L) and added dropwise to a mechanically stirred suspension of anhydrous aluminum chloride (526.9 g, 3.96 mol, 1.65 equiv) in dichloromethane (1 L) while keeping the reaction temperature below 27° C. The reaction was stirred at room temperature for three hours before being quenched into a five gallon bucket which was half-full of ice. The resulting mixture was extracted with dichloromethane (3*3 L). The combined organic layers were washed sequentially with saturated brine (2 L) and saturated sodium bicarbonate (2 L). The organic layer was dried over sodium sulfate, and concentrated under reduced pressure. The resulting solid was dried overnight in a vacuum oven at 30° C. to give compound 2 (435 g, 86% yield) as an off-white solid.
86%
Stage #1: 3-(2-bromophenyl)propionic acid With thionyl chloride In 1,2-dichloro-ethane for 24h; Reflux;
Stage #2: With aluminum (III) chloride In dichloromethane at 20 - 27℃;
[00161] 4-Bromo-2,3-dihydro-lH-inden-l-one (b):
[00161] 4-Bromo-2,3-dihydro-lH-inden-l-one (b): 3-(2-Bromophenyl)propanoic acid (a) (550 g, 2.4 mol, 1 equiv) was dissolved in 1 ,2-dichloroethane (5.5 L). Thionyl chloride (437.8 niL, 6 mol, 2.5 equiv) was added to the solution and the mixture was refluxed for 24 hours. The reaction was cooled to room temperature and concentrated under reduced pressure. The residue was dissolved in methylene chloride (1 L) and added dropwise to a mechanically stirred suspension of anhydrous aluminum chloride (526.9 g, 3.96 mol, 1.65 equiv) in dichloromethane (1 L) while keeping the reaction temperature below 27°C. The reaction was stirred at room temperature for three hours before being quenched into a five gallon bucket which was half-full of ice. The resulting mixture was extracted with dichloromethane (3 x 3 L). The combined organic layers were washed sequentially with saturated brine (2 L) and saturated sodium bicarbonate (2 L). The organic layer was dried over sodium sulfate, and concentrated under reduced pressure. The resulting solid was dried overnight in a vacuum oven at 30°C to give compound b (435 g, 86% yield) as an off-white solid.
76%
Stage #1: 3-(2-bromophenyl)propionic acid With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 0℃;
Stage #2: With aluminum (III) chloride at 0℃; for 1h;
NINETEEN-1 Example NINETEEN-1 (Compound 149)
Example NINETEEN-1 (Compound 149) [4-BROMO-INDAN-1-ONE] was obtained by the following procedure: A solution of [3- (2-BROMO-PHENYL)-PROPIONIC] acid (commercially available from Oakwood Products) (15.0 g, 65.5 mmol) in [CHUCK] at [0 °C] was reacted with oxalyl chloride (7.2 mL, 1.5 eq) followed by 2-3 drops of DMF. The mixture was stirred until no more gas evolution was observed. As the mixture was concentrated and the residue was dissolved in [CH2C12,] cooled to [0 °C,] and treated with [AIDS] (9.6 g, 1.1 eq). After 1 h the mixture was quenched with water and the layers were separated. The aqueous layer was extracted with Et2O (3 x 150 [ML)] and the combined organic extracts were washed with H20 (3 x 100 mL), saturated NaHCO3 (3 x 100 mL), brine (1 x 100 [ML),] dried over MgS04 and concentrated. [4-BROMOINDAN-L-ONE,] 10.5 g (76%) was obtained by chromatography using 10 % EtOAc: hexane as eluant. Use of 4-bromo- [INDAN-1-ONE] in Method NINETEEN produced [4- (4-BROMO-INDAN-2-YL)-1,] 3-dihydro- imidazole-2-thione (Compound 149). 1H NMR (300 MHz, DMSO-d6) 8 12.0 (s, 1H), 11.7 (s, 1H), 7.34 (d, J= 7.8 Hz, 1H), 7.22 (d, [J=] 7.5 Hz, 1H), 7.09 (t, J= 7.5 Hz, [1H),] 6.63 (s, [1H),] 3.50-3. 40 (m, [1H),] 3.30-3. 12 (m, 2H), 3.06-2. 85 (m, 2H).
(i) SOCl2, (ii) AlCl3, CS2; Multistep reaction;
Multi-step reaction with 2 steps
1: thionyl chloride / 0.75 h / Heating
2: 93 percent / AlCl3 / CS2 / 3 h / Heating
Multi-step reaction with 2 steps
1: SOCl2
2: AlCl3
Multi-step reaction with 2 steps
1: thionyl chloride / 1,2-dichloro-ethane / 24 h / Reflux
2: aluminum (III) chloride / dichloromethane / 3 h / 23 - 27 °C
Multi-step reaction with 2 steps
1: thionyl chloride / N,N-dimethyl-formamide / benzene / 3 h / Heating / reflux
2: aluminum (III) chloride / dichloromethane / 1 h / 0 °C
With hydrogenchloride; bromine In tetrahydrofuran; hexane; water
6-Bromoindan-1-one (VIIb)and 4-bromoindan-1-one (VIIa).
6-Bromoindan-1-one (VIIb)and 4-bromoindan-1-one (VIIa). A 250 mL three-necked round bottom flask was equipped with a water cooled Liebig condenser, a mechanical stirrer and a pressure-equalized addition funnel protected by means of drying tubes. The flask was charged with anhydrous aluminum chloride (16.6 g, 0.125 moles) and stirred while 1-Indanone (VI) (6.60 g, 0.05 moles), which had been ground to a fine powder with a mortar and pestle was added in two portions over 3 minutes. Copious evolution of HCl gas was accompanied by a moderate exotherm and the mixture rapidly became a dark brown, homogenous slurry which was stirred for an additional 10 minutes. Bromine (3.1 ml, 0.06 moles) was added dropwise to the well stirred mixture over a 10 minute period. After all the bromine had been added, the molten mixture was heated in a water bath to 80° C. for 5 minutes. While still hot, the mixture was poured into 100 g of crushed ice and 20 ml of concentrated hydrochloric acid. The ice mixture was then stirred for 10 minutes, diluted with 100 ml of water and extracted with ether (2*200 mL). The combined ether extracts were washed with water (2*100 mL) and dried over anhydrous sodium sulfate and concentrated to give 10.6 g of a red oil which crystallized on standing at room temperature. Product analysis by GC-MS showed a 1:1 mixture of monobromo isomers. The material was chromatographed on a silica gel column (45*10 cm) using n-hexane/THF (8:1) thereby separating the isomers. The separated isomers were each recrystallized from hexane to give 3.8 g of 6-bromoindan-1-one (VIIa) and 4.0 g of 4-bromoindan-1-one (VIIb).
With sodium azide; sulfuric acid; In chloroform; at 20 - 45℃; for 24h;
[0105] A triphasic mixture of sodium azide (18.5 g, 284 mmol), sulfuric acid (18.8 M, 4.8 mL, 90 mmol), water (36 mL) and chloroform (144 mL) was stirred at 0C for 2.5 h. The layers were separated and the organic layer was dried over sodium sulfate and filtered. The filtrate was added to a solution of 4-bromoindan-l-one (12.0 g, 56.9 mmol) in chloroform (215 mL). To this solution was added sulfuric acid (18.8 M, 18.7 mL, 351.6 mmol) dropwise over 10 min. The reaction mixture was stirred at 45C for 4 h, then cooled to room temperature and stirred for 20 h. The mixture was poured onto ice (200 g) and neutralized by addition of 10% aqueous sodium hydroxide (50 mL). The layers were separated and the aqueous layer was extracted with chloroform (100 mL). The combined organic layers were dried over sodium sulfate and evaporated. The crude product was recrystallized from ethanol (55 mL) to give the title compound (8.65 g, 38.2 mmol, 67%) as an off-white powder. LCMS: 98%, Rt 1.290, ESMS m/z 226 (M+H)+.
To a stirred mixture of sodium azide (862 mg, 13.3 mmol) in H2O (1.7 mL) and CHCl3 (6.5 mL) was added cone. H2SO4 (0.23 mL, 4.14 mmol). The reaction mixture was stirred vigorously at 0 C for 2.5 h. The CHCl3 layer was separated, dried over Na2SO4, and added to a solution of 4-bromo-l-indanone (1.12 g, 5.31 mmol) in CHCl3 (20 mL). To this mixture was added cone. H2SO4 (1.75 mL, 31.5 mmol) dropwise over 10 min. The reaction mixture was heated at 45 C for 1 h then poured into ice-water, neutralized with 20% aqueous NaOH, and extracted with CH2Cl2 (100 mL). The organic extract was dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by silica gel chromatography, eluting with a gradient of CH2C12:EtOAc - 100:0 to 80:20, to give the title compound. MS: mlz = 226 (M + 1).
With sodium azide; sulfuric acid; In chloroform; at 45℃; for 1.16667h;
To a stirred mixture of sodium azide (862 mg, 13.3 mmol) in H2O (1.7 mL) and CHCl3 (6.5 mL) was added cone. H2SO4 (0.23 mL, 4.14 mmol). The reaction mixture was stirred vigorously at 0 0C for 2.5 h. The CHCl3 layer was separated, dried over Na2SO4, and added to a solution of 4-bromo- 1-indanone (1.12 g, 5.31 mmol) in CHCb (20 mL). To this mixture was added cone. H2SO4 (1.75 mL, 31.5 mmol) dropwise over 10 min. The reaction mixture was heated at 45 C for 1 h then poured into ice- water, neutralized with 20% aqueous NaOH, and extracted with CH2Cl2 (100 mL). The organic extract was dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by silica gel chromatography, eluting with a gradient of CH2C12: EtOAc - 100:0 to 80:20, to give the title compound. MS: ml z = 226 (M + 1).
12.61 g of copper cyanide are added to a solution of 10 g of 4-bromo-1-indanone in 94 ml of dimethylformamide and the reaction mixture is then brought to reflux for 6 hours and stirred at a temperature in the region of 20 C. overnight. The solution is then poured into an aqueous sodium cyanide solution (5%), stirred for 15 minutes and extracted with three times 500 ml of ethyl acetate. The organic phases are dried over sodium sulphate and concentrated under reduced pressure (15 mm Hg; 2 kPa) at 40 C. The residue thus obtained is purified by flash chromatography on a silica column, under a nitrogen stream at medium pressure (0.5 bar), with an ethyl acetate/cyclohexane (20/80 by volume) mixture as eluent. 5.9 g of 4-cyano-1-indanone are thus obtained, which product melts at 122 C. 4-Bromo-1-indanone can be prepared as described by F. G. Holliman, F. G. Manne and D. A. Thornton, J. Chem. Soc., 9 (1960).
In N-methyl-acetamide;
12.61 g of copper cyanide are added to a solution of 10 g of 4-bromo-1-indanone in 94 ml of dimethylformamide, the reaction mixture is then brought to reflux for 6 hours and stirred at a temperature in the region of 20 C. overnight. The solution is then poured into an aqueous sodium cyanide solution (5%), stirred for 15 minutes and extracted with three times 500 ml of ethyl acetate. The organic phases are dried over sodium sulphate and concentrated under reduced pressure (15 mm Hg, 2 kPa) at 40 C. The residue thus obtained is purified by flash chromatography on a silica column, under a stream of nitrogen at moderate pressure (0.5 bar) with an ethyl acetate/cyclohexane (20/80 by volume) mixture as eluent. There are thus obtained 5.9 g of 4-cyano-1-indanone, melting at 122 C. 4-Bromo-1-indanone can be prepared as described by F. G. Holliman, F. G. Manne and D. A. Thornton, J. Chem. Soc., 9 (1960).
With potassium hydroxide; sodium tetrahydroborate; methyl iodide; In THF-methanol; water; dimethyl sulfoxide;
4-Bromo-1-methoxyindane To a solution of 100 g (0.474 mol) of 4-bromo-1-indanone in 530 ml of THF-methanol (2:1, vol.), 26.8 g (0.709 mol) of NaBH4 was added in small portions at vigorous stirring for 1.5 hours at 5 C. This mixture was stirred at room temperature for 12 hours and then added to 1 L of cold water. The hydrogenation product was extracted with 4*200 ml of dichloromethane, and the combined extract was evaporated to dryness. To 108 g (1.93 mol) of KOH in 700 ml of DMSO, 135 g (59.2 ml, 0.948 mol) MeI, and a solution of crude <strong>[16657-10-6]4-bromoindan-1-ol</strong> in 300 ml of DMSO were added. This mixture was stirred for 2 hours at ambient temperature; then, 67.2 g (29.5 ml, 0.473 mol) of MeI was added, and the mixture was additionally stirred for 2 hours. The resulting mixture was added to 2 L of cold water. The crude product was extracted with 5*200 ml of dichloromethane. The combined extract was dried over Na2SO4 and, then, evaporated to dryness. Fractional distillation gave 4-bromo-1-methoxyindane, b.p. 94 C./2 mm Hg. Yield, 100 g (93%) of a colorless oil. Anal. calc. for C10H11BrO: C, 52.89; H, 4.88. Found: C, 52.74; H, 4.81. 1H NMR (300 MHz, CDCl3): δ 4.88 (dd, J=6.7 Hz, J=4.3 Hz, CHOMe), 3.40 (s, 3H, OMe), 3.07 (ddd, J=16.6 Hz, J=8.5 Hz, J=5.9 Hz, 1H, CHH'CHH'CHOMe), 2.83 (ddd, J=16.6 Hz, J=8.6 Hz, J=5.3 Hz, 1H, CHH'CHH'CHOMe), 2.35 (m, 1H, CHH'CHOMe), 2.09 (m, 1H, CHH'CHOMe). 13C{1H} NMR (75 MHz, CDCl3): δ 144.4, 144.2, 131.4, 128.1, 123.8, 120.2, 85.1, 56.1, 31.6, 30.9.
Compound 149
Example NINETEEN-1 Compound 149 4-Bromo-indan-1-one was obtained by the following procedure: A solution of 3-(2-bromo-phenyl)-propionic acid (commercially available from Oakwood Products) (15.0 g, 65.5 mmol) in CH2Cl2 at 0° C. was reacted with oxalyl chloride (7.2 mL, 1.5 eq) followed by 2-3 drops of DMF. The mixture was stirred until no more gas evolution was observed. As the mixture was concentrated and the residue was dissolved in CH2Cl2, cooled to 0° C., and treated with AlCl3 (9.6 g, 1.1 eq). After 1 h the mixture was quenched with water and the layers were separated. The aqueous layer was extracted with Et2O (3*150 mL) and the combined organic extracts were washed with H2O (3*100 mL), saturated NaHCO3 (3*100 mL), brine (1*100 mL), dried over MgSO4 and concentrated. 4-Bromoindan-1-one, 10.5 g (76%) was obtained by chromatography using 10% EtOAc:hexane as eluant. Use of 4-bromo-indan-1-one in Method NINETEEN produced 4-(4-bromo-indan-2-yl)-1,3-dihydroimidazole-2-thione (Compound 149). 1H NMR (300 MHz, DMSO-d6) δ 12.0 (s, 1H), 11.7 (s, 1H), 7.34 (d, J=7.8 Hz, 1H), 7.22 (d, J=7.5 Hz, 1H), 7.09 (t, J=7.5 Hz, 1H), 6.63 (s, 1H), 3.50-3.40 (m, 1H), 3.30-3.12 (m, 2H), 3.06-2.85 (m, 2H).
tetrakis(triphenylphosphine) palladium(0); In 1-methyl-pyrrolidin-2-one; at 95℃; for 7h;Inert atmosphere;
[0435] l-oxo-2,3-dihydro-lH-indene-4-carbonitrile (INT-45)[0436] To a stirring solution of 4-bromo-2,3-dihydro-lH-inden-l-one (100.0 g, 0.48 mol) in150 mL of l-methy-2-pyrrolidine (NMP) was added zinc cyanide (111.8 g, 0.95 mol) and tetrakis(triphenylphosphine)palladium [Pd(PPh3)4] (2.75 g, 0.024 mol). The solution was degassed with N2 and the reaction mixture heated at 95C for 7 h. Upon cooling, the reaction mixture was poured onto ice water (3.5 L). The compound and inorganic Zn salts precipitated. The solid was collected and partitioned between DCM and water. The organic layers were filtered to remove the Zn salts, and the filtrate was concentrated and crystallized from a 4:1 mixture of EtOH and MeOH (400 mL) to give 45.5 g (60 %) of l-oxo-2,3- dihydro-lH-indene-4-carbonitrile INT-45 as a light yellow solid. LCMS-ESI (m/z) calculated for C10H7NO: 157.2; found 158.1 [M+H]+, tR = 2.67 min. 1H NMR (400 MHz, CDC13) 6 8.00 - 7.90 (m, 1H), 7.86 (dd, J = 7.5, 1.1, 1H), 7.50 (t, J = 7.6, 1H), 3.40 - 3.19 (m, 2H), 2.90 - 2.61 (m, 2H). 13C NMR (101 MHz, CDC13) delta 204.70, 157.90, 138.38, 137.88, 128.44, 128.28, 116.31, 111.70, 36.01, 25.49.
60%
With tetrakis(triphenylphosphine) palladium(0); In 1-methyl-pyrrolidin-2-one; at 95℃; for 7h;Inert atmosphere;
To a stirred solution of 4-bromo-2,3-dihydro-lH-inden-l-one (100.0 g, 0.48 mol) in 150 mL of l-methy-2-pyrrolidine (NMP) was added zinc cyanide (111.8 g, 0.95 mol) and tetrakis(triphenylphosphine)palladium [Pd(PPh3)4] (2.75 g, 0.024 mol). The solution was degassed with N2 and the reaction mixture heated at 95C for 7 h. Upon cooling, the reaction mixture was poured onto ice water (3.5 L). The compound and inorganic Zn salts precipitated. The solid was collected and partitioned between DCM (3 X 100 mL) and water. The organic layers were filtered to remove the Zn salts, and the filtrate was concentrated and crystallized from a 4: 1 mixture of EtOH and MeOH (400 mL) to give 45.5 g (60 %) of l-oxo-2,3-dihydro-7H-indene-4-carbonitrile INT-1 as a light yellow solid. LCMS-ESI (m/z) calculated for Ci0H7NO: 157.2; found 158.1 [M+H]+, tR = 2.67 min. 1H NMR (400 MHz, CDC13) delta 8.00 - 7.90 (m, 1H), 7.86 (dd, J = 7.5, 1.1, 1H), 7.50 (t, J = 7.6, 1H), 3.40 - 3.19 (m, 2H), 2.90 - 2.61 (m, 2H). 13C NMPv (101 MHz, CDC13) delta 204.70, 157.90, 138.38, 137.88, 128.44, 128.28, 116.31, 111.70, 36.01, 25.49.
60%
With tetrakis(triphenylphosphine) palladium(0); In N,N-dimethyl-formamide; at 165℃; for 1h;Microwave irradiation;
To a solution of 4-bromo-2,3-dihydroinden-1-one SM1 (1 g, 4.74 mol) in DMF (5 mL) were added Zn(CN)2 (0.55 g, 4.74 mmol) and Pd(PPh3)4 (0.14 g, 0.12 mmol), the reaction mixture was under microwave irradiation for 1 hour at 165 C . The solvent was removed under reduced pressure. The cmde was purified by flash chromatography on silica gel using a mixture of (PE/EtOAc 5:1) as eluent to afford compound 1 (0.45 g, 60%) as an solid. LC-MS: m/z =158[M+H]
60%
With tetrakis(triphenylphosphine) palladium(0); In 1-methyl-pyrrolidin-2-one; at 95℃; for 7h;Inert atmosphere;
To a stirred solution of 4-bromo-2,3-dihydro-1H-inden-1-one (100.0 g, 0.48 mol) in 150 mL of 1-methy-2-pyrrolidine (NMP)was added zinc cyanide (111.8 g,0.95 mol) and tetrakis(triphenylphosphine)palladium [Pd(PPh3)4] (2.75 g, 0.024 mol).The solution was degassed with N2 and the reaction mixture heated at 95C for 7 h. Upon cooling, the reaction mixture was poured onto ice water (3.5 L). The compound and inorganic Zn salts precipitated. The solid was collected and partitioned between DCM (3 X 100 mL) and water. The organic layers were filtered to remove the Zn salts, and the filtrate was concentrated and crystallized from a 4:1 mixture of EtOH andMeOH (400 mL) to give 45.5 g (60 %) of 1-oxo-2,3-dihydro-JH-indene-4-carbonitrile INT-1 as a light yellow solid. LCMS-ESI (m/z) calculated for C,0H7N0: 157.2; found 158.1 [M+H], tR = 2.67 mm. ?H NIVIR (400 MHz, CDC13) 8.00 - 7.90 (m, 1H), 7.86 (dd, J 7.5, 1.1, 1H), 7.50 (t, J= 7.6, 1H), 3.40-3.19 (m, 2H), 2.90-2.61 (m, 2H).?3CNIVIR(1o1 MHz, CDCl3)204.70, 157.90, 138.38, 137.88, 128.44, 128.28, 116.31,111.70, 36.01, 25.49.
49%
With tetrakis(triphenylphosphine) palladium(0); In 1-methyl-pyrrolidin-2-one; at 100℃; for 7h;Inert atmosphere;
To a stirred solution of 4-bromo-1-indanone (10 g, 47.37 mmol) in NMP (30 mL) were added zinc cyanide (11.12 g, 94.74 mmol, 2.0 eq) and [Pd(PPh3)4] (2.7 g, 2.37 mmol, 0.05 eq). The solution was degassed with Ar for 10 mm and the reaction mixture was heated at 100 C for 7 h. Upon cooling, the reaction mixture was poured onto ice water. The compound and inorganic Zn salts precipitated. The solid was collected and partitioned between DCM (x3) and water. The organic layers were filtered to remove the Zn salts, and the filtrate was concentrated and precipitated from a 4:1 mixture of EtOH and MeOH to give 3.7 g (49%) of compound 39 as a yellow solid.
tetrakis(triphenylphosphine) palladium(0); In N,N-dimethyl-formamide; at 165℃; for 1h;Microwave irradiation;
To a solution of 4~bromo~2,3-dihydro-lH-inden-l-one (1.00 g, 4.74 mmol) in 5 mL of DMF was added Zn(CN)2 (556 mg, 4.74 mmol) and Pd(PPh3)4 (77 mg, 0.14 mmol), and the reaction mixture was stirred under microwave irradiation for 1 h at 165 0C. The solvent was removed in vacuum to afford the crude compound, which was purified via column chromatography to afford 1 ~oxo-2,3-dihydro-l H-indene-4-carbonitrile
With tetrakis(triphenylphosphine) palladium(0); In N,N-dimethyl-formamide; at 165℃; for 1h;Microwave irradiation;
To a solution of 4-bromo-2,3-dihydro-1H-inden-1-one (1.00 g, 4.74 mmol) in 5 mL of DMF was added Zn(CN)2 (556 mg, 4.74 mmol) and Pd(PPh3)4 (77 mg, 0.14 mmol), and the reaction mixture was stirred under microwave irradiation for 1 h at 165 C. The solvent was removed in vacuum to afford the crude compound, which was purified via column chromatography to afford 1-oxo-2,3-dihydro-1H-indene-4-carbonitrile
0.18 g
With tetrakis(triphenylphosphine) palladium(0); dimethyl amine; at 150℃; for 1h;Microwave irradiation;
To a solution of 4-bromo-2,3-dihydro-1H-inden-1-one (300 mg, 1.4214 mmol) in dimethylamine (3 mL) was added dicyanozinc (0.1669 g, 1.4214 mmol) and Pd(PPh3)4 (0.0822 g, 0.0711 mmol). The reaction was stirred at 150 C in microwave for 1 h. Water (20 mL) was added to the reaction vessel and the resulting biphasic mixture was transferred to a separatory funnel. The layers were separated and the aqueous phase was extracted with EtOAc (3 x 20 mL). The combined organics were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The resulting oil was purified by flash column chromatography with a gradient elution of EtOAc (5%) and hexane (95%) to EtOAc (10%) and EtOAc (90%) to provide 1-oxo-2,3-dihydro-1H-indene-4-carbonitrile (0.18 g, 1.1453 mmol) as a white solid.
With formic acid; triethylamine In dichloromethane at 20℃; Inert atmosphere;
59.A
(59A) (1S)-4-Bromoindan-1-ol To a mixture of formic acid (1.20 mL, 31.5 mmol) and triethylamine (3.76 mL, 27.0 mmol), a dichloromethane (6.0 mL) solution of 4-bromoindan-1-one (1.90 g, 9.00 mmol) was added, and chloro[(1S,2S)-N-(p-toluenesulfonyl)-1,2-diphenyl ethanediamine](mesitylene)ruthenium(II) (168 mg, 0.270 mmol) was added thereto at room temperature, and then, the resulting mixture was stirred under a nitrogen atmosphere at room temperature for 2 hours. Water was added to the reaction solution, and the organic matter was extracted with ethyl acetate. The organic layer was washed with a saturated sodium bicarbonate solution and a saturated sodium chloride solution, then dried over anhydrous sodium sulfate and filtered. Then, the solvent was distilled off under reduced pressure, whereby a crude product was obtained. This crude product was purified by silica gel column chromatography (hexane:ethyl acetate=95:5 to 65:35 (v/v)), whereby the objective title compound was obtained as a white solid (1.90 g, yield: 99%). 1H NMR (CDCl3, 400 MHz): δ1.77 (1H, d, J=7.4 Hz), 1.93-2.01 (1H, m), 2.49-2.57 (1H, m), 2.83 (1H, dt, J=8.2, 16.4 Hz), 3.08 (1H, ddd, J=4.3, 8.6, 16.5 Hz), 5.32 (1H, dd, J=7.0, 12.5 Hz), 7.13 (1H, dd, J=7.8, 7.8 Hz), 7.36 (1H, d, J=7.4 Hz), 7.43 (1H, d, J=7.8 Hz)
90%
With dimethylsulfide borane complex; (R)-tetrahydro-1-methyl-3,3-diphenyl-1H,3H-pyrrolo[1,2-c]-[1,3,2]oxazaborole In dichloromethane; toluene at -20℃; for 0.3h; Inert atmosphere;
68.1
4-Bromo-1-indanone (5.0mmol) was dissolved in 50mL of dichloromethane solution,And under the protection of nitrogen, the temperature is reduced to -20,Then add (R)-2-methyl-CBS-oxazoborolidine (0.5mmol),The toluene solution (2M, 12.5 mL) of the borane dimethyl sulfide complex was slowly added dropwise through the dropping funnel, and the reaction was continued for about 3 hours after the addition was completed. Then add water to quench the reaction,Extract with dichloromethane, separate the organic phase and dry with anhydrous sodium sulfate. After filtration, the filtrate was concentrated under reduced pressure, and purified by silica gel column chromatography to obtain compound (S)-4-bromo-2,3-dihydro-1H-inden-1-ol (0.96 g) with a yield of 90%.
62%
With dimethylsulfide borane complex; (R)-tetrahydro-1-methyl-3,3-diphenyl-1H,3H-pyrrolo[1,2-c]-[1,3,2]oxazaborole In dichloromethane; toluene at -20℃; for 2h; Inert atmosphere;
(S)-4-bromo-2,3-dihydro-M-inden-1-ol (IND INT-1)
(S)-4-bromo-2,3-dihydro-M-inden-1-ol (IND INT-1) To a 100 mL 3-neck flask equipped with an internal thermometer and an addition funnel was added (R)-(+)-2-methyl-CBS-oxazaborolidine (1.6 mL, 1M solution in toluene) and borane-dimethylsulfide (150 uL) under N2. The reaction was stirred at room temperature for 10 min then diluted with DCM (10 mL) Borane-dimethylsulfide (6.0 mL) was added and the reaction cooled to -20° C. A solution of 4-Bromo-2,3-dihydro-1H-inden-1-one (2.5 g, 11.8 mmol) in DCM (10 mL) was added dropwise over 20 min while maintaining the reaction temperature at -20+-5° C. The reaction was stirred for 2 h after the addition was complete, then quenched by the dropwise addition of MeOH (10 mL). The reaction mixture was diluted with MeOH (20 mL) and the solvent distilled at atmospheric pressure. MeOH (30 mL) was added in two portions and the distillation was repeated twice. All the solvent was evaporated to give a solid which was purified by silica gel column chromatography (EA/hexanes) and recrystallization from 5:1 hexane/EA (30 mL) to provide 1.56 g (62%) of (S)-4-bromo-2,3-dihydro-1H-inden-1-ol as a white powder IND INT-1. LCMS-ESI (m/z) calculated for C9H9BrO: 213.1. found 196.9 [M-OH]+, tR=3.06 min. 1H NMR (400 MHz, CDCl3) δ 7.40 (d, J=7.9 Hz, 1H), 7.33 (d, J=7.5 Hz, 1H), 7.10 (t, J=7.7 Hz, 1H), 5.29 (dd, J=12.6, 6.9 Hz, 1H), 3.05 (ddd, J=16.6, 8.7, 4.6 Hz, 1H), 2.87-2.71 (m, 1H), 2.50 (dddd, J=13.2, 8.4, 7.0, 4.6 Hz, 1H), 1.94 (dddd, J=13.5, 8.8, 6.6, 5.5 Hz, 1H), 1.80 (d, J=7.1 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 146.82, 143.50, 131.24, 128.58, 123.21, 120.25, 76.83, 34.69, 31.19. Chiral HPLC: (S)-4-bromo-2,3-dihydro-1H-inden-1-ol was eluted using 10% IPA in hexanes: >99.9% % ee, tR=6.27 min. (R)-4-bromo-2,3-dihydro-1H-inden-1-ol IND INT-2 was prepared in an analogous manner using (S)-(-)-2-methyl-CBS-oxazaborolidine: 97.6% ee, tR for (R)-enantiomer=5.83 min.
4.5 g
With formic acid; chloro[(1S,2S)-(-)-2-amino-1,2-diphenylethyl](4-toluenesulfonyl)amido}-(mesitylene)ruthenium(ll); triethylamine In dichloromethane at 20℃; for 16h; Cooling with ice; Inert atmosphere;
1 Step 1 : (S)-4-Bromo-2,3-dihydro-1 H-inden-1 -ol
Formic acid (3.2 mL) is added to a solution of triethylamine (10.5 mL) in dichloromethane (50 mL) chilled in an ice bath. 4-Bromo-2,3-dihydro-1 H-inden-1 -one (5 g) is added and the flask is purged with argon for 5 minutes. Chloro[(1 S,2S)-(-)-2- amino-1 ,2-diphenylethyl](4-toluenesulfonyl)amido}-(mesitylene)ruthenium(l l) (700 mg; alternatively, the catalyst is formed in situ from N-[(1 S,2S)-2-amino-1 ,2- diphenylethyl]-4-methylbenzenesulfonamide and dichloro(p-cymene)-ruthenium(ll) dimer) is added and the mixture is stirred at room temperature for 16 hours. Water is added and the resulting mixture is extracted with dichloromethane. The combined extract is washed with saturated aqueous NaHCO3 solution and dried (MgSO4). The solvent is evaporated and the residue is chromatographed on silica gel (cyclohexane/ethyl acetate 99:1→50:50) to give the title compound. Yield: 4.5 g; LC (method 1 ): tR = 1 .04 min; Mass spectrum (ESI+): m/z = 195 [M+H-H2O]+
With [N-[(1S,2S)-2-(amino-κN)-1,2-diphenylethyl]-4-methylbenzenesulfonamidato-κN]chloro[(1,2,3,4,5,6-η)-1-methyl-4-(1-methylethyl)benzene]ruthenium; formic acid; triethylamine In dichloromethane at 20℃; for 16h; Inert atmosphere; Cooling with ice;
1.1 Step 1: Preparation of (S)-4-bromo-2,3-dihydro-1H- inden-1-ol
Formic acid (3.18 mL) is added to a solution of triethylamine (9.9 mL) in dichloromethane (0.2 M) cooled in an ice bath. 4-Bromo-2,3-dihydro-1H-inden-l-one (5 g) is added and the flask is purged with nitrogen for 5 minutes. Chloro [(1S, 2S) - (-) - 2-amino-1,2-diphenylethyl](4-toluenesulfonyl) amido} - (mesitylene) ruthenium (II)(412 mg; alternatively, the catalyst is formed from the N - [(1S, 2S) -2-amino-1,2-diphenylethyl] -4-methylbenzenesulfonamide and dichloro (p-cymene) -ruthenium (ll) ),were added and the mixture was stirred at room temperature for 16 hours under a nitrogen atmosphere. The reaction was terminated by addition of water and the resulting mixture was extracted with dichloromethane to remove moisture with magnesium sulfate. After filtration, the solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography (hexane: ethyl acetate = 90: 10?> 60:40) to obtain the title compound.
With dimethylsulfide borane complex; (R)-tetrahydro-1-methyl-3,3-diphenyl-1H,3H-pyrrolo[1,2-c]-[1,3,2]oxazaborole In dichloromethane; toluene at -20℃; for 2.66667h; Inert atmosphere;
1 Step 1:
To a 500 mL round bottom flask was added (R)-(+)-2-methyl-CBS-oxazaborolidine (985 mg, 3.55 mmol), toluene (5 mL) and borane-dimethylsulfide (12.36 mL, 130 mmol) under N2. Thereaction was stirred at room temperature for 10 mm then diluted with DCM (20 mL) and cooled to -20° C. A solution of 4-Bromo-2,3-dihydro-1H-inden-1-one (5 g, 23.69 mmol) in DCM (20 mL) was added dropwise over 30 mm while maintaining the reaction temperature at -20±5° C. The reaction was stirred for 2 h after the addition was complete, then quenched by the dropwise addition of MeOH (50 mL). The reaction mixture was diluted with an additional MeOH (60 mL) and the solvent distilled at atmosphericpressure. MeOH (60 mL) was added in two portions and the distillation was repeated twice. Finally all the solvent was evaporated under reduced pressure to give a solid which was purified by silica gel column chromatography (EAlhexanes) provided (S)-4-bromo-2,3-dihydro-1H-inden-1-ol as a solid. Product can be recrystallized from 5:1 hexanes-EtOAc.‘H NMR (400 MHz, DMSO-d6) 7.47 - 7.37 (m, 1H), 7.31 (d, J = 7.4 Hz, 1H), 7.23 - 7.05 (m,1H), 5.36 (dd, J = 6.1, 2.0 Hz, 1H), 5.10 (q, J = 6.2 Hz, 1H), 2.97-2.79 (m, 1H), 2.68 (dt, J = 15.6, 7.6Hz, 1H), 2.41 -2.22 (m, 1H), 1.77 (dt, J = 19.0, 7.0 Hz, 1H).
97.5 % ee
With di-μ-chlorobis-[(η6-p-cymene)chlororuthenium(II)]; anhydrous sodium formate In water monomer; isopropanol for 5h; Milling;
With sodium azide; methanesulfonic acid; In dichloromethane; at 0 - 20℃; for 16.0h;
[0107] To a mixture of 4-bromoindan-l-one (4.00g, 18.9 mmol) and methanesulfonic acid (20.2 mL, 310 mmol) in dichloromethane (180 mL) was added sodium azide (2.46 g, 37.9 mmol) at 0C. The mixture was warmed to room temperature and stirred for 16 h. The reaction mixture was poured into 10% aqueous sodium hydroxide (200 mL) and extracted with dichloromethane (100 mL). The combined organic layers were dried over sodium sulfate and evaporated. The crude product was recrystallized from ethyl acetate (40 mL) to give the title compound (3.98 g, 17.6 mmol, 93%) as a white powder. LCMS: 96%, Rt 1.225, ESMS m/z 226 (M+H)+.
93%
With sodium azide; methanesulfonic acid; In dichloromethane; at 0 - 20℃; for 16.0h;
2H-isoquinolin-l-one. [00112] To a mixture of 4-bromoindan-l-one (4.00g, 18.9 mmol) and methanesulfonic acid (20.2 mL, 310 mmol) in dichloromethane (180 mL) was added sodium azide (2.46 g, 37.9 mmol) at 0 C. The mixture was warmed to room temperature and stirred for 16 h. The reaction mixture was poured into 10% aqueous sodium hydroxide (200 mL) and extracted with dichloromethane (100 mL). The combined organic layers were dried over sodium sulfate and evaporated. The crude product was recrystallized from ethyl acetate (40 mL) to give the title compound (3.98 g, 17.6 mmol, 93%) as a white powder. LCMS: 96%, t 1.225, ESMS m/z 226 (M+H)+.
93%
With sodium azide; methanesulfonic acid; In dichloromethane; at 0 - 20℃; for 16.0h;
To a mixture of 4-bromoindan-1-one (4.OOg, 18.9 mmol) and methanesulfonic acid (20.2 mL, 310 mmol) in dichloromethane (180 mL) was added sodium azide (2.46 g, 37.9 mmol) at 0 C. The mixture was warmed to room temperature and stirred for 16 h. The reaction mixture was poured into 10% aqueous sodium hydroxide (200 mL) and extracted with dichloromethane (100 mL). The combined organic layers were dried over sodium sulfate and evaporated. The crude product was recrystallized from ethyl acetate (40 mL) to give the title compound (3.98 g, 17.6 mmol, 93%) as a white powder. LCMS: 96%, Rt 1.225, ESMS m/z 226 (M+H).
93%
With sodium azide; methanesulfonic acid; In dichloromethane; at 0 - 20℃; for 16.0h;
100106] To a mixture of 4-bromoindan-1-one (4.OOg, 18.9 mmol) and methanesulfonic acid (20.2 mL, 310 mmol) in dichloromethane (180 mL) was added sodium azide (2.46 g, 37.9 mmol) at 0 C. The mixture was warmed to room temperature and stirred for 16 h. The reaction mixture was poured into 10% aqueous sodium hydroxide (200 mL) and extracted with dichloromethane (100 mL). The combined organic layers were dried over sodium sulfate and evaporated. The crude product was recrystallized from ethyl acetate (40 mL) to give the title compound (3.98 g, 17.6 mmol, 93%) as a white powder. LCMS: 96%, Rt 1.225, ESMS m/z 226 (M+H).
46%
With sodium azide; In methanesulfonic acid; dichloromethane; at 0 - 20℃;
General Procedure: To an ice-cooled solution of the benzocycloketone (1.4 mmol) in 1:1 CH2Cl2/methanesulfonic acid (10 mL) (except for compound 17, where concentrated HCl was used as solvent), sodium azide (0.2g, 2.8 mmol) was added in portions. The resulting mixture was allowed to stir at 0 C for 1h, and then warmed up to room temperature while stirring overnight. Then, the reaction mixture was poured into ice-water (20 mL), basified with 5M NaOH (aq) until pH 9, and extracted with CH2Cl2 (3 × 10 mL). The organic layers were combined, dried over anhydrous Na2SO4, and concentrated under reduced pressure to give the crude lactam. Purification on silicagel chromathography (1:1 AcOEt/Hexane ? AcOEt) provided the desired 2H-benzolactam. The isomeric 1H benzolactam was also isolated in lower yield.
43%
With sodium azide; methanesulfonic acid; In dichloromethane; at -10℃;
4-bromo-1-indanone in a 50 mL single-mouth bottle (6.00 g, 28.40 mmol)Dissolved in dichloromethane (50 mL) and methanesulfonic acid (25 mL).At -10 C,Slowly add sodium azide (1.02, 42.60 mmol),Stirring was continued at low temperature.TLC monitored the reaction completely. Extracted with EA (50 mL × 3), dried over anhydrous sodium sulfate, concentrated, silica gel column chromatography, eluting with PE/EA=40/1-10/1,Obtained 2.76 g of a white solid, yield 43%.
With sodium carbonate In 1,4-dioxane; water at 85℃; for 2.5h;
8.1
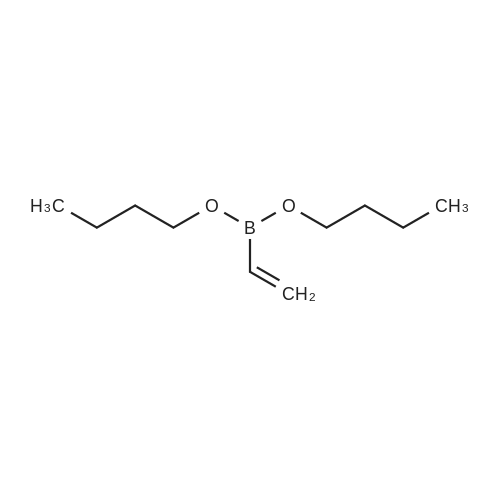
To a solution of 4-bromo-2,3-dihydro-1H-inden-1-one (3.58 g) in dioxane (71.6 ml), triphenylphosphine (1.33 g), dibutyl vinylboronate (3.54 g), palladium acetate (381 mg) and an aqueous solution of 2 M sodium carbonate (42.5 ml) were added and the mixture was stirred for 2.5 hours at 85°C. After adding ethyl acetate, the insolubles were filtered off and the filtrate was washed with brine. After drying over anhydrous sodium sulfate, the desiccant was filtered off and the solvent was removed in vacuo. The resulting residue was purified by silica gel column chromatography (eluent: n-hexane/ethyl acetate/chloroform = 8:1:1 to 7:1:2) to give the titled compound (2.35 g) as a pale yellow powder. MS(EI): 158(M+) 1H NMR (300 MHz, CHLOROFORM-d) δppm 2.64 - 2.83 (m, 2 H), 3.10 - 3.28 (m, 2 H), 5.37 - 5.54 (m, 1 H), 5.73 - 5.94 (m, 1 H), 6.80 - 6.99 (m, 1H), 7.33 - 7.49 (m, 1 H), 7.62 - 7.83 (m, 2 H)
With palladium (II) [1,1'-bis(diphenylphosphanyl)ferrocene] dichloride; anhydrous potassium acetate In N,N-dimethyl acetamide at 100℃; for 2h; Inert atmosphere;
10.1 [0173] Step 1. Compound 10a-1. 4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-indan-1-one.
To a solution of 4-bromo-l-indanone (1.0 g, 4.74 mmol) in NN- dimethylacetamide (25 mL) was added te(pinacolato)diboron (2.41 g, 9.48 mmol), dichloro[l, l '-/?(diphenyl-phosphino)ferrocene] palladium(II) (346 mg, 0.47 mmol) and potassium acetate (1.40 g, 14.22 mmol) and the reaction mixture was stirred at 100 °C for 2 h under nitrogen. The mixture was evaporated and the residue was diluted with water (50 mL) and extracted with dichloromethane (3 x 50 mL). The combined organic layers were dried over sodium sulfate and evaporated. The residue was purified by column chromatography eluting with hexane:ethyl acetate (100:0-^4: 1) to give the title compound (1.8 g, 7.00 mmol, ca. 100%) as an off-white powder. LCMS : 97%, Rt 1.81 1, ESMS m/z 259 (M+H-CH4)+.
100%
With palladium (II) [1,1'-bis(diphenylphosphanyl)ferrocene] dichloride; anhydrous potassium acetate In N,N-dimethyl acetamide at 100℃; for 2h; Inert atmosphere;
7-1 Preparation 7-1. 4-(4,4,5,5-Tetramethyl-l,3,2-dioxaborolan-2-yl)-indan-l- one.
Preparation 7-1. 4-(4,4,5,5-Tetramethyl-l,3,2-dioxaborolan-2-yl)-indan-l- one. [00122] To a solution of 4-bromo-l-indanone (1.0 g, 4.74 mmol) in N,N- dimethylacetamide (25 mL) was added bw(pinacolato)diboron (2.41 g, 9.48 mmol), 1,1 '- ^w(diphenylphosphino)ferrocene palladium(II) dichloride (346 mg, 0.47 mmol) and potassium acetate (1.40 g, 14.22 mmol) and the reaction mixture was stirred at 100 °C for 2 h under nitrogen. The mixture was evaporated and the residue was diluted with water (50 mL) and extracted with dichloromethane (3 x 50 mL). The combined organic layers were dried over sodium sulfate and evaporated. The residue was purified by column chromatography eluting with hexane: ethyl acetate (100:0->4: 1) to give the title compound (1.8 g, 7.00 mmol, ca. 100%) as an off-white powder. LCMS: 97%, Rt 1.811 , ESMS m/z 259 (M+H-CH4)+.
100%
With palladium (II) [1,1'-bis(diphenylphosphanyl)ferrocene] dichloride; anhydrous potassium acetate In N,N-dimethyl acetamide at 100℃; for 2h; Inert atmosphere;
7-1 Preparation 7-1. 4-(4,4,5,5-Tetramethyl- 1,3 ,2-dioxaborolan-2-yl)-indan- 1-one.
General procedure: To a solution of 4-bromo-l-indanone (1.0 g, 4.74 mmol) in N,Ndimethylacetamide (25 mL) was added bis(pinacolato)diboron (2.41 g, 9.48 mmol), 1,1’-bis(diphenylphosphino)ferrocene palladium(11) dichloride (346 mg, 0.47 mmol) and potassiumacetate (1.40 g, 14.22 mmol) and the reaction mixture was stirred at 100 °C for 2 h undernitrogen. The mixture was evaporated and the residue was diluted with water (50 mL) andextracted with dichloromethane (3 x 50 mL). The combined organic layers were dried oversodium sulfate and evaporated. The residue was purified by column chromatography eluting withhexane:ethyl acetate (100:0-4:1) to give the title compound (1.8 g, 7.00 mmol, Ca. 100%) as anoff-white powder. LCMS: 97%, Rt 1.811, ESMS m/z 259 (M+H-CH4).
100%
With palladium (II) [1,1'-bis(diphenylphosphanyl)ferrocene] dichloride; anhydrous potassium acetate In N,N-dimethyl acetamide at 100℃; for 2h; Inert atmosphere;
7-1 Preparation 7-1. 4-(4,4,5,5-Tetramethyl- 1 ,3,2-dioxaborolan-2-yI)-indan- 1-one.
[00116] To a solution of 4-bromo-1-indanone (1.0 g, 4.74 mmol) in N,Ndimethylacetamide (25 mL) was added bis(pinacolato)diboron (2.41 g, 9.48 mmol), 1,1’- bis(diphenylphosphino)ferrocene palladium(II) dichloride (346 mg, 0.47 mmol) and potassium acetate (1.40 g, 14.22 mmol) and the reaction mixture was stirred at 100 °C for 2 h under nitrogen. The mixture was evaporated and the residue was diluted with water (50 mL) and extracted with dichloromethane (3 x 50 mE). The combined organic layers were dried over sodium sulfate and evaporated. The residue was purified by column chromatography eluting with hexane:ethyl acetate (100:0-)4:1) to give the title compound (1.8 g, 7.00 mmol, Ca. 100%) as an off-white powder. LCMS: 97%, Rt 1.811, ESMS m/z 259 (M+H-CH4).
90%
With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II) dichloromethane adduct; anhydrous potassium acetate In 1,4-dioxane at 80℃; for 7h; Inert atmosphere;
1.H
Under nitrogen gas, 4-bromo-l-indanone (8g, 37.9mmol), bis(pinacolato)diboron (11.55g, 45.4mmol), potassium acetate (11.16g, 113.7mmol), l, l'-bis(diphenylphosphino)ferrocene-palladium(II) dichloride dichloromethane complex (1.548g, 1.89mmol), and 180mL 1,4-dioxane were added into a 500 mL 3-neck flask. The mixture was heated to reflux at 80 °C and stirred for 7 hr, then cooled to room temperature, and concentrated by rotary evaporation. 120mL water was added and the mixture was extracted by 100ml X 3 ethyl acetate. The combined organic phase was washed by saturated NaCl solution, dried by anhydrous Na2S04 and evaporated under vacuum to remove solvent, to give a crude product. The product was purified by column chromatography (ethyl acetate: petroleum ether = 10:90) to obtain a white solid product 11 (8.8g, yield 90%).
84%
With palladium (II) [1,1'-bis(diphenylphosphanyl)ferrocene] dichloride; anhydrous potassium acetate In 1,4-dioxane at 80℃; for 20h; Inert atmosphere; Schlenk technique;
1 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,3-dihydro-1H-inden-1-one (FW-220)
In an oven dried schlenk flask were combined 767 mg (3.63 mmol) 4-bromo-2,3- dihydro-1 H-inden-1-one, 969 mg bis(pinacolato)diboron (3.82 mmol), 1070 mg KOAc (10.9 mmol) and 79 mg Pd(dppf)CI2(0.11 mmol) under inert atmosphere and 10 ml degassed dioxane were added. The mixture was heated to 80 °C oil bath temperature for 20 hours. The brown suspension was diluted with EtOAc and filtered over celite. The filtrate was concentrated under reduced pressure and the residue purified via flash chromatography (SiO2, Hex Hex/EtOAc 7:3). Yield: 790 mg (84 %) as yellow solid.1H NMR (200 MHz, CDCI3) δ 8.04 (d, J = 6.9 Hz, 1 H), 7.84 (d, J = 7.8 Hz, 1 H), 7.37 (t, J = 7.4 Hz, 1 H), 3.42 - 3.28 (m, 2H), 2.74 - 2.61 (m, 2H), 1.36 (s, 12H).13C NMR (50 MHz, CDCI3) δ 208.00,162.35, 141.94, 136.58, 126.76, 126.51 , 84.03, 36.41 , 27.49, 25.09.
84%
With palladium (II) [1,1'-bis(diphenylphosphanyl)ferrocene] dichloride; anhydrous potassium acetate In 1,4-dioxane at 80℃; for 20h; Inert atmosphere; Schlenk technique;
1 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,3-dihydro-1H-inden-1-one (FW-220)
In an oven dried schlenk flask were combined 767 mg (3.63 mmol) 4-bromo-2,3- dihydro-1 H-inden-1-one, 969 mg bis(pinacolato)diboron (3.82 mmol), 1070 mg KOAc (10.9 mmol) and 79 mg Pd(dppf)CI2(0.11 mmol) under inert atmosphere and 10 ml degassed dioxane were added. The mixture was heated to 80 °C oil bath temperature for 20 hours. The brown suspension was diluted with EtOAc and filtered over celite. The filtrate was concentrated under reduced pressure and the residue purified via flash chromatography (SiO2, Hex Hex/EtOAc 7:3). Yield: 790 mg (84 %) as yellow solid.1H NMR (200 MHz, CDCI3) δ 8.04 (d, J = 6.9 Hz, 1 H), 7.84 (d, J = 7.8 Hz, 1 H), 7.37 (t, J = 7.4 Hz, 1 H), 3.42 - 3.28 (m, 2H), 2.74 - 2.61 (m, 2H), 1.36 (s, 12H).13C NMR (50 MHz, CDCI3) δ 208.00,162.35, 141.94, 136.58, 126.76, 126.51 , 84.03, 36.41 , 27.49, 25.09.
76%
With palladium (II) [1,1'-bis(diphenylphosphanyl)ferrocene] dichloride; anhydrous potassium acetate In 1,4-dioxane at 120℃; for 0.333333h; Microwave irradiation;
4.1.5. Synthesis of 4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-2,3-dihydro-1H-inden-1-one (29)
The mixture of 4-Bromo-1-indanone 25 (1.0 g, 4.7 mmol, 1.0equiv.), bis(pinacolato)diboron (1.8 g, 7.1 mmol, 1.5 equiv.), KOAc(0.93 g, 9.4 mmol, 2.0 equiv.), dioxane (15 mL) and PdCl2(dppf)2(0.1 g) were microwaved at 120 °C for 20 min. The suspension wasfiltered and washed with EtOAc. The solvent was evaporated andthe residue was diluted with EtOAc and washed with brine, organiclayer was dried over Na2SO4, filtered and concentrated. The crudeproduct was purified by flash column chromatography (EtOAc/hexane; 1:9) to afford desired compound 29 (0.93 g, 3.6 mmol, 76%)as a yellow solid. 1H NMR (CDCl3, 600 MHz) δ 8.04 (d, J 6.6 Hz, 1H),7.84 (d, J 7.8 Hz, 1H), 7.37 (t, J 7.2 Hz, 1H), 3.34-3.33 (m, 2H),2.73-2.66 (m, 2H), 1.29 (s, 12H).
23%
With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II) dichloromethane adduct; anhydrous potassium acetate In methanol at 60℃; for 10h; Inert atmosphere;
With anhydrous potassium acetate In 1,4-dioxane at 80℃; for 4h; Inert atmosphere;
8
In a screw cap vessel 4-Bromo-l-indanone (1.0 g, 4.9 mmol) was dissolved in 1,4- dioxan (40 ml_). Argon was purged through the solution. PdCI2(dppf)2*CH2Cl2 (0.16g, 0.2 mmol) and bis(pinacolato)diborane (1.3g, 5.1 mmol) was added followed by addition of KOAc (l,4g, 14.6 mmol). The vessel was closed and the mixture was stirred at 80°C for 4 h. The mixture was cooled to RT and diluted with EtOAc (50ml_).The suspension was filtered and washed with EtOAc. The filtrate was concentrated and the crude product was purified by flash chromatography (heptane: EtOAc 100 : 0 -> 93 : 7) to afford the title compound as a solid.1H NMR (300 MHz, CDCI3) δ 8.04 (d, J = 7.2 Hz, 1H), 7.84 (d, J = 7.7 Hz, 1H), 7.37 (t, J = 7.4 Hz, 1H), 3.39 - 3.29 (m, 2H), 2.70 - 2.63 (m, 2H), 1.36 (s, 12H).
With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II) dichloromethane adduct; anhydrous potassium acetate In N,N-dimethyl-formamide at 80℃; for 12h;
4 Preparation Example 4: Preparation of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,3-dihydro-1H-inden-1-one (compound of Formula 4-2)
4-bromo-2,3-dihydro-1H-inden-1-one (Formula 4-1, 1 g, 4.74 mmol),Potassium acetate (1.37 g, 14.22 mmol, 3 eq), Pd (dppf) Cl 2 -CH 2 Cl 2 (0.384 g, 0.47 mmol, 0.1 eq), B 2 Pin 2 (2.41 g, 9.48 mmol, 3 eq) was dissolved in DMF (25 ml).The temperature was raised to 80 ° C and the reaction mixture was stirred for 12 hours.After completion of the reaction, the mixture was extracted with dichloromethane. The organic layer was washed sequentially with H 2 O and brine, then dried over anhydrous Na 2 SO 4,The solvent was removed under reduced pressure.Thereafter, the reaction mixture was separated and purified by MPLC (hexane / diethyl acetate; 70:30) to obtain a solid compound (Formula 4-2).
With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II) dichloromethane adduct; anhydrous potassium acetate In dimethyl sulfoxide at 80℃; for 12h;
4 Preparation Example 4: 4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -2,3-dihydro-1H-inden-1-one (Formula Preparation of 4-2)
4-Bromo-2,3-dihydro-1H-inden-1-one (Formula 4-1, 1 g, 4.74 mmol), potassium acetate (1.37 g, 14.22 mmol, 3 eq), Pd (dppf) Cl2 -CH2Cl2 (0.384 g, 0.47 mmol, 0.1 eq), B2Pin2 (2.41 g, 9.48 mmol, 3 eq) was dissolved in DMF (25 ml). The reaction mixture was stirred for 12 hours by raising the temperature to 80 ° C. After completion of the reaction, extraction was performed with dichloromethane. The organic layer was washed sequentially with H2O and brine, then dried over anhydrous Na2SO4, and the solvent was removed under reduced pressure. Then, the reaction mixture was separated and purified by MPLC (hexane / diethyl acetate; 70:30) to obtain a solid compound (Formula 4-2).
General procedure: To a suspension of benzyltriphenylphosphonium bromide (1.63 g, 3.76 mmol) in THF (40 mL) was dropwise added n-BuLi (2.35 mL, 1.6 M in hexanes). After the system was stirred at room temperature for 2 h, indanone (7a, 0.25 g, 1.89 mmol) or its derivative (8a-17a, 1.89 mmol) in THF (10 mL) was added. The reaction mixture was stirred at reflux for 24 h, then, cooled to room temperature, quenched with water (20 mL), and extracted with n-hexane (3 × 30 mL). The organic layers were combined, washed with water, dried (Na2SO4) and filtered. The filtrate was concentrated under reduced pressure. The residue was purified with silica gel column chromatography (n-hexane) to give the corresponding products (7b-17b).
With triethylamine;bis-triphenylphosphine-palladium(II) chloride; under 18751.9 Torr; for 48h;
Preparative Example - Methyl l-oxo-2,3-dihydro-lH-indene-4-carboxylate (PrepEx-11)PrepEx-11[00203] To a solution of 4-bromo-2,3-dihydro-lH-inden-l -one (5 g, 24 mmol) in MeOH (120 mL) and Et3N (40 mL) was added Pd(PPh3)2Cl2 (200 mg) and the mixture was stirred under 2.5 MPa of CO for 48 hours. The reaction solution was concentrated under reduced pressure and the residue was purified by silica gel chromatography using 2:1 petroleum ether:EtOAc to afford methyl l-oxo-2,3-dihydro-lH-indene-4-carboxylate (2.9 g, 64 %). MS ESI calcd for CnHi0O3 [M + H]+ 191, found 191.
63%
With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; triethylamine; at 100℃; under 375038.0 Torr; for 24h;
Into a 1000-mL pressure tank reactor was placed 4-bromo-2,3-dihydro-lH-inden-1-one (30 g, 142 mmol, 1 equiv), MeOH (450 mL), Pd(dppf)Cl2'CH2Cl2 (25 g, 0.1 equiv), and Et3N (150 mL). The resulting mixture was stirred for 24 h at 100 C under an atmosphere of 50 MPa CO (g). After cooling to room temperature, the mixture was filtered through a pad of celite and then concentrated under vacuum. The residue was purified by normal phase chromatography on silica gel using EtO Ac/petroleum ether (1 : 10). The collected fractions were concentrated under vacuum to afford 17 g (63% yield) of the title compound as a white solid. MS: (ES, m/z): 191 [M+H]+.
1-(2-(7-bromo-1H-inden-3-yl)ethyl)-4-methylpiperazine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
51%
To a solution of 1 -(4-methylpiperazin-1 -yl)ethanone (1 .68 g, 1 1 .8 mmol) in THF (25 mL) cooled to -78 C, a solution of LDA (1 .5 M in THF, 9.8 mL, 14.75 mmol) was added, and the resulting mixture was stirred at -78 ^ for 1 h under argon atmosphere. Finally, a solution of 4-bromoindan-1 -one (1 .25 g, 5.9 mmol) in THF (50 mL) was added, and the resulting mixture was kept at -78 ' for 4 h. The reaction mixture was diluted with water and extracted with EtOAc. The organic extracts were dried over Na2S04 and evaporated to dryness. A solution of the previous residue in AcOH:H2S04:H20 (79 mL, 85:10:5) was stirred for 6 h. The reaction mixture was poured into water, basified with 50% NaOH and extracted with EtOAc. The organic extract, after being dried over Na2S04, was evaporated to dryness. To a solution of the previous residue in THF (75 mL) cooled to 0 C, AIH3- NMe2Et (0.5 M in toluene, 20 mL, 10.34 mmol) was added and the resulting mixture was stirred for 5 h. EtOAc:H20 (90 mL, 1 :1 ) was added to the reaction mixture and the resulting suspension was filtered through Celite. The layers were separated and the aqueous phase was extracted with EtOAc. The organic extract was dried over Na2S04 and evaporated to dryness. A solution of the previous residue in 37% HCLEtOH (150 mL, 1 :1 ) was refluxed overnight. The reaction mixture was evaporated to dryness. Purification of the residue by silica gel column chromatography (EtOAc/NH3:MeOH mixtures of increasing polarity as eluent) afforded the desired product (981 mg, 51 %). 1 H-NMR (CDCI3, 300 MHz) delta: 7.32 (m, 2H), 7.18 (t, J= 7.6 Hz, 1 H), 6.30 (s, 1 H), 3.31 (d, J= 1 .8 Hz, 2H), 2.72 (m, 4H), 2.60 (m, 4H), 2.51 (m, 4H), 2.30 (s, 3H) ppm. EI-MS m/z: 320.1 (M).
51%
b) Synthesis of 1-(2-(7-bromo-1H-inden-3-yl)ethyl)-4-methylpiperazine To a solution of <strong>[60787-05-5]1-(4-methylpiperazin-1-yl)ethanone</strong> (1.68 g, 11.8 mmol) in THF (25 mL) cooled to -78 C, a solution of LDA (1.5 M in THF, 9.8 mL, 14.75 mmol) was added, and the resulting mixture was stirred at -78 C for 1 h under argon atmosphere. Finally, a solution of 4-bromoindan-1-one (1.25 g, 5.9 mmol) in THF (50 mL) was added, and the resulting mixture was kept at -78 C for 4 h. The reaction mixture was diluted with water and extracted with EtOAc. The organic extracts were dried over Na2SO4 and evaporated to dryness. A solution of the previous residue in AcOH:H2SO4:H2O (79 mL, 85:10:5) was stirred for 6 h. The reaction mixture was poured into water, basified with 50% NaOH and extracted with EtOAc. The organic extract, after being dried over Na2SO4, was evaporated to dryness. To a solution of the previous residue in THF (75 mL) cooled to 0 C, AlH3-NMe2Et (0.5 M in toluene, 20 mL, 10.34 mmol) was added and the resulting mixture was stirred for 5 h. EtOAc:H2O (90 mL, 1:1) was added to the reaction mixture and the resulting suspension was filtered through Celite. The layers were separated and the aqueous phase was extracted with EtOAc. The organic extract was dried over Na2SO4 and evaporated to dryness. A solution of the previous residue in 37% HCl:EtOH (150 mL, 1:1) was refluxed overnight. The reaction mixture was evaporated to dryness. Purification of the residue by silica gel column chromatography (EtOAc/NH3:MeOH mixtures of increasing polarity as eluent) afforded the desired product (981 mg, 51%). 1H-NMR (CDCl3, 300 MHz) delta: 7.32 (m, 2H), 7.18 (t, J= 7.6 Hz, 1H), 6.30 (s, 1H), 3.31 (d, J= 1.8 Hz, 2H), 2.72 (m, 4H), 2.60 (m, 4H), 2.51 (m, 4H), 2.30 (s, 3H) ppm. EI-MS m/z: 320.1 (M).
With copper(l) iodide; In N,N-dimethyl-formamide; at 120℃; for 16h;
l-oxo-2,3-dihydro-lH-indene-4-carbonitrile: To a solution of 4-bromo-2,3- dihydro-lH-inden-l-one (50 g, 236.91 mmol, 1.00 equiv) in N,N-dimethylformamide (250 mL) was added CuCN (63.67 g, 710.92 mmol, 3.00 equiv), Cul (4.5 g, 23.63 mmol, 0.10 equiv). The resulting solution was stirred for 16 h at 120 C. The reaction mixture was cooled. The resulting solution was diluted with water (700 mL). The solids were filtered out. The resulting solution was extracted with ethyl acetate (4 x 300 mL) and the organic layers were combined, dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by a silica gel column eluting with ethyl acetate/petroleum ether (1:2) to afford 24 g (64%) of l-oxo-2,3-dihydro-lH-indene-4-carbonitrile as a light brown solid.
60%
In N,N-dimethyl-formamide; at 20℃; for 22h;Reflux;
To a solution of 4-bromo-1-indanone (10 g, 47.37 mmol) in DMF (100 mL) was added CuCN (12.72 g, 142.11 mmol) and the reaction mixture heated under reflux for 6h. The reaction mixture was cooled to RT and stirred for 16h. The reaction mixture was filtered through Celite, and washed with EtOAc. The combined filtrate was diluted with EtOAc(500 mL) and washed with ice-water (3 x 300 mL) and the combined organic layers were washed with brine, dried over Na2504 and concentrated in vacuo. The crude material was purified by silica gel column chromatography, eluting with 10-20% EtOAc/pet. ether to afford 1-oxo-indan-4-carbonitrile (4.5 g, 60%) as a yellow solid.Rt: 0.6 (30% EtOAc/pet ether).1H NMR (400MHz, CDCI3): O 7.97 (d, J= 7.6 Hz, 1H), 7.89 (dd, J= 6.0, 1.2 Hz, 1H), 7.52 (dd, J= 4.4, 3.6 Hz, 1H), 3.33 (t, J= 6.0 Hz, 2H), 2.82-2.79 (m, 2H).
With potassium acetate In 1,4-dioxane; dichloromethane
4 4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)indan-1-one (34)
Example 4 4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)indan-1-one (34) Potassium acetate (17.8 g, 181.6 mmol) and Pd(dppf)Cl2(3.3 g, 4.5 mmol) were added to a solution of 4-bromo-1-indanone (19.3 g, 91.5 mmol) and bis(pinacolato)diboron (23.2 g, 91.3 mmol) in dioxane (300 mL). The mixture was heated to 80° C. under argon, kept at this temperature for 16 h, cooled, filtered through Celite, and evaporated. The residue was dissolved in CH2Cl2. The product was purified on a short thick column with silica gel (eluent: hexane-ethyl acetate 100:0→50:50). Yield of 34: 23.4 g. The product containing bis(pinacolato)diboron (about 7 molar %) was used in the next step without additional purification.
4-phenylethylthio-2,3-dihydro-1H-inden-1-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
39.65%
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene In 1,4-dioxane at 95 - 100℃;
32.1 In the first step, 4-phenylethylthio-2,3-dihydro-1H-indan-1-one
The compound 4-bromo-1-indanone (1 g, 4.74 mmol)Phenylethyl mercaptan(0.87 g, 6.11 mmol), Pd2 (dba) 3 (0.522 g, 0.57 mmol), ligands (cas: 161265-03-8, 0.549 g, 0.95 mmol) and cesium carbonate (2.3 g, 7.1 mmol)Dissolved in 1,4-dioxane (20mL), 95 -100 reaction 10h-22h, after the end of the reaction to cool to room temperature, the reactionThe filtrate was concentrated under reduced pressure, distilled water was added, extracted with ethyl acetate (50 mL x 2), washed with saturated brine, combined with organic phase,Dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure by column chromatography (petroleum ether: ethyl acetate = 25: 1)The intermediate product (503 mg, 1.87 mmol, white solid, yield 39.65%) was obtained.
0.53 g (21.8 mmol) of magnesium piece was charged in a 200 mL reactor thoroughly dried and purged with argon, and stirred vigorously for 30 minutes while heating under reduced pressure. After cooling to room temperature, a piece of iodine and 20 mL of tetrahydrofuran were charged and stirred. To this solution, a diluted solution of 30 mL tetrahydrofuran of 5.99 g (20.0 mmol) of 1-bromo-3, 5- di- tert-butyl- 4-methoxy benzene synthesized by the method described above in J.Am. Chem. Soc. 2006, 128, 16486 was slowly added and heated under reflux in an oil bath at 80 C for 1 hour. After cooling the reaction solution to -78 C, 2.50 mL (22.5 mmol) of trimethoxy borane was slowly added and stirring was continued for 21 hours while gently returning to room temperature. 1.0 M aqueous hydrochloric acid solution was added, & the soluble matter was extracted with diethyl ether, and the obtained fraction was washed with water and saturated brine, and dried over anhydrous magnesium sulfate. After filtration of magnesium sulfate, the filtrate was distilled off, and 3.67 g (17.4 mmol) of 4- bromo-1-indanone, 8.54 g (40.2 mmol) of tripotassium phosphate, 0.04 g (0.18 mmol) of palladium acetate, 0.11 g (0.26 mmol) of 2-dicyclo hexyl phosphino-2 ', 6'- dimethoxy biphenyl (S-Phos), 45 mL of tetrahydrofuran and 9 mL of distilled water were charged to the obtained residue, and heated under reflux for 2 hours in an oil bath. After the reaction solution was cooled to room temperature, a saturated aqueous solution of ammonium chloride was added and the soluble matter was extracted with ethyl acetate. The obtained fraction was washed with saturated brine and dried over anhydrous magnesium sulfate. After filtration of the magnesium sulfate, the filtrate was distilled off and the resulting residue was purified by silica gel column chromatography to obtain 5.86 g of the desired product (hereinafter referred to as the compound (2a)) represented by the following formula (2a) (Yield 96%).
Compound e (1.05 g, 5 mmol) was dissolved in methanol (14 ml) and hydroxylamine hydrochloride (1.4 g, 20 mmol) was added and refluxed for 1 h.Concentrate in vacuo, add dichloromethane and 50% NaHCO3 for 20 minutes, dry the organic layer and concentrate to a white solid. Yield: 99%.The resulting white solid (5 mmol) was dissolved in methylene chloride (30 ml), cooled to 0 C, AlH (Bu-i) 2 (20 ml, 30 mmol, 1.5 M in toluene) was slowly added, and the mixture was stirred for 30 min.Move to room temperature and stir overnight. Quenching with water under ice bath conditions, dichloromethane extraction, saturated brine,After drying over anhydrous sodium sulfate, it was concentrated and purified by silica gel column (PE:EA=3:1) to give 600 mg of pale yellow solid f, yield: 56.6%.
1‘-benzyl-4-bromospiro[indene-2,4’-piperidin]-1(3H)-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
38%
With sodium hydride; In N,N-dimethyl-formamide; mineral oil; at 0℃; for 1h;
Into a 500-mL 3-necked round-bottom flask was placed 4-bromo-2,3-dihydro-lH- inden-l-one (7.23 g, 34.3 mmol, 1 equiv), DMF (70 mL), and N-benzyl-2-bromo-N-(2- bromoethyl)ethanamine (16.5 g, 53.8 mmol, 1.57 equiv). This was followed by the addition of NaH (60% dispersion in oil, 5.72 g, 143 mmol, 4.17 equiv), in portions at 0 C. The resulting solution was stirred for 1 h at 0 C. The reaction mixture was quenched by the addition of 80 mL of ice-water and the resulting solution was extracted with 2x100 mL of EtO Ac. The combined organic layers were dried over anhydrous Na2S04, filtered, and concentrated under vacuum. The residue was purified by normal phase chromatography on silica gel using EtO Ac/petroleum ether (1 :5). The collected fractions were concentrated under vacuum to afford 4.87 g (38% yield) of the title compound as a brown solid. MS: (ES, m/z): 370 [M+H]+.
With hydrogenchloride; sodium azide; In water; at -10℃; for 1.0h;
In a 100 mL single-mouth bottle, 4-bromo-1-indanone (5.00 g, 23.70 mmol) was dissolved in concentrated hydrochloric acid (50 mL). The temperature was lowered to -10 C, NaN3 (3.08 g, 47.40 mmol) was slowly added, and stirred at low temperature for 1.0 h. Rise to room temperature, continueThe reaction was stirred continuously and the solution was white suspension. The reaction was monitored by TLC, extracted with EA (50mL×3), dried over anhydrous sodium sulfate and concentrated.Silica gel column chromatography,Rinse with PE/EA=40/1-10/1,5-bromo-3,4-dihydroisoquinolin-1(2H)-one 1.22 g of a white solid was obtained, yield 23%.
4-((4-fluorobenzyl)thio)-2,3-dihydro-1H-inden-1-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
51.9%
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene In 1,4-dioxane at 109℃; for 13h;
1.1 First step, 4-((4-fluorobenzyl)thio)-2,3-dihydro-1H-inden-1-one
The compound 4-bromo-1-indolone (1 g, 4.74 mmol), 4-fluorobenzyl mercaptan (0.88 g, 6.16 mmol), tris(dibenzylideneacetone) dipalladium (Pd2(dba)3, 0.522 g, 0.57mmol),Ligand (Xantphos, CAS: 161265-03-8, 0.549g,0.95 mmol) and cesium carbonate (Cs2CO3, 2.3 g, 7.1 mmol) were dissolved in 1,4-dioxane (30 mL), reacted at 109 ° C for 13 h, and cooled to room temperature after completion of the reaction.The reaction solution was suction filtered, and the filtrate was concentrated under reduced pressure, and distilled water was added.Ethyl acetate extraction (50 mL × 2), washed with saturated brine.The organic phases were combined, dried over anhydrous sodium sulfate and filtered.The filtrate was concentrated under reduced pressure and purified by column chromatography (ethyl ether: ethyl acetate = 25:1) to afford Intermediate 1a (670mg, 2.46mmol, white solid, yield 51.9%).
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene In 1,4-dioxane at 100℃;
1.2.2 General procedure for the preparation of compounds b7-b9
General procedure: To a solution of compound 4 (4-bromo-1-indanone, 1.0 equiv), benzyl mercaptan derivatives (1.3 equiv), Pd2(dba)3 (0.12 equiv), xantphos (0.2 equiv) and cesium carbonate (1.5 equiv) in 1,4-dioxane (20 mL), the mixture was stirred for 14 h at 100 °C. Then, the mixture was cooled to room temperature and the mixture was washed with saturated salt water and extracted with ethyl acetate (3×15 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated under vacuum. The residues were purified by chromatography on a silica gel column to give the corresponding target product (b7-b9).
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene In 1,4-dioxane at 100℃;
1.2.2 General procedure for the preparation of compounds b7-b9
General procedure: To a solution of compound 4 (4-bromo-1-indanone, 1.0 equiv), benzyl mercaptan derivatives (1.3 equiv), Pd2(dba)3 (0.12 equiv), xantphos (0.2 equiv) and cesium carbonate (1.5 equiv) in 1,4-dioxane (20 mL), the mixture was stirred for 14 h at 100 °C. Then, the mixture was cooled to room temperature and the mixture was washed with saturated salt water and extracted with ethyl acetate (3×15 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated under vacuum. The residues were purified by chromatography on a silica gel column to give the corresponding target product (b7-b9).
To a solution of 4-Bromo-1-indanone (1.05 g, 5.0 mmol) in MeOH (14.0 ml) hydroxylamine hydrochloride (1.4 g, 20.0 mmol)was added. The resulting mixture was heated to reflux for 1 h andthen cooled to room temperature. The mixture was concentratedunder vacuum. DCM (15.0 ml) and 50% NaHCO3 (15.0 ml) wereadded. The mixture was vigorously stirred for another 20min andthe organic phase was separated, dried over anhydrous Na2SO4,filtered and concentrated under reduced pressure. The obtainedwhite solid (5.0 mmol) in DCM (30.0 ml) at 0 C was treated withAlH(Bu-i)2 (20.0 ml, 30.0 mmol, 1.5M in toluene) dropwise andstirred for 30 min. The reaction mixture was warmed to roomtemperature, stirred for another 12 h and quenched with coldwaterand extracted with DCM. The organic phase was dried over anhydrousNa2SO4, filtered and concentrated under vacuum. Pure 5-bromo-tetrahydroquinoline (3) was obtained as a yellow solid(600.0 mg, yield 56.6%) after flash column chromatography using asolvent gradient of 5-25% ethyl acetate in hexanes.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping