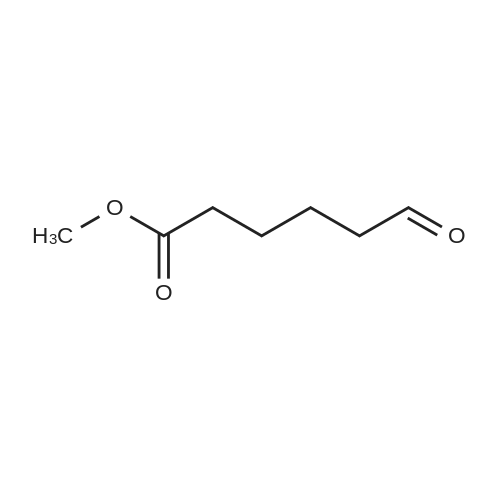
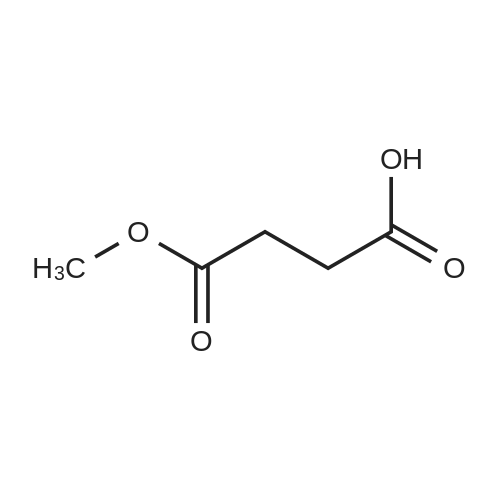
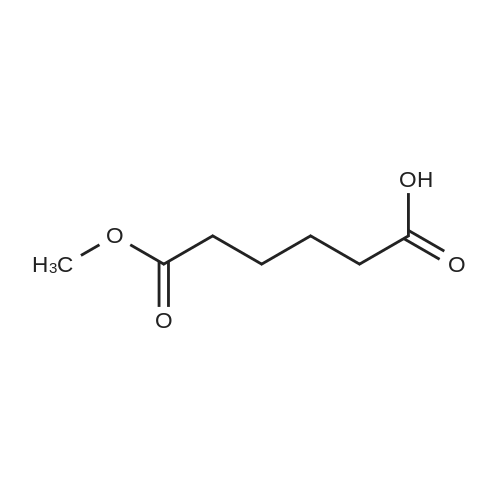
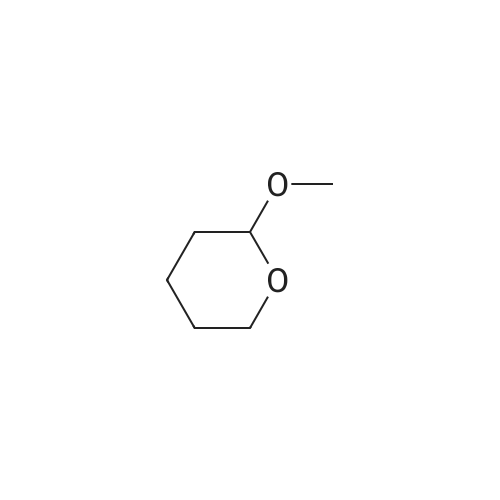
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: With potassium hydroxide In methanol at 20℃; for 4 h; Stage #2: With hydrogenchloride In water
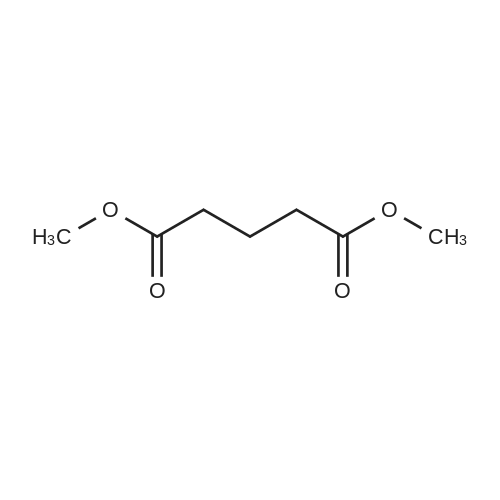
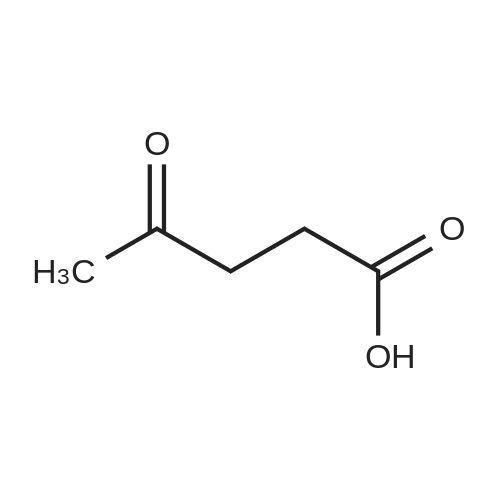
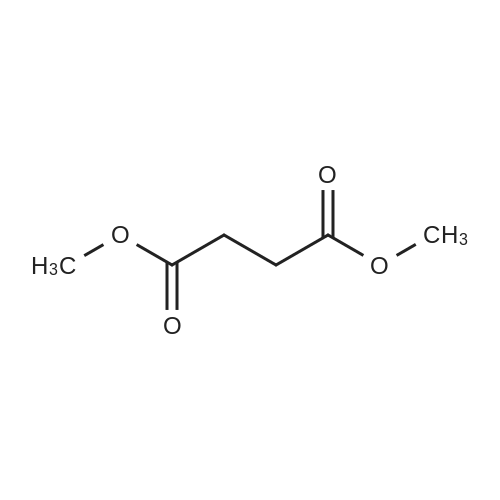
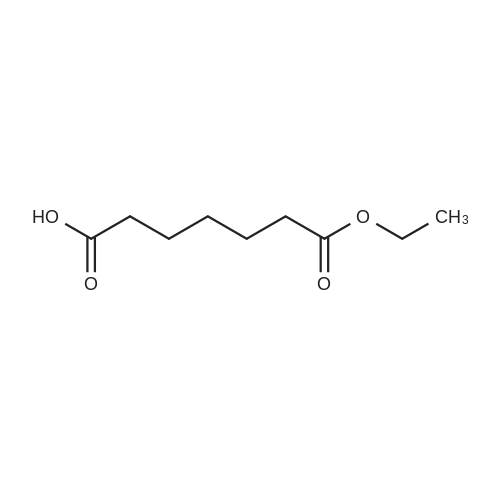
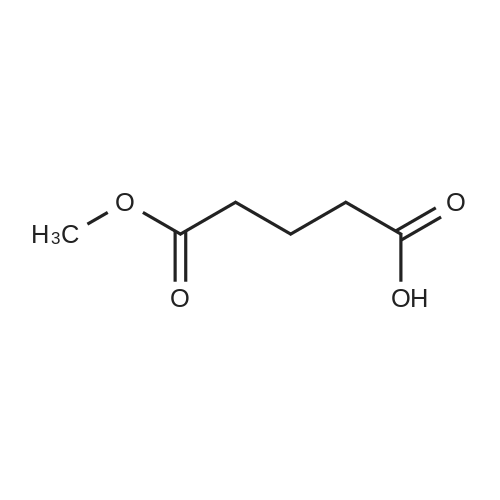
General procedure: A solution of KOH (5.87 g, 104.65 mmol) in MeOH (150 ml) was added to dimethyl glutarate (13.15 g, 90 mmol), and the mixture was stirred for 4 h at rt. The solvent was then removed, and Et2O (100 ml) and H2O (200 ml) were added. The organic layer was separated, washed with brine, dried (MgSO4), and concentrated under reduced pressure to afford 3a as a yellow oil (4.61 g, 32percent). The aqueous layer was acidified with concentrated HCl to pH 3, and extracted with Et2O (3 .x. 100 ml). The combined organic phase was washed with brine (3 .x. 100 ml) and dried (MgSO4). The solvent was removed to give a mixture of a white solid and an oil. Filtration and concentration in vacuum and purification with silica gel column chromatography gave 5.79 g (44percent) of 4a as a colorless oil.
Reference:
[1] Bioorganic and Medicinal Chemistry, 2012, vol. 20, # 12, p. 3865 - 3872
[2] Chemistry Letters, 1995, # 7, p. 539 - 540
2
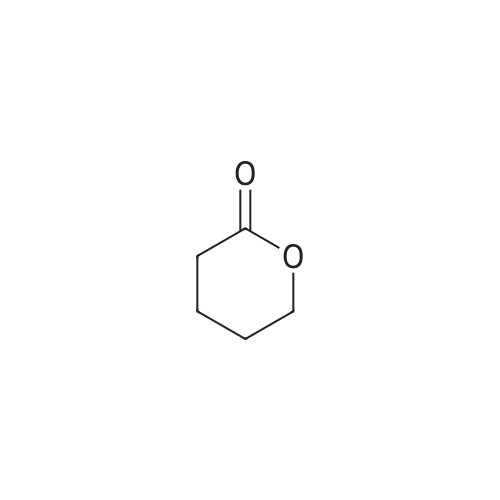
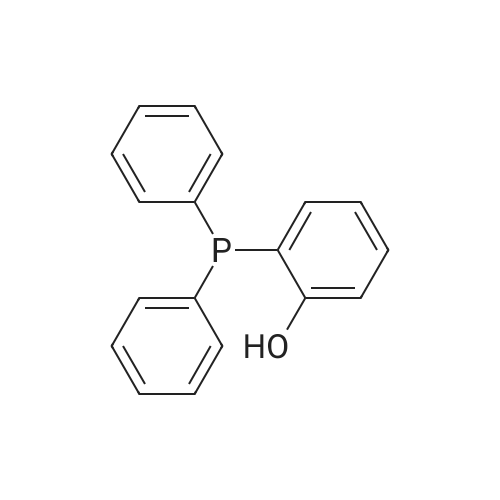
[ 67-56-1 ]
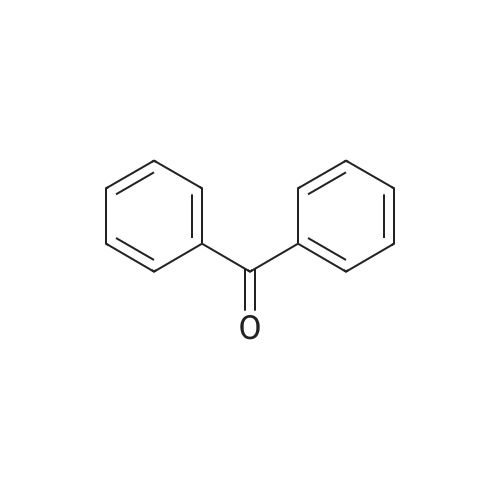
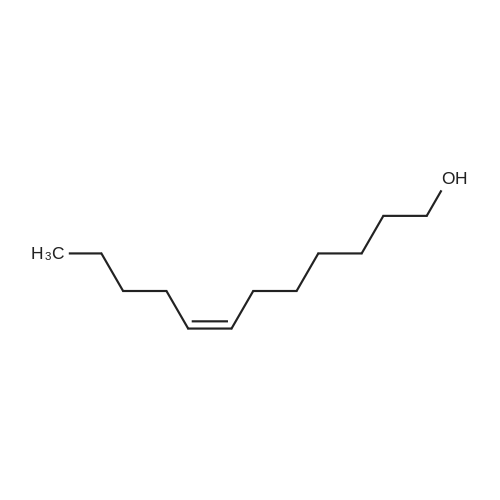
[ 108-55-4 ]
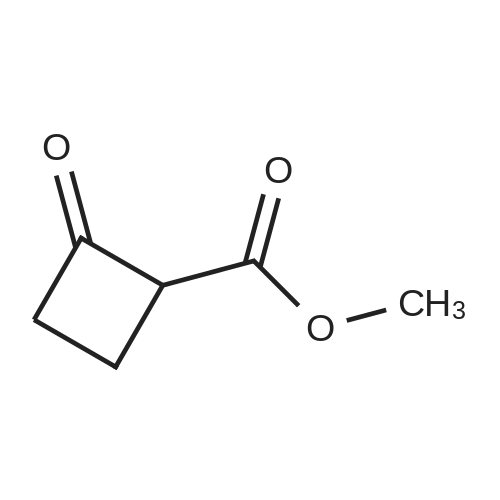
[ 1501-27-5 ]
Reference:
[1] Advanced Synthesis and Catalysis, 2007, vol. 349, # 3, p. 432 - 440
[2] Chemical Communications, 2016, vol. 52, # 49, p. 7715 - 7718
[3] Biochemical Journal, 1925, vol. 19, p. 393
[4] Journal of the Chemical Society, 1947, p. 1108
[5] Journal of the American Chemical Society, 1965, vol. 87, p. 1984 - 1990
[6] Tetrahedron, 1995, vol. 51, # 3, p. 695 - 702
[7] Bioorganic and Medicinal Chemistry Letters, 2004, vol. 14, # 6, p. 1543 - 1546
[8] Journal of Organic Chemistry, 2006, vol. 71, # 12, p. 4565 - 4577
[9] European Journal of Medicinal Chemistry, 2009, vol. 44, # 4, p. 1638 - 1643
[10] Patent: WO2012/25474, 2012, A1, . Location in patent: Page/Page column 29
[11] Letters in Drug Design and Discovery, 2011, vol. 8, # 6, p. 500 - 505
[12] Chemistry and Physics of Lipids, 2014, vol. 184, p. 105 - 118
[13] European Journal of Medicinal Chemistry, 2016, vol. 116, p. 126 - 135
3
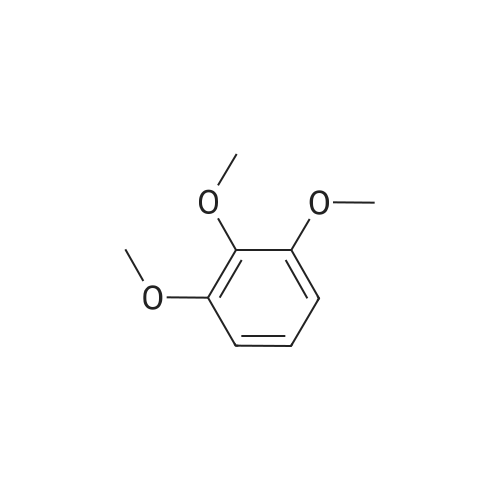
[ 110-94-1 ]
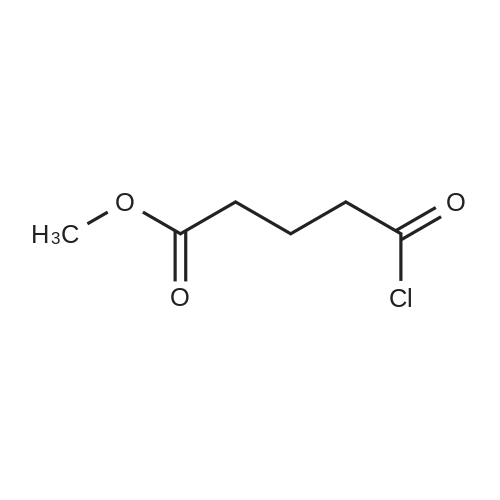
[ 18107-18-1 ]
[ 1501-27-5 ]
Yield
Reaction Conditions
Operation in experiment
39%
With methanol In tetrahydrofuran; hexane
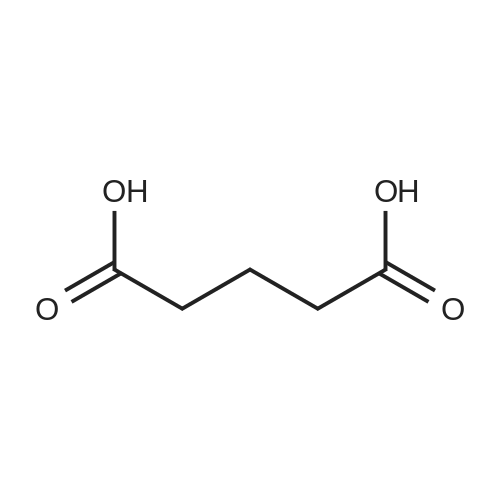
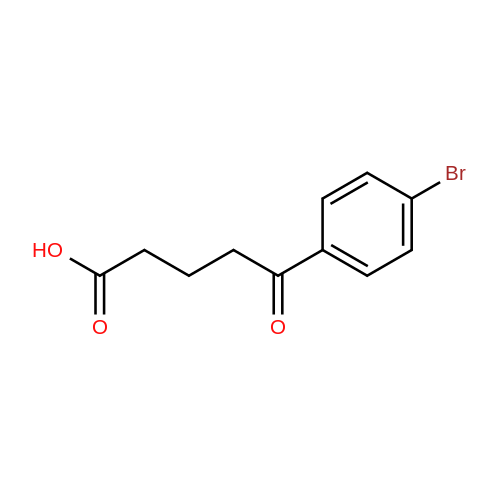
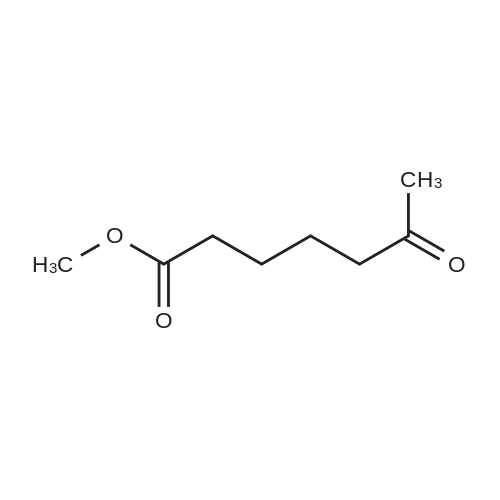
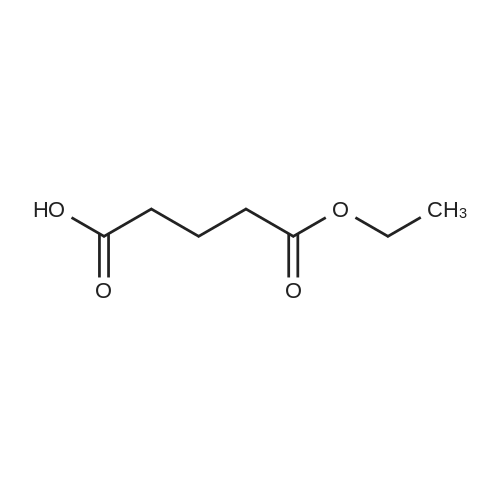
Example 21; Synthesis of 5-(4-((3,5-bis(trifluoromethyl)phenyl)(2-methyl-2H-tetrazol-5-yl)methyl)-2-ethyl-6-(trifluoromethyl)-3,4-dihydroquinoxalin-1(2H)-yl)pentan-1-ol (58); The synthetic scheme for the synthesis of compound (58) according to Example 21 is shown in FIG. 12. Step A; 5-Methoxy-5-oxopentanoic acid (54); TMSCHN2 (2.0 M hexanes, 3.8 mL) was added to a solution of glutaric acid (1.0 g, 7.6 mmol) in 4:1 THF/MeOH (75 mL). The solution was stirred and then the volatiles were removed in vacuo. The crude product was purified was purified by column chromatography (Biotage 40M, with 5:9 EtOAc/hexanes) to afford 5-methoxy-5-oxopentanoic acid (54) as a colorless oil (430 mg, 39percent).
Stage #1: Pentanedioic acid, monomethyl ester With borane-THF In tetrahydrofuran at 0 - 20℃;
Stage #2: With water; sodium hydrogencarbonate In tetrahydrofuran
21.B
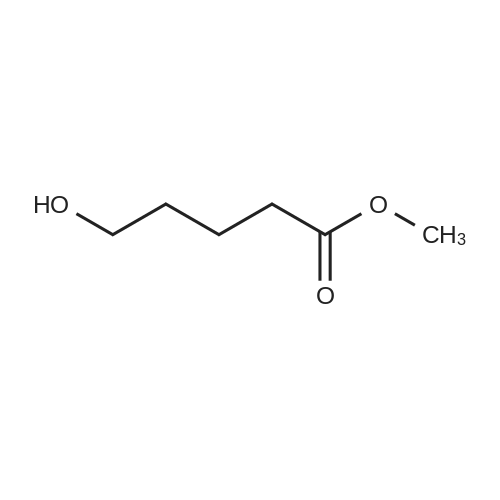
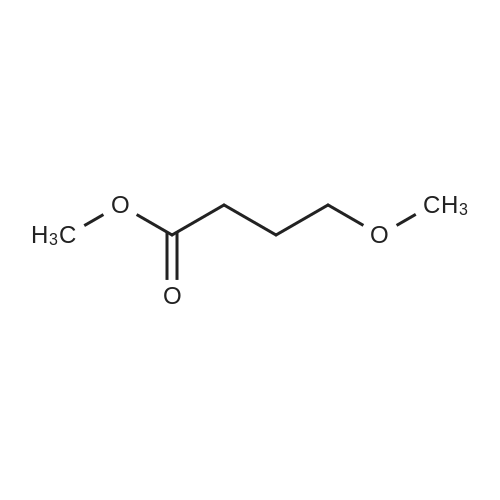
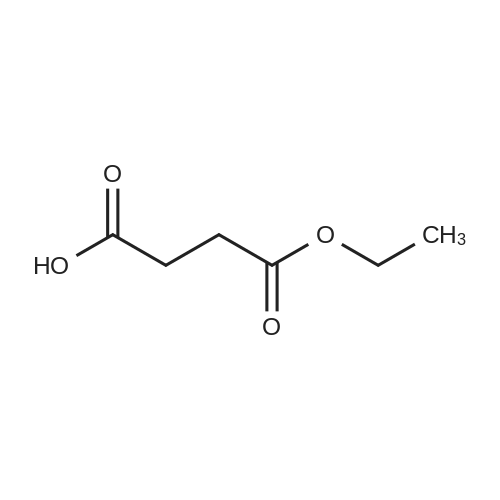
Step B; Methyl 5-hydroxypentanoate (55); BH3 (1 M THF, 38 mL) was added to a 0° C. solution of 5-methoxy-5-oxopentanoic acid (5.0 g, 34 mmol) in THF (280 mL). The solution was stirred at 0° C. to room temperature overnight. The reaction was quenched via the addition of 2:1 saturated NaHCO3/H2O (300 mL). The mixture was diluted with 300 mL Et2O and the phases were separated. The aqueous phase was extracted twice more with Et2O (200 mL each). The organics were then combined and washed with brine (300 mL). The organics were dried over MgSO4 and concentrated in vacuo to afford methyl 5-hydroxypentanoate (55) as a colorless oil (4.15 g, 92%).
With PPA; Polyphosphoric acid (PPA) at 45℃; for 2.5h;
2 5-Oxo-5-(2,3,4-trimethoxy-phenyl)-pentanoic acid methyl ester (60)
5-Oxo-5-(2,3,4-trimethoxy-phenyl)-pentanoic acid methyl ester (60) 75 g of polyphosphoric acid (Acros) was charged in a 250 mL round bottom flask, followed by addition of 1,2,3-trimethoxybenzene (5.0 g, 29.73 mmol), and mono-methylglutarate (6.516 g, 44.60 mmol). The reaction mixture was stirred mechanically for 2.5 h at 45° C. The reaction mixture was then poured into a 1000 mL beaker containing around 250 mL of ice, and stirred well until all the product precipitated out. The tan colored product was then filtered and washed with water and dried under vacuum. No, purification required at this stage. 6.78 g (77%) of product was obtained, which was pure by NMR. 1H NMR (CDCl, 300 MHz): δ 7.48 (d, J=8.86 Hz, 1H), δ 6.71 (d, J=8.98 Hz, 1 H), δ 3.96 (s, 3H), δ 3.91 (s, 3H), δ 3.87 (s, 3H), δ 3.67 (s, 3H), δ 3.02 (t, J=7.14, 2H), δ 2.41 (t, J=7.40 Hz, 2H), δ 2.03 (p, J=7.16 Hz, 2H).
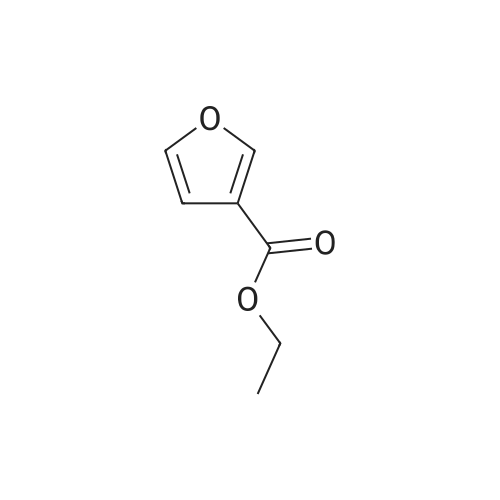
5-(4-carboxybutyryl)furan-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
64%
Stage #1: ethyl 3-furancarboxylate; Pentanedioic acid, monomethyl ester With phosphoric acid; trifluoroacetic anhydride In water; acetonitrile at 30℃; for 0.5h;
Stage #2: With hydrogenchloride; sodium hydroxide In water; acetonitrile
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; triethylamine; In dichloromethane; at 20℃;Inert atmosphere;
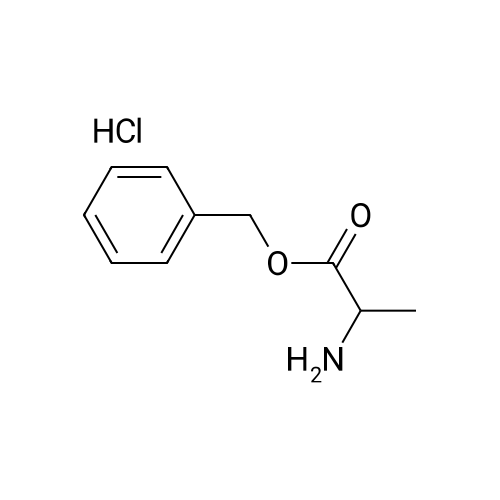
General procedure: Typical Procedure for Amide-Bond Formation of Carboxylic Acid with L-Alanine Benzyl Ester To a mixture of the corresponding carboxylic acid (1.2 mmol) and <strong>[5557-81-3]L-alanine benzyl ester hydrochloride</strong> (1.0 mmol) in CH2Cl2(4.4 mL), Et3N (3.0 mmol), DMAP (0.35 mmol) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (1.44 mmol) were added successively at r.t. The mixture was stirred for several hours to few days at r.t. until the starting material was consumed on TLC. Water was added and the mixture was extracted with ether. The extract was washed with water, brine and dried over MgSO4. Solvent was removed and the crude product was purified by column chromatography on silica gel eluted with an appropriate ratio of ethyl acetate and hexane. In the case of 10?and 11?EtOAc was used for the extraction instead of ether.
General procedure: To a round bottom flask was added the acid (5 mmol) and dry CH2Cl2 (20 mL). Thenthe flask was submerged in a brine ice bath, N-methylmorpholine (6 mmol) was addedvia syringe and the solution stirred for 15 min. After that, isobutylchloroformate (5.5mmol) was added dropwise over 20 minutes. After stirring for 6 h at ambienttemperature, the resulting mixture was washed with sat. Na2CO3, and brine, dried overMgSO4, filtered and concentrated under reduced pressure. The residue was purified byflash chromatography to give the desired product
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; at 25℃;Alkaline conditions;
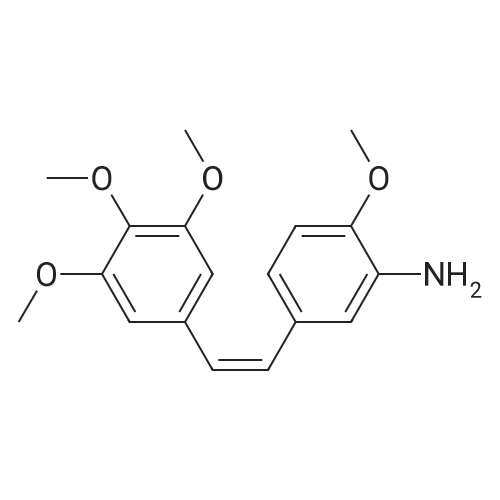
To a solution of mono-methyl glutarate (1.0g, 3.8 mmol) in dry CH2Cl2 (30 mL)was added EDCI (730 mg, 3.8 mmol), DMAP (465 mg, 3.8 mmol) and compound 7 (800 mg, 2.5 mmol). The resultingmixture was stirred at 25 C forovernight and monitored by TLC. After completion of reaction, the mixture wasdiluted with CH2Cl2 (100 mL) and washed with water (100mL), and the organic layer was dried over anhydrousNa2SO4, filtered, and concentrated under reducedpressure. The crude product which was purified bychromatography on silica gel eluted with petroleum ether/ethyl acetate(V:V=5:1, 4:1) to give the desired compound as yellow oil (yield: 850 mg, 87.9%).1HNMR (300 MHz, CDCl3) delta 8.34 (s, 1H), 7.71 (s, 1H), 6.98 (d, J= 10.5 Hz, 1H), 6.70 (d, J = 8.5 Hz, 1H), 6.54 - 6.48 (m, 3H), 6.43 (d, J= 12.2 Hz, 1H), 3.84 (d, J = 4.4 Hz, 6H), 3.68 (s, 9H), 2.48 - 2.42 (m,4H), 2.09 - 2.02 (m, 2H). HR-MS(m/z) (ESI): calcd for C24H29NO7[M+H]+:444.2020;found: 444.2027.
Stage #1: Pentanedioic acid, monomethyl ester; tartaric acid With thionyl chloride; N,N-dimethyl-formamide at 20℃;
Stage #2: With sulfuric acid at 70℃;
Stage #3: With ammonia In methanol at 20℃;
2 Example 2
A reactor was charged with 5-methoxy-5-oxopentanoic acid (25.0 g) and a catalytic amount of DMF. Thionyl chloride (40.7 g) was dropwise added thereto using a dropping funnel at room temperature. After the stirring was completed, O,O'-(1,4-dichloro-1,4-dioxobutane-2,3-diyl) dimethyl diglutarate was synthesized at a yield of 90% using an evaporator. Then, O,O′-(1,4-dichloro-1,4-dioxobutane-2,3-diyl) dimethyl diglutarate (5.22 g), tartaric acid (2.38 g), and sulfuric acid were added using the reactor, and the components were stirred at 70° C. After the stirring, the product was purified. Thereby, 2,3-bis((5-methoxy-5-oxopentanoyl)oxy)succinic acid, which is the target product, was obtained at a yield of 52%. Then, 2,3-bis((5-methoxy-5-oxopentanoyl)oxy)succinic acid (3.23 g) and MeOH were added to the reactor and stirred, and 2 M NH3 in MeOH (7.95 mL) was dropwise added thereto at room temperature. After stirring, the product was dried. Thereby, the target ammonium salt was obtained at a yield of 90%.
Stage #1: Pentanedioic acid, monomethyl ester; 2-methyl-3-phenylpropanoic acid With 4-dimethylaminopyridine; NHPI; diisopropyl-carbodiimide In dichloromethane at 20℃; for 1h;
Stage #2: With nickel(II) chloride ethylene glycol dimethyl ether complex; sodium iodide; 2,6-di(4,5-dihydro-1,3-oxazol-2-yl)-4-methoxypyridine In dichloromethane; N,N-dimethyl-formamide at 20℃; for 2.5h; Electrochemical reaction;
General procedure
General procedure: The general procedure proceeds as follows: to a mixture of acid components (the less expensive of which is used in 3 equiv.) in CH2Cl2 are added diisopropylcarbodiimide (DIC) and N-hydroxyphthalimide (NHPI) (1.1 equiv. each relative to total acid quantity, 4.4 equiv. total) along with catalytic amount of 4-dimethylaminopyridine (10 mol% to total acid quantity, 0.4 equiv. total). After stirring for 1 h, without any solvent removal, the solution is diluted with N,N-dimethylformamide and NiCl2•dme along with L4 are added (roughly 5 mol% each relative to total acid quantity, 20 mol% total), followed by the addition of NaI (0.2 M). Electrolysis using a standard ElectraSyn2.0 potentiostat (Zn anode and Ni foam cathode) for about 2.5 h (0.1 mmol scale) followed by standard workup and purification delivers the coupled product.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping