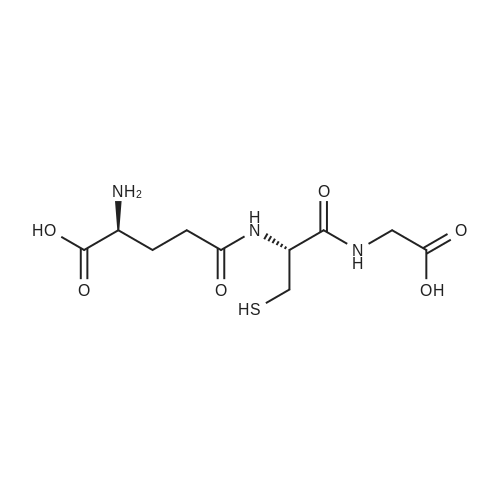
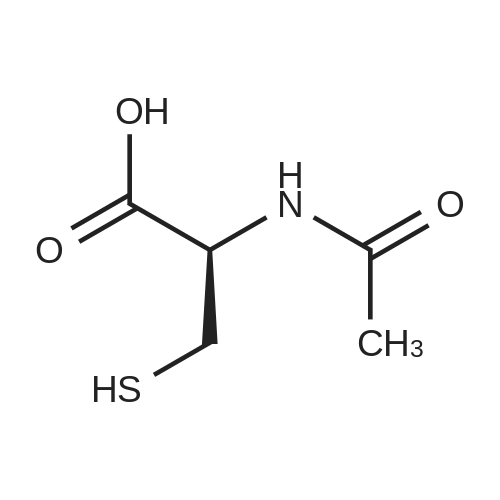
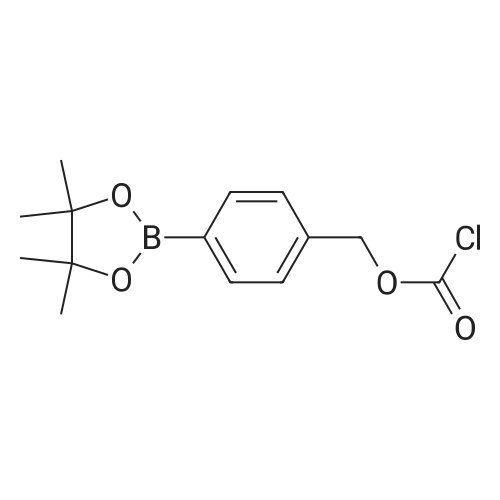
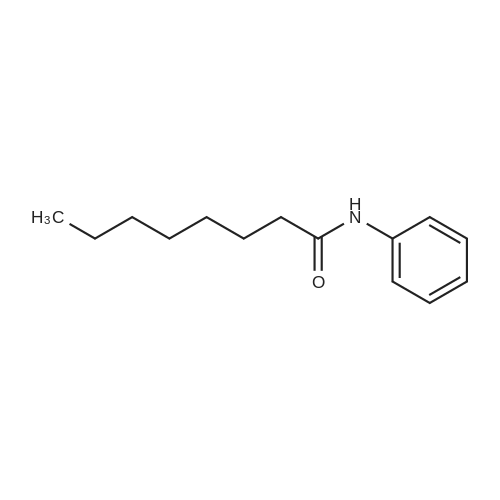
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
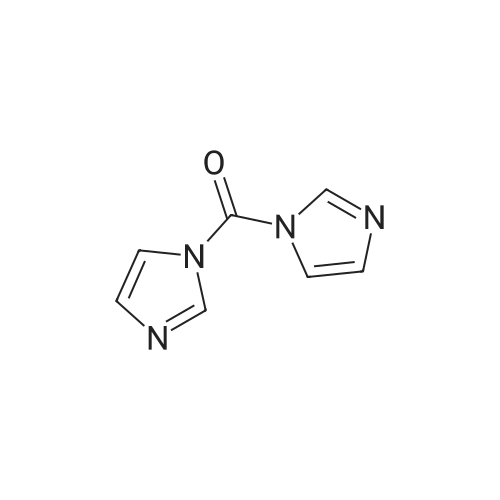
Stage #1: With 1,1'-carbonyldiimidazole In N,N-dimethyl-formamide at 25 - 30℃; for 0.5 h; Stage #2: With hydroxylamine hydrochloride In N,N-dimethyl-formamide for 0.5 h;
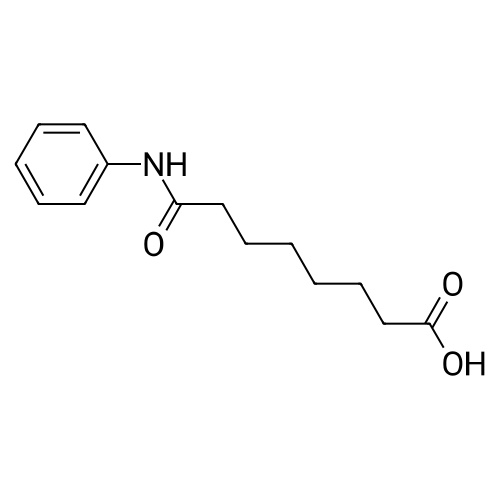
The suberanilic acid (leq) obtained in stage 1 was dissolved in DMF (5 vol) and CDI (2eq) was added at 25-3O0C and maintained for 30 minutes under stirring. Hydroxylamine hydrochloride (4eq) was added and stirring continued for 30 minutes. Water (25 vol) was then added and the mixture stirred for 2 hours. The precipitated solid was filtered, washed with water (2x5 vol) and dried under vacuum at 500C. Molar Yield = 70-75percent Purity by HPLC = 99percent
75%
Stage #1: With 1,1'-carbonyldiimidazole In N,N-dimethyl-formamide at 25 - 30℃; for 0.5 h; Stage #2: With hydroxylamine hydrochloride In N,N-dimethyl-formamide for 0.5 h;
The suberanilic acid (1 eq) obtained in stage 1 was dissolved in DMF (5 vol) and CDI (2 eq) was added at 25-30° C. and maintained for 30 minutes under stirring. Hydroxylamine hydrochloride (4 eq) was added and stirring continued for 30 minutes. Water (25 vol) was then added and the mixture stirred for 2 hours. The precipitated solid was filtered, washed with water (2.x.5 vol) and dried under vacuum at 50° C.Molar Yield=70-75percentPurity by HPLC=99percent
68%
Stage #1: With dmap; ethyl 2-(tert-butoxycarbonyloxyimino)-2-cyanoacetate; N-ethyl-N,N-diisopropylamine In tetrahydrofuran at 0 - 5℃; for 0.5 h; Stage #2: With hydroxylamine hydrochloride In tetrahydrofuran; N,N-dimethyl-formamide at 20℃;
General procedure: Ethyl 2-(tert-butoxycarbonyloxyimino)-2-cyanoacetate(Boc-Oxyma,I)(1 mmol) was added to a stirred solution of suberic acid (2mmol) and DIPEA in DCM (2 mL) at 0-5 °C temperature. Then the reaction mixturewas stirred for 1 h followed by the addition of aniline (1 mmol). The progressof the reaction was monitored by TLC. After completion of the reaction, thereaction mixture was acidified with 5percent HCl and extracted with dichloromethane(2×15 mL), and dried under anhydrous CaCl2 and the evaporation ofthe solvent gave a solid powder.Ethyl 2-(tert-butoxycarbonyloxyimino)-2-cyanoacetate(Boc-Oxyma, I)(1 mmol) was added to a stirred solution of previous carboxylic acid derivative(1 mmol), DIPEA (1 mmol) and DMAP (0.1 mmol) in THF (2 mL) at room temperature.Then the reaction mixture was stirred for 30 min followed by the addition ofhydroxylamine hydrochloride in DMF (0.5 mL), DIPEA (1.5 mmol). The progress ofthe reaction was monitored by TLC at room temperature. After completion of thereaction, the reaction mixture was concentrated using rotary evaporator andthen diluted with 15 mL of ethyl acetate and washed with 5percent HCl (2×10 mL), 5percentNaHCO3 (2×10 mL), saturated NaCl solution (2×10 mL) and dried overanhydrous Na2SO4 and the evaporationof the solvent gave a residue that was purified on silica gel columnchromatography using hexane and ethyl acetate.
Reference:
[1] Nature Chemistry, 2017, vol. 9, # 6, p. 571 - 577
[2] Patent: WO2010/43904, 2010, A2, . Location in patent: Page/Page column 18
[3] Patent: US2011/263712, 2011, A1, . Location in patent: Page/Page column 7
[4] Tetrahedron Letters, 2015, vol. 56, # 44, p. 6108 - 6111
[5] ChemSusChem, 2017, vol. 10, # 9, p. 1969 - 1975
[6] Organic Preparations and Procedures International, 2001, vol. 33, # 4, p. 391 - 394
[7] Chemistry - A European Journal, 2016, vol. 22, # 20, p. 6964 - 6967
[8] Patent: CN106008267, 2016, A, . Location in patent: Paragraph 0053 - 0056
[9] ACS Medicinal Chemistry Letters, 2018, vol. 9, # 7, p. 635 - 640
[10] ChemMedChem, 2018, vol. 13, # 19, p. 2073 - 2079
[11] Patent: WO2005/23179, 2005, A2,
2
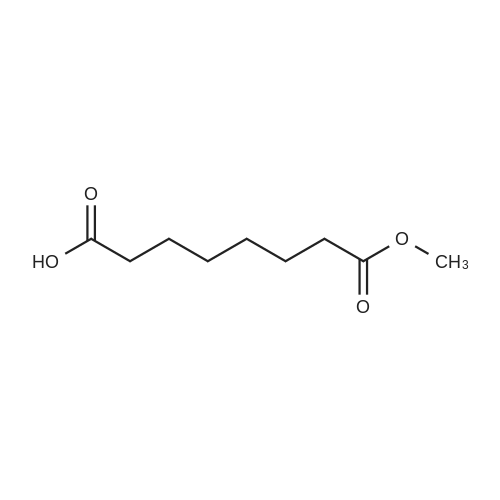
[ 149647-86-9 ]
[ 62-53-3 ]
[ 149647-78-9 ]
Yield
Reaction Conditions
Operation in experiment
82.4%
With dicyclohexyl-carbodiimide; potassium hydroxide In ethanol at 50℃; for 0.5 h;
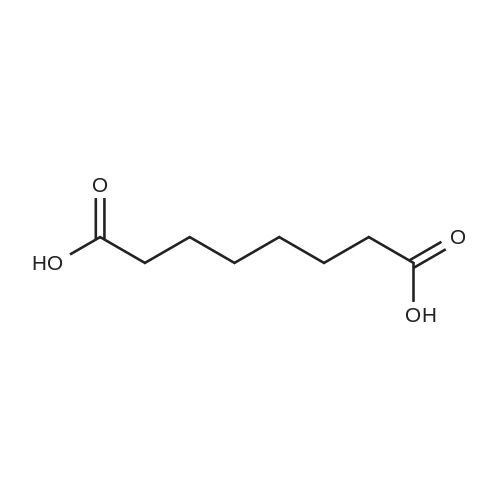
3.8 g (20 mmol) of N-hydroxy-7-carboxy-heptanoamide was dissolved in 20 ml of absolute ethanol and then 1,3-dicyclohexyl(100 mmol) of potassium hydroxide, heated to 50 ° C and stirred for 30 min under heat. After completion of the reaction, the reaction solution was concentrated and poured into water, Filtered, washed with water, and dried in vacuum to give 4 · 4g of fuloxitopine, with a yield of 82.4percent and a purity of 99.52percent.
Reference:
[1] Patent: CN106008267, 2016, A, . Location in patent: Paragraph 0047; 0048
[2] Patent: US2011/263712, 2011, A1, . Location in patent: Page/Page column 7
3
[ 162853-41-0 ]
[ 149647-78-9 ]
Yield
Reaction Conditions
Operation in experiment
95.6%
Stage #1: With hydroxylamine hydrochloride; sodium methylate In methanol; water for 16.1667 h; Stage #2: With water; acetic acid In methanol for 0.166667 h;
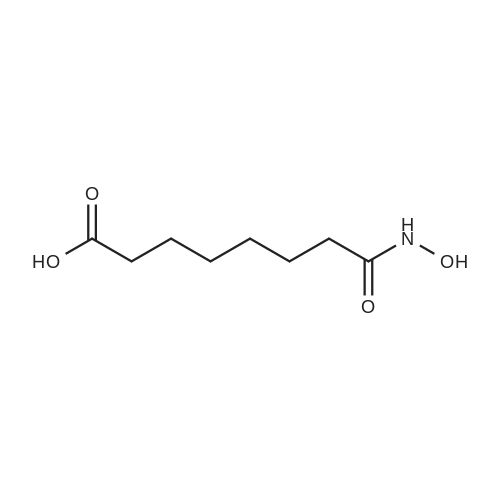
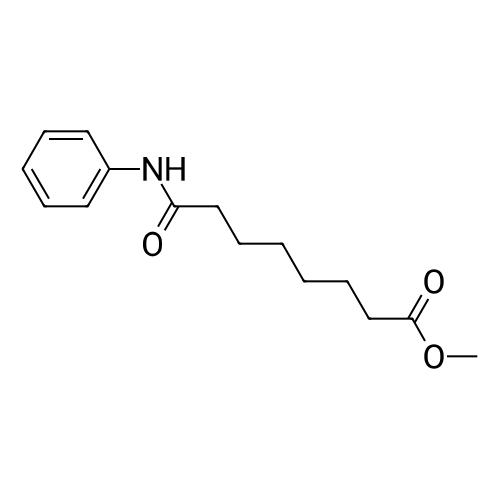
To a 50 L flask with a mechanical stirrer, thermocouple, and inlet for inert atmosphere was added 1,451.9 g of hydroxylamine hydrochloride, 19 L of anhydrous methanol, and a 3.93 L of a 30percent sodium methoxide solution in methanol. The flask was then charged with 2,748.0 g of methyl suberanilate, followed by 1.9 L of a 30percent sodium methoxide solution in methanol. The mixture was allowed to stir for 16 hr and 10 minutes. Approximately one half of the reaction mixture was transferred from the reaction flask (flask 1) to a 50 L flask (flask 2) fitted with a mechanical stirrer. Then 27 L of deionized water was added to flask 1 and the mixture was stirrer for 10 minutes. The pH was taken using a pH meter; the pH was 11.56. The pH of the mixture was adjusted to 12.02 by the addition of 100 ml of the 30percent sodium methoxide solution in methanol; this gave a clear solution (the reaction mixture at this time contained a small amount of solid. The pH was adjusted to give a clear solution from which the precipitation the product would be precipitated). The reaction mixture in flask 2 was diluted in the same manner; 27 L of deionized water was added, and the pH adjusted by the addition of 100 ml of a 30percent sodium methoxide solution to the mixture, to give a pH of 12.01 (clear solution). The reaction mixture in each flask was acidified by the addition of glacial acetic acid to precipitate the product. Flask 1 had a final pH of 8.98, and Flask 2 had a final pH of 8.70. The product from both flasks was isolated by filtration using a Buchner funnel and filter cloth. The filter cake was washed with 15 L of deionized water, and the funnel was covered and the product was partially dried on the funnel under vacuum for 15.5 hr. The product was removed and placed into five glass trays. The trays were placed in a vacuum oven and the product was dried to constant weight. The first drying period was for 22 hours at 60° C. using a Nash pump as the vacuum source with an argon bleed. The trays were removed from the vacuum oven and weighed. The trays were returned to the oven and the product dried for an additional 4 hr and 10 minutes using an oil pump as the vacuum source and with no argon bleed. The material was packaged in double 4-mill polyethylene bags, and placed in a plastic outer container. The final weight after sampling was 2633.4 g (95.6percent).
95.6%
With hydroxylamine hydrochloride; sodium methylate In methanol for 16.1667 h;
To a 50 L flask with a mechanical stirrer, thermocouple, and inlet for inert atmosphere was added 1,451.9 g of hydroxylamine hydrochloride, 19 L of anhydrous methanol, and a 3.93 L of a 30percent sodium methoxide solution in methanol. The flask was then charged with 2,748.0 g of methyl suberanilate, followed by 1.9 L of a 30percent sodium methoxide solution in methanol. The mixture was allowed to stir for 16 hr and 10 minutes. Approximately one half of the reaction mixture was transferred from the reaction flask (flask 1) to a 50 L flask (flask 2) fitted with a mechanical stirrer. Then 27 L of deionized water was added to flask 1 and the mixture was <n="63"/>stirrer for 10 minutes. The pH was taken using a pH meter; the pH was 11.56. The pH of the mixture was adjusted to 12.02 by the addition of 100 ml of the 30percent sodium methoxide solution in methanol; this gave a clear solution (the reaction mixture at this time contained a small amount of solid. The pH was adjusted to give a clear solution from which the precipitation the product would be precipitated). The reaction mixture in flask 2 was diluted in the same manner; 27 L of deionized water was added, and the pH adjusted by the addition of 100 ml of a 30 percent sodium methoxide solution to the mixture, to give a pH of 12.01 (clear solution).The reaction mixture in each flask was acidified by the addition of glacial acetic acid to precipitate the product. Flask 1 had a final pH of 8.98, and Flask 2 had a final pH of 8.70. The product from both flasks was isolated by filtration using a Buchner funnel and filter cloth. The filter cake was washed with 15 L of deionized water, and the funnel was covered and the product was partially dried on the funnel under vacuum for 15.5 hr. The product was removed and placed into five glass trays. The trays were placed in a vacuum oven and the product was dried to constant weight. The first drying period was for 22 hours at 6O0C using a Nash pump as the vacuum source with an argon bleed. The trays were removed from the vacuum oven and weighed. The trays were returned to the oven and the product dried for an additional 4 hr and 10 minutes using an oil pump as the vacuum source and with no argon bleed. The material was packaged in double 4-mill polyethylene bags, and placed in a plastic outer container. The final weight after sampling was 2633.4 g (95.6percent).
95.6%
Stage #1: With hydroxylamine hydrochloride; sodium methylate In methanol for 16.3333 h; Stage #2: With sodium methylate In methanol; water for 0.166667 h; Stage #3: With acetic acid In methanol; water
To a 50 L flask with a mechanical stirrer, thermocouple, and inlet for inert atmosphere was added 1,451.9 g of hydroxylamine hydrochloride, 19 L of anhydrous methanol, and a 3.93 L of a 30percent sodium methoxide solution in methanol. The flask was then charged with 2,748.0 g of methyl suberanilate, followed by 1.9 L of a 30percent sodium methoxide solution in methanol. The mixture was allowed to stir for 16 hr and 10 minutes. Approximately one half of the reaction mixture was transferred from the reaction flask (flask 1) to a 50 L flask (flask 2) fitted with a mechanical stirrer. Then 27 L of deionized water was added to flask 1 and the mixture was stirred for 10 minutes. The pH was taken using a pH meter; the pH was 11.56. The pH of the mixture was adjusted to 12.02 by the addition of 100 ml of the 30percent sodium methoxide solution in methanol; this gave a clear solution (the reaction mixture at this time contained a small amount of solid. The pH was adjusted to give a clear solution from which the precipitation the product would be precipitated). The reaction mixture in flask 2 was diluted in the same manner; 27 L of deionized water was added, and the pH adjusted by the addition of 100 ml of a 30percent sodium methoxide solution to the mixture, to give a pH of 12.01 (clear solution).The reaction mixture in each flask was acidified by the addition of glacial acetic acid to precipitate the product. Flask 1 had a final pH of 8.98, and Flask 2 had a final pH of 8.70. The product from both flasks was isolated by filtration using a Buchner funnel and filter cloth. The filter cake was washed with 15 L of deionized water, and the funnel was covered and the product was partially dried on the funnel under vacuum for 15.5 hr. The product was removed and placed into five glass trays. The trays were placed in a vacuum oven and the product was dried to constant weight. The first drying period was for 22 hours at 60° C. using a Nash pump as the vacuum source with an argon bleed. The trays were removed from the vacuum oven and weighed. The trays were returned to the oven and the product dried for an additional 4 hr and 10 minutes using an oil pump as the vacuum source and with no argon bleed. The material was packaged in double 4-mill polyethylene bags, and placed in a plastic outer container. The final weight after sampling was 2633.4 g (95.6percent).
95.6%
With hydroxylamine hydrochloride; sodium methylate In methanol
To a 50 L flask with a mechanical stirrer, thermocouple, and inlet for inert atmosphere was added 1,451. 9 g of hydroxylamine hydrochloride, 19 L of anhydrous methanol, and a 3.93 L of a 30percent sodium methoxide solution in methanol. The flask was then charged with 2,748. 0 g of methyl suberanilate, followed by 1.9 L of a 30percent sodium methoxide solution in methanol. The mixture was allowed to stir for 16 hr and 10 minutes. Approximately one half of the reaction mixture was transferred from the reaction flask (flask 1) to a 50 L flask (flask 2) fitted with a mechanical stirrer. Then 27 L of deionized water was added to flask 1 and the mixture was stirrer for 10 minutes. The pH was taken using a pH meter; the pH was 11.56. The pH of the mixture was adjusted to 12.02 by the addition of 100 ml of the 30percent sodium methoxide solution in methanol; this gave a clear solution (the reaction mixture at this time contained a small amount of solid. The pH was adjusted to give a clear solution from which the precipitation the product would be precipitated). The reaction mixture in flask 2 was diluted in the same manner; 27 L of deionized water was added, and the pH adjusted by the addition of 100 ml of a 30 percent sodium methoxide solution to the mixture, to give a pH of 12.01 (clear solution). The reaction mixture in each flask was acidified by the addition of glacial acetic acid to precipitate the product. Flask 1 had a final pH of 8.98, and Flask 2 had a final pH of 8.70. The product from both flasks was isolated by filtration using a Buchner funnel and filter cloth. The filter cake was washed with 15 L of deionized water, and the funnel was covered and the product was partially dried on the funnel under vacuum for 15. 5 hr. The product was removed and placed into five glass trays. The trays were placed in a vacuum oven and the product was dried to constant weight. The first drying period was for 22 hours at 60°C using a Nash pump as the vacuum source with an argon bleed. The trays were removed from the vacuum oven and weighed. The trays were returned to the oven and the product dried for an additional 4 hr and 10 minutes using an oil pump as the vacuum source and with no argon bleed. The material was packaged in double 4-mill polyethylene bags, and placed in a plastic outer container. The final weight after sampling was 2633.4 g (95.6percent).
95%
With hydroxylamine hydrochloride; sodium hydroxide In methanol at 50℃; for 0.5 h; Green chemistry
A. 3.96 g of hydroxylamine hydrochloride was dissolved in a single-necked flask with 40 mL of methanol, and the flask was placed on a thermostatically heated thermostat. Magnetic stirrer, heated to 50 ° C; B, 3.80 g of sodium hydroxide was added to a solution of hydroxylamine hydrochloride in methanol, mixed for 5 minutes, maintaining a reaction environment of pH=9; C, adding 5g (0.019mol) methyl 7-(anilinoformyl)heptanoate; D. After 30 minutes of reaction, the solvent was removed by rotary evaporation and the residue was thoroughly mixed with 120 mL of 5 mol/L diluted hydrochloric acid. With suction filtration, multiple washings and vacuum drying of white solids, high-purity vorinostat is obtained without the need for subsequent recrystallization treatment. Vorinostat was obtained by the above method with a yield of 95.0percent and a purity of 99.0percent.
95.6%
With hydroxylamine hydrochloride; sodium methylate In methanol
To a 50 L flask with a mechanical stirrer, thermocouple, and inlet for inert atmosphere was added 1,451. 9 g of hydroxylamine hydrochloride, 19 L of anhydrous methanol, and a 3.93 L of a 30percent sodium methoxide solution in methanol. The flask was then charged with 2,748. 0 g of methyl suberanilate, followed by 1.9 L of a 30percent sodium methoxide solution in methanol. The mixture was allowed to stir for 16 hr and 10 minutes. Approximately one half of the reaction mixture was transferred from the reaction flask (flask 1) to a 50 L flask (flask 2) fitted with a mechanical stirrer. Then 27 L of deionized water was added to flask 1 and the mixture was stirrer for 10 minutes. The pH was taken using a pH meter; the pH was 11.56. The pH of the mixture was adjusted to 12.02 by the addition of 100 ml of the 30percent sodium methoxide solution in methanol; this gave a clear solution (the reaction mixture at this time contained a small amount of solid. The pH was adjusted to give a clear solution from which the precipitation the product would be precipitated). The reaction mixture in flask 2 was diluted in the same manner; 27 L of deionized water was added, and the pH adjusted by the addition of 100 ml of a 30 percent sodium methoxide solution to the mixture, to give a pH of 12.01 (clear solution). The reaction mixture in each flask was acidified by the addition of glacial acetic acid to precipitate the product. Flask 1 had a final pH of 8.98, and Flask 2 had a final pH of 8.70. The product from both flasks was isolated by filtration using a Buchner funnel and filter cloth. The filter cake was washed with 15 L of deionized water, and the funnel was covered and the product was partially dried on the funnel under vacuum for 15. 5 hr. The product was removed and placed into five glass trays. The trays were placed in a vacuum oven and the product was dried to constant weight. The first drying period was for 22 hours at 60°C using a Nash pump as the vacuum source with an argon bleed. The trays were removed from the vacuum oven and weighed. The trays were returned to the oven and the product dried for an additional 4 hr and 10 minutes using an oil pump as the vacuum source and with no argon bleed. The material was packaged in double 4-mill polyethylene bags, and placed in a plastic outer container. The final weight after sampling was 2633.4 g (95.6percent).
92.1%
With hydroxylamine hydrochloride; sodium methylate In methanol at 40℃; for 3 h;
3) Hydroxylamine hydrochloride 6.5g (100mmol) of sodium methoxide and 6.5g (120mmol) was stirred in anhydrous methanol reaction 0.5h, filtered and the suberanilic acid methyl ester was added to the filtrate, the reaction deg.] C 40 3 hours, cooled to room temperature, adjusted pH = 7, filtered off with suction, the filter cake was washed with water, and recrystallized from ethanol to give 21.7 g of vorinostat, yield 92.1percent, purity 99.87percent.
90%
With hydroxylamine; potassium hydroxide In methanol at 20℃; for 1.5 h;
Step (b): Hydroxylamine hydrochloride (5.84 g, 0.084 mol) in methanol (15 mL) was mixed with KOH (4.70 g, 0.084 mol) at 40 °C in methanol (25 mL), cooled to 0 °C, and filtered. The suberanilic acid methyl ester (1.2 g, 0.004 mol) was then added to the filtrate followed by addition (over 30 min) of KOH (380 mg, 0.06 mol). The mixture was stirred at room temperature for 1 h. The mixture was added to stirring cold water (150 mL), and the pH was adjusted to seven by adding acetic acid. The precipitate was filtered off, and the resulting product was dried in a vacuum oven at 40 °C overnight to yield 1.05 g (yield 90.0 percent) of SAHA [2]: mp 158–161 °C.
48.6%
With hydroxylamine hydrochloride; potassium hydroxide In methanol at 20 - 40℃; for 0.5 h; Inert atmosphere
(B) To a 100-mL beaker equipped with a Teflon-coatedmagnetic stirring bar were sequentially placed potassiumhydroxide (5.05g, 90 mmol) and methanol (20mL). The mixturewas stirred until a complete dissolution was obtained.Hydroxylamine hydrochloride (6.95g, 100 mmol) was added. After an additional 30 min of stirring at 40oC, the reactionmixture was cooled to ambient temperature. Methyl 8-(phenyl amino)-8-oxooctanoate (1.32g, 5mmol, obtainedfrom the part A) was added. Stirring was continued for 30min. TLC was employed to monitor the progress of the reaction.After the complete reaction, the reaction mixture waspoured into 600mL of ice-water mixture with vigorous stirringleading to the formation of a solid. The resulting solidwas collected by vacuum filtration, and successively washedwith water (2 X 10mL) and ether (2 X 10mL). After havingbeen dried in vacuo, the crude product was further purifiedby recrystallization from acetonitrile to afford N-hydroxy-N'-phenyloctane-diamide as a white solid (0.64 g, 48.6percent);HPLC Rt=14.26min; mp: 159-161oC; 1HNMR (400 MHz,DMSO-d6): = 10.32(s,1H), 9.83(s,1H), 8.64(s,1H), 7.58(d,J=7.6 Hz, 2H), 7.27(t, J=7.4 Hz, 2H), 7.01(t, J=7.2 Hz, 1H),2.28(t, J=7.2Hz, 2H), 1.93(t, J=7.1Hz, 2H), 1.53(m, 4H),1.28ppm (m, 4H); 13C NMR (400 MHz, DMSO-d6):=25.00, 28.38, 32.22, 36.35, 119.01, 122.87, 128.58,139.31, 169.11, 171.20ppm. C14H20N2O3, MS (ES+) m/z:265.1560 [M+H]+.
48.6%
With hydroxylamine hydrochloride; potassium hydroxide In methanol at 40℃; for 1 h; Green chemistry
(A) To an oven-dried, 100-mL, three-necked, round bottomed flask equipped with a thermometer, a vacuum jacketed Dean-Stark trap topped with a reflux condenser fitted with a T-joint inlet, glass stoppers, and a Teflon-coated magnetic stirring bar was sequentially charged with 8-methoxy-8-oxooctanoic acid (3.76g, 20 mmol, 1 equiv),30mL of toluene, and boric acid (0.62g, 10 mmol, 0.5 equiv).While stirring vigorously under nitrogen, aniline (1.86g, 20mmol, 1 equiv) was added in one portion. The reaction was heated at reflux for 10h in an oil bath. TLC analysis, using ether: petroleum ether (2:1) as the eluent, indicated the complete reaction. The reaction mixture was allowed to cool to room temperature and then poured into 300mL of hexane with vigorous stirring leading to the precipitation of a solid.Stirring was continued for an additional 30min, and the resultingsolid was then filtered off with suction through a sintered glass filter funnel. After having been successively washed with water and hexane, the collected solid was further dried in vacuo at ambient temperature for 12h to afford methyl 8-(phenyl amino)-8-oxooctanoate as a white solid(3.82g, 72.5percent). (B) To a 100-mL beaker equipped with a Teflon-coated magnetic stirring bar were sequentially placed potassium hydroxide (5.05g, 90 mmol) and methanol (20mL). The mixture was stirred until a complete dissolution was obtained. Hydroxylamine hydrochloride (6.95g, 100 mmol) was added. After an additional 30 min of stirring at 40oC, the reaction mixture was cooled to ambient temperature. Methyl 8-(phenylamino)-8-oxooctanoate (1.32g, 5mmol, obtained from the part A) was added. Stirring was continued for 30 min. TLC was employed to monitor the progress of the reaction. After the complete reaction, the reaction mixture was poured into 600mL of ice-water mixture with vigorous stirring leading to the formation of a solid. The resulting solid was collected by vacuum filtration, and successively washed with water (2 X 10mL) and ether (2 X 10mL). After having been dried in vacuo, the crude product was further purified by recrystallization from acetonitrile to afford N-hydroxy-N'-phenyloctane-diamide as a white solid (0.64 g, 48.6percent);HPLC Rt=14.26min; mp: 159-161oC; 1HNMR (400 MHz,[D6]DMSO): = 10.32(s,1H), 9.83(s,1H), 8.64(s,1H), 7.58(d,J=7.6 Hz, 2H), 7.27(t, J=7.4 Hz, 2H), 7.01(t, J=7.2 Hz, 1H),2.28(t, J=7.2Hz, 2H), 1.93(t, J=7.1Hz, 2H), 1.53(m, 4H),1.28ppm (m, 4H); 13C NMR (400 MHz [D6]DMSO):=25.00, 28.38, 32.22, 36.35, 119.01, 122.87, 128.58,139.31, 169.11, 171.20ppm. C14H20N2O3, MS (ES+) m/z:265.1560 [M+H]+.
Reference:
[1] Patent: US2007/117815, 2007, A1, . Location in patent: Page/Page column 29
[2] Patent: WO2007/56244, 2007, A2, . Location in patent: Page/Page column 60-61
[3] Patent: US2008/132575, 2008, A1, . Location in patent: Page/Page column 14
[4] Patent: WO2005/23179, 2005, A2, . Location in patent: Page/Page column 84-85
[5] Patent: CN108069878, 2018, A, . Location in patent: Paragraph 0011; 0032; 0058-0064; 0067; 0074; 0081; 0087
[6] Patent: WO2005/23179, 2005, A2, . Location in patent: Page/Page column 84-85
[7] Journal of Medicinal Chemistry, 2010, vol. 53, # 8, p. 3038 - 3047
[8] Patent: CN105777582, 2016, A, . Location in patent: Paragraph 0023; 0027
[9] Journal of Medicinal Chemistry, 1995, vol. 38, # 8, p. 1411 - 1413
[10] Journal of Medicinal Chemistry, 2005, vol. 48, # 15, p. 5047 - 5051
[11] Journal of Natural Products, 2013, vol. 76, # 7, p. 1260 - 1266
[12] Apoptosis, 2016, vol. 21, # 11, p. 1249 - 1264
[13] ChemMedChem, 2018, vol. 13, # 19, p. 2073 - 2079
[14] Angewandte Chemie - International Edition, 2007, vol. 46, # 42, p. 7960 - 7964
[15] Journal of Medicinal Chemistry, 2013, vol. 56, # 20, p. 7997 - 8007
[16] Medicinal Chemistry, 2015, vol. 11, # 7, p. 636 - 648
[17] Medicinal Chemistry, 2016, vol. 12, # 8, p. 767 - 774
[18] Patent: WO2008/39421, 2008, A2, . Location in patent: Page/Page column 33-34
[19] Patent: US2008/132575, 2008, A1, . Location in patent: Page/Page column 15
[20] Patent: WO2010/62333, 2010, A1, . Location in patent: Page/Page column 10
[21] Journal of Medicinal Chemistry, 2011, vol. 54, # 13, p. 4694 - 4720
[22] Journal of Drug Targeting, 2018, vol. 26, # 5-6, p. 448 - 457
[23] ACS Medicinal Chemistry Letters, 2018, vol. 9, # 7, p. 635 - 640
To a 50 L flask with a mechanical stirrer, thermocouple, and inlet for inert atmosphere was added 1,451.9 g of hydroxylamine hydrochloride, 19 L of anhydrous methanol, and a 3.93 L of a 30% sodium methoxide solution in methanol. The flask was then charged with 2,748.0 g of methyl suberanilate, followed by 1.9 L of a 30% sodium methoxide solution in methanol. The mixture was allowed to stir for 16 hr and 10 minutes. Approximately one half of the reaction mixture was transferred from the reaction flask (flask 1) to a 50 L flask (flask 2) fitted with a mechanical stirrer. Then 27 L of deionized water was added to flask 1 and the mixture was stirrer for 10 minutes. The pH was taken using a pH meter; the pH was 11.56. The pH of the mixture was adjusted to 12.02 by the addition of 100 ml of the 30% sodium methoxide solution in methanol; this gave a clear solution (the reaction mixture at this time contained a small amount of solid. The pH was adjusted to give a clear solution from which the precipitation the product would be precipitated). The reaction mixture in flask 2 was diluted in the same manner; 27 L of deionized water was added, and the pH adjusted by the addition of 100 ml of a 30% sodium methoxide solution to the mixture, to give a pH of 12.01 (clear solution). The reaction mixture in each flask was acidified by the addition of glacial acetic acid to precipitate the product. Flask 1 had a final pH of 8.98, and Flask 2 had a final pH of 8.70. The product from both flasks was isolated by filtration using a Buchner funnel and filter cloth. The filter cake was washed with 15 L of deionized water, and the funnel was covered and the product was partially dried on the funnel under vacuum for 15.5 hr. The product was removed and placed into five glass trays. The trays were placed in a vacuum oven and the product was dried to constant weight. The first drying period was for 22 hours at 60 C. using a Nash pump as the vacuum source with an argon bleed. The trays were removed from the vacuum oven and weighed. The trays were returned to the oven and the product dried for an additional 4 hr and 10 minutes using an oil pump as the vacuum source and with no argon bleed. The material was packaged in double 4-mill polyethylene bags, and placed in a plastic outer container. The final weight after sampling was 2633.4 g (95.6%).
95.6%
With hydroxylamine hydrochloride; sodium methylate; In methanol; for 16.1667h;
To a 50 L flask with a mechanical stirrer, thermocouple, and inlet for inert atmosphere was added 1,451.9 g of hydroxylamine hydrochloride, 19 L of anhydrous methanol, and a 3.93 L of a 30% sodium methoxide solution in methanol. The flask was then charged with 2,748.0 g of methyl suberanilate, followed by 1.9 L of a 30% sodium methoxide solution in methanol. The mixture was allowed to stir for 16 hr and 10 minutes. Approximately one half of the reaction mixture was transferred from the reaction flask (flask 1) to a 50 L flask (flask 2) fitted with a mechanical stirrer. Then 27 L of deionized water was added to flask 1 and the mixture was <n="63"/>stirrer for 10 minutes. The pH was taken using a pH meter; the pH was 11.56. The pH of the mixture was adjusted to 12.02 by the addition of 100 ml of the 30% sodium methoxide solution in methanol; this gave a clear solution (the reaction mixture at this time contained a small amount of solid. The pH was adjusted to give a clear solution from which the precipitation the product would be precipitated). The reaction mixture in flask 2 was diluted in the same manner; 27 L of deionized water was added, and the pH adjusted by the addition of 100 ml of a 30 % sodium methoxide solution to the mixture, to give a pH of 12.01 (clear solution).The reaction mixture in each flask was acidified by the addition of glacial acetic acid to precipitate the product. Flask 1 had a final pH of 8.98, and Flask 2 had a final pH of 8.70. The product from both flasks was isolated by filtration using a Buchner funnel and filter cloth. The filter cake was washed with 15 L of deionized water, and the funnel was covered and the product was partially dried on the funnel under vacuum for 15.5 hr. The product was removed and placed into five glass trays. The trays were placed in a vacuum oven and the product was dried to constant weight. The first drying period was for 22 hours at 6O0C using a Nash pump as the vacuum source with an argon bleed. The trays were removed from the vacuum oven and weighed. The trays were returned to the oven and the product dried for an additional 4 hr and 10 minutes using an oil pump as the vacuum source and with no argon bleed. The material was packaged in double 4-mill polyethylene bags, and placed in a plastic outer container. The final weight after sampling was 2633.4 g (95.6%).
95.6%
To a 50 L flask with a mechanical stirrer, thermocouple, and inlet for inert atmosphere was added 1,451.9 g of hydroxylamine hydrochloride, 19 L of anhydrous methanol, and a 3.93 L of a 30% sodium methoxide solution in methanol. The flask was then charged with 2,748.0 g of methyl suberanilate, followed by 1.9 L of a 30% sodium methoxide solution in methanol. The mixture was allowed to stir for 16 hr and 10 minutes. Approximately one half of the reaction mixture was transferred from the reaction flask (flask 1) to a 50 L flask (flask 2) fitted with a mechanical stirrer. Then 27 L of deionized water was added to flask 1 and the mixture was stirred for 10 minutes. The pH was taken using a pH meter; the pH was 11.56. The pH of the mixture was adjusted to 12.02 by the addition of 100 ml of the 30% sodium methoxide solution in methanol; this gave a clear solution (the reaction mixture at this time contained a small amount of solid. The pH was adjusted to give a clear solution from which the precipitation the product would be precipitated). The reaction mixture in flask 2 was diluted in the same manner; 27 L of deionized water was added, and the pH adjusted by the addition of 100 ml of a 30% sodium methoxide solution to the mixture, to give a pH of 12.01 (clear solution).The reaction mixture in each flask was acidified by the addition of glacial acetic acid to precipitate the product. Flask 1 had a final pH of 8.98, and Flask 2 had a final pH of 8.70. The product from both flasks was isolated by filtration using a Buchner funnel and filter cloth. The filter cake was washed with 15 L of deionized water, and the funnel was covered and the product was partially dried on the funnel under vacuum for 15.5 hr. The product was removed and placed into five glass trays. The trays were placed in a vacuum oven and the product was dried to constant weight. The first drying period was for 22 hours at 60 C. using a Nash pump as the vacuum source with an argon bleed. The trays were removed from the vacuum oven and weighed. The trays were returned to the oven and the product dried for an additional 4 hr and 10 minutes using an oil pump as the vacuum source and with no argon bleed. The material was packaged in double 4-mill polyethylene bags, and placed in a plastic outer container. The final weight after sampling was 2633.4 g (95.6%).
95%
With hydroxylamine hydrochloride; sodium hydroxide; In methanol; at 50℃; for 0.5h;pH 9;Green chemistry;
A. 3.96 g of hydroxylamine hydrochloride was dissolved in a single-necked flask with 40 mL of methanol, and the flask was placed on a thermostatically heated thermostat. Magnetic stirrer, heated to 50 C; B, 3.80 g of sodium hydroxide was added to a solution of hydroxylamine hydrochloride in methanol, mixed for 5 minutes, maintaining a reaction environment of pH=9; C, adding 5g (0.019mol) methyl 7-(anilinoformyl)heptanoate; D. After 30 minutes of reaction, the solvent was removed by rotary evaporation and the residue was thoroughly mixed with 120 mL of 5 mol/L diluted hydrochloric acid. With suction filtration, multiple washings and vacuum drying of white solids, high-purity vorinostat is obtained without the need for subsequent recrystallization treatment. Vorinostat was obtained by the above method with a yield of 95.0% and a purity of 99.0%.
95.6%
With hydroxylamine hydrochloride; sodium methylate; In methanol;
To a 50 L flask with a mechanical stirrer, thermocouple, and inlet for inert atmosphere was added 1,451. 9 g of hydroxylamine hydrochloride, 19 L of anhydrous methanol, and a 3.93 L of a 30% sodium methoxide solution in methanol. The flask was then charged with 2,748. 0 g of methyl suberanilate, followed by 1.9 L of a 30% sodium methoxide solution in methanol. The mixture was allowed to stir for 16 hr and 10 minutes. Approximately one half of the reaction mixture was transferred from the reaction flask (flask 1) to a 50 L flask (flask 2) fitted with a mechanical stirrer. Then 27 L of deionized water was added to flask 1 and the mixture was stirrer for 10 minutes. The pH was taken using a pH meter; the pH was 11.56. The pH of the mixture was adjusted to 12.02 by the addition of 100 ml of the 30% sodium methoxide solution in methanol; this gave a clear solution (the reaction mixture at this time contained a small amount of solid. The pH was adjusted to give a clear solution from which the precipitation the product would be precipitated). The reaction mixture in flask 2 was diluted in the same manner; 27 L of deionized water was added, and the pH adjusted by the addition of 100 ml of a 30 % sodium methoxide solution to the mixture, to give a pH of 12.01 (clear solution). The reaction mixture in each flask was acidified by the addition of glacial acetic acid to precipitate the product. Flask 1 had a final pH of 8.98, and Flask 2 had a final pH of 8.70. The product from both flasks was isolated by filtration using a Buchner funnel and filter cloth. The filter cake was washed with 15 L of deionized water, and the funnel was covered and the product was partially dried on the funnel under vacuum for 15. 5 hr. The product was removed and placed into five glass trays. The trays were placed in a vacuum oven and the product was dried to constant weight. The first drying period was for 22 hours at 60C using a Nash pump as the vacuum source with an argon bleed. The trays were removed from the vacuum oven and weighed. The trays were returned to the oven and the product dried for an additional 4 hr and 10 minutes using an oil pump as the vacuum source and with no argon bleed. The material was packaged in double 4-mill polyethylene bags, and placed in a plastic outer container. The final weight after sampling was 2633.4 g (95.6%).
92.1%
With hydroxylamine hydrochloride; sodium methylate; In methanol; at 40℃; for 3h;
3) Hydroxylamine hydrochloride 6.5g (100mmol) of sodium methoxide and 6.5g (120mmol) was stirred in anhydrous methanol reaction 0.5h, filtered and the suberanilic acid methyl ester was added to the filtrate, the reaction deg.] C 40 3 hours, cooled to room temperature, adjusted pH = 7, filtered off with suction, the filter cake was washed with water, and recrystallized from ethanol to give 21.7 g of vorinostat, yield 92.1%, purity 99.87%.
90%
With hydroxylamine; potassium hydroxide; In methanol; at 20℃; for 1.5h;
Step (b): Hydroxylamine hydrochloride (5.84 g, 0.084 mol) in methanol (15 mL) was mixed with KOH (4.70 g, 0.084 mol) at 40 C in methanol (25 mL), cooled to 0 C, and filtered. The suberanilic acid methyl ester (1.2 g, 0.004 mol) was then added to the filtrate followed by addition (over 30 min) of KOH (380 mg, 0.06 mol). The mixture was stirred at room temperature for 1 h. The mixture was added to stirring cold water (150 mL), and the pH was adjusted to seven by adding acetic acid. The precipitate was filtered off, and the resulting product was dried in a vacuum oven at 40 C overnight to yield 1.05 g (yield 90.0 %) of SAHA [2]: mp 158-161 C.
48.6%
With hydroxylamine hydrochloride; potassium hydroxide; In methanol; at 20 - 40℃; for 0.5h;Inert atmosphere;
(B) To a 100-mL beaker equipped with a Teflon-coatedmagnetic stirring bar were sequentially placed potassiumhydroxide (5.05g, 90 mmol) and methanol (20mL). The mixturewas stirred until a complete dissolution was obtained.Hydroxylamine hydrochloride (6.95g, 100 mmol) was added. After an additional 30 min of stirring at 40oC, the reactionmixture was cooled to ambient temperature. Methyl 8-(phenyl amino)-8-oxooctanoate (1.32g, 5mmol, obtainedfrom the part A) was added. Stirring was continued for 30min. TLC was employed to monitor the progress of the reaction.After the complete reaction, the reaction mixture waspoured into 600mL of ice-water mixture with vigorous stirringleading to the formation of a solid. The resulting solidwas collected by vacuum filtration, and successively washedwith water (2 X 10mL) and ether (2 X 10mL). After havingbeen dried in vacuo, the crude product was further purifiedby recrystallization from acetonitrile to afford N-hydroxy-N'-phenyloctane-diamide as a white solid (0.64 g, 48.6%);HPLC Rt=14.26min; mp: 159-161oC; 1HNMR (400 MHz,DMSO-d6): = 10.32(s,1H), 9.83(s,1H), 8.64(s,1H), 7.58(d,J=7.6 Hz, 2H), 7.27(t, J=7.4 Hz, 2H), 7.01(t, J=7.2 Hz, 1H),2.28(t, J=7.2Hz, 2H), 1.93(t, J=7.1Hz, 2H), 1.53(m, 4H),1.28ppm (m, 4H); 13C NMR (400 MHz, DMSO-d6):=25.00, 28.38, 32.22, 36.35, 119.01, 122.87, 128.58,139.31, 169.11, 171.20ppm. C14H20N2O3, MS (ES+) m/z:265.1560 [M+H]+.
48.6%
With hydroxylamine hydrochloride; potassium hydroxide; In methanol; at 40℃; for 1h;Green chemistry;
(A) To an oven-dried, 100-mL, three-necked, round bottomed flask equipped with a thermometer, a vacuum jacketed Dean-Stark trap topped with a reflux condenser fitted with a T-joint inlet, glass stoppers, and a Teflon-coated magnetic stirring bar was sequentially charged with 8-methoxy-8-oxooctanoic acid (3.76g, 20 mmol, 1 equiv),30mL of toluene, and boric acid (0.62g, 10 mmol, 0.5 equiv).While stirring vigorously under nitrogen, aniline (1.86g, 20mmol, 1 equiv) was added in one portion. The reaction was heated at reflux for 10h in an oil bath. TLC analysis, using ether: petroleum ether (2:1) as the eluent, indicated the complete reaction. The reaction mixture was allowed to cool to room temperature and then poured into 300mL of hexane with vigorous stirring leading to the precipitation of a solid.Stirring was continued for an additional 30min, and the resultingsolid was then filtered off with suction through a sintered glass filter funnel. After having been successively washed with water and hexane, the collected solid was further dried in vacuo at ambient temperature for 12h to afford methyl 8-(phenyl amino)-8-oxooctanoate as a white solid(3.82g, 72.5%). (B) To a 100-mL beaker equipped with a Teflon-coated magnetic stirring bar were sequentially placed potassium hydroxide (5.05g, 90 mmol) and methanol (20mL). The mixture was stirred until a complete dissolution was obtained. Hydroxylamine hydrochloride (6.95g, 100 mmol) was added. After an additional 30 min of stirring at 40oC, the reaction mixture was cooled to ambient temperature. Methyl 8-(phenylamino)-8-oxooctanoate (1.32g, 5mmol, obtained from the part A) was added. Stirring was continued for 30 min. TLC was employed to monitor the progress of the reaction. After the complete reaction, the reaction mixture was poured into 600mL of ice-water mixture with vigorous stirring leading to the formation of a solid. The resulting solid was collected by vacuum filtration, and successively washed with water (2 X 10mL) and ether (2 X 10mL). After having been dried in vacuo, the crude product was further purified by recrystallization from acetonitrile to afford N-hydroxy-N'-phenyloctane-diamide as a white solid (0.64 g, 48.6%);HPLC Rt=14.26min; mp: 159-161oC; 1HNMR (400 MHz,[D6]DMSO): = 10.32(s,1H), 9.83(s,1H), 8.64(s,1H), 7.58(d,J=7.6 Hz, 2H), 7.27(t, J=7.4 Hz, 2H), 7.01(t, J=7.2 Hz, 1H),2.28(t, J=7.2Hz, 2H), 1.93(t, J=7.1Hz, 2H), 1.53(m, 4H),1.28ppm (m, 4H); 13C NMR (400 MHz [D6]DMSO):=25.00, 28.38, 32.22, 36.35, 119.01, 122.87, 128.58,139.31, 169.11, 171.20ppm. C14H20N2O3, MS (ES+) m/z:265.1560 [M+H]+.
Methyl suberanilate (Compound 4, 263.3 g, 1.0 mole) and 2M hydroxylamine freebase solution (0.8-1.0 L) are combined. While maintaining the batch at a maximum of 2O0C, the apparent pH is adjusted to > 10.5 with sodium methoxide in methanol as needed. While maintaining the batch at maximum 200C and apparent pH > 10.5 using sodium methoxide in methanol, the batch is aged. During the age, hydroxylamine freebase solution (0.5-0.6 L) is added, and the batch is maintained at maximum 2O0C and apparent pH > 10.5 until the reaction is complete. The reaction is quenched by adding the batch to water (0.9-1.1 L) while maintaining the batch temperature between 20-350C, and the water content of the batch is adjusted to 35-45%. The pH is adjusted to 8.8-9.2 using glacial acetic acid and sodium carbonate as needed. The batch is cooled to 0-100C over 5-10 hours. The batch is filtered and the cake washed with 55:45 (v/v) methanol/water (0.45-0.6 L) at 0-100C. The wet cake is vacuum conditioned until the water content is < 35%. The vorinostat crude (264.32 g, 1.0 mole) wet cake is combined with denatured ethanol (1308-1599 g) and water (167-204 g). Hydroxylamine hydrochloride (> 9 mEquiv) and sodium methoxide in methanol (> 9 mEquiv) are added to the slurry, and the batch is heated to 70-800C. The solution is filtered and then crystallized by slowly cooling to 0-100C. The batch is <n="35"/>filtered and the cake washed with cold 4:1 (v/v) denatured ethanol/water. The wet cake is dried at a maximum of 450C under vacuum.
Methyl suberanilate (Compound 4, 263.3 g, 1.0 mole) and 2M hydroxylamine freebase solution (0.8-1.0 L) are combined. While maintaining the batch at a maximum of 20 C., the apparent pH is adjusted to 10.5 with sodium methoxide in methanol as needed. While maintaining the batch at maximum 20 C. and apparent pH10.5 using sodium methoxide in methanol, the batch is aged. During the age, hydroxylamine freebase solution (0.5-0.6 L) is added, and the batch is maintained at maximum 20 C. and apparent pH10.5 until the reaction is complete. The reaction is quenched by adding the batch to water (0.9-1.1 L) while maintaining the batch temperature between 20-35 C., and the water content of the batch is adjusted to 35-45%. The pH is adjusted to 8.8-9.2 using glacial acetic acid and sodium carbonate as needed. The batch is cooled to 0-10 C. over 5-10 hours. The batch is filtered and the cake washed with 55:45 (v/v) methanol/water (0.45-0.6 L) at 0-10 C. The wet cake is vacuum conditioned until the water content is 35%.
Example 2: Preparation of Vorinostat form IIIA solution of Potassium hydroxide (2.7 g, 0.048 mol, 2.5 eq.) in methanol (10 mL, 2 vol.) (flask A) was added to a stirred solution of Hydroxylamine hydrochloride (3.3 g, 0.048 mol, 2.5 eq.) in methanol (20 mL, 4 vol) at 0C-5C (flask B). The flask A was rinsed with Methanol (5 mL, 1 vol.) and added to the suspension in flask B. The suspension was stirred for 15-30 min at 0C-5C; the salts were filtered and washed with Methanol (5 mL, 1 vol.). 8- oxo-8-(phenylamino)octanoic acid methyl ester ("OPO") (5 g, 0.019 mol, 1.0 eq.) was added to the filtrate followed by a solution of Potassium hydroxide (1.3 g, 0.023 mol, 1.5 eq.) in Methanol (10 mL, 2 vol.) maintaining temperature at 0C-5C. The flask was rinsed with Methanol (5 mL, 1 vol.) and the reaction mixture was stirred at the same temperature until completion. The suspension was added to cold water (75 mL, 15 vol.) at 0-50C over 1-2 min and was stirred for 10-15 min at 18-220C. IN Hydrochloric acid was slowly added until pH 7.4, the suspension was cooled to 0-50C and was stirred for 0.5-1.5 h at the same temperature. The solid was filtered and washed twice with water, (10 mL, 10 vol. each) The white to light orange solid was allowed to drain under vacuum for 1 hour
suberoylanilide hydroxamic acid iron complex[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
96%
With iron(III) chloride; N-ethyl-N,N-diisopropylamine; In ethanol; at 20℃; for 16.0833h;
EXAMPLE 6 Preparation of Iron SAHA Metal Chelate Complex Chemical Reaction SchemeExperimental At ambient temperature, SAHA (1.0 g, 3.78 mmol) and ethanol (10.0 mL) were added to an erlenmyer flask, and N,N-diisopropylethylamine was then added dropwise (0.66 mL, <n="44"/>3.78 mmol) with stirring. To the resultant slurry, a solution of ferric chloride in ethanol (0.205 g, 1.26 mmol in 10.0 mL of ethanol) was added. The mixture immediately took on a deep red color as the ferric chloride solution was added. Ethanol (5.0 mL) was used to wash the remaining ferric chloride solution into the reaction flask. After 5 min of stirring at ambient temperature, the reaction mixture became a clear, dark red solution. The reaction mixture was allowed to stir at ambient temperature for 16 h. After this time, a dark orange precipitate was observed. The orange solid was collected via filtration and washed with ethanol (3 x 3.0 mL). The filtrate was colorless. The orange solid was allowed to dry for 24 h (mass 1.02 g, 96 % yield).
The suberanilic acid (leq) obtained in stage 1 was dissolved in DMF (5 vol) and CDI (2eq) was added at 25-3O0C and maintained for 30 minutes under stirring. Hydroxylamine hydrochloride (4eq) was added and stirring continued for 30 minutes. Water (25 vol) was then added and the mixture stirred for 2 hours. The precipitated solid was filtered, washed with water (2x5 vol) and dried under vacuum at 500C. Molar Yield = 70-75% Purity by HPLC = 99%
75%
The suberanilic acid (1 eq) obtained in stage 1 was dissolved in DMF (5 vol) and CDI (2 eq) was added at 25-30 C. and maintained for 30 minutes under stirring. Hydroxylamine hydrochloride (4 eq) was added and stirring continued for 30 minutes. Water (25 vol) was then added and the mixture stirred for 2 hours. The precipitated solid was filtered, washed with water (2×5 vol) and dried under vacuum at 50 C.Molar Yield=70-75%Purity by HPLC=99%
68%
General procedure: Ethyl 2-(tert-butoxycarbonyloxyimino)-2-cyanoacetate(Boc-Oxyma,I)(1 mmol) was added to a stirred solution of suberic acid (2mmol) and DIPEA in DCM (2 mL) at 0-5 C temperature. Then the reaction mixturewas stirred for 1 h followed by the addition of aniline (1 mmol). The progressof the reaction was monitored by TLC. After completion of the reaction, thereaction mixture was acidified with 5% HCl and extracted with dichloromethane(2×15 mL), and dried under anhydrous CaCl2 and the evaporation ofthe solvent gave a solid powder.Ethyl 2-(tert-butoxycarbonyloxyimino)-2-cyanoacetate(Boc-Oxyma, I)(1 mmol) was added to a stirred solution of previous carboxylic acid derivative(1 mmol), DIPEA (1 mmol) and DMAP (0.1 mmol) in THF (2 mL) at room temperature.Then the reaction mixture was stirred for 30 min followed by the addition ofhydroxylamine hydrochloride in DMF (0.5 mL), DIPEA (1.5 mmol). The progress ofthe reaction was monitored by TLC at room temperature. After completion of thereaction, the reaction mixture was concentrated using rotary evaporator andthen diluted with 15 mL of ethyl acetate and washed with 5% HCl (2×10 mL), 5%NaHCO3 (2×10 mL), saturated NaCl solution (2×10 mL) and dried overanhydrous Na2SO4 and the evaporationof the solvent gave a residue that was purified on silica gel columnchromatography using hexane and ethyl acetate.
The octanoyl aniline acid (leq) obtained in the first stage was dissolved in DMF (5 vol) at 25 C and CDI (2 eq) was added, followed by incubation for 30 minutes with stirring.Hydroxylamine hydrochloride (4 eq) was added and stirring was continued for 30 minutes.Water (25 vol) was then added and the mixture was stirred for 2 hours.The precipitated solid was filtered, washed with water (2 x 5 vol) and then dried in vacuo at 50 & lt; 0 & gt; C.Molar yield = 71.2%, HPLC purity = 98.75%.Phase 3: Roughing the promise of his purificationAmmonia water (2.5 vol) was added to the crude oxo (l eq) in acetonitrile (l5 vol) at 30 & lt; 0 & gt; C.The mixture was then incubated at 55 C for 1 hour before cooling to 25 C and stirring for an additional 1 hour.The obtained solid was filtered, washed with acetonitrile (2 x 0.5 vol) and vacuum dried at 50 C for 5 hours.Molar yield = 52.1%, HPLC purity ?99.8%.
N-[(8-anilino-8-oxooctanoyl)oxy]-N'-phenyloctanediamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
77%
To a stirring solution of suberanilic acid (3.0 g, 12.0 minol) in DMF (60 mL) was added 1- hydroxybenzotriazole (1.79 g, 13.2 mmol), 1-(3-dimethylaminopropyl)-3- ethylcarbodiimide hydrochloride (EDCI) (2.54 g, 13.2 mmol). After 5 mins, a solution of SAHA (3.18 g, 12.0 mmol) in DMF (30 mL) was added to the mixture with stirring. After 24 h, the solvent was removed and the residue was triturated with EtOAc (50 mL). The slurry was filtered to yield an off-white solid, which was triturated in CH2C12 (50 mL). The slurry was filtered yielding a white solid (4.6 g, 77%). Half of the material was purified further by repeating the washing procedures with an addition of H20 (20 mL) in the EtOAc wash. The white solid was filtered to yield the desired material (2.1 g). MP: 151-153 C. 'H NMR (DMSO-d6) No. 11.51 (s, 1H), 9.82 (s, 2H), 7.54 (d, J= 7.9 Hz, 4H), 7.23 (dd, J= 7.9,7.6 Hz, 4H), 6.97 (t, J= 7.6 Hz, 2H), 2.39 (t, J= 7.2 Hz, 2H), 2.24 (t, J= 6.8 Hz, 4H), 1.98 (t, J = 6.8 Hz, 2H), 1.66-1.38 (m, 8H), 1.38-1.16 (m, 8H). ¹3C NMR (DMSO-d6) No. 171.33,169.94, 139.49,128.72, 123.02, 119.21, 36.49,32.02, 31.06, 28.50, 28.37, 28.14, 25.12,25.05, 24.79,24.38. MS (EI): cal'd (MH(at)) 496, exp (MH+) 496.
With methanol; water; at -5 - 60℃; for 72.5h;Heating / reflux;Purification / work up;
A 50 L flask with a mechanical stirrer, thermocouple, condenser, and inlet for inert atmosphere was charged with the crude SAHA to be crystallized (2,525.7 g), followed by 2,625 ml of deionized water and 15,755 ml of methanol. The material was heated to reflux to give a solution. Then 5,250 ml of deionized water was added to the reaction mixture. The heat was turned off, and the mixture was allowed to cool. When the mixture had cooled sufficiently so that the flask could be safely handled (28 C.), the flask was removed from the heating mantle, and placed in a tub for use as a cooling bath. Ice/water was added to the tub to cool the mixture to -5 C. The mixture was held below that temperature for 2 hours. The product was isolated by filtration, and the filter cake washed with 1.5 L of cold methanol/water (2:1). The funnel was covered, and the product was partially dried under vacuum for 1.75 hr. The product was removed from the funnel and placed in 6 glass trays. The trays were placed in a vacuum oven, and the product was dried for 64.75 hr at 60 C. using a Nash pump as the vacuum source, and using an argon bleed. The trays were removed for weighing, and then returned to the oven and dried for an additional 4 hours at 60 C. to give a constant weight. The vacuum source for the second drying period was an oil pump, and no argon bleed was used. The material was packaged in double 4-mill polyethylene bags, and placed in a plastic outer container. The final weight after sampling was 2,540.9 g (92.5%).
Octanedioic Acid Hydroxyamide Phenylamide (48) The title compound 48 was obtained as a brown gum (9 mg) by a series of steps analogous to the preparation of 47. TLC Rf 0.20 (5% MeOH/CH2Cl2); 1H-NMR (400 MHz, CD3OD) delta 7.51 (t, 2H), 7.41 (t, 1H), 7.30 (d, 2H), 3.29 (s, 3H), 2.11 (m, 4H), 1.58 (m, 4H), 1.22 (m, 4H).
With water; In ethanol; for 2h;Agitation;Purification / work up;
In other experiments, crude SAHA was crystallized using the following conditions:All these reaction conditions produced SAHA Polymorph I.
With water; In ethanol; for 72h;Agitation;Purification / work up;
In other experiments, crude SAHA was crystallized using the following conditions:All these reaction conditions produced SAHA Polymorph I.
With water; In ethanol; for 72h;Purification / work up;
In other experiments, crude SAHA was crystallized using the following conditions:All these reaction conditions produced SAHA Polymorph I.
With water; In methanol; for 72h;Agitation;Purification / work up;
In other experiments, crude SAHA was crystallized using the following conditions:All these reaction conditions produced SAHA Polymorph I.
With water; In methanol; for 72h;Purification / work up;
In other experiments, crude SAHA was crystallized using the following conditions:All these reaction conditions produced SAHA Polymorph I.
In ethanol; for 72h;Agitation;Purification / work up;
In other experiments, crude SAHA was crystallized using the following conditions:All these reaction conditions produced SAHA Polymorph I.
In propan-1-ol; for 72h;Purification / work up;
In other experiments, crude SAHA was crystallized using the following conditions:All these reaction conditions produced SAHA Polymorph I.
In methanol; for 2h;Purification / work up;
In other experiments, crude SAHA was crystallized using the following conditions:All these reaction conditions produced SAHA Polymorph I.
In methanol; for 72h;Agitation;Purification / work up;
In other experiments, crude SAHA was crystallized using the following conditions:All these reaction conditions produced SAHA Polymorph I.
[2] the intraocular pressure-lowering agent according to [1], wherein the compound having an HDAC inhibitory effect is a compound selected from the following compounds or a salt thereof ·(2E,4E,6R)-7-(4-Dimethylaminophenyl)-N-hydroxy-4,6-dimethyl-7-oxo-2,4-heptadienamide (Trichostatin A), · N-Hydroxy-N'-phenyloctanediamide (SAHA), · 4-Dimethylamino-N-(6-hydroxycarbamoylhexyl)benzamide (M 344), · (E)-N-Hydroxy-5-[3-(phenylsulfonylamino)phenyl]-2-penten-4-ynamide (Oxamflatin), · N-Hydroxy-3-[3-hydroxyamino-3-oxo-1-propenyl]benzamide (CBHA), · (E)-N-Hydroxy-3-[1-methyl-4-(4-methylbenzoyl)-1H-pyrrol-2-yl]-2-propenamide (MC1293), · (1R)-[(2S,3S)-3-[(E)-2-[(2S,3S)-3-[(1R)-Hydroxyethyl]oxiran-2-yl] vinyl]oxiran-2-yl]-2-propen-1-ol (Depudecin), · N-(2-Aminophenyl)-4-(pyridin-3-ylmethyloxycarbonylaminomethyl)benzamide (MS 275), · cyclo[L-(2-Amino-8-oxodecanoyl)-(1'-methoxy-L-tryptophyl)-L-isoleucyl-D-pipecolinyl] (Apicidin); and ...
The intraocular pressure-lowering agent according to claim 1, wherein the compound having a histone deacetylase inhibitory effect is a compound selected from the following compounds or a salt thereof: ·(2E,4E,6R)-7-(4-Dimethylaminophenyl)-N-hydroxy-4,6-dimethyl-7-oxo-2,4-heptadienamide (Trichostatin A), ·N-Hydroxy-N'-phenyloctanediamide (SAHA), ·4-Dimethylamino-N-(6-hydroxycarbamoylhexyl)benzamide (M 344), .(E)-N-Hydroxy-5-[3-(phenylsulfonylamino)phenyl]-2-penten-4-ynamide (Oxamflatin), ·N-Hydroxy-3-[3-hydroxyamino-3-oxo-1-propenyl]benzamide (CBHA), ·(E)-N-Hydroxy-3-[1-methyl-4-(4-methylbenzoyl)-1H-pyrrol-2-yl]-2-propenamide (MC1293), ·(1R)-[(2S,3S)-3-[(E)-2-[(2S,3S)-3-[(1R)-Hydroxyethyl]oxiran-2-yl] vinyl]oxiran-2-yl]-2-propen-1-ol (Depudecin), ·N-(2-Aminophenyl)-4-(pyridin-3-ylmethyloxycarbonylaminomethyl) benzamide (MS 275), cyclo[L-(2-Amino-8-oxodecanoyl)-(1'-methoxy-L-tryptophyl)-L-isoleucyl-D-pipecolinyl] (Apicidin); and ...
Yield
Reaction Conditions
Operation in experiment
60%
With ammonia; In water; acetonitrile; at 20 - 60℃;Purification / work up;
Aqueous ammonia (2.5 vol) was added to the crude <strong>[149647-78-9]vorinostat</strong> (leq) in acetonitrile (15 vol) at 25-30C. The mixture was then maintained at 55-60C for 1 hour before being cooled to 20-25C and being stirred for a further hour. The resulting solid was filtered, washed with acetonitrile (2x0.5 vol) and dried under vacuum at 45-5O0C for 5 hours. Molar Yield = 55-60% Purity by HPLC > 99.8%
With hydroxylamine; sodium methylate; In methanol; ethanol; water; at 70 - 80℃;Purification / work up;
The <strong>[149647-78-9]vorinostat</strong> crude (264.32 g, 1.0 mole) wet cake is combined with denatured ethanol (1308-1599 g) and water (167-204 g). Hydroxylamine hydrochloride (>9 mEquiv) and sodium methoxide in methanol (>9 mEquiv) are added to the slurry, and the batch is heated to 70-80 C. The solution is filtered and then crystallized by slowly cooling to 0-10 C. The batch is filtered and the cake washed with cold 4:1 (v/v) denatured ethanol/water. The wet cake is dried at a maximum of 45 C. under vacuum.
In ethanol; water; at 60 - 66℃; under 775.743 Torr; for 0.5h;Purification / work up;
The seed slurry is prepared by combining <strong>[149647-78-9]vorinostat</strong>-fine dry cake (97.8-116.3 g, 0.37-0.44 mol) and 50:50 (v/v) ethanol/water solution (1.0-1.2 L). Under a minimum of 15 psig pressure, the seed slurry is heated to 62-66 C., aged for about 0.5 hours and then cooled to 60-64 C.
In ethanol; water; at -5 - 70℃; under 775.743 Torr; for 3h;Purification / work up;
<strong>[149647-78-9]Vorinostat</strong> (Compound 5, 264.3 g, 1.0 mole) is slurried in a 50:50 (v/v) ethanol/water solution (4.9-5.5 L). Under a minimum of 15 psig pressure, the slurry is heated to 65-70 C. to dissolve and then cooled to 60-64 C. A seed slurry is transferred into the batch while maintaining the batch temperature. The batch is aged for a minimum of 2 hours at 61-63 C. The batch is cooled in three steps by controlling the jacket temperature: (1) to 55 C. at 0.35-0.78 C./hour, (2) to 45 C. at 0.83-2.00 C./hour, and (3) to -5 to 25 C. at 2.00-4.44 C./hour. The final slurry is aged at -5 to 25 C. for about 1 hour and then filtered. The wet cake is washed with water (minimum 0.8 L). The wet cake is dried at a maximum of 55 C. under vacuum to yield <strong>[149647-78-9]vorinostat</strong>-fine drug substance.
In ethanol; water; at 7 - 30℃;Purification / work up;
<strong>[149647-78-9]Vorinostat</strong> (Compound 5, 264.3 g, 1.0 mole) is slurried in a 50:50 (v/v) ethanol/water solution (minimum 2.8 L). The <strong>[149647-78-9]vorinostat</strong> slurry is wet-milled to a mean size of 25-45 mum while maintaining the batch temperature at 7-30 C. The final slurry is filtered and the wet cake is washed with 0-40 C. water (minimum 0.8 L). The wet cake is dried at a maximum of 55 C. under vacuum to a maximum water content of 0.2% (w/w) to yield <strong>[149647-78-9]vorinostat</strong>-fine drug substance.
In methanol; at 60℃; for 1h;Product distribution / selectivity;
Example 2: Preparation of Crystalline <strong>[149647-78-9]Vorinostat</strong> Form I<strong>[149647-78-9]Vorinostat</strong> (1Og) was charged to a reaction flask containing alcohol (50ml) (organic solvent). The resulting suspension was heated at 60C for one hour under stirring. The resulting clear solution was poured into water (50ml) at 20-25C. White solid precipitated out. The solid product was filtered and dried at 60C under vacuum until a constant weight was obtained.
at 135℃; for 1h;Purification / work up;
Example 3: Preparation of <strong>[149647-78-9]Vorinostat</strong> form VI<strong>[149647-78-9]Vorinostat</strong> form III was heated at 135 +/- 2 C for 1 hour in a drying oven.
With ammonia; In water; acetonitrile; at 60℃; for 1h;Product distribution / selectivity;
Example 3: Preparation of Crystalline <strong>[149647-78-9]Vorinostat</strong> Form VI <strong>[149647-78-9]Vorinostat</strong> (1Og) was charged to a reaction flask containing acetonitrile (300ml) and aqueous ammonia (~25% w/w, 90ml) and the suspension was heated at 600C for one hour under stirring. The resultant clear solution was cooled to 250C and filtered to obtain a solid product, which was dried at 600C under vacuum until a constant weight was obtained. Yield = 8.3g (83%) Chemical purity > 99.9% (as measured by HPLC)
With ammonia; In water; acetonitrile; at 25 - 60℃;Purification / work up;
Aqueous ammonia (2.5 vol) was added to the crude <strong>[149647-78-9]vorinostat</strong> (1 eq) in acetonitrile (15 vol) at 25-30 C. The mixture was then maintained at 55-60 C. for 1 hour before being cooled to 20-25 C. and being stirred for a further hour. The resulting solid was filtered, washed with acetonitrile (2×0.5 vol) and dried under vacuum at 45-50 C. for 5 hours.Molar Yield=55-60%Purity by HPLC99.8%
In N,N-dimethyl-formamide; at 60℃; for 1h;Purification / work up;
<strong>[149647-78-9]Vorinostat</strong> (10 g) was charged to a reaction flask containing amide (50 ml) (organic solvent). The resulting suspension was heated at 60 C. for one hour under stirring. The resulting clear solution was poured into water (250 ml) at 20-25 C. White solid precipitated out. The solid product was filtered and dried at 60 C. under vacuum until a constant weight was obtained.
Yield
Reaction Conditions
Operation in experiment
In water; at 50℃;Reactivity;
Preparation of the inclusion complex SAHA-Methyl-beta-CD in a ratio of 1-2To 5 mg of SAHA (0.0189 mmol) are added 2 ml of an aqueous solution of cyclodextrin prepared by dissolving 0.045 g. of methyl-beta-cyclo- dextrin (0.0378 mmol) in double -distilled water. The solution is left to stir at approximately 5O0C for a few days until a perfectly clear solution is obtained; after filtration of the resulting solution, the water is removed by lyophilization. 40 mg of white lyophilized product are obtained.1H-NMR analysis in D2O confirms that the complexation has taken place.
Yield
Reaction Conditions
Operation in experiment
In water; at 20 - 50℃;Reactivity;
Solubility tests on SAHA by means of UV spectrophotomety .Five aqueous solutions of SAHA in cyclodextrin were prepared, maintaining a constant amount of drug and gradually increasing the cyclodextrin concentraion.Two mg of SAHA (0.0075 mmol) are weighed in five assay tubes and to each is added 1 ml of a solution of 2-hydroxypropyl-beta-cyclodextrin (HPbetaCD) in double-distilled water; we started with a molar ratio of 1:1 corresponding to 0.0116 g of cyclodextrin, proceeding then with molar ratios of 1:2 corresponding to 0.0232 g, 1:3 corresponding to 0.035 g, 1:4 corresponding to 0.046, up to a final ratio of 1:5, corresponding to 0.058 g of cyclodextrin.The tubes thus prepared and well sealed are left for one week under stirring in a mechanical stirrer at room temperature.At the end of this period, the solutions are filtered with a 0.5 mum diameter membrane filter in five 10 ml round-bottomed flasks brought to volume with double-distilled water, and the spectrophotometric readings are taken at 240 nm. <n="27"/>Table 2Absorbance values measured for solutions of SAHA and HPbetaCD in increasing molar ratiosMolar ratio Assorbance (AU) Concentration (mM)1:1 1.6862 0.1261:2 2,5591 0.1911:3 2.6196 0.1961:4 2.7752 0.2071:5 2.8932 0.216The data reported in Table 2 show an increase in absorbance values as a function of the increase in cyclodextrin concentration: the greatest increase was obtained on going over from the 1:1 to the 1:2 molar ratio.From the mean value of epsilon calculated previously, and using Lambert and Beer's law, the millimolar concentrations of the five solutions can be calculated, as shown in the table.An increase in solubility was therefore observed with the increase in cyclodextrin concentration, especially when going over from the 1:1 to the 1:2 solution, as reflected by an increase in absorbance.With the concentration values thus calculated, a solubility curve for SAHA could be plotted as a function of the cyclodextrin concentration. Lastly, the solutions were lyophilized to eliminate the water and submitted to ESI-FT-ICR analysis, confirming that formation of the complexes had taken place.On the basis of the solubility data obtained we decided to prepare the SAHA-HPbetaCD complexes in a molar ratio of 1:5 in that, in these conditions, maximum solubility was observed and the increases in solubility as a result of the addition of higher concentrations of HPbetaCD proved to be minimal. <n="28"/>Preparation of the inclusion complex SAHA-HPbetaCD in a molar ratio of 1:5.To 5 mg of SAHA (0.0189 mmol) are added 2 ml of an aqueous solution of cyclodextrin, dissolving 0.145 g of 2-hydroxypropyl-beta-cyclodextrin (0.094 mmol) in double-distilled water.The solution is left to stir at approximately 500C for a few days until a perfectly clear solution is obtained; at this point the water is removed by lyophilization.140 mg of white lyophilized product are obtained.This product was submitted to 1H-NMR analysis in D2O as is and following the addition of cyclodextrin up to a molar ratio of 1:5 corresponding to 0.116 g of cyclodextrin (0.075 mmol). The respective signals are indicated in Table 3.SAHATable 31H chemical shifts in DsO of aromatic protons of SAHA alone and of inclusion complexes with SAHA-HPbetaCD in a molar ratio of 1:5 prepared a) with the classic method, b) with microwavesHb.Hb1 Hc.Hc' HaSAHA 7.45 7.45 7.30SAHA-HPbetaCD 1:5 7.48 7.31 7.14 Classic method SAHA-HPbetaCD 1:5 7.57 7.40 7.22 Microwaves
21
[ 149647-78-9 ]
[ 127588-56-1 ]
[ 1155413-90-3 ]
Yield
Reaction Conditions
Operation in experiment
With benzotriazol-1-ol; N-(3-dimethylaminopropyl)-N-ethylcarbodiimide; In N,N-dimethyl-formamide; at 20℃; for 24h;
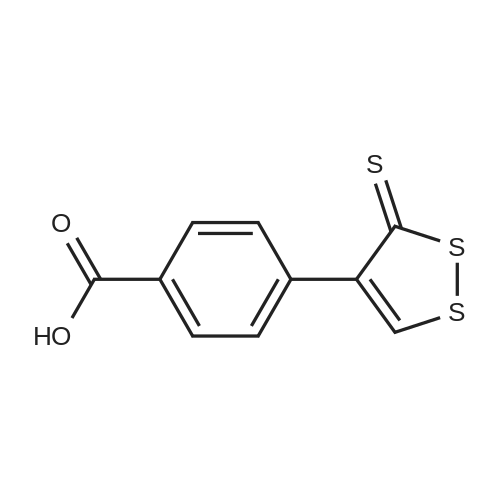
Step 2: Synthesis of N1-phenyl -N8- (4- (3-thioxo-3H-l, 2- di thiol -4-yl )benzoyloxy) octanediamide; A solution of N-hydroxy-N-phenyl octanediamide (SAHA) (100 mg; 0.38 mmol) in 1 ml of dimethylformamide (DMF) is added to a solution of the compound prepared in step 1 (96 mg; 0.38 mmol), N-hydroxybenzotriazole (HOBt) (41.5 mg) 1- [3- (dimethylamino) propyl] -3- ethylcarbodiimide (EDAC) (0.38 mmol) in 2 ml of anhydrous DMF . <n="25"/>The reaction is stirred under nitrogen at room temperature for 24 hours. At the end of the reaction DMF is evaporated and the crude product filtered is washed with water, CH2Cl2 and THF at room temperature. The obtained product has a melting point of 139.5-140 0C.
With benzotriazol-1-ol; N-(3-dimethylaminopropyl)-N-ethylcarbodiimide; In N,N-dimethyl-formamide; at 20℃; for 24h;
EXAMPLE 13. Synthesis of m-phenyl-N8-(2-thioxo-1,3- dithiole-4-carbonyloxy)octanediamide; A solution of N-hydroxy-N-phenyl octanediamide (SAHA, 100 mg; 0.38 mmol) in anhydrous DMF (1 ml) is added to a solution of EDAC (86.8 mg, 0.45 mmol), HOBT <n="34"/>(69.5; 0.45 mmol) and 2-thioxo-l , 3-dithiole-4- carboxylic acid (60 mg; 0.38 mmol) in anhydrous DMF (2 ml) and the mixture is stirred under nitrogen, at room temperature, for 24 hours. At the end of the reaction, DMF is evaporated and the residue dissolved in ethyl acetate. The organic phase is washed with water, dried on anhydrous sodium sulphate and evaporated to dryness. The product is washed with ethyl acetate and ethyl ether and after crystallization with THF has melting point 138-141 0C (decomposition) .
With hydroxylamine; In tetrahydrofuran; methanol; at 0 - 5℃; for 0.25h;
Vorinostat <n="14"/>Suberic acid (l.Oeq) was dissolved in tetrahydrofuran (15vol) and the clear solution was chilled to 0-5C. Methyl chloro formate (l.leq) and triethylamine (1.1 eq) were added to the solution at the same temperature and the mixture was stirred for 15 minutes. The triethylamine.HCl salt formed was filtered off, then aniline (leq) was added to the reaction mixture at 0-50C and stirring was continued for 15 minutes. Methyl chloroformate (l.leq) and triethylamine (l.leq) were added to the clear solution and stirring was continued for a further 15 minutes at 0-5C. This chilled reaction mixture was added to a freshly prepared hydroxylamine solution in methanol (*see below) chilled to 0-5C and stirred for 15 minutes at 0-5C. The solvent was removed under vacuum at 40C and the residue obtained was taken in methylene dichloride and the organic solution was washed with water and dried over anhydrous sodium sulfate. Methylene dichloride was removed under vacuum at 40C and acetonitrile was added to the residue. This mixture was stirred for 15 minutes before the solid was filtered under vacuum and dried under vacuum at 60C to afford the product as a white solid. Molar yield = 35-41%; HPLC purity = 99.90%.1H-NMR (DMSO-d6): 1.27 (m, 4H, 2 x -CH2-), 1.53 (m, 4H, 2 x -CH2-), 1.94 (t, J = 7.3 Hz, 2H, -CH2-), 2.29 (t, J = 7.4 Hz, 2H, -CH2-), 7.03 (t, J = 7.35 Hz, IH, aromatic para position), 7.27 (t, J = 7.90 Hz, 2H, aromatic meta position), 7.58 (t, J = 7.65 Hz, 2H, aromatic ortho position), 8.66 (s, IH, -OH, D2O exchangeable), 9.85 (s, IH, amide -NH-, D2O exchangeable), 10.33 (s, IH, -NH-OH, D2O exchangeable).13C-NMR (DMSO-d6): 25.04 (2C, 2 x -CH2-), 28.43 (2C, 2 x -CH2-), 32.24 (1C, -CH2-), 36.34 (1C, -CH2-), 119.01 (2C, Ar-C), 122.96 (1C, Ar-C), 128.68 (2C, Ar-C), 139.24 (1C, Ar- C, =CNH-), 169.23 (1C, -CO-), 171.50 (1C, -CO-).
With hydroxylamine; dicyclohexyl-carbodiimide; 1,1'-carbonyldiimidazole; In tetrahydrofuran; at 25 - 30℃;
A mixture of CDI (0.5eq) and DCC (0.8eq) in THF (15 vol) was stirred for 1 hour at 25- 30C. Suberic acid (leq) and hydroxylamine (leq) in THF (1 vol) was added and the mixture stirred for a further 1 hour. Then CDI (0.5eq), DCC (0.8eq) and aniline (leq) were added to the mixture and the mixture was stirred for a further 16-20 hours. The solid byproduct was removed by filtration and the filtrate was concentrated in vacuo at 50C to obtain crude vorinostat. Molar Yield = 55-60% Purity by HPLC > 95.8%
With dicyclohexyl-carbodiimide; potassium hydroxide; In ethanol; at 50℃; for 0.5h;
3.8 g (20 mmol) of N-hydroxy-7-carboxy-heptanoamide was dissolved in 20 ml of absolute ethanol and then 1,3-dicyclohexyl(100 mmol) of potassium hydroxide, heated to 50 C and stirred for 30 min under heat. After completion of the reaction, the reaction solution was concentrated and poured into water, Filtered, washed with water, and dried in vacuum to give 4 · 4g of fuloxitopine, with a yield of 82.4% and a purity of 99.52%.
With dicyclohexyl-carbodiimide; 1,1'-carbonyldiimidazole; In tetrahydrofuran;
A mixture of CDI (0.5 eq) and DCC (0.8 eq) in THF (15 vol) was stirred for 1 hour at 25-30 C. Suberic acid (1 eq) and hydroxylamine (1 eq) in THF (1 vol) was added and the mixture stirred for a further 1 hour. Then CDI (0.5 eq), DCC (0.8 eq) and aniline (1 eq) were added to the mixture and the mixture was stirred for a further 16-20 hours. The solid by-product was removed by filtration and the filtrate was concentrated in vacuo at 50 C. to obtain crude vorinostat.Molar Yield=55-60%Purity by HPLC95.8%
With triethylamine; In diethyl ether; dichloromethane; acetonitrile; at 20℃; for 12h;Inert atmosphere;
In this example, R3 is an epigenetic modulator: SAHA The following are added to 5 mL of dry DCM under a nitrogen atmosphere: 7c (0.20 g; 0.43 mmol; 1 eq.) and a 2M HCl solution in ether (0.43 mL; 0.86 mmol, 2 eq.). The solution is refluxed for 2 hours and then the solvent is co-evaporated with toluene in order to give the intermediate chloride in the form of an oil. The preceding chloride dissolved in a minimum of ACN is added at ambient temperature to a solution of SAHA (0.340 g; 0.129 mmol, 3 eq.) in dry NEt3 (0.24 mL, 1.72 mmol, 4 eq.) in 5 mL of ACN. The solution is stirred for 12 hours at ambient temperature. The solution is then filtered and washed with ACN to recover the remaining SAHA and the filtrate is concentrated under vacuum. Purification of the concentrate (automatic flash chromatography, silica gel, eluent DCM/EtOH/Et3N) gives the prodrug 8c in the form of a colourless oil (0.052 g; 0.073 mmol; 17%). The alcohol 7c that remains is also recovered. Rf (DCM/EtOH/Et3N 94:5:1)=0.33 1H-NMR (acetone-d6, 400 MHz): delta=9.75 (s, 1H), 9.08 (s, 1H), 7.69-7.65 (m, 3H), 7.46-7.41 (m, 4H), 7.34-7.25 (m, 8H), 7.04-7.00 (m, 1H), 4.57 (t, 2H, J=5.1 Hz), 4.16 (d, 2H, J=2.4 Hz), 3.88 (t, 2H, J=5.2 Hz), 3.62-3.55 (m, 6H), 3.53-3.49 (m, 6H), 2.93 (t, 2H, J=2.4 Hz), 2.31 (t, 2H, J=7.5 Hz), 1.93 (t, 2H, J=7.2 Hz), 1.60 (m, 2H), 1.39 (m, 2H), 1.25 (m, 2H), 1.12 (m, 2H).
0.075 g
With triethylamine; In acetonitrile; at 20℃;Inert atmosphere;
General procedure: N-phenyl-N0-[[1-[2-[2-[2-(2-prop-2-ynoxyethoxy) ethoxy] ethoxy]ethyl] triazol-4-yl]-bis(phenyl) methoxy] octanediamide 4a.Method A: To a solution of 3a (0.20 g, 0.43 mmol) in DCM (5 mL)was added HCl (2 M in Et2O, 0.3 mL, 0.86 mmol). After 2 h reflux,the solution was co-evaporated with toluene to give the crudechloride 10a as oil. To a solution of SAHA (0.340 g, 0.129 mmol)and dry NEt3 (0.24 mL, 1.72 mmol) in ACN (5 mL) was added thecrude chloride 10a dissolved in a minimum of ACN at ambienttemperature. The resulting solution was stirred overnight at roomtemperature, then filtered, and washed with ACN to recover unreactedSAHA as a solid. The concentrated filtrate was purified byCombi Flash (DCM/EtOH/Et3N 94:5:1) to give 4a as a colorless oil (0.052 g, 0.073 mmol, 17%). The unreacted alcohol 3a is also recovered.Method B: To a solution of 3a (0.11 g, 0.25 mmol, 1 eq.) in drytoluene under nitrogen atmosphere was added AcCl (0.09 mL,1.25 mmol, 5 eq.). After 2 h reflux, the solution was evaporated togive the crude chloride 10a as oil. The addition of SAHA, workup,and purification was performed as for method A. Compound4a was obtained in better yields with method B (0.075 g,0.107 mmol, 42%). The unreacted alcohol 3a (0.010 g, 10%) andSAHA (0.124 g, 36%)) were also recovered.TLC MeOH/CH2Cl2/Et3N 5:94:1 Rf = 0.33; 1H NMR (400 MHz,Acetone-D6): d = 9.75 (s, 1H), 9.08 (s, 1H), 7.69-7.65 (m, 3H),7.46-7.41 (m, 4H), 7.34-7.25 (m, 8H), 7.04-7.00 (m, 1H), 4.57 (t,2H, J = 5.1 Hz), 4.16 (d, 2H, J = 2.4 Hz), 3.88 (t, 2H, J = 5.2 Hz),3.62-3.55 (m, 6H), 3.53-3.49 (m, 6H), 2.93 (t, 2H, J = 2.4 Hz), 2.31(t, 2H, J = 7.5 Hz), 1.93 (t, 2H, J = 7.2 Hz), 1.60 (m, 2H), 1.39 (m,2H), 1.25 (m, 2H), 1.12 (m, 2H); 13C NMR (100 MHz, Acetone-D6): d = 26.0, 33.7, 37.7., 50.8, 58.6, 69.8, 70.1, 70.9, 71.1, 71.16,71.2, 71.22, 75.8, 80.9, 115.7, 119.9, 123.8, 126.7, 128.5, 128.6,129.2, 129.4, 133.6, 140.8, 161,0, 172,0; HRESI-MS: calcd. for[M+Na]+ (C40H49N5O7Na): 734.35242, found: 734.3524.
With triethylamine; In acetonitrile; at 20℃;Inert atmosphere;
General procedure: N-phenyl-N0-[[1-[2-[2-[2-(2-prop-2-ynoxyethoxy) ethoxy] ethoxy]ethyl] triazol-4-yl]-bis(phenyl) methoxy] octanediamide 4a.Method A: To a solution of 3a (0.20 g, 0.43 mmol) in DCM (5 mL)was added HCl (2 M in Et2O, 0.3 mL, 0.86 mmol). After 2 h reflux,the solution was co-evaporated with toluene to give the crudechloride 10a as oil. To a solution of SAHA (0.340 g, 0.129 mmol)and dry NEt3 (0.24 mL, 1.72 mmol) in ACN (5 mL) was added thecrude chloride 10a dissolved in a minimum of ACN at ambienttemperature. The resulting solution was stirred overnight at roomtemperature, then filtered, and washed with ACN to recover unreactedSAHA as a solid. The concentrated filtrate was purified byCombi Flash (DCM/EtOH/Et3N 94:5:1) to give 4a as a colorless oil (0.052 g, 0.073 mmol, 17%). The unreacted alcohol 3a is also recovered.Method B: To a solution of 3a (0.11 g, 0.25 mmol, 1 eq.) in drytoluene under nitrogen atmosphere was added AcCl (0.09 mL,1.25 mmol, 5 eq.). After 2 h reflux, the solution was evaporated togive the crude chloride 10a as oil. The addition of SAHA, workup,and purification was performed as for method A. Compound4a was obtained in better yields with method B (0.075 g,0.107 mmol, 42%). The unreacted alcohol 3a (0.010 g, 10%) andSAHA (0.124 g, 36%)) were also recovered.TLC MeOH/CH2Cl2/Et3N 5:94:1 Rf = 0.33; 1H NMR (400 MHz,Acetone-D6): d = 9.75 (s, 1H), 9.08 (s, 1H), 7.69-7.65 (m, 3H),7.46-7.41 (m, 4H), 7.34-7.25 (m, 8H), 7.04-7.00 (m, 1H), 4.57 (t,2H, J = 5.1 Hz), 4.16 (d, 2H, J = 2.4 Hz), 3.88 (t, 2H, J = 5.2 Hz),3.62-3.55 (m, 6H), 3.53-3.49 (m, 6H), 2.93 (t, 2H, J = 2.4 Hz), 2.31(t, 2H, J = 7.5 Hz), 1.93 (t, 2H, J = 7.2 Hz), 1.60 (m, 2H), 1.39 (m,2H), 1.25 (m, 2H), 1.12 (m, 2H); 13C NMR (100 MHz, Acetone-D6): d = 26.0, 33.7, 37.7., 50.8, 58.6, 69.8, 70.1, 70.9, 71.1, 71.16,71.2, 71.22, 75.8, 80.9, 115.7, 119.9, 123.8, 126.7, 128.5, 128.6,129.2, 129.4, 133.6, 140.8, 161,0, 172,0; HRESI-MS: calcd. for[M+Na]+ (C40H49N5O7Na): 734.35242, found: 734.3524.
With triethylamine; In acetonitrile; at 20℃; for 12h;
The following are added to 5 mE of dry DCM under a nitrogen atmosphere: 7d 0.20 g (0.38 mmol; 1 eq.) and a 2M RC1 solution in ether (0.38 mE; 0.76 mmol; 2 eq.). The solution turns red and is refluxed for 2 hours. The solution is then co-evaporated with toluene to give the intermediate chloride in the form of a red oil. The preceding chloride dissolved in a minimum of ACN is added at ambient temperature to a solution of SARA (0.302 g; 1.14 mmol; 3 eq.) and dry NEt3 (0.21 mE; 1.52 mmol; 4 eq.) in 5 mE of dry ACN. The red colour disappears after a few minutes and the solution is stirred for 12 hours. The solution is filtered and washed with CAN to recover the remaining SARA and the filtrate is concentrated under vacuum. The concentrate is purified (automatic flash chromatography, silica gel, eluent DCMJEtOR/ Et3N) in order to give the prodrug 8d in the form of a viscous oil (0.054 g; 0.070 mmol; 18%). The alcohol 7d that remains is also recovered. 10486] Rf (DCM/EtOR/Et3N 94:5:1 )=0.2910487] ?R-NMR (acetone-d5, 400 MRz): oe=9.75 (s, 1R),9.08 (s, 1R), 7.68-7.67 (m, 3R), 7.32-7.25 (m, 6R), 7.04-7.00 (m, 1R), 6.87-7.85 (m, 4R), 4.57 (t, 2R, J=5.1 Rz), 4.16 (d, 2R, J=2.410488] Rz), 3.88 (t, 2R, J=5.2 Rz), 3.79 (s, 6R), 3.62-3.56 (m, 6R), 3.54-3.50 (m, 6R), 2.93 (t, 1R, J=2.4 Rz), 2.31 (t, 2R, J=7.5 Rz), 1.93 (t, 2R, J=7.2 Rz), 1.60 (m, 2R), 1.39 (m, 2R), 1.25 (m, 2R), 1.12 (m, 2R).
With triethylamine; In acetonitrile; at 20℃;Inert atmosphere;
General procedure: N-phenyl-N0-[[1-[2-[2-[2-(2-prop-2-ynoxyethoxy) ethoxy] ethoxy]ethyl] triazol-4-yl]-bis(phenyl) methoxy] octanediamide 4a.Method A: To a solution of 3a (0.20 g, 0.43 mmol) in DCM (5 mL)was added HCl (2 M in Et2O, 0.3 mL, 0.86 mmol). After 2 h reflux,the solution was co-evaporated with toluene to give the crudechloride 10a as oil. To a solution of SAHA (0.340 g, 0.129 mmol)and dry NEt3 (0.24 mL, 1.72 mmol) in ACN (5 mL) was added thecrude chloride 10a dissolved in a minimum of ACN at ambienttemperature. The resulting solution was stirred overnight at roomtemperature, then filtered, and washed with ACN to recover unreactedSAHA as a solid. The concentrated filtrate was purified byCombi Flash (DCM/EtOH/Et3N 94:5:1) to give 4a as a colorless oil (0.052 g, 0.073 mmol, 17%). The unreacted alcohol 3a is also recovered.Method B: To a solution of 3a (0.11 g, 0.25 mmol, 1 eq.) in drytoluene under nitrogen atmosphere was added AcCl (0.09 mL,1.25 mmol, 5 eq.). After 2 h reflux, the solution was evaporated togive the crude chloride 10a as oil. The addition of SAHA, workup,and purification was performed as for method A. Compound4a was obtained in better yields with method B (0.075 g,0.107 mmol, 42%). The unreacted alcohol 3a (0.010 g, 10%) andSAHA (0.124 g, 36%)) were also recovered.TLC MeOH/CH2Cl2/Et3N 5:94:1 Rf = 0.33; 1H NMR (400 MHz,Acetone-D6): d = 9.75 (s, 1H), 9.08 (s, 1H), 7.69-7.65 (m, 3H),7.46-7.41 (m, 4H), 7.34-7.25 (m, 8H), 7.04-7.00 (m, 1H), 4.57 (t,2H, J = 5.1 Hz), 4.16 (d, 2H, J = 2.4 Hz), 3.88 (t, 2H, J = 5.2 Hz),3.62-3.55 (m, 6H), 3.53-3.49 (m, 6H), 2.93 (t, 2H, J = 2.4 Hz), 2.31(t, 2H, J = 7.5 Hz), 1.93 (t, 2H, J = 7.2 Hz), 1.60 (m, 2H), 1.39 (m,2H), 1.25 (m, 2H), 1.12 (m, 2H); 13C NMR (100 MHz, Acetone-D6): d = 26.0, 33.7, 37.7., 50.8, 58.6, 69.8, 70.1, 70.9, 71.1, 71.16,71.2, 71.22, 75.8, 80.9, 115.7, 119.9, 123.8, 126.7, 128.5, 128.6,129.2, 129.4, 133.6, 140.8, 161,0, 172,0; HRESI-MS: calcd. for[M+Na]+ (C40H49N5O7Na): 734.35242, found: 734.3524.
With triethylamine; In acetonitrile; at 20℃; for 12h;
Then, evaporate the toluene in the mixture under vacuum.10493] The intermediate chloride obtained is diluted in a minimum of distilled ACN and is added to a mixture of 44.00 mg of SARA (0.17 mmol; 2 eq.) in 2 ml of distilled ACN. Then add 47.00 jii of distilled Et3N (0.34 mmol; 4 eq.) and stirthe mixture for 12 h at ambient temperature. The unreacted SARA is filtered with ACN. The mixture is evaporated under vacuum and then purified by combi flash using dry solvents (DCM/MeOR: from 0% to 5% in MeOR).10495] 24.00mg of product (8g) is obtained in the form ofa yellow solid.10496] The starting alcohol (7g) is also recovered.j0497] Yield: 38%10498] Rf(DCM/MeOR:95/5): 0.4110499] MP: 132.1 C.j0500] ?R NMR (ACETONE D6, 400 MRz) oe (ppm): 1.25(m, 6R); 1.61 (t, 2R, J=8 Rz); 1.94 (t, 2R, J=8 Rz); 2.31 (t, 2R,J=8 Rz); 2.93 (s, 1R); 3.52 (m, 12R); 3.89 (t, 2R, J=8 Rz);4.15 (s, 2R); 4.57 (t, 2R, J=8 Rz); 7.02 (t, 3R, J=8 Rz); 7.11(t, 3R, J=8 Rz); 7.45 (m, 4R); 7.65 (d, 2R, J=8 Rz); 7.79 (s,1R); 9.08 (s, 1R); 9.74 (s, 1R).j0501] ?3C NMR (ACETONE D6, 75.4 MRz) oe (ppm):25.9; 33.3;37.6; 50.8; 58.5; 69.7; 69.9;70.9; 71.0; 71.1; 71.2;71.3; 75.8; 80.8; 115.1; 115.3; 119.9; 123.8; 126.6; 129.4;131.4; 131.5; 139.5; 140.6; 162.0; 171.9j0502] ?9F NMR (ACETONE D6, 400 MRz) oe (ppm):-116.1
hydrotris(3,5-methylphenylpyrazolyl)boratozinc(II) hydroxide[ No CAS ]
[ 149647-78-9 ]
C44H47BN8O3Zn[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
35.1%
In methanol; dichloromethane; at 20℃;Inert atmosphere;
(TpPh,Me)ZnOH (56.7mg, 0.10mmol) was dissolved in 15mL of CH2Cl2. To this solution, suberanilohydroxamic acid (28.8mg, 0.11mmol) dissolved in 10mL of MeOH was added. The reaction was allowed to stir at room temperature overnight under a nitrogen atmosphere. The solution was then evaporated to dryness on a rotary evaporator. The solid was dissolved in a small amount of toluene (5mL) and the material was recrystallized by vapor diffusion of pentane into the toluene solution. Clear crystals were produced (28.6mg, yield: 35.1%). 1H NMR (CDCl3, 300MHz): delta=8.18 (s, 1H, NH), 7.69 (d, 6H, JH-H=6.9Hz, ArH), 7.06-7.36 (m, 9 H, ArH), 6.18 (s, 3H, pyrazole C-H), 2.51 (s, 9H, CH3), 2.20 (t, 2H, JH-H=7.5Hz, CH2), 1.65 (s, 1H, NH). 1.52 (m, 2H, CH2), 1.02 (m, 4H, CH2), 0.79 (m, 2H, CH2), 0.54 (m, 2H, CH2). IR (solid ATR): 2543, 1683, 1603cm-1. Anal. Calc. for C44H47O3N8BZn: C, 65.07; H, 5.83; N, 13.86. Found: C, 65.41; H, 6.03; N, 13.69%.
N1,N2-dimethyl-N1-(2-phenylcyclopropyl)ethane-1,2-diamine dihydrochloride[ No CAS ]
[ 530-62-1 ]
[ 149647-78-9 ]
N1-((methyl(2-(methyl(2-phenylcyclopropyl)amino)ethyl)carbamoyl)oxy)-N8-phenyloctanediamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
82.7%
SAHA (50.0 mg) was suspended in acetonitrile (1.5 mL), the reaction solution was cooled to 10 C., carbonyl diimidazole (33.7 mg) was added, and the mixture was stirred for 20 minutes. Compound (11) (57.7 mg) and diisopropylethylamine (78 muL) were added to the reaction solution, and the mixture was stirred at room temperature for 12 hours.The reaction solution was diluted with ethyl acetate, washed with water and saturated brine, and dried over anhydrous sodium sulfate. After filtration and concentration under reduced pressure, the obtained residue was purified by silica gel flash column chromatography (developing solvent n-hexane: ethyl acetate = 3: 2) to give the title compound (PCPA-SAHA-b, Example 4) , Yield 82.7%) as a pale yellow solid.Further, the obtained white solid was recrystallized from ethyl acetate to give the title compound (PCPA-SAHA-b, Example 4) (32.7 mg) as a white solid.
4-(hydroxymethyl)phenylmethyl(2-(methyl(2-phenylcyclopropyl)amino)ethyl)carbamate[ No CAS ]
[ 149647-78-9 ]
4-(((8-oxo-8-(phenylamino)octanamido)oxy)methyl)phenylmethyl(2-(methyl(2-phenylcyclopropyl)amino)ethyl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
58.8%
Compound (53) (210 mg) obtained in Step 10-3 was dissolved in dichloromethane (6 mL), thionyl chloride (86.4 muL) was added under ice-cooling, the mixture was stirred for 5 minutes and then stirred at room temperature for 3 hours . A saturated aqueous sodium hydrogen carbonate solution was added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure.[0226]60% sodium hydride (27.0 mg) was suspended in N, N-dimethylformamide (4 mL), SAHA (142 mg) was added under ice cooling, and the mixture was stirred for 30 minutes. The residue obtained above was dissolved in N, N-dimethylformamide (2 mL), added dropwise to the reaction solution, and stirred at room temperature for 19 hours. Ice water was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was separated, washed with water and saturated brine, and dried over anhydrous sodium sulfate. After filtration and concentration under reduced pressure, the resulting residue was purified by silica gel flash column chromatography (developing solvent ethyl acetate) to give the title compound (PCPA-SAHA-c, Example 11) (190 mg, 58.8% yield) As a pale yellow solid. Further, the obtained solid was recrystallized from ethyl acetate to give the title compound (PCPA-SAHA-c, Example 11) (84.5 mg) as a white powder.
N1-{{[N-methyl-N-(trans-2-phenylcyclopropyl)amino]ethyl}carbamoyl}oxy}-N8-phenyloctanediamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With triethylamine; In N,N-dimethyl-formamide; at 20℃; for 14h;
The compound (13) (220 mg) obtained in the step 3-1 was dissolved in toluene (5 mL), and triethylamine (166 uL) and diphenylphosphoryl azide (260 muL) were added under ice cooling. The reaction solution was stirred at room temperature for 30 minutes and then heated under reflux for 1 hour. The reaction solution was returned to room temperature, and SAHA (317 mg) dissolved in triethylamine (166 muL) and N, N-dimethylformamide (2 mL) was added under ice cooling, and the reaction solution was stirred at room temperature for 14 hours.Water was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine. After drying with anhydrous sodium sulfate, filtration and concentration under reduced pressure were carried out. The obtained residue was purified by silica gel flash column chromatography (developing solvent: chloroform: methanol = 20: 1) to give the title compound (PCPA-SAHA-a, Example 3) (224 mg, yield 46.5%) as a white solid . Further purification by RP-HPLC (Gradient (I)) gave the title compound (PCPA-SAHA-a, Example 3) (22.8 mg) as a white solid.
cisplatin and 169.87mg 300mg silver nitrate dissolved in 10mL DMF and the reaction is removed after precipitation of silver chloride 24h, a solution to obtain a supernatant; and 88.1mg 13.3mg <strong>[149647-78-9]vorinostat</strong> with a sodium hydroxide dispersion in 5mL of methanol, insolubles were removed after the reaction to give a supernatant solution II; a solution and mixing two reaction solution 48h, the organic solvent is removed, dissolved in methanol to remove excess cisplatin, recrystallized amphiphilic drugs - drug complexes having the formula (a) below
[(8-anilino-8-oxo-octanoyl)amino] N-isopropylcarbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
93%
In N,N-dimethyl-formamide; acetone; at -15 - 20℃;
General procedure: A solution of 1.1 eq SAHA (synthesized as described in the literature [10,11] or bufexamac(purchased from Sigma Aldrich, St. Louis, MO, USA) in DMF (1 mL/mmol) and acetone (2 mL/mmol)was cooled to -15 C. After 1 eq of the substituted isocyanate was slowly added, the batch was stirred over night at room temperature. The solvent was removed under reduced pressure, and the desired carbamate was precipitated by the addition of water. The raw product was purified by recrystallization from ethyl acetate. [(8-Anilino-8-oxo-octanoyl)amino]N-isopropylcarbamate (2a). Remained as a white powder (0.324 g, 93%yield); 1H NMR (400 MHz, DMSO-d6): delta (ppm) = 11.24 (s, 1H, NH), 9.84 (s, 1H, NH), 7.61-7.54 (m, 3H,NH, Ar2,6-H), 7.31-7.25 (m, 2H, Ar3,5-H), 7.04-6.99 (m, 1H, Ar4-H), 3.64-3.53 (m, 1H, CH-(CH3)2),2.29 (t, 3J = 7.4, 2H, Ar-NH-CO-CH2), 2.06 (t, 3J = 7.9, 2H, NH-CO-CH2), 1.63-1.48 (m, 4H, (CH2)2),1.34-1.27 (m, 4H, (CH2)2), 1.08 (d, 3J = 6.9, 6H, CH-(CH3)2); 13C NMR (100 MHz, DMSO-d6): delta (ppm)= 171.62 (CO), 170.42 (CO), 154.51 (COO), 139.77 (Ar-C1), 129.04 (Ar-C3,5), 123.33 (Ar-C4), 119.42 (Ar-C2,6), 43.46 (CH(CH3)2), 36.80 (Ar-NH-CO-CH2), 32.36 (NH-CO-CH2), 28.80 (CH2), 28.68 (CH2),25.42 (CH2), 25.09 (CH2), 22.80 ((CH3)2); MS-ESI+: m/z 350 [M + H]+, 372 [M + Na]+.
[(8-anilino-8-oxo-octanoyl)amino] N-benzylcarbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
94%
In N,N-dimethyl-formamide; acetone; at -15 - 20℃;
General procedure: A solution of 1.1 eq SAHA (synthesized as described in the literature [10,11] or bufexamac(purchased from Sigma Aldrich, St. Louis, MO, USA) in DMF (1 mL/mmol) and acetone (2 mL/mmol)was cooled to -15 C. After 1 eq of the substituted isocyanate was slowly added, the batch was stirredover night at room temperature. The solvent was removed under reduced pressure, and the desired carbamate was precipitated by the addition of water. The raw product was purified by recrystallization from ethyl acetate.
[(8-anilino-8-oxo-octanoyl)amino] N-ethylcarbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
92%
In N,N-dimethyl-formamide; acetone; at -15 - 20℃;
General procedure: A solution of 1.1 eq SAHA (synthesized as described in the literature [10,11] or bufexamac(purchased from Sigma Aldrich, St. Louis, MO, USA) in DMF (1 mL/mmol) and acetone (2 mL/mmol)was cooled to -15 C. After 1 eq of the substituted isocyanate was slowly added, the batch was stirredover night at room temperature. The solvent was removed under reduced pressure, and the desired carbamate was precipitated by the addition of water. The raw product was purified by recrystallization from ethyl acetate.
[(8-anilino-8-oxo-octanoyl)amino] N-phenylcarbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
96%
In N,N-dimethyl-formamide; acetone; at -15 - 20℃;
General procedure: A solution of 1.1 eq SAHA (synthesized as described in the literature [10,11] or bufexamac(purchased from Sigma Aldrich, St. Louis, MO, USA) in DMF (1 mL/mmol) and acetone (2 mL/mmol)was cooled to -15 C. After 1 eq of the substituted isocyanate was slowly added, the batch was stirredover night at room temperature. The solvent was removed under reduced pressure, and the desired carbamate was precipitated by the addition of water. The raw product was purified by recrystallization from ethyl acetate.
O4-[(8-anilino-8-oxo-octanoyl)amino] O1-tert-butyl piperazine-1,4-dicarboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
34%
In N,N-dimethyl-formamide; acetone; at 20℃;
One-point-six-threemillimoles of the carbamoyl imidazolium salt (5) were dissolved in acetone and added dropwiseto a solution of 1.5 mmol SAHA (1) in DMF (1 mL/mmol). After stirring over night at roomtemperature, the solvent was removed under reduced pressure, and water was added to the remainingsolution. The raw product was extracted with dichloromethane, and the combined organic phaseswere dried over sodium sulfate. After removal of the solvent, the product was purified via flashchromatography (ethyl acetate/cyclohexane 9:1) to result in 0.240 g of a white solid (34% yield).1H NMR (400 MHz, CDCl3): delta (ppm) = 9.29 (s, 1H, NH), 7.78 (s, 1H, NH), 7.57-7.51 (m, 2H,Ar2,6-H), 7.34-7.29 (m, 2H, Ar3,5-H), 7.13-7.07 (m, 1H, Ar4-H), 3.57-3.41 (m, 8H, N-(CH2)2-(CH2)2),2.37 (t, 3J = 7.4, 2H, Ar-NH-CO-CH2), 2.25 (t, 3J = 7.4, 2H, NH-CO-CH2), 1.79-1.66 (m, 4H, (CH2)2),1.48 (s, 9H, C(CH3)3), 1.43-1.39 (m, 4H, (CH2)2). 13C NMR (100 MHz, CDCl3): delta (ppm) = 171.98(CO), 171.72 (CO), 154.42 (COO), 154.11 (CO), 138.08 (Ar-C1), 128.91 (Ar-C3,5), 124.08 (Ar-C4),119.74 (Ar-C2,6), 80.43 (C-CH3), 44.17 (N-(CH2)2), 43.10 (N-(CH2)2), 37.03 (Ar-NH-CO-CH2), 32.49(NH-CO-CH2), 28.31 (C(CH3)3), 28.12 (CH2), 27.95 (CH2), 25.04 (CH2), 24.59 (CH2). MS-ESI+: m/z477.2 [M + H]+, 499.3 [M + Na]+.
With potassium hydroxide; In methanol; water; at 25℃; for 0.5h;
General procedure: Copper(ll) perchlorate hexahydrate (Cu(CI04)2-6H20) (100 mg, 0.270 mmol) was dissolved in a minimum volume of deionised water (1 ml). Warm solutions of SAHA (71 .4 mg, 0.270 mmol) and nitrogen donor ligand of either Bipy (42.5 mg, 0.270 mmol), Phen (48.7 mg, 0.270 mmol) or DPQ (46.7 mg, 0.270 mmol) in 1 ml of methanol were added sequentially. A solution of potassium hydroxide (15.1 mg, 0.270 mmol) in 1 ml of deionised water was immediately added and the resulting suspension stirred at room temperature for 30 min. The solid product was filtered, washed with cold deionised water , methanol and dried under vacuum. [Cu(SAHA.iH)(Bipy)]CIO4 0.5 H20 (Cu-SAHA-Bipy): Green solid, Yield 107.3 mg, 67.7 %. C24H28CICUN4O8 requires C - 48.08 %; H - 4.71 %; N - 9.35 %; Cu - 10.60 %; CI - 5.91 %; found C - 48.39 %; H - 4.38 %; N - 9.30 %; Cu - 10.67 %; CI - 5.73 %. ESI-MS ([M-H]+) (MeOH) mass-to-charge ratio (m/z): 483 ([Cu(SAHA.iH)(BIPY)]+) 265.2 (SAHA) 219.8 ([Cu(BIPY)]+]). IR (ATR) cm"1: 3567.39 (s,s), 2931 .19, 1660.60 (s,s) 1595.55. (s,s) 1071.57 (s,br). [Cu(SAHA.iH)(Phen)]CIO4 0.5 H20 (Cu-SAHA-Phen): Blue solid, Yield 90 mg, 80 %. C26H28CICUN4O9 requires C - 48.83 %; H - 4.41 %; N - 8.76 %; Cu - 9.94 %; CI - 5.54 %; found C - 49.25 %; H - 4.56 %; N - 8.62 %; Cu - 10.36 %; CI - 5.44 %. IR (ATR) cm"1: 3495.44, 3065.47, 1650.33 1535.02 1070.18. ESI-MS ([M-H]+) (MeOH) m/z: 506.1 ([Cu(SAHA-iH)(Phen)]+) 265.1 (SAHA) 243.9 ([Cu(Phen)]+). [Cu(SAHA. IH)(DPQ)]CI04 (CU-SAHA-DPQ): Grey solid, Yield 96.8 mg, 53.01 %. C28H27CICUN6O7 requires C - 51.07 %; H - 4.13 %; N - 12.76 %; Cu - 9.65 %; CI - 5.38 %; found C - 50.65 %; H - 3.81 %; N - 12.57 %; Cu - 9.63 %; CI - 4.97 %. ESI-MS ([M-H]+) (MeOH) m/z: 558.1 ([CU(SAHA.IH)(DPQ)]+) 294.8 ([Cu(DPQ)]+) 265.1 (SAHA). IR (ATR) cm-1: 3350.21 ,3050.31 , 1658.57, 1595.07, 1087.07.
Cu(ClO4)2-6H20 (106.4 mg, 0.287 mmol) was dissolved in 1 ml of deionised water . To the Cu solution, warm solutions of SAHA (75.9 mg, 0.287 mmol) and DPPZ (81.04 mg, 0.287 mmol) in methanol (3ml) were added sequentially. The resulting suspension was stirred at room temperature for 30 min. The solid green product (Cu-SAHA-DPPZ) was filtered, washed with cold deionised water, methanol and dried under vacuum, Scheme 1 . Yield 127 mg, 60.5%. C32H31CICUN6O7 requires C - 54.09 %; H - 4.40 %; N - 1 1 .83 %; Cu - 8.94 %; CI - 4.99 % ; found C - 53.70 %; H - 3.83 %; N - 1 1.65 %; Cu - 9.05 %; CI 4.80 %. ESI- MS ([M-H]+) (MeOH) m/z: 609 ([Cu(SAHA.iH)(DPPZ)]+) 265.2 (SAHA). IR (KBr) cm"1: 3346.37, 3071.09, 1652.96, 1592.59, 1073.17.
With potassium hydroxide; In methanol; ethanol; water; at 25℃; for 0.5h;
Cu(ClO4)2-6H20 (108.6 mg, 0.293 mmol) was dissolved in 1 ml of deionised water. Warm solutions of SAHA (77.7 mg, 0.293 mmol) in 1 ml of methanol and 1 ,10-phenanthroline- 5,6-dione (Phendio) (61 .7 mg, 0.293 mmol) in 4 ml of ethanol were added sequentially. A solution of potassium hydroxide (16.4 mg, 0.293 mmol) in 1 ml of deionised water was added and the solution left to stir at room temperature for 30 minutes. The filtrate was left to slowly evaporate at room temperature for -5 days. A green solid by-product was filtered and the solvent from the filtrate was concentrated in vacuo. The waxy solid was suspended in deionised water and agitated. A brown solid (Cu-SAHA-Phendio) was filtered, washed with cold deionised water, methanol and dried under vacuum, Scheme 2. Yield 105 mg, 67%. [Cu(SAHA.iH)(Phendio)]CIO4 0.5 H20 (Cu-SAHA-Phendio): C26H26CICU N4O10 requires C - 47.79 %; H - 4.01 %; N - 8.57 %; Cu - 9.72 %; CI - 5.43 %; found C - 47.55 %; H - 3.99 %; N - 8.37 %; Cu - 9.51 %; CI - 5.21 %. IR (ATR) cm-1: 3377.67, 3222.67, 1693.41 , 1598.73, 1070.18.
<strong>[149647-78-9]Vorinostat</strong> 913 mg (3.46 mmol) was dissolved in 20 mL pyridine and cooled at 0C for 5 minutes. 3 g (3.14 mmol) of compound (ii) was added to the above solution at 0C to give a red suspension.The suspension was warmed to 25[deg.] C. and stirred for 5 hours while warming, after which 100 mL of water was added to quench the reaction.The reaction solution was extracted with ethyl acetate (50 mL×3 times), and the organic layers were combined, sequentially extracted with 0.5 M HCl (30 mL×4 times) and saturated brine (30 mL×2 times), and dried over anhydrous sodium sulfate.Concentration on a rotary evaporator, preparation of chromatography (dichloromethane: methanol = 50: 1) was isolated to give intermediate iii 3.4g, the yield was 92%.
orinostat 913 mg (3.46 mmol) was dissolved in 20 mL pyridine,Cool at 0C for 5 minutes.Compound (ii) 3 g (3.14 mmol) was added to the above solution at 0C.Become a red suspension.The suspension is warmed to 25C,Stir and stir for 5 hoursAfter adding 100 mL of water, the reaction was quenched.The reaction solution was extracted with ethyl acetate (50 mL×3 times), and the organic layers were combined, sequentially extracted with 0.5 M HCl (30 mL×4 times) and saturated brine (30 mL×2 times), and dried over anhydrous sodium sulfate.Concentrate on a rotary evaporator,Preparative chromatography (dichloromethane:methanol=50:1) gave 3.4 g of intermediate compound (iii) in 92% yield.
In dichloromethane; at 20℃; for 1h;Inert atmosphere;
1 g (3.78 mmol) of compound II was added to a 100 mL two-necked flask under nitrogen.Add 20ml of DCM to it,Subsequently, 0.61 g (3.78 mmol) of N,N'-carbonyldiimidazole was added.Stir at room temperature for 1 h and then add0.5 g (3.78 mmol) of glycine tert-butyl ester,Continue to react for 10 h.Diluted with dichloromethane (20 mL) and methanol (10 mL).0.2M HCl wash (15mL x 2),Dry over anhydrous sodium sulfate,Distilling the solvent under reduced pressure,The crude product was subjected to column chromatography (dichloromethane:methanol=50:1?30:1)Compound IV was obtained as a white solid, 1.05 g.The yield was 65.90%.
In dichloromethane; at 20℃; for 1h;Inert atmosphere;
Under the protection of nitrogen, Compound 2 (SAHA) (1 g,3.78 mmol) was dispersed in CH2Cl2 (20 mL), and then CDI (0.61 g,3.78 mmol) was added. After stirring for 1 h at room temperature toform active intermediate 3, BOC-glycine (0.5 g, 3.78 mmol) was addedand stirred for 10 h continually. The reaction mixture was diluted withCH2Cl2 and washed with 0.2M HCl, dried Na2SO4, filtered. The filtratewas concentrated under reduced pressure. The residue was purifiedusing silica gel column chromatography with CH2Cl2 and MeOH to give 4 (1.05 g, 66%) as a white solid.
With dibutyltin dilaurate; In toluene; at 110℃; for 8h;Reflux;
To a solution of 1 (0.1 mmol) in dry toluene (10 mL) is addedSAHA (0.09 mmol) and DBTL (0.001 mmol). The reaction is heated to reflux at 110 C for8h. Upon completion, the reaction is cooled to rt and concentrated under reduced pressure.The crude residue is purified by flash chromatography (2% MeOH:DCM) to yield 2.
ethyl 7-(((S)-1-(((S)-1-((4-(bromomethyl)phenyl)amino)-1-oxo-5-ureidopentan-2-yl)amino)-3-methyl-1-oxobutan-2-yl)amino)-7-oxoheptanoate[ No CAS ]
[ 149647-78-9 ]
ethyl 7-(((S)-3-methyl-1-oxo-1-(((S)-1-oxo-1-((4-(((8-oxo-8-(phenylamino)octanamido)oxy)methyl)phenyl)amino)-5-ureidopentan-2-yl)amino)butan-2-yl)amino)-7-oxoheptanoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
68%
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 20℃; for 12h;Inert atmosphere;
In a vial under N2 atmosphere, bromide [18] (60 mg, 0.09 mmol), SAHA (28 mg, 0.10 mmol) and 1 mL of DMF dry are mixed at room temperature. Freshly distilled DIPEA (25 mg, 0.20 mmol) is added dropwise and the solution stirred at room temperature for 12 h. The solvent is then removed via rotator evaporation and high vacuum. The residue is purified by flash column chromatography with a gradient 0- 20% methanol in dichloromethane to provide the product [31 ] as a white solid 48 mg, 68% of yield. MS: m/z 795.9 [M+H]+, 818.0 [M+Na]+.1H NMR (400 MHz, DMSO) delta 7.63-7.598 (m, 2H), 7.57 (d, J= 8 Hz, 2H), 7.39 (d, J= 8.4 Hz,2H), 7.313 (t, J= 8 Hz, 2H), 7.09 (t, J= 8; 1 H), 4,89 (s, 2H), 4.58 (s, 2H), 4.20 (d, J= 7.2, 1 H), 4.14 (t, J= 7.2 Hz, 2H), 4.10 (t, J= 7.4 Hz, 1 H),3,56-3,45(m, 1 H), 3,34 (d, J= 7.8 Hz, 2H), 2,41 -2,37 (m, 1 H), 2,34-2,30 (m, 2H), 2,13-2,06 (m, 2H), 1 ,79-1 ,60 (m, 4H), 1 ,42-1 ,36 (m, 4H), 1 ,28-1 ,24 (m, 3H), 1 ,01 -0,97 (m, 6H).
methyl (1R,4R)-4-((6-(3-((4-(bromomethyl)phenyl)thio)-2,5-dioxopyrrolidin-1-yl)hexanamido)methyl)cyclohexane-1-carboxylate[ No CAS ]
[ 149647-78-9 ]
C40H54N4O8S[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
45%
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 20℃; for 12h;Inert atmosphere;
In a vial under N2 atmosphere, bromide [29] (78 mg, 0.14 mmol), SAHA (50 mg, 0.18 mmol) and 1 ml_ of DMF dry are mixed at room temperature. Freshly distilled DIPEA (45 mg, 0.36 mmol) is added dropwise and the solution is stirred at room temperature for 12 h. The solvent is then removed via rotator evaporation and high vacuum. The residue is purified by flash column chromatography with a gradient 0- 20% methanol in dichloromethane to provide the product [30] as a white solid 48 mg, 45% of yield. MS: m/z 751 .4 [M+H]+.
With hydroxylamine hydrochloride; In N,N-dimethyl-formamide; at 40℃; for 1h;
Dissolve 0.1 mol of the above-obtained octanoylaniline in 200 ml of DMF at room temperature.25 g of modified mesoporous silica was added, then stirred and mixed uniformly, and 0.3 mol of hydroxylamine hydrochloride was added thereto, and stirring was continued at 40 C for 60 min.After the reaction is completed, the reaction solution is concentrated, and then poured into 600 ml of deionized water, stirred and mixed for 15 min, and filtered.The solid was washed twice with deionized water and dried under vacuum at 50 C to obtain vorinostat. The HPLC purity was 99.5%.The yield was 88.5% based on octanoyl aniline.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping