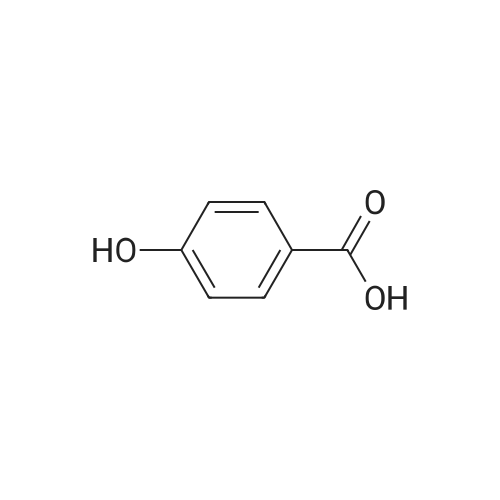
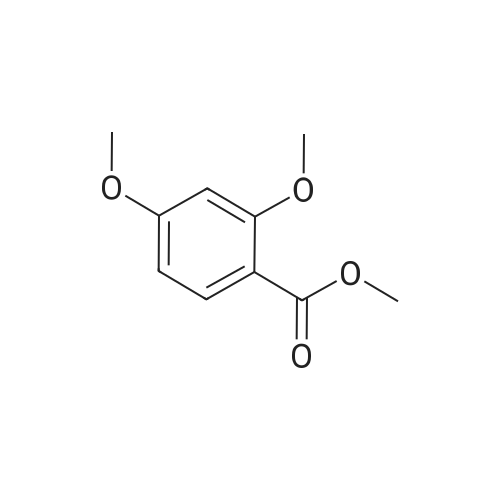
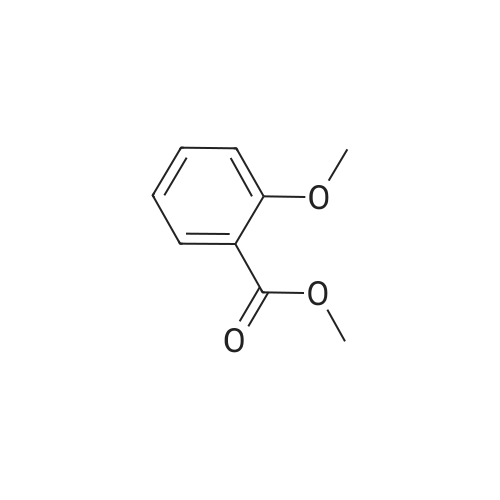
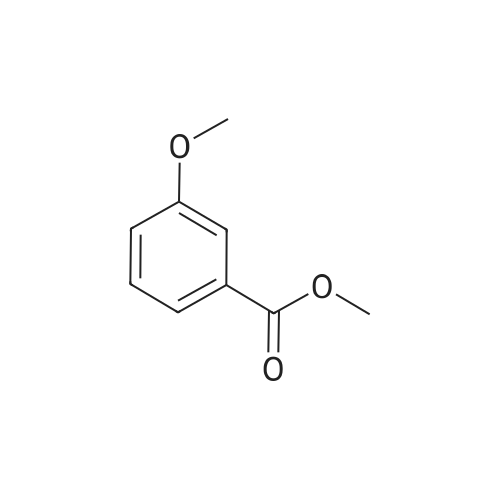
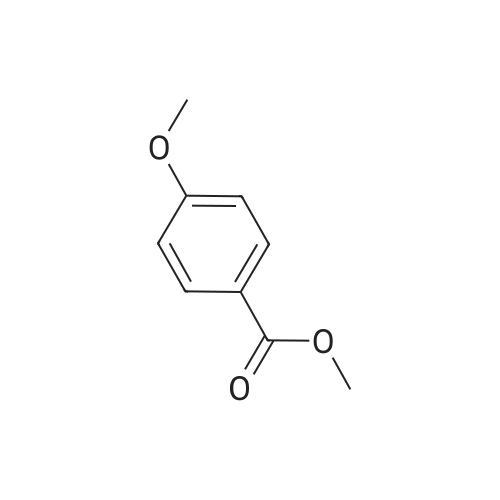
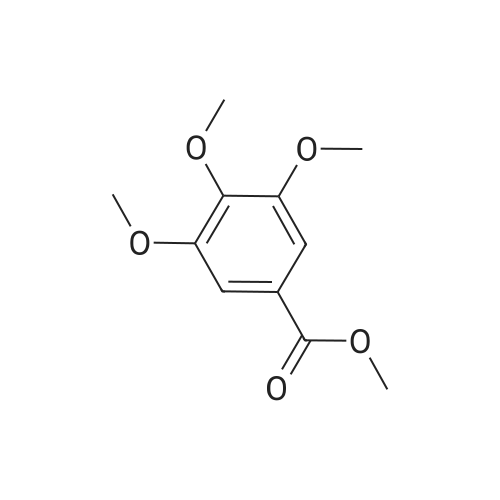
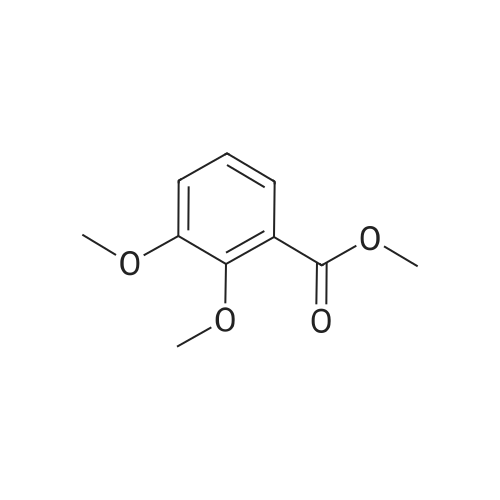
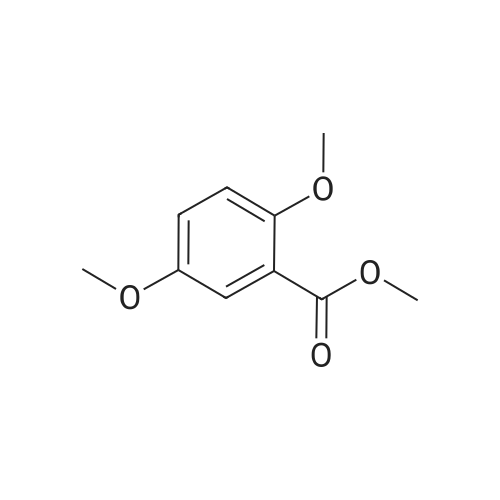
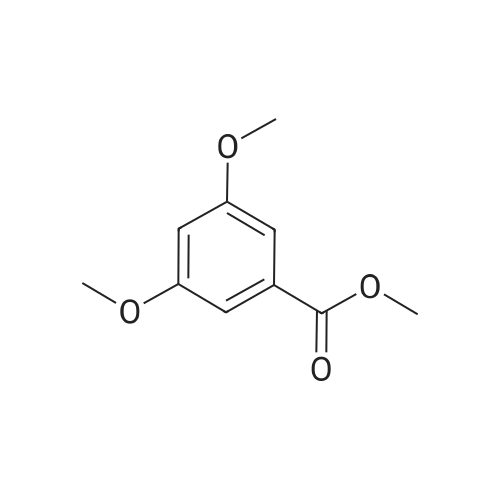
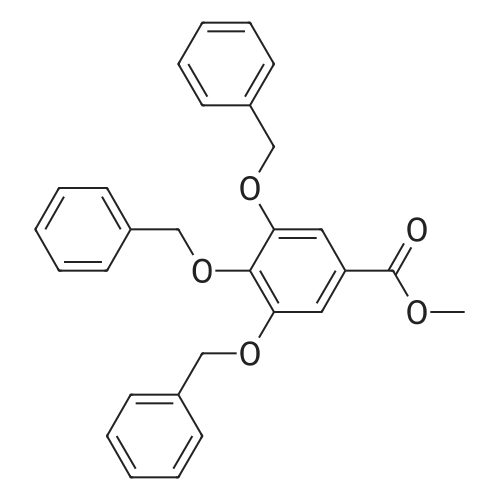
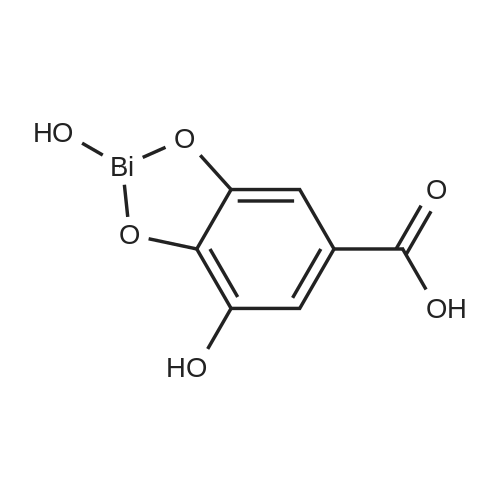
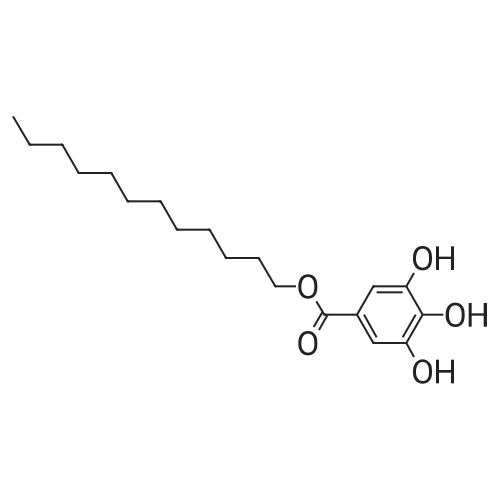
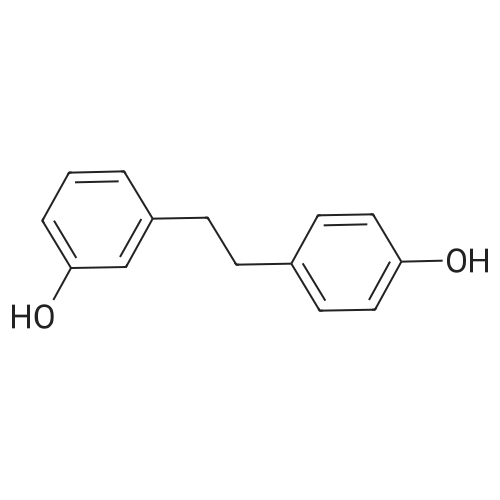
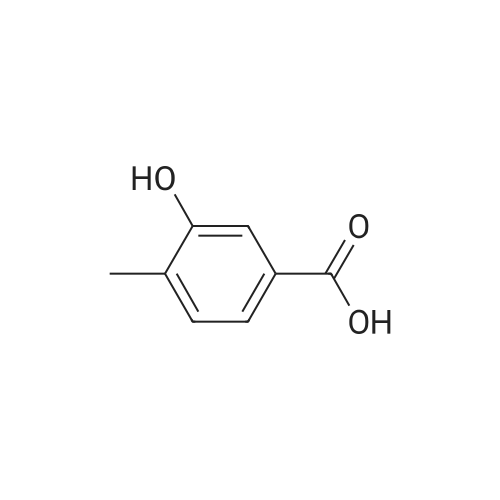
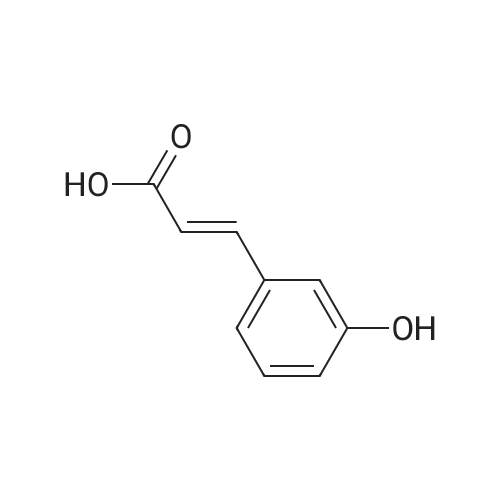
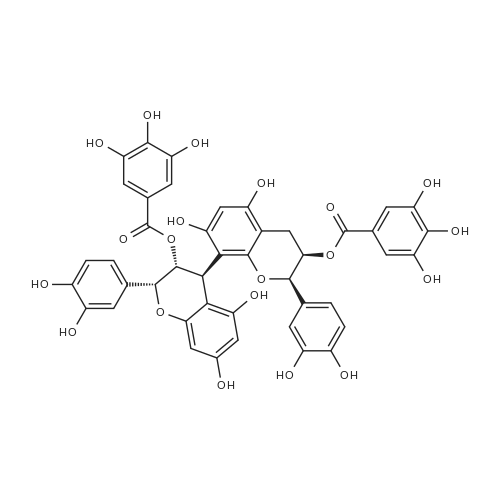
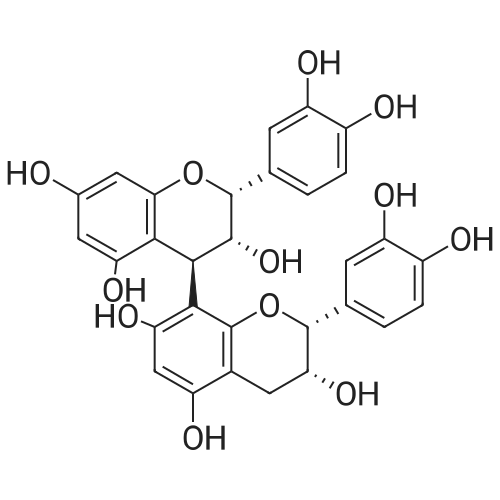
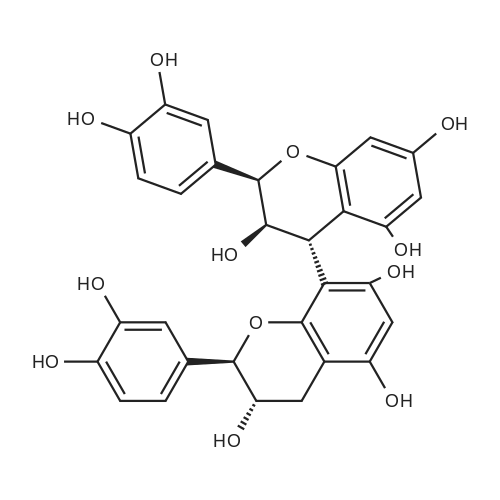
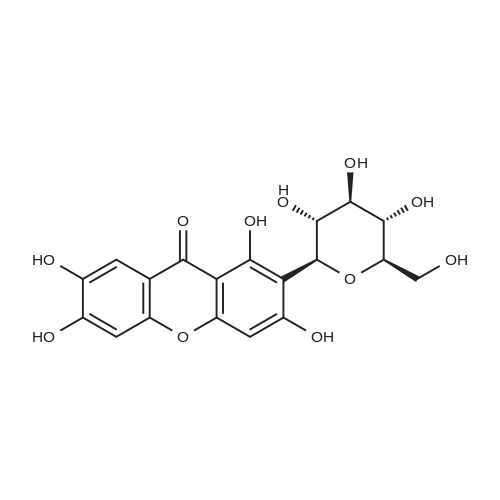
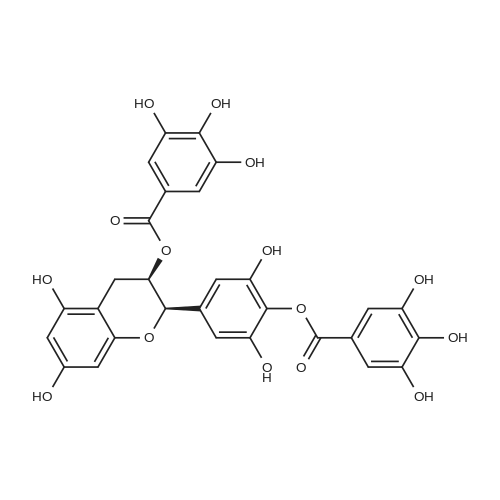
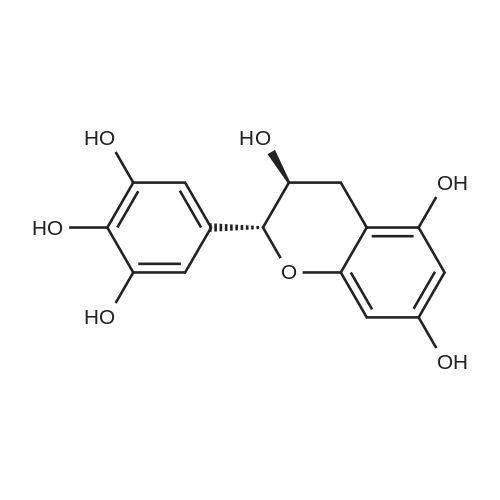
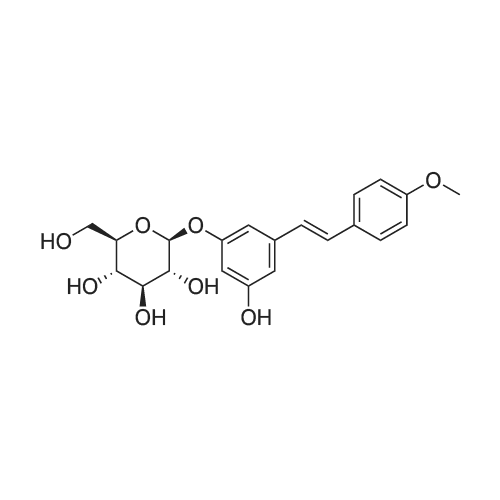
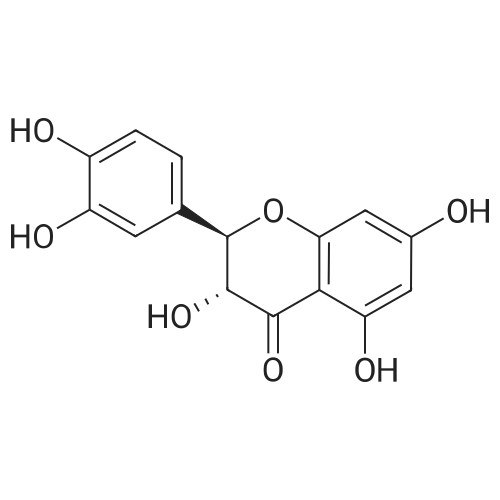
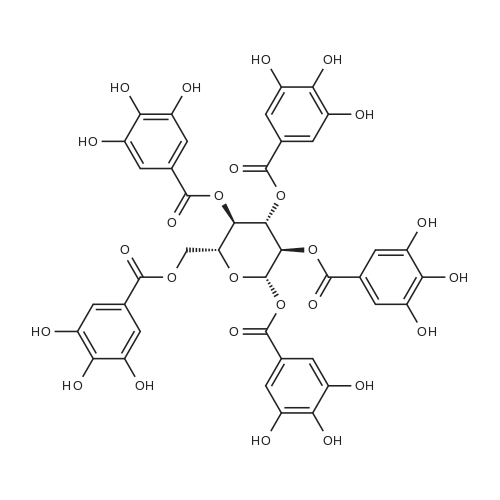
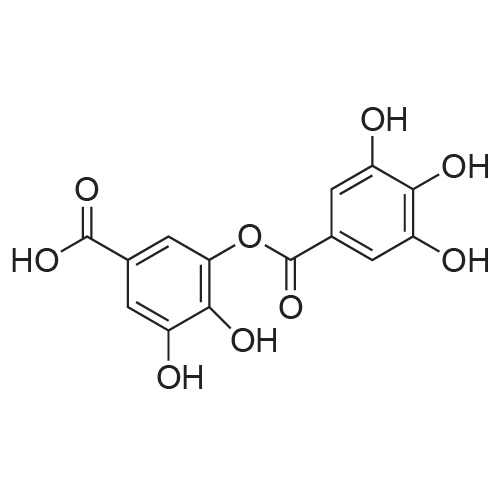
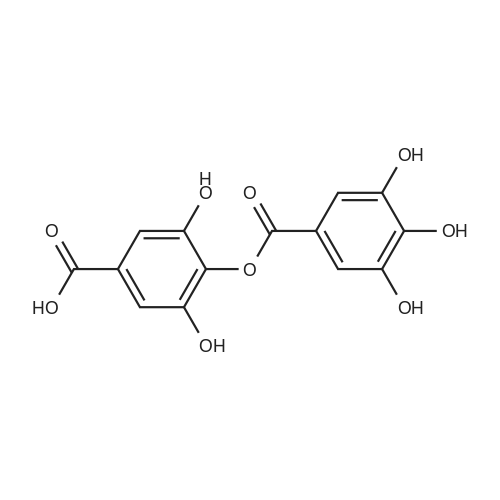
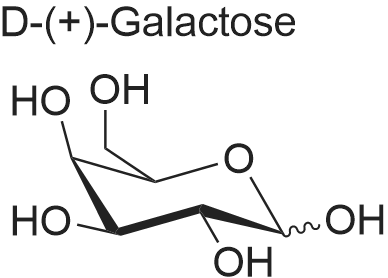
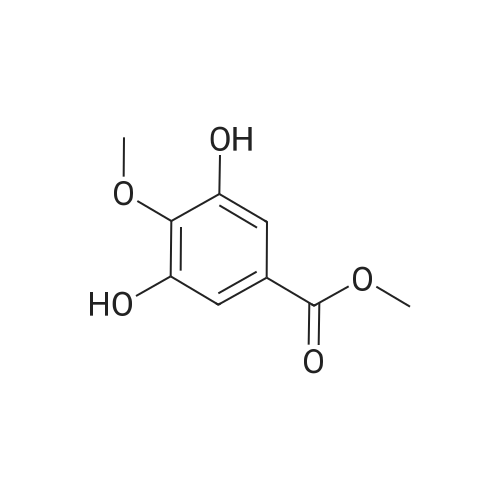
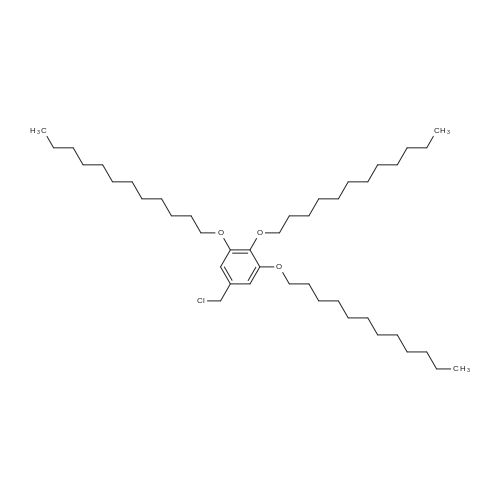
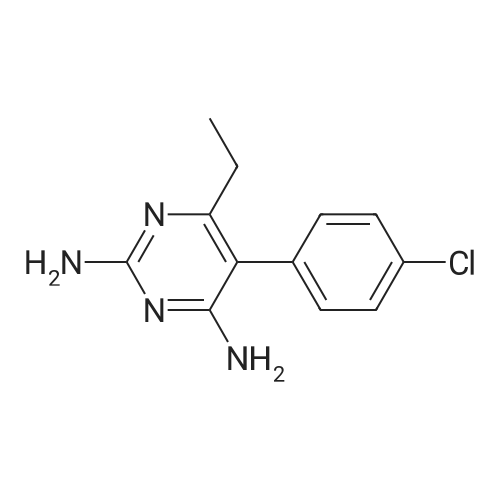
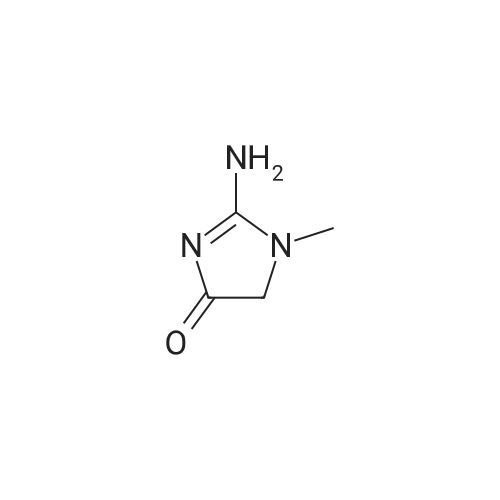
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
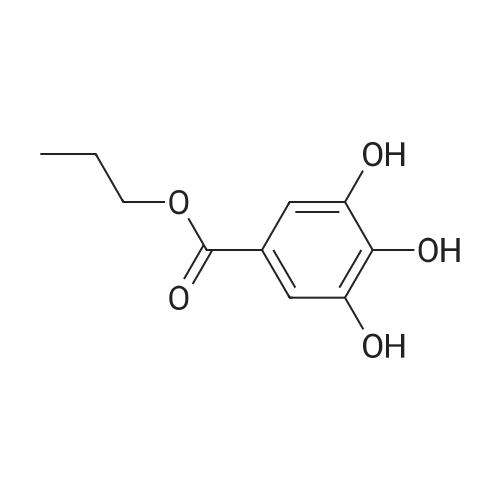
100 g of gallic acid and 200 g of n-propanol were weighed into a reactor,20 g of the brominated modified sulfonic acid resin obtained in Reference Example 2 was added,Electric stirring,Heating to 100 DEG C to react for 5 h, filtering to obtain the filtrate, and recovering brominated modified sulfonic acid resin;the filtrate obtained in step (1) is distilled off excess alcohol to obtain the crude product, then recrystallization, dewatering and drying are carried out by deionized water to obtain n-propyl gallate,The yield was 98percent and the product purity was 99.9percent.
66%
Stage #1: With diisopropyl-carbodiimide In tetrahydrofuran at 0℃; for 1 h; Stage #2: With dmap In tetrahydrofuran at 0℃; for 6 h;
General procedure: To the solution containing gallic acid (250 mg, 1.47 mmol) in THF solvent at 0° C is added alcohol (2.94 mmol) and the DIC (0.34 mL, 2.205 mmol) as an activator. The reaction mixture was stirred for 1 h at 0° C, then added DMAP catalyst(18 mg, 0.147 mmol), and stirred again for the next 6 h at 0° C, then allowed to reach room temperature.The reaction was terminated when the TLC analysis showed no spot of the remaining gallic acid. After the reaction is complete, the reaction mixture is diluted with ether, filtered, evaporated,and purified by column silica gel chromatography. Pure compounds were analyzed by Thin Layer Chromatography (TLC), Nuclear Magnetic Resonance Spectrometer (NMR), and High Resolution Mass Spectrometer (HRMS)
2.40 g
at 50 - 70℃; for 5 h; Molecular sieve
Weighed gallic acid (2.0g) and n-propanol (40mL) were mixed, stirred, heated to 50 ,Product A (100 mg) was added and heating was continued to 70 ° C for 5 hours. The product A was removed by filtration,The filtrate was concentrated under reduced pressure to give propyl gallate (2.40 g, 96.2percent conversion, HPLC purity of about 98.5percent) The propyl gallate obtained in Example 5 or 6 was dissolved in hot ethanol (60 ° C) to be recrystallized once to obtain whiteCrystals (HPLC purity 99.95percent).
Reference:
[1] Patent: CN105294433, 2016, A, . Location in patent: Paragraph 0028
[2] Asian Journal of Chemistry, 2015, vol. 27, # 4, p. 1351 - 1354
[3] Journal of Molecular Catalysis B: Enzymatic, 2010, vol. 67, # 3-4, p. 242 - 250
[4] Arzneimittel-Forschung/Drug Research, 2005, vol. 55, # 1, p. 66 - 75
[5] Oriental Journal of Chemistry, 2018, vol. 34, # 2, p. 655 - 662
[6] Journal of Chemical Thermodynamics, 1996, vol. 28, # 2, p. 171 - 185
[7] Patent: US2616042, 1951, ,
[8] Patent: US2616042, 1951, ,
[9] Patent: US2616042, 1951, ,
[10] Archiv der Pharmazie (Weinheim, Germany), 1931, vol. 269, p. 545,560
[11] Journal of the Chemical Society, 1927, p. 2648
[12] Bioorganic and Medicinal Chemistry Letters, 2001, vol. 11, # 3, p. 347 - 350
[13] Planta Medica, 2002, vol. 68, # 8, p. 690 - 693
[14] Natural Product Research, 2010, vol. 24, # 18, p. 1758 - 1765
[15] Journal of Agricultural and Food Chemistry, 2010, vol. 58, # 9, p. 5355 - 5362
[16] Current Medicinal Chemistry, 2012, vol. 19, # 26, p. 4534 - 4540,7
[17] Preparative Biochemistry and Biotechnology, 2013, vol. 43, # 5, p. 445 - 455
[18] Bioorganic and Medicinal Chemistry Letters, 2014, vol. 24, # 7, p. 1812 - 1814
[19] Patent: CN106117052, 2016, A, . Location in patent: Paragraph 0009
[20] Patent: CN107488111, 2017, A, . Location in patent: Paragraph 0042; 0043; 0045; 0053
2
[ 149-91-7 ]
[ 71-36-3 ]
[ 121-79-9 ]
Yield
Reaction Conditions
Operation in experiment
75%
With sulfuric acid In propan-1-ol; 2-methyl-propan-1-ol; water
EXAMPLE 1 Preparation of N-propyl gallate A mixture of 500 g of tara (theoretical gallic acid content about 1.5 moles), 2,000 g (33.3 moles) of n-propanol and 50 g of anhydrous sulfuric acid was refluxed at 100° C. for 25 hours, the water of reaction being removed continuously in the form of a propanol/water azeotrope. When water was no longer eliminated, the mixture was neutralized with dilute sodium hydroxide solution and freed from excess propanol by distillation. The residue was taken up in a mixture of 900 ml of isobutanol and 600 ml of n-butanol, and this solution was then washed with a total of 6 l of water. The organic phase was evaporated down, after which 400 g of a crude n-propyl gallate having a purity of 60percent (according to HPLC analysis) were obtained in the form of an oil. The yield was 75percent, based on the gallic acid content of the tara used, and converted to pure ester. Recrystallization from water gave the ester in a yield of 60percent and with a purity of 95percent.
Reference:
[1] Journal of Natural Products, 2012, vol. 75, # 10, p. 1798 - 1802
6
[ 64-17-5 ]
[ 149-91-7 ]
[ 831-61-8 ]
Yield
Reaction Conditions
Operation in experiment
98%
at 80℃; for 5 h;
100 g of gallic acid and 150 g of ethanol were weighed into a reactor, 35 g of the brominated modified sulfonic acid resin obtained in Referential Example 2 was weighed, heated to 80 ° C for 5 h, filtered to obtain a filtrate, and bromine Modified sulfonic acid resin;the filtrate obtained in step (1) is distilled off excess alcohol to obtain crude product, and then recrystallization, dewatering and drying are carried out by deionized water to obtain ethyl gallate in 98percent yield and purity of 99.9percent .
92%
at 70℃; for 24 h;
To a 50 mL round bottom flask, the gallic acid (2.0 g, 11.8 mmol), ethanol (30 mL, 1.94 mol) and 10percent of concentrated sulfuric acid (0.2 mL) were vigorously stirred at 70°C in reflux apparatus. The reaction was monitored by TLC until end of reaction (24 h). The product was extracted using 20 mL of saturated solution of sodium bicarbonate (NaHCO3), 20 mL of distilled water and finally extracted for three times with 20 mL of ethyl acetate. The collected organic phase was evaporated and Na2SO4 anhydrous was added. After extraction ethyl gallate G was obtained already purified (92percent) and it was characterized by IR, GC-MS and NMR analyses.
70%
at 100℃; for 0.833333 h; Microwave irradiation
General procedure: A microwave vial was loaded with gallic acid 1 (0.3mmol, 50mg), the corresponding alcohol (0.9mmol), and concentrated H2SO4 (0.07mL). The reaction vessel was sealed and irradiated in a microwave reactor at 100°C for 50min. After cooling, volatiles were evaporated to dryness and the residue was dissolved in ethyl acetate (30mL) and washed successively with saturated solutions of NaHCO3 (3×20mL) and NaCl (1×20mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was then purified by column chromatography using hexane/EtOAc, (3:7) as the eluent. Melting points and characterization of compounds 2–7 were consistent with those found in the literature [25d]. In our case, the following yields were obtained: 2 (70percent), 3 (97percent), 4 (83percent), 5 (98percent), 6 (96percent), 7 (82percent).
69%
Stage #1: With diisopropyl-carbodiimide In tetrahydrofuran at 0℃; for 1 h; Stage #2: With dmap In tetrahydrofuran at 0℃; for 6 h;
General procedure: To the solution containing gallic acid (250 mg, 1.47 mmol) in THF solvent at 0° C is added alcohol (2.94 mmol) and the DIC (0.34 mL, 2.205 mmol) as an activator. The reaction mixture was stirred for 1 h at 0° C, then added DMAP catalyst(18 mg, 0.147 mmol), and stirred again for the next 6 h at 0° C, then allowed to reach room temperature.The reaction was terminated when the TLC analysis showed no spot of the remaining gallic acid. After the reaction is complete, the reaction mixture is diluted with ether, filtered, evaporated,and purified by column silica gel chromatography. Pure compounds were analyzed by Thin Layer Chromatography (TLC), Nuclear Magnetic Resonance Spectrometer (NMR), and High Resolution Mass Spectrometer (HRMS)
51.5%
Reflux
General procedure: In 50 ml_ of the alcohol of interest, 3,4,5-trihydroxybenzoic acid (1 ) (1 .5 g, 8.85 mmol) was dissolved, followed by addition of 10 drops of sulfuric acid. The solution was refluxed overnight. Afterwards, the solution was allowed to cool to room temperature. The residual alcohol was evaporated, yielding the gallic acid ester derivatives 22, 23, 24 as a solid compound.
47.2%
for 40 h; Heating / reflux
In a flask was charged 15.1 g of 3,4,5-trihydroxybenzoic acid (1), and 160 ml of ethanol and 5 ml of sulfuric acid were added thereto in the order cited and heating with stirring was performed for 40 hours under reflux. Thereafter, the solvent was distilled off, the residue was extracted with ether and the organic layer was dried over magnesium sulfate. After the magnesium sulfate was filtered off, and the organic solvent was evaporated to afford a white solid (2) (yield of the reaction: 17.6 g, percent yield:47.2percent).
Reference:
[1] Patent: CN105294433, 2016, A, . Location in patent: Paragraph 0026
[2] Asian Journal of Chemistry, 2015, vol. 27, # 4, p. 1351 - 1354
[3] New Journal of Chemistry, 2018, vol. 42, # 20, p. 16999 - 17008
[4] ChemPhysChem, 2016, p. 2225 - 2237
[5] Tetrahedron, 2009, vol. 65, # 40, p. 8354 - 8361
[6] Tetrahedron Letters, 2017, vol. 58, # 9, p. 825 - 828
[7] Angewandte Chemie - International Edition, 2012, vol. 51, # 49, p. 12246 - 12249[8] Angew. Chem., 2012, vol. 124, # 49, p. 12412 - 12415,4
[9] Arzneimittel-Forschung/Drug Research, 2005, vol. 55, # 1, p. 66 - 75
[10] Journal of Organic Chemistry, 2013, vol. 78, # 2, p. 527 - 544
[11] European Journal of Medicinal Chemistry, 2015, vol. 92, p. 656 - 671
[12] New Journal of Chemistry, 2018, vol. 42, # 7, p. 5382 - 5394
[13] Oriental Journal of Chemistry, 2018, vol. 34, # 2, p. 655 - 662
[14] Patent: WO2018/96088, 2018, A1, . Location in patent: Page/Page column 33; 40
[15] Patent: EP1389605, 2004, A1, . Location in patent: Page 4
[16] Justus Liebigs Annalen der Chemie, 1872, vol. 163, p. 218[17] Chemische Berichte, 1879, vol. 12, p. 1533
[18] Bulletin de la Societe Chimique de France, 1864, vol. <2> 2, p. 94[19] Jahresbericht ueber die Fortschritte der Chemie und Verwandter Theile Anderer Wissenschaften, 1864, p. 404
[20] Chemische Berichte, 1905, vol. 38, p. 3348
[21] Patent: US2615042, 1951, ,
[22] Chemische Berichte, 1905, vol. 38, p. 3348
[23] Tetrahedron, 1997, vol. 53, # 6, p. 2163 - 2176
[24] Planta Medica, 2002, vol. 68, # 8, p. 690 - 693
[25] Chemical Communications, 2003, # 18, p. 2306 - 2307
[26] Tetrahedron Letters, 2009, vol. 50, # 49, p. 6901 - 6905
[27] Natural Product Research, 2010, vol. 24, # 18, p. 1758 - 1765
[28] Molecular Crystals and Liquid Crystals, 2010, vol. 518, p. 160 - 167
[29] Current Medicinal Chemistry, 2012, vol. 19, # 26, p. 4534 - 4540,7
[30] European Journal of Medicinal Chemistry, 2013, vol. 62, p. 289 - 296
[31] Bioorganic and Medicinal Chemistry Letters, 2014, vol. 24, # 7, p. 1812 - 1814
[32] Dyes and Pigments, 2015, vol. 119, p. 108 - 115
[33] Bulletin of the Korean Chemical Society, 2015, vol. 36, # 11, p. 2682 - 2687
[34] Carbohydrate Polymers, 2016, vol. 140, p. 171 - 180
[35] Bulletin of the Korean Chemical Society, 2015, vol. 36, # 11, p. 2740 - 2745
[36] Chemistry - A European Journal, 2018,
General procedure: A microwave vial was loaded with gallic acid 1 (0.3mmol, 50mg), the corresponding alcohol (0.9mmol), and concentrated H2SO4 (0.07mL). The reaction vessel was sealed and irradiated in a microwave reactor at 100°C for 50min. After cooling, volatiles were evaporated to dryness and the residue was dissolved in ethyl acetate (30mL) and washed successively with saturated solutions of NaHCO3 (3×20mL) and NaCl (1×20mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was then purified by column chromatography using hexane/EtOAc, (3:7) as the eluent. Melting points and characterization of compounds 2–7 were consistent with those found in the literature [25d]. In our case, the following yields were obtained: 2 (70percent), 3 (97percent), 4 (83percent), 5 (98percent), 6 (96percent), 7 (82percent).
51%
Stage #1: With diisopropyl-carbodiimide In tetrahydrofuran at 0℃; for 1 h; Stage #2: With dmap In tetrahydrofuran at 0℃; for 6 h;
General procedure: To the solution containing gallic acid (250 mg, 1.47 mmol) in THF solvent at 0° C is added alcohol (2.94 mmol) and the DIC (0.34 mL, 2.205 mmol) as an activator. The reaction mixture was stirred for 1 h at 0° C, then added DMAP catalyst(18 mg, 0.147 mmol), and stirred again for the next 6 h at 0° C, then allowed to reach room temperature.The reaction was terminated when the TLC analysis showed no spot of the remaining gallic acid. After the reaction is complete, the reaction mixture is diluted with ether, filtered, evaporated,and purified by column silica gel chromatography. Pure compounds were analyzed by Thin Layer Chromatography (TLC), Nuclear Magnetic Resonance Spectrometer (NMR), and High Resolution Mass Spectrometer (HRMS)
Reference:
[1] European Journal of Medicinal Chemistry, 2015, vol. 92, p. 656 - 671
[2] Journal of Agricultural and Food Chemistry, 2002, vol. 50, # 14, p. 3992 - 3998
[3] Journal of Agricultural and Food Chemistry, 2002, vol. 50, # 23, p. 6692 - 6696
[4] Journal of Agricultural and Food Chemistry, 2000, vol. 48, # 4, p. 1393 - 1399
[5] Arzneimittel-Forschung/Drug Research, 2005, vol. 55, # 1, p. 66 - 75
[6] Oriental Journal of Chemistry, 2018, vol. 34, # 2, p. 655 - 662
[7] Angewandte Chemie - International Edition, 2012, vol. 51, # 49, p. 12246 - 12249[8] Angew. Chem., 2012, vol. 124, # 49, p. 12412 - 12415,4
[9] ACS Medicinal Chemistry Letters, 2018, vol. 9, # 1, p. 51 - 55
[10] DRP/DRBP Org.Chem.,
[11] Patent: US2623897, 1949, ,
[12] Journal of the American Chemical Society, 1947, vol. 69, p. 2003
[13] Recueil des Travaux Chimiques des Pays-Bas, 1951, vol. 70, p. 277,281
[14] DRP/DRBP Org.Chem.,
[15] Patent: US2623897, 1949, ,
[16] Bioorganic and Medicinal Chemistry Letters, 2001, vol. 11, # 3, p. 347 - 350
[17] Current Medicinal Chemistry, 2012, vol. 19, # 26, p. 4534 - 4540,7
[18] Bioorganic and Medicinal Chemistry Letters, 2014, vol. 24, # 7, p. 1812 - 1814
[19] Journal of Enzyme Inhibition and Medicinal Chemistry, 2015, vol. 30, # 2, p. 299 - 307
9
[ 6906-38-3 ]
[ 149-91-7 ]
[ 487-70-7 ]
Reference:
[1] Journal of Agricultural and Food Chemistry, 2009, vol. 57, # 12, p. 5271 - 5278
10
[ 149-91-7 ]
[ 21852-50-6 ]
Reference:
[1] Patent: CN108503561, 2018, A,
[2] Journal of Enzyme Inhibition and Medicinal Chemistry, 2018, vol. 33, # 1, p. 1554 - 1564
[3] Patent: CN108727222, 2018, A,
11
[ 99-50-3 ]
[ 89-86-1 ]
[ 149-91-7 ]
[ 99-10-5 ]
[ 303-38-8 ]
[ 490-79-9 ]
[ 99-06-9 ]
[ 69-72-7 ]
[ 74-88-4 ]
[ 99-96-7 ]
[ 2150-41-6 ]
[ 606-45-1 ]
[ 5368-81-0 ]
[ 121-98-2 ]
[ 2150-38-1 ]
[ 1916-07-0 ]
[ 2150-42-7 ]
[ 2150-40-5 ]
[ 2150-37-0 ]
Reference:
[1] Chemical Communications, 2014, vol. 50, # 14, p. 1694 - 1697
12
[ 149-91-7 ]
[ 70424-94-1 ]
Reference:
[1] Chemical and Pharmaceutical Bulletin, 2003, vol. 51, # 9, p. 1085 - 1088
[2] Synthetic Communications, 2002, vol. 32, # 20, p. 3149 - 3158
[3] Organic and Biomolecular Chemistry, 2014, vol. 12, # 1, p. 73 - 85
[4] Tetrahedron Letters, 2016, vol. 57, # 24, p. 2652 - 2654
[5] Heterocycles, 2014, vol. 8, # 2, p. 1371 - 1396
[6] Tetrahedron Letters, 2016, vol. 57, # 49, p. 5469 - 5474
[7] Journal of Medicinal Chemistry, 2018, vol. 61, # 4, p. 1609 - 1621
[8] Monatshefte fur Chemie, 2018, vol. 149, # 7, p. 1293 - 1300
With brominated modified sulfonic acid resin; at 100℃; for 5h;
100 g of gallic acid and 200 g of n-propanol were weighed into a reactor,20 g of the brominated modified sulfonic acid resin obtained in Reference Example 2 was added,Electric stirring,Heating to 100 DEG C to react for 5 h, filtering to obtain the filtrate, and recovering brominated modified sulfonic acid resin;the filtrate obtained in step (1) is distilled off excess alcohol to obtain the crude product, then recrystallization, dewatering and drying are carried out by deionized water to obtain n-propyl gallate,The yield was 98% and the product purity was 99.9%.
91.6%
With thionyl chloride; at 35 - 65℃; for 1h;
400 g of n-propanol and gallic acid 85 g (0.5 mol) were placed in the reaction flask, and83.3g (0.7 mol) ofthionyl chloride was slowly added dropwise at room temperature tomaintain the temperature at 35-40 C. After the completion of the dropwise addition, the temperature was raised to 60- The reaction was carried out at 65 C for 1 hour, and the sampling test(HPLC) was completed. The ratio of the product to the raw material was 50:1 to determine the end point of the reaction. After the reaction was completed, it was concentrated to a non-slip liquid, and the mixture was droppedinto 400 g of water at room temperature for 1 hour, filtered, and dried.97 g of product were obtained, HPLC: 96.9%, yield: 91.6%.
81.6%
With perchloric acid; In benzene; at 80 - 90℃;
Accurately weigh 37.6 g of monohydrate gallic acid (0.2 mol), add 500 mL three-necked flask, measure 3.2 mL (0.04 mol) of perchloric acid, 40 mL of benzene, and take 45 mL (0.6 mol) of n-propanol into a three-necked flask. The three-necked flask is placed in a 1L electric heating jacket. One port of the three-necked flask is inserted into the thermometer, the other port is connected to the water separator, the upper end of the water separator is connected to the return pipe, the middle mouth is clamped with a long handle, and the PTFE stirring paddle is connected to start the stirring. The motor, the electric heating jacket and the condensed water are kept at a temperature of 80 C to 90 C and stirred for reflux reaction, and the reaction is carried out for about 12 hours to 14 hours.When the trap collected approximately 7.2mL (0.4mol) condensate, was added 0.97g (0.01mol) sulfamic acid, and stirring was continued concentrated to half the total volume of the reaction mixture was refluxed for 1 hour under reduced pressure to obtain a crude product of propyl gallate concentrate . After the propyl propyl ester concentrate was crystallized from the purified water, the raw material of gallic acid was not detected in the crude crystal. The crude propyl propyl ester was washed with purified water to neutrality, decolorized by activated carbon, recrystallized from purified water, filtered and dried to obtain a fine propylene. The ester was 34.6 g, the yield was about 81.60 %, and the purity was 99.9 %
73%
With thionyl chloride;Inert atmosphere;
Propyl 3,4,5-trihydroxybenzoate was prepared bydissolving GA (1) (1.0 g, 5.8 mmol) in propyl alcohol (15.0ml) in a single neck round bottom flask. Thionyl chloride(0.9 ml, 2.4 mmol) was added dropwise along with stirringat 0C. R eaction m ixture w as r efluxed a t 7 0C for 5-6 hunder nitrogen atmosphere and monitored by TLC. Solidproduct was obtained by removing solvent with the help ofrotary evaporator. It was redissolved in EtOAc and washedwith NaHCO3 solution (3×20.0 ml), brine solution (2×20.0ml) and finally with water. EtOAc layer was dried oversodium sulphate and then under nitrogen at rt. Pure propyl 3, 4, 5-trihydroxy benzoate was obtained by a column washusing Pet ether :EtOAc (1 : 1) solvent mixture. m.p. 146-148C; yield: 73 %; IR (KBr), nu/cm-1: 3445 (OH), 3030 (sp2 CH),1740 (C=O), 1625, 1495 (C=C), 1240 (C-O); 1H NMR(methanol-d6, 500 MHz) delta/ppm: 1.04 (t, 3H, J = 7.4 Hz, CH3),1.77 (m, 2H, CH2), 4.20 (t, 2H, J = 6.6 Hz, OCH2), 7.08 (s, 2H,H2 & H6 ); 13C NMR (methanol-d6, 125 MHz ) delta/ppm: 10.12(C10), 22.59 (C9), 67.13 (C8), 110.67(C2/C6), 122.45 (C1),139.54 (C4), 146.81 (C3/C5), 167.26 (C7);HR-ESI-MS: m/z =213.40[M+H]+.
66%
General procedure: To the solution containing gallic acid (250 mg, 1.47 mmol) in THF solvent at 0 C is added alcohol (2.94 mmol) and the DIC (0.34 mL, 2.205 mmol) as an activator. The reaction mixture was stirred for 1 h at 0 C, then added DMAP catalyst(18 mg, 0.147 mmol), and stirred again for the next 6 h at 0 C, then allowed to reach room temperature.The reaction was terminated when the TLC analysis showed no spot of the remaining gallic acid. After the reaction is complete, the reaction mixture is diluted with ether, filtered, evaporated,and purified by column silica gel chromatography. Pure compounds were analyzed by Thin Layer Chromatography (TLC), Nuclear Magnetic Resonance Spectrometer (NMR), and High Resolution Mass Spectrometer (HRMS)
With citric acid; at 60℃; for 2h;
Finally, 12 g of the above-mentioned gallic acid crystals and 5 g of citric acid were weighed, added to a planetary ball mill, then 20mL pre-heated to 60 deg. C n-propanol, start the ball mill, wet milling at 720r / min for 2h, discharging, the resulting material was transferred to a rotary evaporator, the mixture was concentrated by steaming at 85 deg. C for 25 min, discharged, the obtained material was placed in a refrigerator at 4 C for 36 h, filtered, the filtrate was removed, the filter cake was washed 5 times with deionized water, the residue was then placed in a 110 C vacuum oven, drying to constant weight, propyl gallate.
2.40 g
With mordenite; at 50 - 70℃; for 5h;Molecular sieve;
Weighed gallic acid (2.0g) and n-propanol (40mL) were mixed, stirred, heated to 50 ,Product A (100 mg) was added and heating was continued to 70 C for 5 hours. The product A was removed by filtration,The filtrate was concentrated under reduced pressure to give propyl gallate (2.40 g, 96.2% conversion, HPLC purity of about 98.5%) The propyl gallate obtained in Example 5 or 6 was dissolved in hot ethanol (60 C) to be recrystallized once to obtain whiteCrystals (HPLC purity 99.95%).
With thionyl chloride; for 5.5h;Reflux;
1.2 Propyl Gallate (Compound 3) To a stirred suspension of gallic acid monohydrate (0.30 g, 1.6 mmol) in 1-propanol (5 mL), thionyl chloride (0.8 mL, 11 mmol) was added dropwisely. The mixture was heated under reflux for 5.5 hr. The reaction mixture was poured into crushed ice and extracted with 70 mL of ethyl acetate for three times. The combined organic layer was washed with water for several times, dried over MgSO4, and evaporated under reduced pressure. The crude product was recrystallized from aqueous ethanol to provide propyl gallate as white crystal, mp 145-148 C. 1H NMR (300 MHz, CD3OD) delta (ppm) 1.03 (t, J=7.5 Hz, 3H), 1.76 (m, 2H), 4.18 (t, J=6.5 Hz, 2H), 7.05 (s, 2H) [39]; 13C NMR (125 MHz, CD3OD) delta (ppm) 11.0, 23.4, 67.4, 110.2, 121.9, 139.9, 146.6, 168.8.
With sulfuric acid; at 100℃; for 0.833333h;Microwave irradiation;
General procedure: A microwave vial was loaded with gallic acid 1 (0.3mmol, 50mg), the corresponding alcohol (0.9mmol), and concentrated H2SO4 (0.07mL). The reaction vessel was sealed and irradiated in a microwave reactor at 100C for 50min. After cooling, volatiles were evaporated to dryness and the residue was dissolved in ethyl acetate (30mL) and washed successively with saturated solutions of NaHCO3 (3×20mL) and NaCl (1×20mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was then purified by column chromatography using hexane/EtOAc, (3:7) as the eluent. Melting points and characterization of compounds 2-7 were consistent with those found in the literature [25d]. In our case, the following yields were obtained: 2 (70%), 3 (97%), 4 (83%), 5 (98%), 6 (96%), 7 (82%).
51%
General procedure: To the solution containing gallic acid (250 mg, 1.47 mmol) in THF solvent at 0 C is added alcohol (2.94 mmol) and the DIC (0.34 mL, 2.205 mmol) as an activator. The reaction mixture was stirred for 1 h at 0 C, then added DMAP catalyst(18 mg, 0.147 mmol), and stirred again for the next 6 h at 0 C, then allowed to reach room temperature.The reaction was terminated when the TLC analysis showed no spot of the remaining gallic acid. After the reaction is complete, the reaction mixture is diluted with ether, filtered, evaporated,and purified by column silica gel chromatography. Pure compounds were analyzed by Thin Layer Chromatography (TLC), Nuclear Magnetic Resonance Spectrometer (NMR), and High Resolution Mass Spectrometer (HRMS)
With sulfuric acid; at 100.0℃; for 0.833333h;Microwave irradiation;
General procedure: A microwave vial was loaded with gallic acid 1 (0.3mmol, 50mg), the corresponding alcohol (0.9mmol), and concentrated H2SO4 (0.07mL). The reaction vessel was sealed and irradiated in a microwave reactor at 100C for 50min. After cooling, volatiles were evaporated to dryness and the residue was dissolved in ethyl acetate (30mL) and washed successively with saturated solutions of NaHCO3 (3×20mL) and NaCl (1×20mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was then purified by column chromatography using hexane/EtOAc, (3:7) as the eluent. Melting points and characterization of compounds 2-7 were consistent with those found in the literature [25d]. In our case, the following yields were obtained: 2 (70%), 3 (97%), 4 (83%), 5 (98%), 6 (96%), 7 (82%).
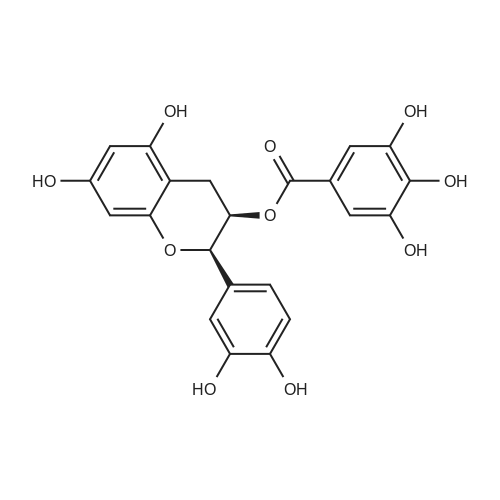
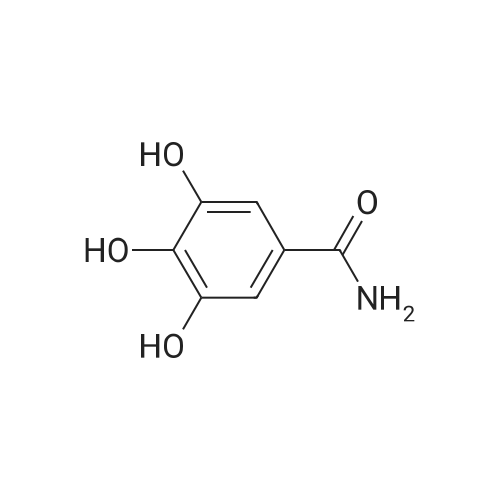
3,4,5-trihydroxy-N-[2-(4-nitrophenyl)ethyl]-benzamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
42%
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; triethylamine; In DMF (N,N-dimethyl-formamide); at 20℃; for 20h;
Example 83,4,5-trihydroxy-N-{4-[2-[(2-thienyl(imino)methyl)-amino]phenyl]ethyl}-benzamide hemi-fumarate: 8 8.1) 3,4,5-trihydroxy-N-[2-(4-nitrophenyl)ethyl]-benzamide: 2 g (11.5 mmoles) of gallic acid, 2.5 g (11.5 mmoles) of 4-nitrophenetylamine hydrochloride, 1.8 g (11.5 mmoles) of hydrated 1-hydroxybenzotriazole, 2.25 g (11.5 mmoles) of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride and 3.3 ml (23 mmoles) of triethylamine are introduced into a 100 ml flask containing 30 ml of anhydrous DMF. The orange-coloured solution obtained is agitated at 20 C. for 20 hours and diluted in a mixture of dichloromethane (50 ml) and water (30 ml). After decanting, the organic phase is washed with a molar solution of hydrochloric acid (20 ml) and with water (3×20 ml) until neutrality is achieved. After drying the organic phase over magnesium sulphate, followed by filtration and concentration under vacuum, the residue is purified on a silica gel column (eluant: dichloromethane/methanol: 9/1). The expected product is obtained in the form of a colourless oil with a yield of 42% (1.57 g). NMR 1H (100 MHz, DMSO d6, delta): 8.95 (m, 3H, 3× OH); 7.85 (m, 4H, Ph-NO2); 6.80 (s, 2H, Ph); 3.36 (m, 2H, CH2-N); 2.97 (t, 2H, CH2-Ph, J=6.0 Hz).
polyphenol oxidase; at 20℃; for 5.0h;pH 5.0;Phosphate-citrate buffer; Enzymatic reaction;
EC (1.160 g, 4.0 mmole) and gallic acid (0.520 g, 4.0 mmole) were dissolved in the 100 mL of phosphate-citrate buffer (50 mM, pH 5.0), and 1.2 g of crude PPO enzyme was added into the reaction solution with stirring. The enzymatic oxidation was carried out at room temperature for 3.5 hour. After the reaction, the solution extracted with the same volume of ethyl acetate with three times. Then, the ethyl acetate extracts was concentrated in vacuo. The resulting residues were then subjected to Sephadex LH-20, eluting with gradient of ethanol to 20% of acetone in ethanol. Among the collected 168 (each c.a. 15 mL) fractions, 4752 fractions were combined, and it was concentrated under reduced pressure. The resulting residue was applied on a RP-18 silica gel column eluting with gradient of 20%50% of aqueous methanol, and it was afforded compound 5. Then, 7285 fractions, isolated from Sephadex LH-20, were combined, and it was subjected to RP-18 column chromatography eluting with gradient of 10%50% of aqueous methanol, and compound 6 was isolated. [0137] ECG (0.66 g) and gallic acid (0.26 g) were dissolved in the 50 mL of phosphate-citrate buffer (50 mM, pH 5.0), and 1.2 g of crude PPO enzyme was dissolved in the reaction solution. The enzymatic oxidation was performed at room temperature for 5 hour. The reaction solution was then extracted with ethyl acetate with three times. Then, the organic layer was concentrated under reduced pressure. The resulting residues were subjected to Sephadex LH-20 column chromatography eluting with gradient of ethanol to 20% of acetone in ethanol. Among the collected 128 fractions (each c.a. 15 mL), 1820 fractions were combined, and concentrated under reduced pressure. The resulting residue was subjected for further purification on a RP-18 silica gel column eluting with gradient of 20%50% of aqueous methanol, and afforded compound 7. The 3748 fractions were combined and subjected to RP-18 column chromatography for further purification eluting with gradient of 10%30% of aqueous methanol, and afforded compound 8. The 90108 fractions were combined and subjected to RP-18 column chromatography eluting with gradient of 40%50% of aqueous methanol, and it was then afforded compound 9.
With sodium hydroxide; sulfuric acid; sodium carbonate; dimethyl sulfate; In methanol; water;
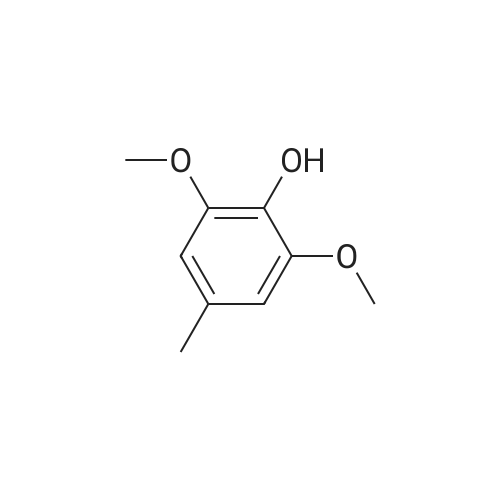
(1) In 600 ml of water, 200 g (1.18 mol) of gallic acid were suspended. While vigorously stirred, the suspension was added dropwise with 720 ml of an aqueous solution of 300 g (7.5 mmol) of sodium hydroxide and 480 ml (3.8 mmol) of dimethyl sulfate over 6 hours. After further stirred at room temperature for 13 hours, the reaction mixture was added with 6N-HCl. The reaction mixture was extracted with ethyl acetate (500 ml*3). The organic layers were combined together, washed with water and then dried over anhydrous magnesium sulfate. The residue obtained by distilling off the solvent from the organic layer was dissolved in 1 l of methanol. The resulting solution was added with 20 ml of concentrated sulfuric acid, followed by heating under reflux for 20 hours. After the reaction mixture was allowed to cool down to room temperature, the solvent was distilled off. The residue was thereafter added with 70 g of sodium carbonate and ice, followed by extraction with ethyl acetate (500 ml*1, 300 ml*2). The organic layers were combined together and then extracted with ice-cooled 4N-NaOH (100 ml*4). Just after the separation, the water layer extracted with the NaOH was made acidic with ice-cooled 6N-HCl (400 ml). The acidified water layer was extracted with ethyl acetate (300 ml*2). The organic layers were combined together, washed with saturated NaHCO3 and then dried over anhydrous magnesium sulfate. The solvent was distilled off from the organic layer, whereby 67.7 g of the residue mainly composed of methyl 3,4-dimethoxy-5-hydroxybenzoate were obtained as brown oil (yield: 27%). 1 H-NMR (CDCl3): 3.90-4.00(9H,m), 5.92(1H,s), 7.20-7.50(2H,m).
With sulfuric acid; In propan-1-ol; 2-methyl-propan-1-ol; water;
EXAMPLE 1 Preparation of N-propyl gallate A mixture of 500 g of tara (theoretical gallic acid content about 1.5 moles), 2,000 g (33.3 moles) of n-propanol and 50 g of anhydrous sulfuric acid was refluxed at 100 C. for 25 hours, the water of reaction being removed continuously in the form of a propanol/water azeotrope. When water was no longer eliminated, the mixture was neutralized with dilute sodium hydroxide solution and freed from excess propanol by distillation. The residue was taken up in a mixture of 900 ml of isobutanol and 600 ml of n-butanol, and this solution was then washed with a total of 6 l of water. The organic phase was evaporated down, after which 400 g of a crude n-propyl gallate having a purity of 60% (according to HPLC analysis) were obtained in the form of an oil. The yield was 75%, based on the gallic acid content of the tara used, and converted to pure ester. Recrystallization from water gave the ester in a yield of 60% and with a purity of 95%.
With sodium hydroxide; sulfuric acid; sodium carbonate; dimethyl sulfate; In methanol; water;
(1) In 600 ml of water, 200 g (1.18 mol) of gallic acid were suspended. While vigorously stirred, the suspension was added dropwise with 720 ml of an aqueous solution of 300 g (7.5 mmol) of sodium hydroxide and 480 ml (3.8 mmol) of dimethyl sulfate over 6 hours. After further stirred at room temperature for 13 hours, the reaction mixture was added with 6N-HCl. The reaction mixture was extracted with ethyl acetate (500 ml x 3). The organic layers were combined together, washed with water and then dried over anhydrous magnesium sulfate. The residue obtained by distilling off the solvent from the organic layer was dissolved in 1 l of methanol. The resulting solution was added with 20 ml of concentrated sulfuric acid, followed by heating under reflux for 20 hours. After the reaction mixture was allowed to cool down to room temperature, the solvent was distilled off. The residue was thereafter added with 70 g of sodium carbonate and ice, followed by extraction with ethyl acetate (500 ml x 1, 300 ml x 2). The organic layers were combined together and then extracted with ice-cooled 4N-NaOH (100 ml x 4). Just after the separation, the water layer extracted with the NaOH was made acidic with ice-cooled 6N-HCl (400 ml). The acidified water layer was extracted with ethyl acetate (300 ml x 2). The organic layers were combined together, washed with saturated NaHCO3 and then dried over anhydrous magnesium sulfate. The solvent was distilled off from the organic layer, whereby 67.7 g of the residue mainly composed of methyl 3,4-dimethoxy-5-hydroxybenzoate were obtained as brown oil (yield: 27%).
With horseradish peroxidase type I; dihydrogen peroxide; In acetone;pH 5.0;aq. buffer; Enzymatic reaction;
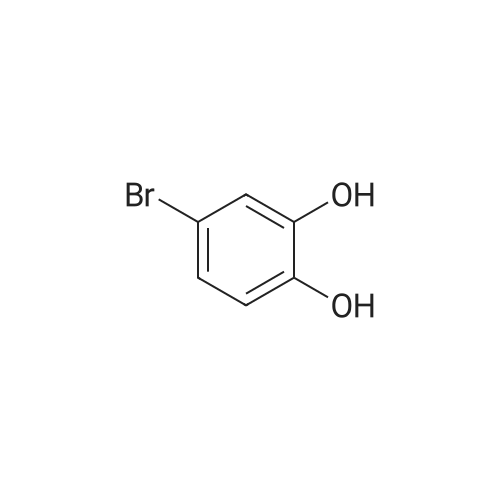
Gallic acid (3, 0.6 g, 3.53 mmol) and <strong>[17345-77-6]4-bromocatechol</strong> (7, 0.96 g, 5.08 mmol) were dissolved in acetone/pH 5.0 phosphate/citrate buffer (1:10 v/v, 50 mL). Horseradish peroxidase type I (25 ku) was added in three portions, 2 mg each time. Additionally, every 15 min and four times per period, 3% H2O2 (0.6 mL) was added to the reaction mixture, which was shaken by hand occasionally. The red precipitate was collected by filtration and washed with water. After drying under high vacuum overnight, 8 (0.86 g, 75%) was obtained as a red powder; dec>300 C (lit.4 mp 303-305 C); IR (neat): 3674 (br), 3385 (br), 3220 (w), 1648 (s), 1520 (s), 1487 (w), 1456 (w), 1401 (w), 1361 (s), 1288 (s), 1150 (w), 1088 (w), 969 (m) cm-1; 1H NMR (400 MHz, DMSOd6): delta 7.57 (d, J=1.4 Hz, 1H), 7.82 (s, 1H), 8.86 (d, J=1.4 Hz, 1H), 9.96 (s, 1H), 10.72 (s, 1H), 12.53 (br, 1H), 14.79 (s, 1H); 13C NMR (100 MHz, DMSOd6): delta 115.6, 118.8, 122.1, 125.3, 126.0, 126.8, 135.9, 149.1, 152.0, 154.5, 168.0, 185.7; HRMS (ESI-) calcd for C12H6BrO6 [M-H]: 324.9348, found: 324.9349.
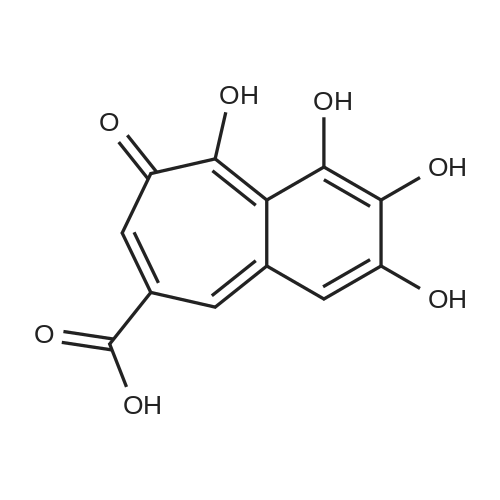
Compound 6 (400 mg) in a flat bottom glass vial (20 mL) washeated at 200 C for 30 min. The mixture was separated by MCI-gel CHP20P CC (2 cm i.d. × 24 cm) with H2O containing increasing proportions of MeOH (0-100%, 10% stepwise) to give gallic acid (53.8 mg), pyrogallol (6.9 mg), meta- and para-digallic acid (15) (10.5 mg) and 6 (66.0 mg), and a mixture of a polymeric substance( 46.8 mg). Gallic acid (2 mg) was heated under similar conditions and HPLC analysis of the reaction mixture showed the generation of a small amount of 15.
With dihydrogen peroxide; horseradish peroxidase; In acetone;pH 5.0;
General procedure: A: Catechol (110 mg, 1.00 mmol ) and pyrogallol (126 mg, 1.00 mmol) were dissolved in a mixture of acetone-pH 5.0 phosphate-citrate (1 : 1=0.2 M Na2HP04: 0.1 M citrate) buffer (1 :5 v/v, 5 lnL), which contained 0.1 mg horseradish peroxidase (cas 9003-99-0, 5KU, Fisher). Four aliquots of 3percent H2O2 (2 mL each) were added eveiy 10 min over 40 min while stirring. After 2-3 h, the resulting orange precipitate was filtered off, washed with water (3 x 6 mL) and dried under high vacuum condition to give a mixture of 2 and NCI35676 (purpurogallin). The mixture was purified by flash chromatography on silica gel, using ethyl acetate/hexane ( 1 :4) as eluent, to give 2 as an orange solid (23 mg, 1 1 percent). 3,4,6-trihydroxy-5H-benzo[7]annulen-5-one (2). Following the general method IA, gave 2 as an orange solid (23 mg, 11 percent). Following the general method IA, using <strong>[363-52-0]3-fluorocatechol</strong> (128 mg, 1.00 mmol) instead of catechol, gave an orange solid. The mixture was purified by flash chromatography on silica gel, using ethyl acetate/hexane (1 :4) as eluent, to give 3 as an orange solid (20 mg, 9 percent): NMR (300 MHz, DMSO) delta 15.45 (s, 1H), 9.81 (s, 2H), 7.53-7.37 (m, 2H), 7.19 (dd, J= 9.6, 0.7 Hz, 1H), 6.86 (dd, J= 11.4, 9.6 Hz, 1H); 13C NMR (75 MHz, DMSO) delta 184.20, 155.77, 154.17, 152.58, 134.70, 132.41, 124.98, 118.61, 118.17, 110.76, 110.50; HRMS (ESI): calcd for: C?H7F04 [M-H]' = 221.0255, obsd [M-H]"= 221.0255.
3,4,5-trihydroxy-N-(prop-2-yn-1-yl)benzamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With N,N-diisopropylcarbodiimide; benzotriazol-1-ol; triethylamine; In N,N-dimethyl-formamide; at 20℃; for 18h;
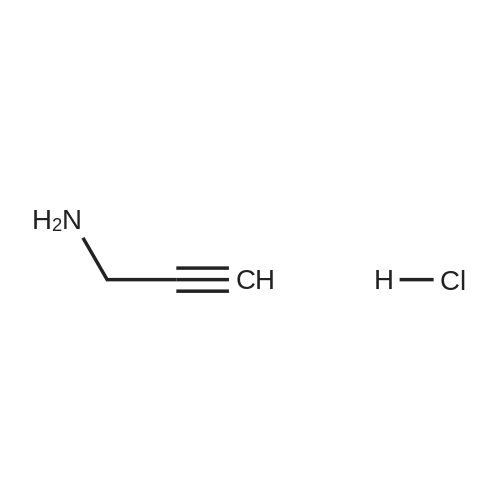
General procedure: A mixture of carboxylic acid (3 mmol) and N-hydroxybenzotriazole(1.5 mmol) was dissolved in DMF (8 mL). Propargylaminehydrochloride (3.1 mmol) and triethylamine (3 mmol) were added.The mixture was stirred at room temperature for 5 min, then N,NdiisopropylcarbodiimideHCl (DIC, 3.7 mmol)was added. After 18 hstirring, water (30 ml) was added to stop the reaction. The aqueoussolution was extracted with methylene chloride (4 8 ml) thecombined extract was placed in the refrigerator. After 12 h theformed precipitate was filtrated of, and the solvent was removed.The obtained alkyne was crystallized from ether or used withoutfurther purification.
the amphoteric surfactant (0.01 mol) and 20 mL ofmethanol were poured into a 50 mL glass reactor containinga magnetic stirring bar. Next, equimolar amount of GA dissolved in20 mL of methanol was added and the reaction was conducted for 1 h at a temperature of 25 C. Afterwards, methanol was evaporated by a rotary evaporator under vacuum and the product was driedunder vacuum for 24 h at a temperature of 25 C.
57.8 g of bismuth oxide was dissolved in 80 ml of concentrated nitric acid having a mass fraction of 68%After bismuth trioxide was dissolved, the mixture was cooled to room temperature and 50 ml of deionized water was added,To obtain bismuth nitrate solution; At 80 ,50 g of gallic acid was dissolved in 200 ml of deionized water,To obtain a gallic acid aqueous solution;To be dissolved in gallic acid,The gallic acid aqueous solution was cooled to 60 C,After which the gallic acid aqueous solution was slowly added to the bismuth nitrate solution,Insulation reaction 20min,That is yellow precipitate;And then filtered yellow to obtain a filter cake,The filter cake was washed with 50 C deionized water until the pH of the deionized water was close to 7,Followed by vacuum drying at 80 C for 3 hoursBasic bivalence of gallic acidOf the powder 100 g, the yield was 98%.
(2-fluoro-3,4-dihydroxyphenyl)(3,4,5-trihydroxyphenyl)methanone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With zinc(II) chloride; trichlorophosphate; at 80℃; for 2h;
3,4,5-trihydroxybenzoic acid, 3-fluorobenzene-l,2-diol, anhydrous zinc chloride and phosphorous oxychloride were combined in a round bottom flask, stirred and heated to 80°C for 2 hours. After cooling, the product was precipitated from the reaction mixture by addition of a mixture of water and ice. The product was washed with water, treated with a solution of sodium bicarbonate, washed again with water and finally recrystallized from aqueous solution to yield the desired product.
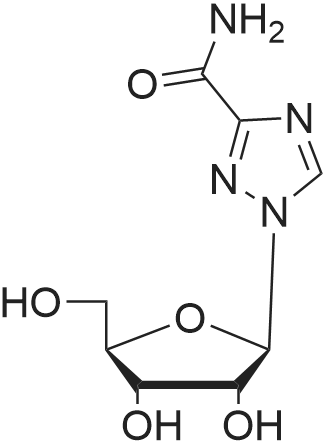
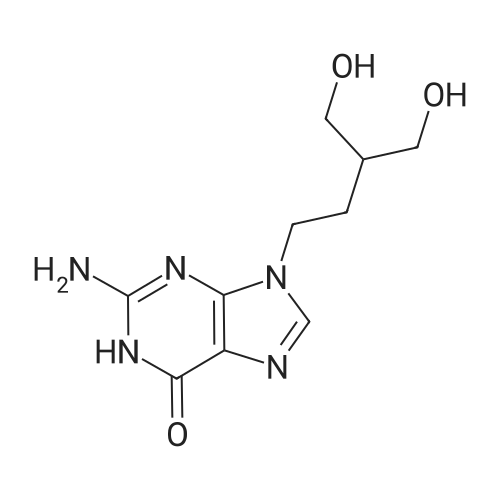
10 mg (0.027 mmol) of (S-isopropyl-2-(((5)-((((R)- 1 -(6-amino-9H-purin-9-yl)propan-2- yl)oxy)methyl)(phenoxy)phospboryl)amino)propanoate was dissolved in 0.6 mL of acetonitrile. 4.7 mg (0.027 mmol) of gallic acid was dissolved in 1.2 mL of acetonitrile. Both the solutions were mixed together and the obtained solution was left at the room temperature.During slow evaporation of the solvent a white crystalline substance was separated. The crystalline product was dried in a vacuum drier (200 mBar) at the room temperature for 2h. Melting point: 125°C (DSC). XRPD pattern: see Figure 25. IR spectrum: see Figure 27.
General procedure: The TN salts/molecular salts was preparedby grinding an equimolar mixture containing 200mg (1 mmol) of TN and 1mmol of correspondingcarboxylic acids/PTSA wetted with few drops of waterwas manually grounded in an agate mortar for 100minutes until a dried powder was obtained.
With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 48h;
A solution of gallic acid (17.65 mmol), and K2CO3 (52,83 mmol) in DMF (25 mL) were added Methyl iodide (52.83 mmol) and stirred at room temperature for 48 hours. The reaction mixture was then added 75 ml ethyl acetate and then washed with 3x25 ml saturated NaHCO3 and 2x25 ml saturated NaCl, sequently. The organic phase was then added Mg2SO4 anhydrate to take out the water residue. The filtrate was then drying to obtain the crude product. The product was purified using chromatography colomn with chloroform as mobile phase and give 3 products : 4-monomethoxymethylgallate, 4,5-dimethoxymethyl gallate, 3,4,5-trimethoxymethyl gallate.
10%; 61%; 14%
With potassium carbonate; In N,N-dimethyl-formamide; at 0 - 20℃;
General procedure: To the gallic acid solution in DMF at 0 C,K2CO3 and MeI were added. The reaction mixture is stirred at 0C and allowed to reach at room temperature overnight, with a total reaction time of 18-24 hours. The reaction mixture is then diluted byaddition of ethylacetate, and washed repeatedly with saturated NaHCO3 solution. The ethylacetate phase is then washed further with a saturated NaCl(Brine) solution to withdraw residual water, then dried with anhydrous MgSO4, filtered, and concentrated, resulting in a crude extract of methylated product. Furthermore, the crude extract is purified by silica gel chromatography column. Pure compounds were analyzed by Thin Layer Chromatography (TLC), Nuclear Magnetic Resonance Spectrometer, and High Resolution Mass Spectrometer
2,4-diamino-5-(4-chlorophenyl)-6-ethylpyrimidin-1-ium 3,4,5-trihydroxybenzoate-methanol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
at 60℃; for 4h;
In a 50mL round bottom flask, to the stir solution of pyrimethamine(300 mg, 1.2 mmol) in 10 mL of dry methanol, wasadded 3,4,5-trihydroxybenzoic acid (184 mg, 1.2 mmol). The temperaturewas increased to 60 C and stirring was continued for 4 h.The resulting reaction mixture was cooled to room temperature,transfer to 25 mL beaker and left-over night to obtain pale yellow rod shape crystals.
The preparation method of the <strong>[107-43-7]betaine</strong> gallic acid ion salt of the present embodiment,Includes the following steps:Under a nitrogen atmosphere,Dissolve 0.01 mol gallic acid in the reactor with 30 ml of methanol.Dissolve 0.01 mol of <strong>[107-43-7]betaine</strong> in 20 ml of methanol,And adding dropwise to the reactor in which gallic acid is dissolved,Heated to 60 C,Ionized to salt reaction for 12 hours;After the reaction is completed,The solution was concentrated under vacuum to 1/10 of the reaction solution, and the crystals were frozen.And filtered,Washing and separating to obtain a <strong>[107-43-7]betaine</strong> gallate ion salt crystal product,Drying in a vacuum oven for 48 hours yielded 2.68 g of <strong>[107-43-7]betaine</strong> gallate ion salt with a purity of more than 99%.Yield 93.32%
100 mg of gallic acid monohydrate was added to 59.9 mg of <strong>[60-27-5]creatinine</strong> to form 1:1 molar ratio. The mixture was subjected to manual liquid-assisted grinding using acetonitrile for 30 min using a mortar-pestle [20]. The ground materials were subjected to PXRD and FTIR to as certain the formation of multicomponent organic solids. As supersaturation,nucleation and crystal growth are the basic three steps in crystallization, about 50 mg of the sample mixture was dissolved in methanol to obtain a supersaturated solution. Thesolution was left for slow evaporation of the solvent (with partial covering) at room temperature. Colorless needles aped crystals suitable for X-ray diffraction are obtainedi n few days.
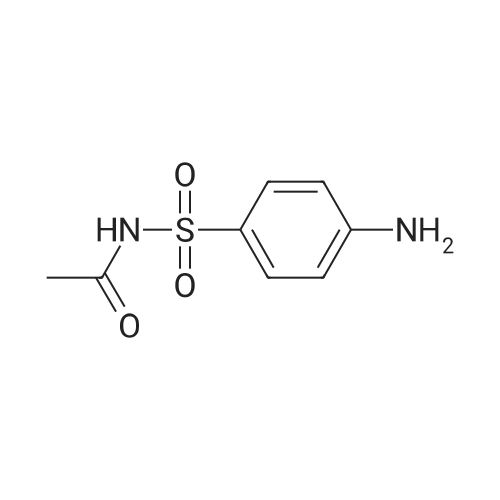
2-((4-(N-acetylsulfamoyl)phenyl)diazenyl)-3,4,5-trihydroxybenzoic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
2.142 g, 0.01 mol of N-(4-aminophenylsulfonyl) acetamidewas dissolved in 10 mL hydrochloric acid (1:1) anddiazotized in ice path at 0-4 C with a solution of sodium nitrite. The resulting diazo-solution was added drop wisewith stirring to an ice cooled solution of 1.701 g, 0.01 molof 3,4,5-trihydroxybenzoic acid [gallic acid] which was dissolved in 10 mL of 2 M sodium hydroxide. The formedprecipitate was then filtered and washed with a (1:1) mixture of ethanol:water several times and left to dry in the air(Scheme 1). A black solid with 88% yield and m.p. 210 C was obtained.
General procedure: For synthesis of I an equimolar ratio (1:1) of pyrimethamine(API) and benzoic acid (co-former) in methanol were refluxed for2 h. Single crystals were obtained after one week, by slow evaporationof solvent at room temperature.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping