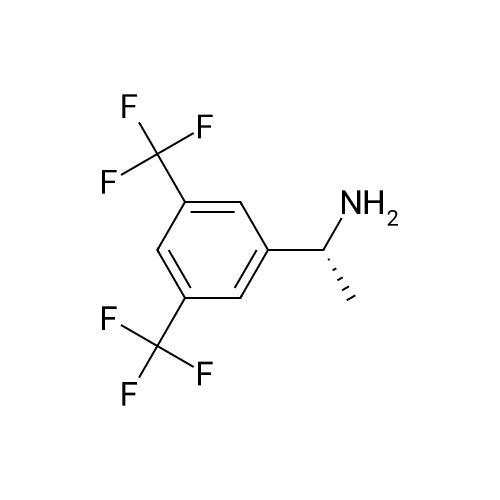
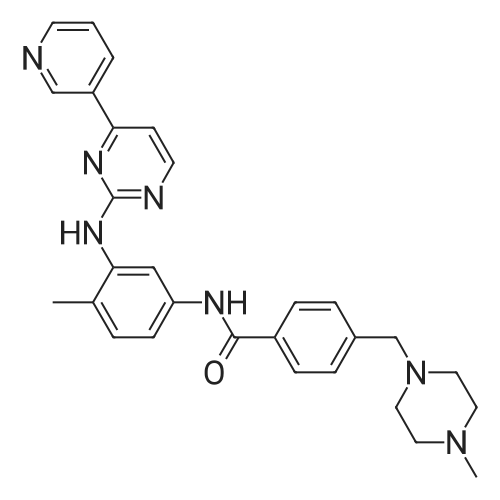
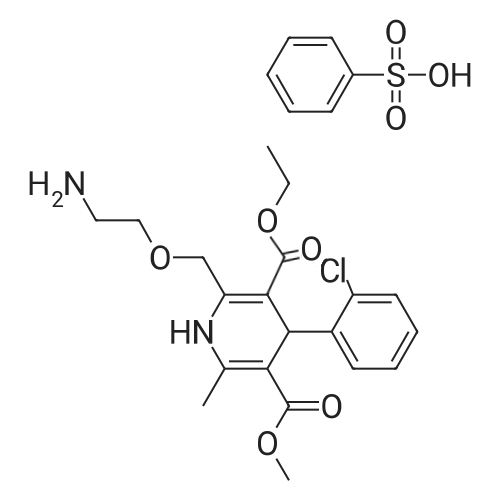
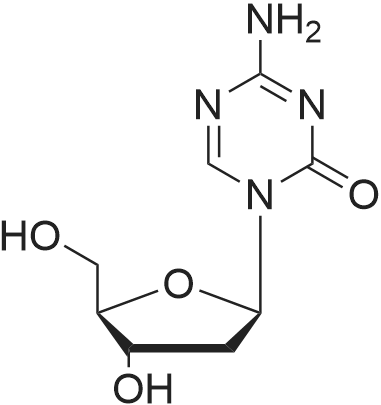
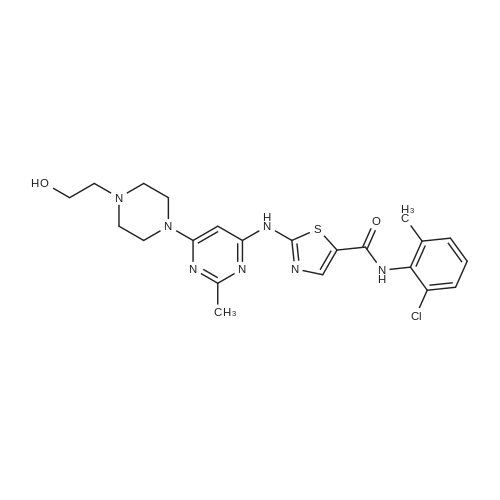
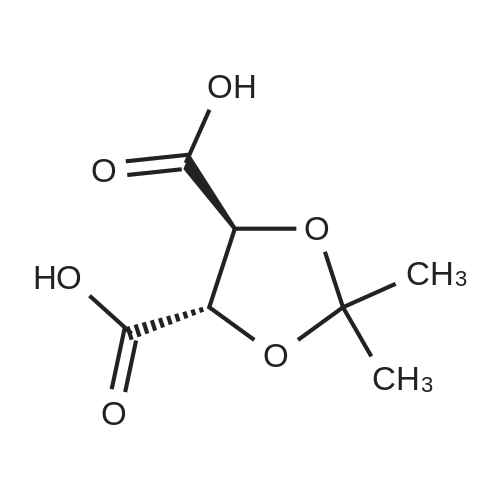
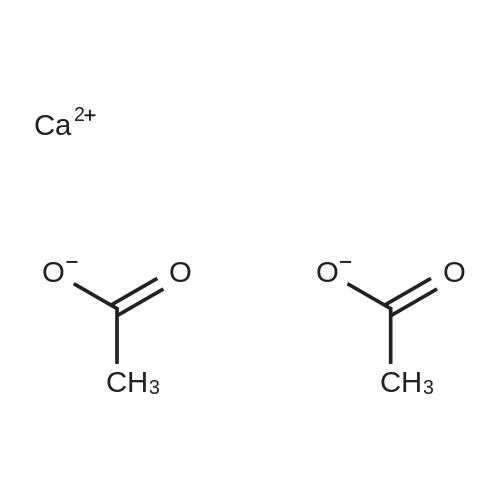
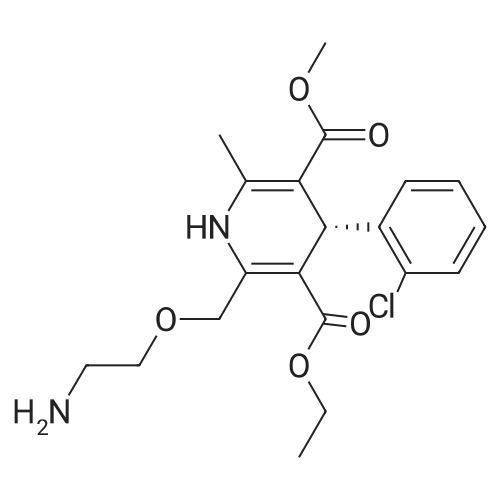
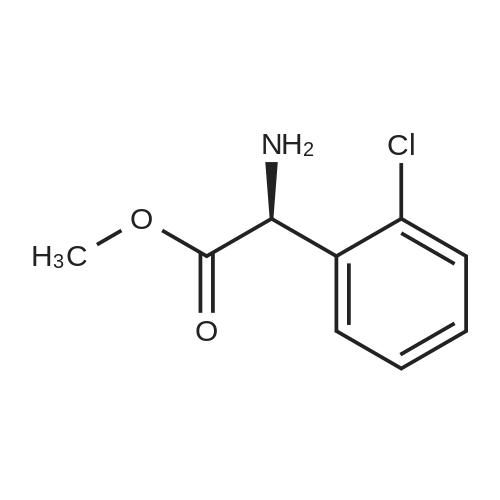
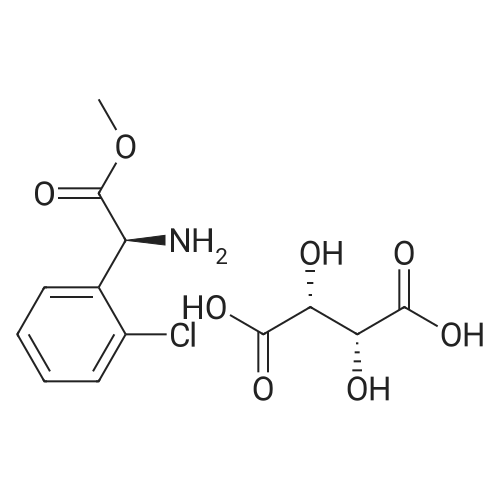
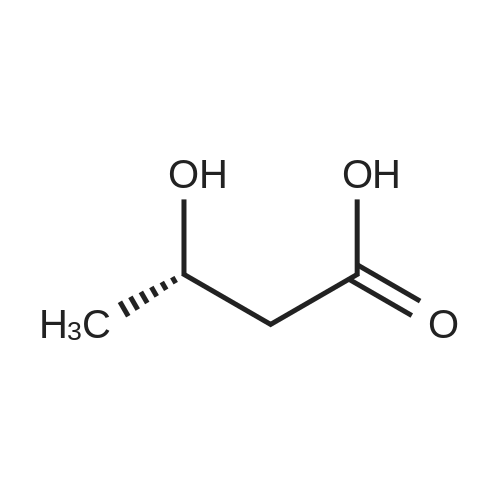
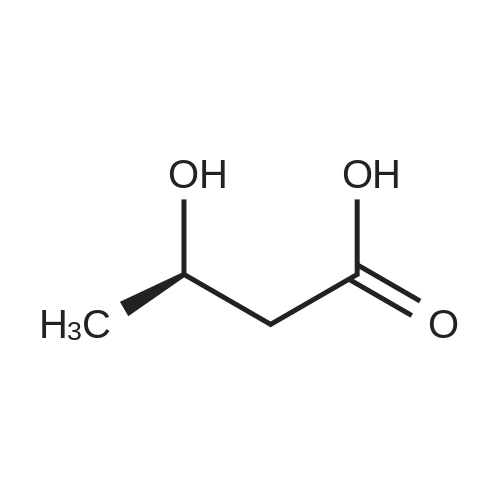
| 55% |
Stage #1: 3-(3-ethoxycarbonylmethoxy-4-methylphenyl)piperidine-1-carboxylic acid benzyl ester With hydrogen In ethanol; water for 2h;
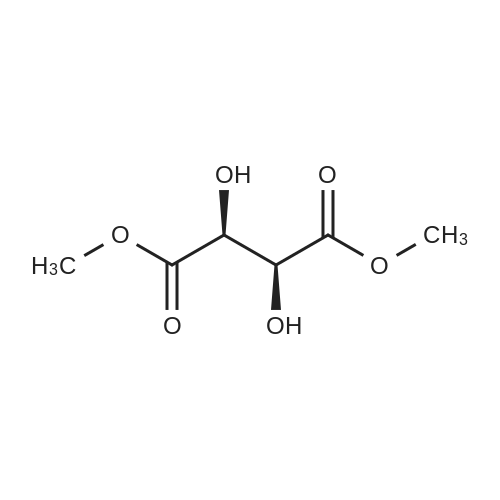
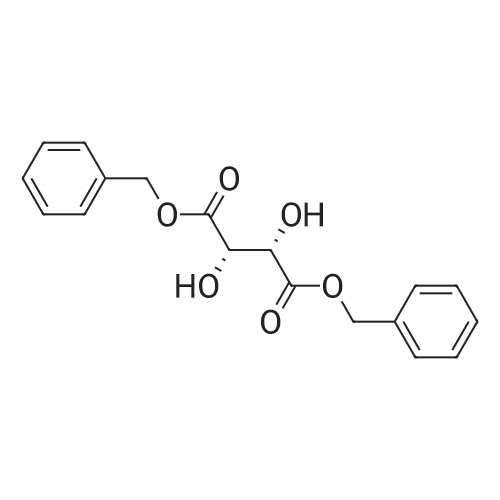
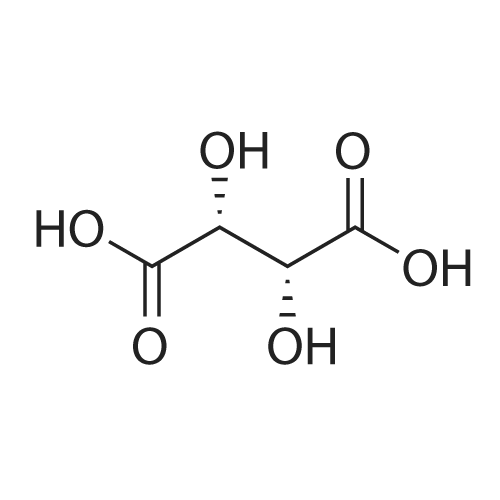
Stage #2: D-tartaric acid In ethanol at 20℃; for 24h; |
12-2 Example 12-2; (R)- (2-METHYL-5- {1- [4-METHYL-2- (4-TRIFLUOROMETHYL-PHENYL)-THIAZOLE-5-CARBONYL]- piperidin-3-yl}-phenoxy)-acetic acid
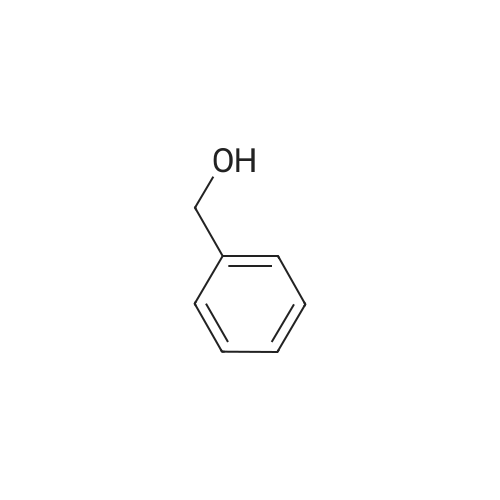
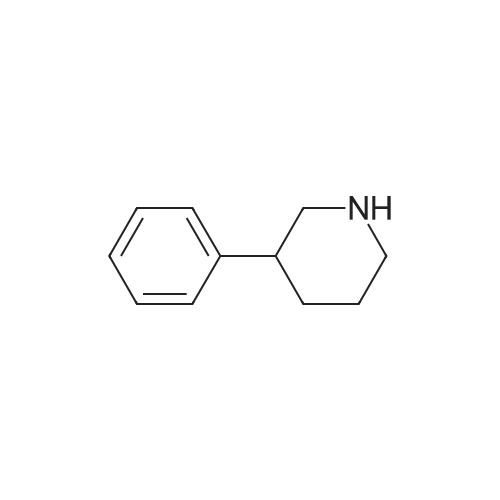
To a solution of 3- (3-hydroxy-4-methyl-phenyl)-piperidine-1-carboxylic acid benzyl ester (Example 11-2; 2.34g, 7.19 mmol) in 15 mL DIMETHYLFORMAMIDE was added cesium carbonate (4.69g, 14.38 mmol) and ethyl bromoacetate (1.60 mL, 14.38 MMOL). The mixture was heated to 60°C under N2 with stirring for 3 h and cooled to ambient temperature. The resultant brown suspension was diluted with 300 mL water and extracted with diethyl ether (2 x 200 mL). The organic phases were combined and dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The resultant oil was flash chromatographed with 15% ethyl acetate/hexanes to yield 1.78 g (60%) of the desired 3- (3-ETHOXYCARBONYLMETHOXY-4- methyl-phenyl)-piperidine-1-carboxylic acid benzyl ester as a clear oil. MS (LC-MS) 462.1 (M + NA) +. H NMR (400 MHz, CDCI3) 8 7.35 (m, 5H), 7.09 (d, 1 H), 6.76 (d, 1 H), 6.56 (s, 1H), 5.14 (m, 2H), 4.61 (s, 2H), 4.23 (m, 2H), 2.78 (q, 2H), 2.62 (m, 1H), 2.25 (s, 3H), 1.98 (m, 1H), 1.76 (m, 1H), 1.56 (m, 3H), 1.29 (t, 3H). A 250 mL Parr bottle was charged with 0.18g of 10% palladium on carbon (50% water) and covered with 20 mL ethanol. 3- (3-ETHOXYCARBONYLMETHOXY-4-METHYL- PHENYL)-PIPERIDINE-1-CARBOXYLIC acid benzyl ester (1.78g, 4.33 mmol) was dissolved in 50 mL ethanol and added to the catalyst suspension. The reaction was hydrogenated at 50 psi for 2 h. The catalyst was filtered through a celite plug. The filter cake was washed with 150 mL ethanol and the filtrated concentrated under reduced pressure. The resultant oil was taken up in 20 mL hot ethanol to which was added D-tartaric acid (650 mg, 4.33 mmol) in 10 mL hot ethanol. The solution was allowed to stir 24 h at ambient temperature. The white crystalline precipitate was collected by filtration to yield 1. 014G (55%) of (2-methyl-5-piperidin-3-yl-phenoxy)- acetic acid ethyl ester D-tartaric acid salt as a white crystalline solid. MS (LC-MS) 278.3 (M + H) +. 1 HNMR (400 MHz, DMSOd6) 8 7. 11 (d, 1 H), 6.78 (d, 1 H), 6.44 (s, 1 H), 4.16 (q, 2H), 3.81 (s, 2H), 3.21 (t, 2H), 2.78 (m, 2H), 2.10 (s, 3H), 1.81 (m, 2H), 1.69 (m, 1H), 1.56 (m, 1H), 1.51 (s, 6H), 1.14 (t, 3H). HPLC analysis : Chiralpak AD, 1 mL/min, 5% isopropanol/heptane 0.2% diethylamine, rt = 3.18. ee = 98.9. (2-METHYL-5-PIPERIDIN-3-YL-PHENOXY)-ACETIC acid ethyl ester D-tartaric acid salt was carried on using procedures analogous to those described in Example 12 to give the title compound, (R)- (2-METHYL-5- {1- [4-METHYL-2- (4-TRIFLUOROMETHYL-PHENYL)-THIAZOLE- 5-CARBONYL]-PIPERIDIN-3-YL}-PHENOXY)-ACETIC acid. MS (LC-MS) 450.1 (M-H)-. 1H NMR (400 MHz, CD30D) 8 8.13 (d, 2H), 7.78 (d, 2H), 7.06 (m, 1H), 6. 75 (brm, 2H), 4.66 (brs, 2H), 2.76 (t, 1 H), 2.48 (s, 3H), 2.20 (s, 3H), 2.04 (d, 1H), 1.83 (m, 2H), 1.67 (m, 1H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping