* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With hydrogenchloride In water; ethyl acetate at 20℃; for 0.25 h;
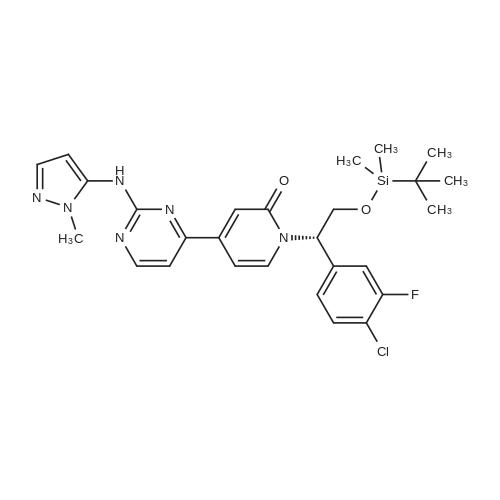
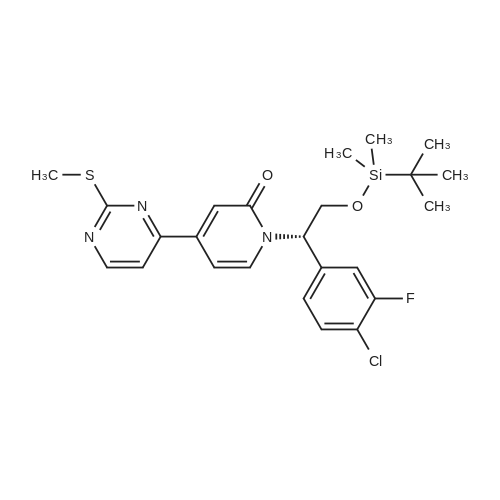
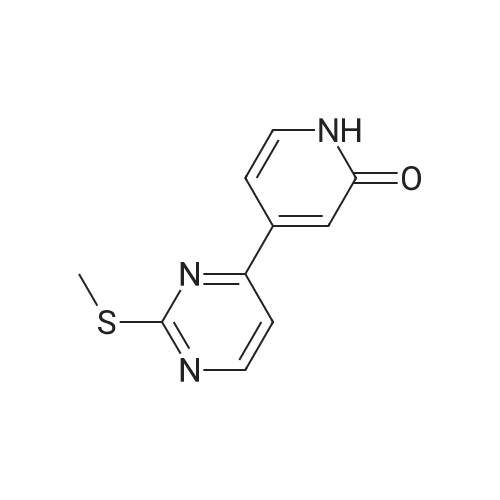
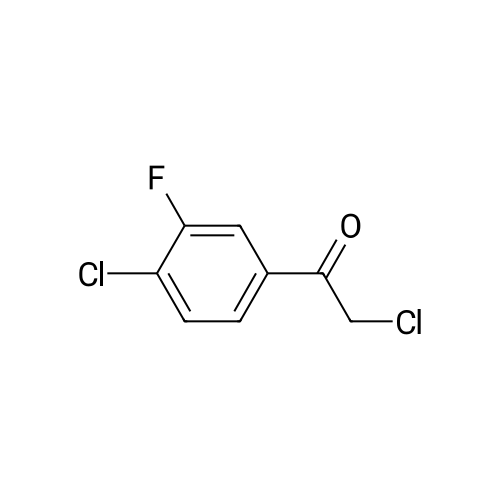
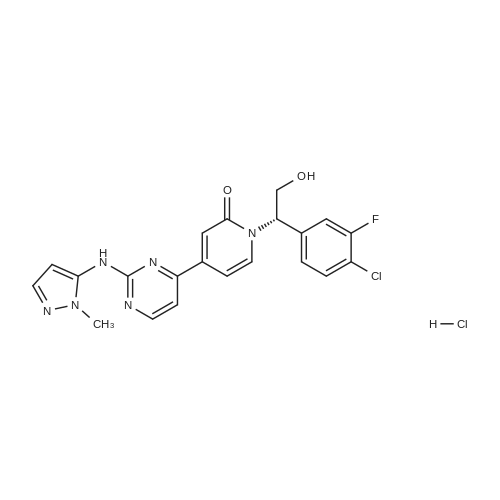
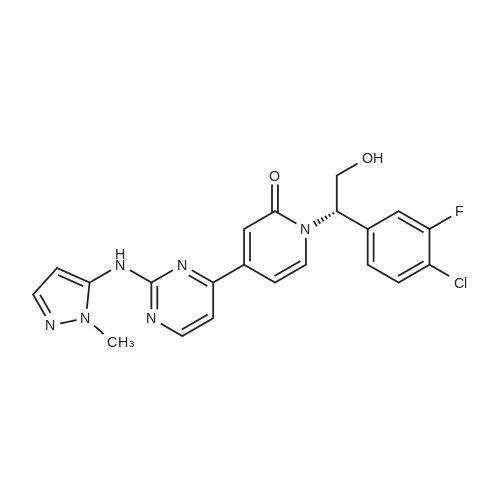
(003991 Step B: Crude (5)-i -(2-((tert-butyldimethylsilyl)oxy)- 1 -(4-chloro-3 -fluorophenyl)ethyl)-4-(2-(( 1-methyl-i H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2( 1 H)one (48 mg) was dissolved in ethyl acetate (4 mL) and treated dropwise slowly (over 2 minutes) with an ethyl acetate solution (1.0 mL, which had been saturated with HCI gas). The reaction stirred at room temperature for 15 minutes, after which time LCMS indicated complete consumption of the starting material. The reaction mixture was concentrated to an oily residue and purified by prep RP HPLC to yield (S)-1-(l-(4-chloro-3-fluorophenyl)-2- hydroxyethyl)-4-(2-(( i-methyl-i H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2( 111)-one (20.8 mg, 54.6percent yield) as a lyophilized powder. ‘H NMR (400 MHz, (CD3)2S0) 6 9.58 (s, iH), 8.60 (d, J = 5.1 Hz, iH), 7.91 (t, J = 9.0 Hz, 1H),7.58 (t, J = 8.1 Hz, iH), 7.52-7.41 (m, 2H), 7.37 (d, J = 1.8 Hz, 1H), 7.14 (dd, J = 10.7,5.1 Hz 2H), 6.86 (dd, J = 7.3, 1.8 Hz, 1H), 6.27(d, J = 1.7 Hz, 1H), 5.97 (dd, J 7.7, 5.7 Hz, 1H), 5.31(t, J 5.2 Hz, ill), 4.15 (m, iH),4.10-3.95 (m,1H), 3.69 (s, 3H); LCMS m/z 441 (M+H)+.
Reference:
[1] Patent: WO2013/130976, 2013, A1, . Location in patent: Paragraph 00399
[2] Journal of Medicinal Chemistry, 2016, vol. 59, # 12, p. 5650 - 5660
[3] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[4] Patent: US2017/22183, 2017, A1, . Location in patent: Paragraph 0143; 0161
2
[ 1453851-69-8 ]
[ 1453848-26-4 ]
Reference:
[1] Patent: WO2013/130976, 2013, A1,
[2] Journal of Medicinal Chemistry, 2016, vol. 59, # 12, p. 5650 - 5660
[3] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[4] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[5] Patent: US2017/22183, 2017, A1,
3
[ 1453854-84-6 ]
[ 1453848-26-4 ]
Reference:
[1] Patent: WO2013/130976, 2013, A1,
[2] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[3] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[4] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[5] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[6] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[7] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[8] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[9] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[10] Patent: US2017/22183, 2017, A1,
4
[ 1395078-43-9 ]
[ 1453848-26-4 ]
Reference:
[1] Patent: WO2013/130976, 2013, A1,
[2] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[3] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[4] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[5] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[6] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[7] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[8] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[9] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[10] Patent: US2017/22183, 2017, A1,
5
[ 1453851-59-6 ]
[ 1453848-26-4 ]
Reference:
[1] Patent: WO2013/130976, 2013, A1,
[2] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[3] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[4] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[5] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[6] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[7] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[8] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[9] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[10] Patent: US2017/22183, 2017, A1,
6
[ 1453851-79-0 ]
[ 1453848-26-4 ]
Reference:
[1] Patent: WO2013/130976, 2013, A1,
[2] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[3] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[4] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[5] Organic Process Research and Development, 2017, vol. 21, # 3, p. 387 - 398
[6] Patent: US2017/22183, 2017, A1,
With hydrogenchloride; In water; ethyl acetate; at 20.0℃; for 0.25h;
(003991 Step B: Crude (5)-i -(2-((tert-butyldimethylsilyl)oxy)- 1 -(4-chloro-3 -fluorophenyl)ethyl)-4-(2-(( 1-methyl-i H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2( 1 H)one (48 mg) was dissolved in ethyl acetate (4 mL) and treated dropwise slowly (over 2 minutes) with an ethyl acetate solution (1.0 mL, which had been saturated with HCI gas). The reaction stirred at room temperature for 15 minutes, after which time LCMS indicated complete consumption of the starting material. The reaction mixture was concentrated to an oily residue and purified by prep RP HPLC to yield (S)-1-(l-(4-chloro-3-fluorophenyl)-2- hydroxyethyl)-4-(2-(( i-methyl-i H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2( 111)-one (20.8 mg, 54.6% yield) as a lyophilized powder. ?H NMR (400 MHz, (CD3)2S0) 6 9.58 (s, iH), 8.60 (d, J = 5.1 Hz, iH), 7.91 (t, J = 9.0 Hz, 1H),7.58 (t, J = 8.1 Hz, iH), 7.52-7.41 (m, 2H), 7.37 (d, J = 1.8 Hz, 1H), 7.14 (dd, J = 10.7,5.1 Hz 2H), 6.86 (dd, J = 7.3, 1.8 Hz, 1H), 6.27(d, J = 1.7 Hz, 1H), 5.97 (dd, J 7.7, 5.7 Hz, 1H), 5.31(t, J 5.2 Hz, ill), 4.15 (m, iH),4.10-3.95 (m,1H), 3.69 (s, 3H); LCMS m/z 441 (M+H)+.
With hydrogenchloride; In methanol; at 45.0℃;Large scale;
Step 2: To the methanolic (S)-1-(2-((tert-butyldimethylsilyl)oxy)-1-(4-chloro-3-fluorophenyl)ethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one (IX) solution in MeOH was added HCl (10.7 kg, 1.25 M in MeOH) at RT. It was slightly exothermic. After the addition was completed, the reaction was heated to 45 C. If the reaction was incomplete after 14 to 16 h, additional HCl (1 kg, 1.25 M in MeOH) was added and agitation at 45 C. was continued for 2 h. The reaction was equipped with a distillation setup with acid scrubber. The reaction was concentrated to between 20 and 30 L under a vacuum below 50 C. To the resulting solution was added MeOH (35 kg) and the reaction was concentrated to 20 to 30 L again under a vacuum below 50 C. The solvent was then switched to EtOAc using 40 kg of EtOAc. The solvent ratio was monitored by Headspace GC and the solvent swap continued until it was less than 1/5. The solution was concentrated to between 20 and 30 L under a vacuum below 50 C. After the solution was cooled below 30 C., aqueous NaHCO3 (1.2 kg of NaHCO3 and 20 kg of water) was added slowly with a medium agitation and followed by EtOAc (40 kg). The organic layer was washed with water (2×10 kg) then concentrated to 20-30 L under a vacuum below 50 C. The solvent was then switched to MEK using 35 kg of MEK. The residue MeOH was monitored by Headspace GC and the solvent swap continued until the MeOH was <0.3%. The solution containing (S)-1-(1-(4-chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one (VIII) was concentrated to 20 to 30 L under a vacuum below 50 C. for the next step.
(S)-1-(1-(4-chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one benzenesulfonate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In water; butanone; at 75.0℃;Large scale;
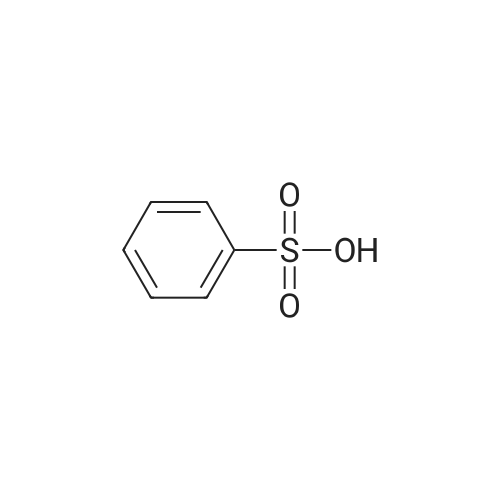
Step 3: The solution of VIII in MEK was transferred to a second 100 L cylindrical reaction vessel through a 1 mum line filter. In a separate container was prepared benezenesulfonic acid solution (1.3 kg of benzenesulfonic acid, 1.4 kg of water and 4.4 kg of MEK). The filtered VIII solution was heated to 75 C. and to the resulting solution was added 0.7 kg of the benzenesulfonic acid solution through a 1 mum line filter. The clear solution was seeded with crystalline benzenesulfonic acid salt of VIII (0.425 kg) as a slurry in MEK (0.025 kg of VIIIb crystalline seed and 0.4 kg of MEK) which produced a thin slurry. The remaining benzenesulfonic acid solution was then added through a 1 mum line filter in 2 h. After addition, the slurry was heated at 75 C. for additional 1 h and then cooled to 18 C. in a minimum of 3 h. The resulting thick slurry was agitated at 20 C. for 14 to 16 h. The solid was filtered using an Aurora dryer. The mother liquor was assayed by HPLC (about 0.3% loss). The solid was then washed with 1 mum line filtered 15.8 kg of MEK and water solution (0.8 kg of water and 15 kg of MEK) and followed by 1 mum line filtered 30 kg of MEK. Washes were assayed by HPLC (<1% loss). The wet cake was dried under a vacuum and a nitrogen sweep at a jacket temperature of 45 C. for a minimum 12 h to afford the benzenesulfonic acid salt of VIII, which is labeled VIIIb
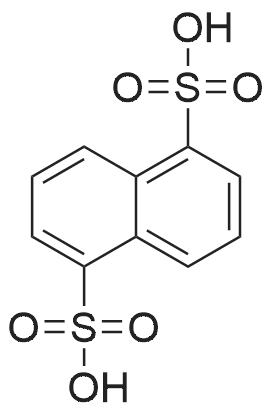
(S)-1-(1-(4-chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one 1,5-naphthalenedisulfonic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In methanol; butanone;
<strong>[1453848-26-4](S)-1-(1-(4-chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one</strong> (21.8 mg, 0.0494 mmol) was dissolved in MEK (0.5 mL). 1,5-naphthalenedisulfonic acid tetrahydrate (25.1 mg, 0.0871 mmol) was dissolved in methanol (1.0 mL) and about 0.36 mL of the solution was added drop wise to the free base solution with stirring. Precipitation occurred. The suspension was allowed to slowly evaporate until only a trace of solvent remained. The solid was vacuum dried at 40 C. using house vacuum.
(S)-1-(1-(4-chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one p-toluenesulfonic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In isopropyl alcohol;Sonication;
Synthesis of <strong>[1453848-26-4](S)-1-(1-(4-chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one</strong>, tosylate IPA solvate and <strong>[1453848-26-4](S)-1-(1-(4-chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one</strong>, tosylate Form A <strong>[1453848-26-4](S)-1-(1-(4-chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one</strong> (105.1 mg, 0.239 mmol) was mostly dissolved in isopropanol (1 mL) using sonication. p-Toluenesulfonic acid monohydrate (97.5% pure, 52.9 mg, 0.271 mmol) was dissolved in isopropanol (1 mL). The toluenesulfonic acid solution was added drop wise to the free base solution with stirring to give a yellow solid. Additional isopropanol was added (1 mL). The the solid was isolated by filtration and the reactor and solids were rinsed with 1 mL isopropanol. The solid was analyzed by XRPD in a holder open to the atmosphere while still wet with solids to give a disordered <strong>[1453848-26-4](S)-1-(1-(4-chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one</strong>, tosylate IPA solvate. TG analysis was conducted on the XRPD sample. The remaining solid was dried at 60 C. under a vacuum for 4 days to give <strong>[1453848-26-4](S)-1-(1-(4-chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one</strong>, tosylate crystalline Form A.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping