* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
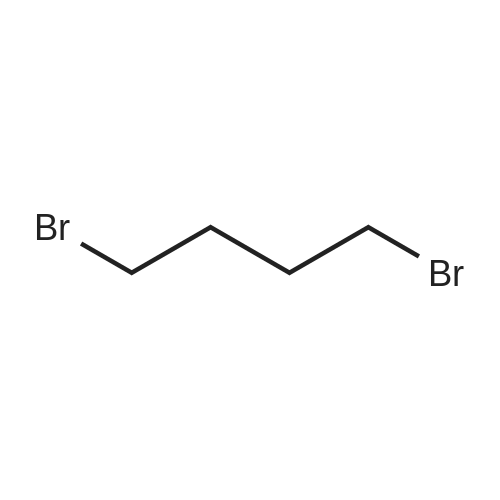
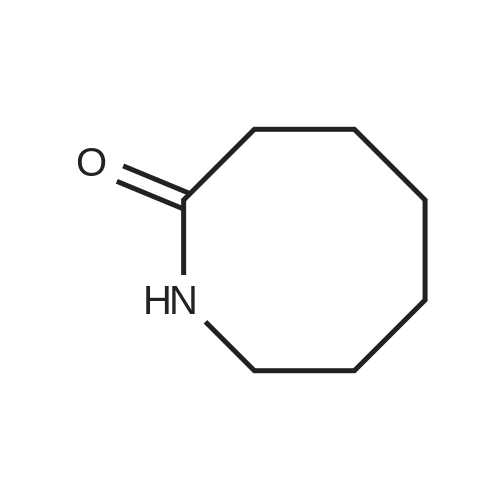
REFERENCE EXAMPLE 5 To a solution of cyclohexane-1,2-dicarboximide (10 g; 65.3 mmol) in anhydrous dimethylformamide (50 ml), there was added 60% sodium hydride (2.6 g; 68.5 mmol) under nitrogen at room temperature, and the resultant mixture was stirred at the same temperature for 3 hours. Tetramethylene bromide (70.5 g; 0.327 mol) was added thereto, and the mixture was stirred for 3 hours. After completion of the reaction, the resultant mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with water twice and with a saturated sodium chloride solution. The organic layer was dried over anhydrous magnesium sulfate, and the solvent and excessive tetramethylene bromide were removed by evaporation to give N-(4-bromobutyl)cyclohexane-1,2-dicarboximide. IR numaxFilm (cm-1): 1770, 1700.
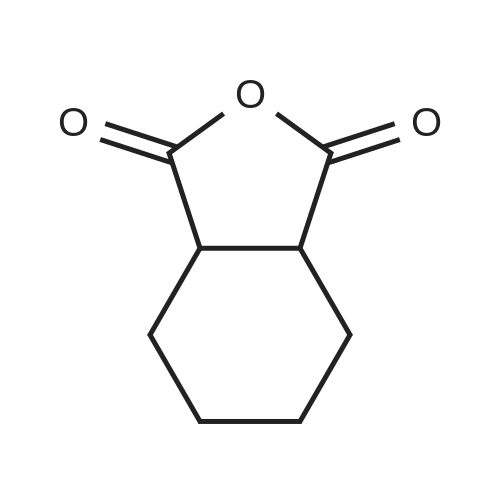
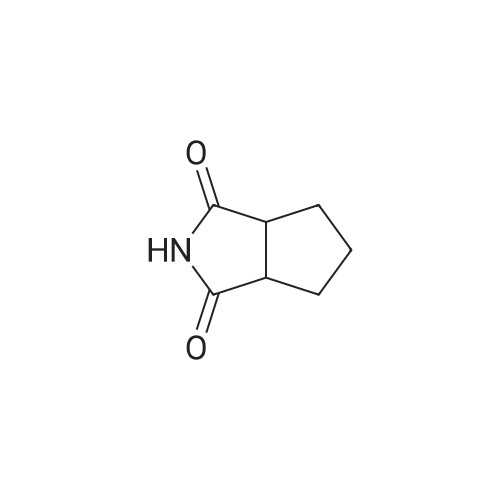
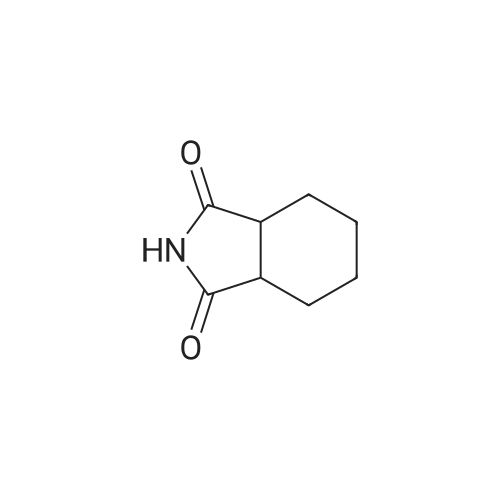
REFERENCE EXAMPLE 4 A mixture of 1,2-cyclohexanedicarboxylic anhydride (3 g; 19.5 mmol) and 29% aqueous ammonia (3.4 g) was heated and kept at an inner temperature of 180-190 C. for 2 hours to give cyclohexane-1,2-dicarboximide. M.P. 132-136 C.
With ammonia; In water; at 180℃; for 1.0h;
General procedure: a) for compounds of formula (V) with G moiety, wherein L represents C=0, E, if present, is different from O, NH or N-(d-C3)alkyl (imide derivatives) To the appropriate dicarboxylic acid (Via) or acid anhydride (VIb) (0.05 mol) 25% aqueous ammonia (equivalent to 0.05 mol ammonia) and distilled water (10 ml) were added. The mixture was placed in a sand bath and heated until the water evaporates, and then it was heated for 1 hour at 180C. Crude products were purified by crystallization from methanol or 2-propanol.
REFERENCE EXAMPLE 3 A mixture of cis- 4-tetrahydrophthalimide (20 g; 0.132 mol), 50% water-containing 5% palladium-carbon (2 g) and tetrahydrofuran (200 ml) was halogenated at room temperature for 8 hours. Precipitates were removed by filtration, and the filtrate was evaporated under reduced pressure to give cyclohexane-1,2-dicarboximide. IR numaxFilm (cm-1): 1760, 1720, 1700.
With hydrogen;palladium 10% on activated carbon; In methanol; under 15514.9 Torr; for 24.0h;
Step A: Hexahydro-1 H-isoindole-1 ,3(2H)-dioneA solution of (3aR,7aS)-3a,4,7,7a-tetrahydro-1 H-isoindole-1 ,3(2H)-dione (3.0 g, 9.9 mmol) and 10% Pd/C (300 mg) in eOH (100 mL) was shaken in an H2 atmosphere (300 psi) in a Parr apparatus for 24h. The reaction mixture was filtrated and the liquids concentrated to dryness, affording the titie compound (2.6 g, 16.99 mmol), that was sued without any further purification, as a white powder.1 H-NMR (CDCI3) delta: 8.16 (br. s, 1 H); 2.73-3.05 (m, 2H); 1.64-1.98 (m, 4H),1.40-1.64 (m, 4H).
With hydrogen;palladium 10% on activated carbon; In methanol; under 15514.9 Torr; for 24.0h;
Step A: Hexahydro-1H-isoindole-1,3(2H)-dione A solution of (3aR,7aS)-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione (3.0 g, 19.9 mmol) and 10% Pd/C (300 mg) in MeOH (100 mL) was shaken in an H2 atmosphere (300 psi) in a Parr apparatus for 24h. The reaction mixture was filtrated and the liquids concentrated to dryness, affording the title compound (2.6 g, 16.99 mmol), that was sued without any further purification, as a white powder. 1H-NMR (CDCl3) delta. 8.16 (br. s, 1H); 2.73-3.05 (m, 2H); 1.64-1.98 (m, 4H), 1.40-1.64 (m, 4H).
N-[(2-chloromethyl)cyclopropylmethyl]cyclohexane-1,2-dicarboximide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With potassium iodide; potassium carbonate; In chloroform; acetonitrile;
A mixture of the compound (8) (0.3 g; 2.2 mmol), cyclohexane-1,2-dicarboximide (253) (66 rag; 0.43 mmol), potassium carbonate (0.3 g; 2.2 mmol), potassium iodide (0.3 g; 1.8 mmol) and acetonitrile (20 ml) is refluxed for 5 hours, and the reaction mixture is, after cooling, concentrated under reduced pressure. Chloroform is added to the residue, which is washed with water, dried, concentrated under reduced pressure and chromatographed on a silica gel column to give the objective compound (Compound No. 202) (0.11 g).
With N,N-dimethylamino-pyridine; In dichloromethane; water; acetone;
EXAMPLE 7 Preparation of 2,4,6-tris (hexahydrophthalimido)-1,3,5-triazine A mixture of <strong>[1444-94-6]hexahydrophthalimide</strong> (1.0 g; 6.54 mmol), methylene chloride (10 ml) and N,N-dimethylaminopyridine (DMAP) (0.8 g; 6.56 mmol) was stirred under nitrogen at room temperature for 0.5 hours. A solution of acetone (5 ml) and cyanuric chloride (0.4 g; 2.17 mmol) was added dropwise to the mixture over a 1 hour period. A thick slurry was formed and stirred overnight. The reaction mixture was then diluted with methylene chloride, followed by the addition of water to dissolve the solids. The biphasic mixture was washed with water in a sepratory funnel. The organic layer was separated, dried over Na2 SO4 and concentrated under reduced pressure. A pale yellow semi-solid was obtained. The solid was subjected to thin layer chromatography (ethyl acetate eluent) and the title compound was separated and identified by 13 C NMR.
STR75 To a mixture of 60% sodium hydride (0.12 g) and dimethylformamide (5 ml), cyclohexane-1,2-dicarboximide (0.5 g) was portionwise added. A solution of 1-(3,4-epoxybutyl)-4-(6-fluoro-1,2-benzisoxazol-3-yl)piperidine (0.8 g) in dimethylformamide (10 ml) was dropwise added thereto at room temperature, and the resultant mixture was kept at an inner temperature of 90 to 100 C. for 16.5 hours. Insoluble materials were removed by filtration, and the residue was combined with water and extracted with ethyl acetate. The organic layer was washed with a saturated sodium chloride solution, dried over magnesium sulfate and concentrated under reduced pressure. The resulting crude crystals were recrystallized from a mixture of isopropyl ether and isopropanol to give the objective compound (0.6 g). Yield, 49% M.P., 128 -130 C.
With hydrogenchloride; In N-methyl-acetamide; mineral oil;
EXAMPLE 2 To a solution of cyclohexane-1,2-dicarboximide (6.50 g) in anhydrous dimethylformamide (65 ml), a 60% mineral oil suspension of sodium hydride (1.60 g) was added at room temperature while stirring, and the resultant mixture was continuously stirred for 30 minutes. A solution of 3-[4-(2-pyrimidinyl)piperazinyl]propyl chloride (11.30 g) in anhydrous dimethylformamide (30 ml) was dropwise added thereto, and the resultant mixture was stirred at room temperature for 4.5 hours. The solvent was removed under reduced pressure, and the residue was purified by chromatography to give an oily substance, which was then treated with hydrogen chloride to give N-[3-{4-(2-pyrimidinyl)-1-piperazinyl}propyl]cyclohexane-1,2-dicarboximide hydrochloride. M.P., 180-182 C.
1-(3,4-epoxybutyl)-4-(4-fluorobenzoyl)piperidine[ No CAS ]
[ 1444-94-6 ]
[ 116363-96-3 ]
Yield
Reaction Conditions
Operation in experiment
10.2%
In N-methyl-acetamide; chloroform;
EXAMPLE 3 N-[4-{4-(4-Fluorobenzoyl)piperidinyl}-2-hydroxybutyl]cyclohexane-1,2-dicarboximide (Compound No. 3) STR81 To a mixture of 60% sodium hydride (0.26 g) and dimethylformamide (20 ml), cyclohexane-1,2-dicarboximide (1.13 g) was gradually added. A mixture of 1-(3,4-epoxybutyl)-4-(4-fluorobenzoyl)piperidine (1.7 g) and dimethylformamide (50 ml) was dropwise added thereto at room temperature, and the resultant mixture was kept at an inner temperature of 90 to 100 C. for 3 hours. Insoluble materials were removed by filtration, and the filtrate was distilled to eliminate dimethylformamide. The residue was dissolved in chloroform, washed with water, dried over magnesium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give the objective compound (0.27 g). Yield, 10.2%. M.P., 129-130 C.
1-(2-hydroxyethyl)-2-methyl-5-nitroimidazole methanesulfonate[ No CAS ]
[ 67-63-0 ]
[ 1444-94-6 ]
[ 59068-40-5 ]
Yield
Reaction Conditions
Operation in experiment
With potassium hydroxide; In N-methyl-acetamide; methanol;
EXAMPLE 18 A solution of 8 parts of potassium hydroxide pellets in 80 parts of warm methanol is added to a warm solution of 20 parts of cyclohexane-1,2-dicarboximide in dimethylformamide. The mixture is diluted with about 160 parts of 2-propanol and then concentrated under reduced pressure to a volume of about 100 parts. This solution is divided into two equal portions and, to one portion, there is added a solution of 16.2 parts of 1-(2-hydroxyethyl)-2-methyl-5-nitroimidazole methylsulfonate in 95 parts of hot dimethylformamide. The mixture darkens and is heated on a steam bath for 16 minutes and then allowed to cool for 30 minutes. The resulting mixture is diluted with cold water to a volume of 500 parts. It is then extracted twice with toluene and the combined toluene extracts are washed once with water and dried over sodium sulfate and the solvent is evaporated under reduced pressure to leave an orange-yellow syrup which crystallizes slowly. This is digested at room temperature with about 50 parts of 2-propanol and the insoluble crystalline material is separated by filtration, washed, and dried. The product thus obtained is N-[2-(2-methyl-5-nitro-1-imidazolyl)ethyl]cyclohexane-1,2-dicarboximide melting at about 88-90 C.
With N,O-bis-(trimethylsilyl)-acetamide; In acetonitrile;
A. A mixture of 30.2 g (198 mmoles) of <strong>[1444-94-6]hexahydrophthalimide</strong>, 60 ml (246 mmoles) of N,O-bis(trimethylsilyl)acetamide and 240 ml of dry acetonitrile was stirred for 16 hours at room temperature and then evaporated to dryness. The residue was crystallized from dry n-heptane; yield: 20.7 g (92 mmoles) of N-trimethylsilyl<strong>[1444-94-6]hexahydrophthalimide</strong> (46%).
Step B: Octahydro-1 H-isoindoleA solution of hexahydro-1 H-isoindole-1 ,3(2H)-dione (1.0 g, 6.5 mmoi) in dry THF (14 mL) was added dropwise to a stirred slurry of LiAIH4 in dry THF (6 mL) at such a rate that the solvent gentle ref.uxed. The resulting reaction mixture was heated to reflux for 20h and then allowed to cool to RT. The reaction mixture was cooled with an ice-bath, and H20 (1 mL), a 20% aqueous solution of NaOH (2.5 mL) and H20 (2 mL) were sequentially added. The mixture was filtered and THF removed under reduced pressure. The aqueous phase was extracted with Et20, and the separated organics were dried over Na2S04, filtered and concentrated to dryness, affording the title compound (690 mg, 5.52 mmol) as a white powder that was used in the next step without any further purification.LC- S m/z (ESI+): 126.1 (MH+), Rt= 0.80 min (method E).
Step B: Octahydro-1 H-isoindole A solution of <strong>[1444-94-6]hexahydro-1H-isoindole-1,3(2H)-dione</strong> (1.0 g, 6.5 mmol) in dry THF (14 mL) was added dropwise to a stirred slurry of LiAIH4 in dry THF (6 mL) at such a rate that the solvent gentle refluxed. The resulting reaction mixture was heated to reflux for 20h and then allowed to cool to RT. The reaction mixture was cooled with an ice-bath, and H2O (1 mL), a 20% aqueous solution of NaOH (2.5 mL) and H2O (2 mL) were sequentially added. The mixture was filtered and THF removed under reduced pressure. The aqueous phase was extracted with Et2O and the separated organics were dried over Na2SO4, filtered and concentrated to dryness, affording the title compound (690 mg, 5.52 mmol) as a white powder that was used in the next step without any further purification. LC-MS m/z (ESI+): 126.1 (MH+), Rt = 0.80 min (method E).
(Z)-2-(2-phenoxy-3-phenylallyl)hexahydro-1H-isoindole-1,3(2H)-dione[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
74%
With 1,1'-bis-(diphenylphosphino)ferrocene; bis(eta3-allyl-mu-chloropalladium(II)); caesium carbonate; In toluene; at 80℃; for 16.0h;Inert atmosphere;
General procedure: The reaction conditions and results are shown in Tables 1-4. A typical procedure isgiven for the reaction of 2-fluorocinnamyl acetate (1a), succinimide (2a), and phenol(3a) (Table 1, Entry 7). To a solution of [Pd(pi-allyl)Cl]2 (2.4 mg, 0.0065 mmol),DPPF (7.2 mg, 0.013 mmol), succinimide (2a) (58 mg, 0.59 mmol), phenol (3a) (49 mg0.52 mmol), and Cs2CO3 (169 mg, 0.52 mmol) in Toluene (0.8 mL) was added(Z)-2-fluoro-3-phenylallyl acetate (1a) (25 mg, 0.13 mmol) at room temperature, thenstirred at 80 C for 16 h. The reaction mixture was quenched with H2O, and extractedwith diethyl ether (3 x 2 mL). The combined organic layers were dried over MgSO4and concentrated in vacuo. The residue was chromatographed on silica gel(hexane/EtOAc = 60/40) to give 34 mg (84%) of 4aa as a mixture of two stereoisomers.The pure major stereoisomer (Z-isomer) was obtained after recrystallization fromCH2Cl2/hexane (1/5) at room temperature.
With potassium carbonate; In N,N-dimethyl-formamide; at 0 - 20℃;
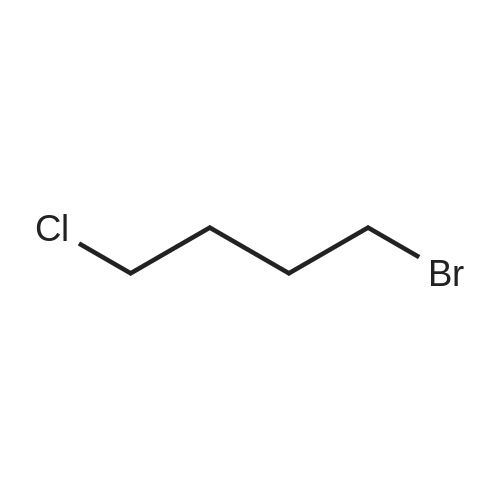
General procedure: To a suspension of corresponding cyclic imide 1a-1k (4mmol), potassium carbonate (4.4mmol) in DMF (5mL), 1-bromo-4-chlorobutane (1,2-dibromoethane (5 eq) for 2l, 1-bromo-3-chloropropane for 2m, 1-bromo-5-chloropentane for 2n) (4.4mmol) was added dropwise at 0C. The mixture was stirred at room temperature overnight and then partitioned between ethyl acetate (EA, 20mL) and water (30mL). The organic layer was washed successively with water (3×30mL), saturated brine, and dried over anhydrous Na2SO4. The solvent was evaporated under vacuum and the residue was purified by column chromatography using petroleum ether(PE): EA (8:1) as eluent to give 2a-2n.
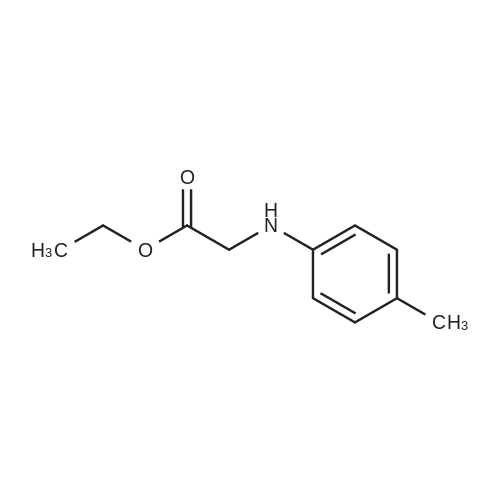
ethyl 2-(1,3-dioxooctahydro-2H-isoindol-2-yl)-2-(p-tolylamino)acetate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
96%
With copper(l) chloride; In acetonitrile; at 60℃; for 24.0h;
General procedure: To a solution of N-arylglycine esters 1 (0.3 mmol) in MeCN(2 mL) were added imides or amides 2 (0.2 mmol) and CuCl(2.0 mg, 0.02 mmol). Then, the reaction mixture was stirred at60 C under air atmosphere until the reaction was completed.Then, the resulting mixture was concentrated under vacuum,and the residue was purified by column chromatography (silicagel, petroleum ether/EtOAc as an eluent) to afford the correspondingproducts 3.(10)
96%
With copper(l) chloride; In acetonitrile; at 60℃; for 24.0h;
N-4-methylphenylglycine ethyl ester (0.3 mmol) under air atmosphere,A phthalimide compound (0.2 mmol) and cuprous chloride (0.02 mmol) were placed in a dry reaction tube with stirring magnets.Then, an acetonitrile solvent (2 mL) was added to the test tube, and the reaction tube was reacted in an air atmosphere at 60 C for 24 hours.After the reaction was completed, it was cooled to room temperature, and the solvent was distilled off under reduced pressure using a rotary evaporator.The residue is purified by column chromatography to give a pure pale yellow solid 3o.The yield was 96%.
With choline chloride; urea; at 140℃; for 1.0h;Green chemistry;
General procedure: DES ChCl/urea was synthesized as described in literature(Abbott et al. 2004). Briefly, choline chloride and urea with the molar ratio of 1:2 were stirred at 80 C for 1 h until a clear and transparent solution was formed (Fig. 1). The obtained DES was used without additional purification. Into a 25-mL round-bottom flask were added ChCl/urea (5.19 g, 20 mmol), phthalic anhydride (1.48 g, 10 mmol) and urea (0.60 g, 10 mmol) in successive, then the mixture was heated at 140 C for 1 h under vigorous stirring. The reaction progress was monitored by GC analysis. After reaction, the reaction mixture was cooled to room temperature, followed by addition of 2 mL of water. The DES was dissolved and the product was precipitated. The solid was collected by filtration and washed thoroughly with water. The white solid was dried thoroughly to afford the product in a yield of 84%. The DES was recovered by evaporation of water under vacuum, and subjected to next run. The product was received in a yield of yield of 95% in the second run.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping