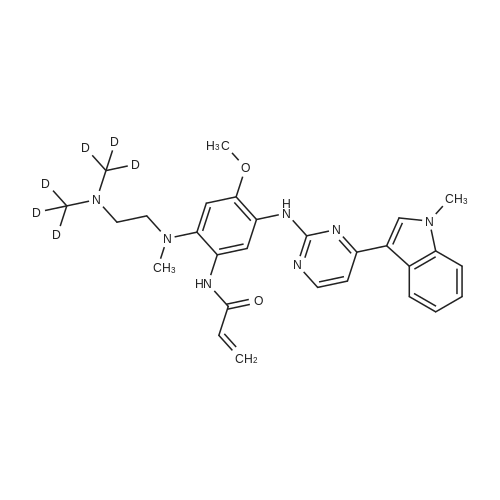
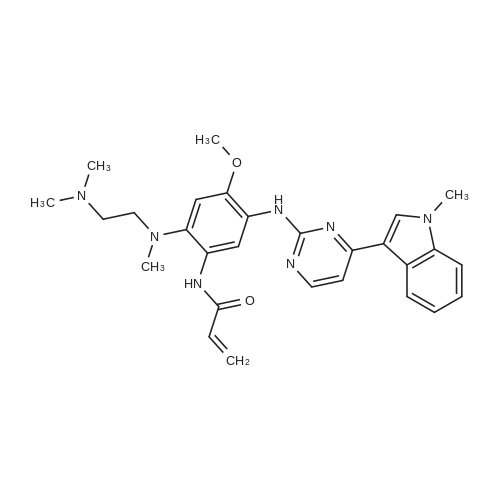
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With triethylamine In acetonitrile at 80℃; for 6 h;
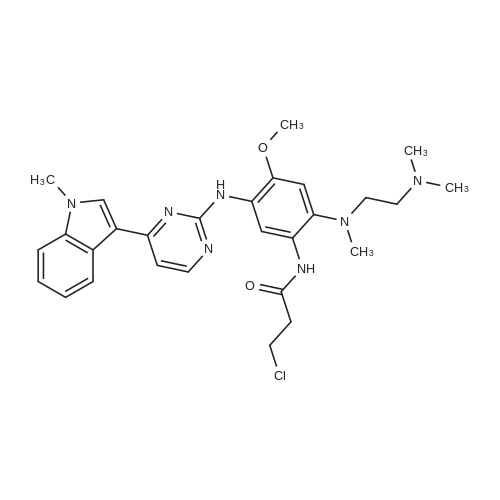
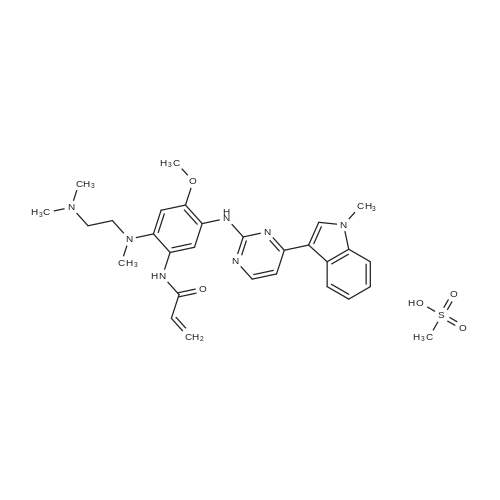
To a stirred solution of 3-chloro-N-[2-[2-dimethylaminoethyl(methyl)amino]-4-methoxy-5-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]phenyl]propanamide (Intermediate 174, 31.5 g, 58.76 mmol) in acetonitrile (310 mL) was added triethylamine (17.84 g, 176.28 mmol) at r.t. The resulting mixture was heated to 80°C for 6h then cooled to r.t.. Water (130 mL) was then added and the mixture stirred for 12h. The mixture was then filtered, washed with a mixture of water and acetonitrile (160 mL, 1:1) and dried at 50°C for overnight to give the title compound (19.2 g, 94percent) as a solid form identified herein as polymorphic form D. 1H NMR: 2.69 (3H, s), 2.83 (6H, d), 3.35 (4H, s), 3.84 (3H, s), 3.91 (3H, s), 5.75 (1H, d), 6.28 (1H, d), 6.67 (1H, dd), 7.05-7.23 (2H, m), 7.29 (1H, t), 7.43 (1H, d), 7.56 (1H, d), 8.21 (2H, s), 8.81 (1H, s), 9.47 (1H, s), 9.52 (1H, s), m/z: ES+ MH+ 500.26.
94%
With sodium hydroxide In tetrahydrofuran; water at 65℃; for 10 h;
(2-dimethylaminoethyl)-5-methoxy-N1-methyl-N4-[4-(1-methylindol-3-yl)pyrimidine at 0 ° C Yl) benzene-1,2,4-triamine (Intermediate 100, 10 g, 21.32 mmol) in THF (95 mL) and water (9.5 mL) was added 3-chloropropionyl chloride (3.28 G, 25.59 mmol). The mixture was stirred at room temperature for 15 minutes and then NaOH (3.48 g, 85.28 mmol) was added. The resulting mixture was heated to 65 ° C and maintained for 10 hours. The mixture was then cooled to room temperature and CH30H (40 mL) and water (70 mL) were added. The resulting mixture was stirred overnight. The resulting solid was collected by filtration, washed with water (25 mL) and dried at 50 ° C for 12 hours to give compound A (7.0 g, 94percent) as a solid.
94%
Stage #1: With triethylamine In acetonitrile at 80℃; for 6 h; Stage #2: at 20℃; for 12 h;
To a solution of 3-chloro-N- (2- [2-dimethylaminoethyl (methyl) amino] -4-methoxy-5 - [4- (1-methylindole- Yl) pyrimidin-2-yl] amino} phenyl) propanamide(31.5 g, 58.76 mmol)In acetonitrile (310 mL)In the stirred solutionTriethylamine (17.84 g, 176.28 mmol).The resulting mixture was heated to 80 & lt; 0 & gt; C for 6 hours,Then cooled to room temperature.Then add water (130 mL),The mixture was stirred for 12 hours.The mixture was then filtered,Washed with a mixture of water and acetonitrile (160 mL, 1: 1), dried at 50 ° C overnight,The title compound was obtained as a title compound (19.2 g, 94percent).
94%
With triethylamine In acetonitrile at 20 - 80℃; for 6 h;
At room temperature,To 3-chloro-N-(2-[2-dimethylaminoethyl(methyl)amino]-4-methoxy-5-[4-(1-methylindol-3-yl)pyrimidine 2-yl]amino}phenyl)propanamide (31.5 g, 58.76 mmol) in acetonitrile (310 mL)Triethylamine (17.84 g, 176.28 mmol) was added to the stirred solution.The resulting mixture was heated to 80 ° C for 6 hours and then cooled to room temperature.Water (130 mL) was then added and the mixture was stirred for 12 h.The mixture was then filtered and washed with a mixture of water and acetonitrile (160 mL, 1:1).Drying at 50 ° C overnight gave the title compound (19.2 g, 94percent).
91.2%
With triethylamine In acetonitrile at 80℃; Large scale
N-[(4-dimethylamino-ethylamino-2-methoxy-5-chloropropionylaminophenyl)]-4-(1-methyl-1H-indol-3-yl)-2-pyrimidinamine3 kg (5.6 mol) was added to 30 L of acetonitrile, and 1.7 kg (16.7 mol) of triethylamine was added thereto with stirring, and the mixture was heated at 80 ° C overnight.The reaction was completely monitored by TLC, then 15 L of water was added, the temperature was lowered, the mixture was stirred overnight, and the mixture was filtered, and the filter cake was rinsed with a mixture of acetonitrile and water (7.5 L / 7.5 L).Drying gave 2.55 kg of an off-white powder with a yield of 91.2percent (HPLC purity: 99.6percent).
70.99 g
With diethylamine; triethylamine In acetonitrile at 70℃; for 10.5 h;
Acetonitrile (700 ml) was added to the compound B obtained in the first step. Triethylamine (79.43 g, 0.785 mol) was added to the system, and the temperature was raised to 70 ° C. The mixture was stirred for 10.5 h and added dropwise with water Finished, cooling and stirring.The filter cake was washed with a mixture of acetonitrile and water and dried in vacuo to give Compound C (70.99 g, 0.142 mol). HPLC purity: 99.76percent. Two-step total yield of 90.5percent.
Reference:
[1] Patent: WO2013/14448, 2013, A1, . Location in patent: Page/Page column 65; 66
[2] Patent: CN106674202, 2017, A, . Location in patent: Paragraph 0072
[3] Patent: CN107043369, 2017, A, . Location in patent: Paragraph 0527; 0528; 0529; 0530; 0531; 0532
[4] Patent: CN108929311, 2018, A, . Location in patent: Paragraph 0244-0247
[5] Patent: CN108623567, 2018, A, . Location in patent: Paragraph 0068; 0069; 0070
[6] Patent: CN107522690, 2017, A, . Location in patent: Paragraph 0030; 0031; 0035; 0036; 0040; 0041; 0045; 0046
[7] Patent: CN108218839, 2018, A, . Location in patent: Paragraph 0031
2
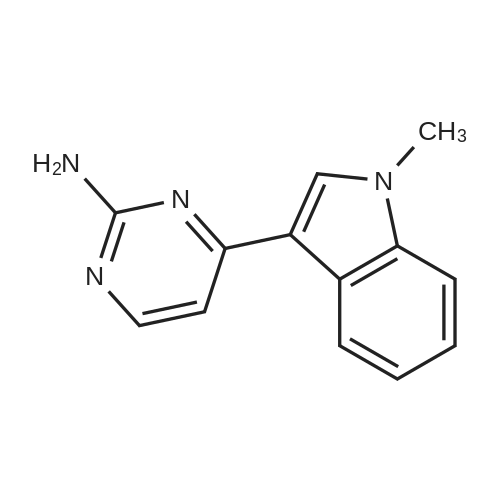
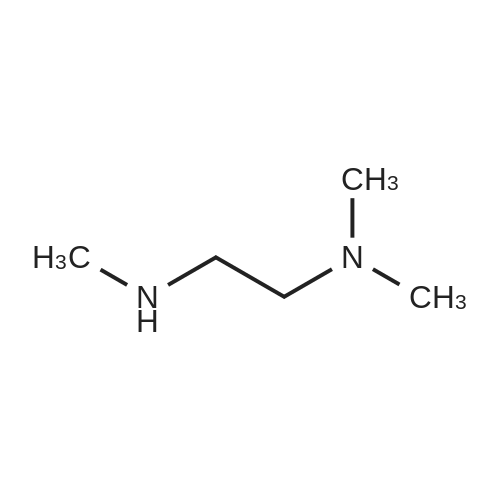
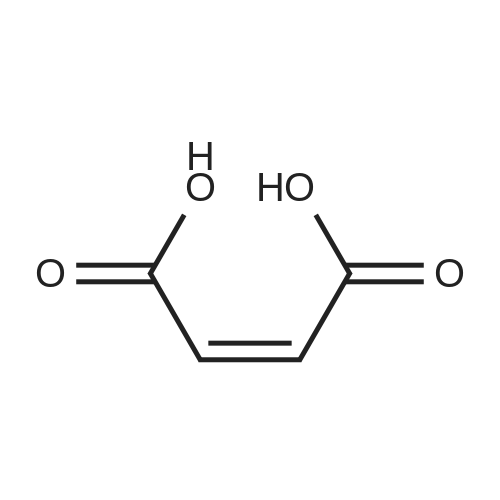
[ 1421372-66-8 ]
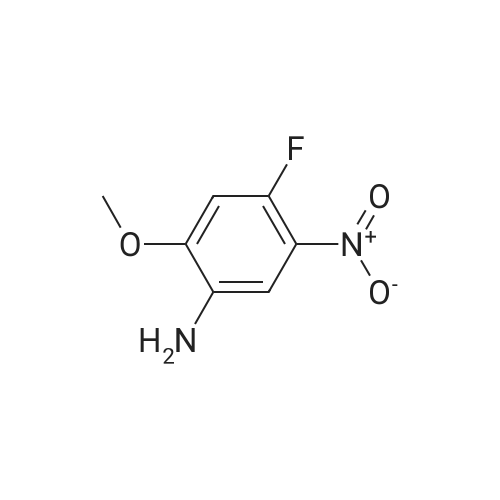
[ 814-68-6 ]
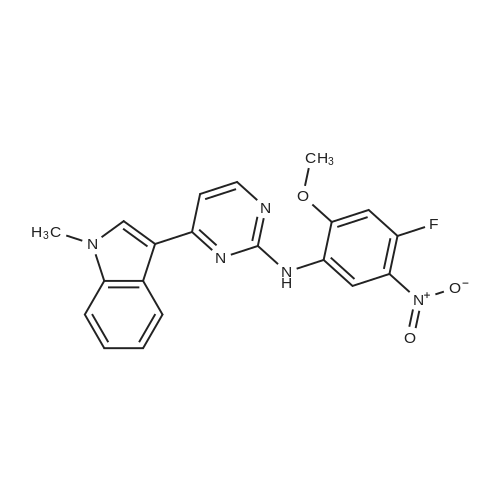
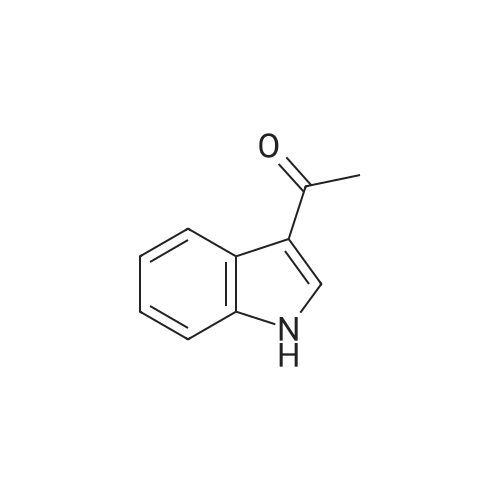
[ 1421373-65-0 ]
Yield
Reaction Conditions
Operation in experiment
95.5%
With sodium carbonate In N,N-dimethyl-formamide at -5℃; for 2 h;
The intermediate (VIII) (13.24 g, 29.74 mmol, 1 eq) was dissolved in 30 ml of N,N-dimethylformamide (DMF), and the mixture was stirred and cooled to -5 °, and acryloyl chloride (3.23 g, 35.69 mmol, 1.2) was added dropwise. Eq), the reaction was stirred at this temperature for 2 h, TLC was followed until the reaction was completed, 100 ml of water was added dropwise, and the mixture was stirred for 1 hour, filtered, and washed with water to give intermediate (X) 14.19 g, yield 95.5percent.
88%
With N-ethyl-N,N-diisopropylamine In tetrahydrofuran at 40℃; for 12 h;
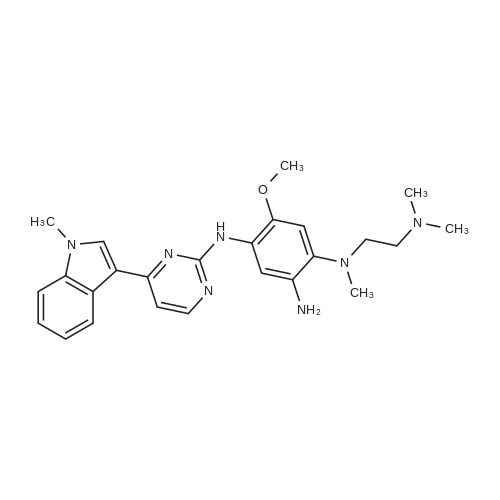
2-(4-(N-(2-(dimethylamino)ethyl)-N-methylamino)-2-methoxy-5-amino)-4-(1-methyl-1H-indole-3-yl) pyrimidine (4.0g, 9 . 0mmol) dissolved in tetrahydrofuran (10 ml), stirring, by adding acryloyl chloride (0.9g, 9 . 9mmol), dropping N,N-diisopropylethylamine (4.1g, 31.4 µM), the reaction mixture 40 °C stirring for 12 hours, TLC board determine the completion of the reaction, the reaction solution and concentration to dry, adding dilute hydrochloric acid is adjusted to neutral, adding the ethyl acetate extraction, magnesium sulfate drying, and concentration to dry, methanol is recrystallized, osimertinib, white solid (4.0g), yield 88.0percent, the step reaction equation is as follows:
86.37%
With triethylamine In ethyl acetate at 5 - 10℃; Inert atmosphere
A 250 ml four-necked flask was taken and Compound G (2.56 g, 5.75 mmol) was added.Triethylamine (1.08 g, 10.67 mmol) was dissolved in ethyl acetate (110 ml), and the mixture was stirred and cooled to 5 ° C under nitrogen.A mixed solution of acryloyl chloride (0.98 g, 10.83 mmol) / ethyl acetate (20 ml) was added dropwise at 5 to 10 °C.After the completion of the dropwise addition, the reaction was stirred, and water (100 ml) was added and stirred for 30 min, and then the organic phase was separated, and the aqueous phase was added to ethyl acetate (100 ml), and the organic phase was combined.The organic phase was washed once with (200 ml) saturated brine, and the organic phase was concentrated to dryness.Purification by column chromatography gave a pale yellow foamy solid 2.48 g.The molar yield was 86.37percent.
83%
With N-ethyl-N,N-diisopropylamine In dichloromethane for 1.5 h; Cooling with ice
Intermediate 3 prepared in Comparative Example 1 in which 3.45 g (38 mmol) of acryloyl chloride in 100 ml of dichloromethane was added dropwise to an ice-cooled water bath was charged with 17 g (38 mmol) and 7.2 ml of N, N-diisopropylethylamine ) In 250 ml of dichloromethane, In, stirring 1.5h. The mixture was then diluted with 250 ml of dichloromethane, washed with 2 × 250 ml of saturated sodium bicarbonate solution and the aqueous layer extracted twice with dichloromethane. The combined organic phases were dried over anhydrous magnesium sulfate and filtered, and the filtrate was concentrated in vacuo at 40 ° C. The crude product was purified by column chromatography to afford 16.01 g of a brown solid in 83.0percent yield with 99.3percent purity
72%
With N-ethyl-N,N-diisopropylamine In chloroform at 5 - 15℃; for 2 h;
A solution of N1- (2- (dimethylamino) ethyl) -5-methoxy-N1-methyl-N4- (4- (1-methylindol-3-yl) pyrimidin- Yl-1,2,4-triamine (8.9 g, 0.02 mol) was added to a 500 mL one-Add 250 mL of chloroform,Was added diisopropylethylamine (3.2g, 0.025mol) at 5 ,Then, acryloyl chloride (2.0 g, 0.022 mol) was added dropwise,Then reacted at 15 ° C for 2 h.After the reaction,The reaction solution was washed successively with 200 mL of water and 200 mL of saturated aqueous NaCl. The organic phase was dried over Na2SO4 and concentrated to give the crude product.The crude product was purified by recrystallization from 90 mL mixture of ethyl acetate and n-heptane (1: 1 by volume of ethyl acetate and n-heptane in the mixture)The resulting white solid product, Osimertinib, 7.2 g, yield 72percent.
72%
With N-ethyl-N,N-diisopropylamine In chloroform at 5 - 15℃; for 2 h;
A solution of N1- (2- (dimethylamino) ethyl) -5-methoxy-N1-methyl-N4- (4- (1-methylindol-3-yl)2-yl) phenyl-1,2,4-triamine (8.9 g, 0.02 mol) was added to 500 mLSingle mouth bottle,A mixture of 250 mL of chloroform was added to dissolve the solid, and then cooled to 5 to 10 ° C by adding diisopropylethylamine (3.2 g, .025 mol)Acryloyl chloride (2.0 g, 0.022 mol) was added and the reaction was carried out at 15 ° C for 2 h after completion of the dropwise addition.After completion of the reaction, the reaction solution was washed successively with 200 mL of water and 200 mL of saturated aqueous NaCl solution,The organic phase was dried with Na2SO4 and concentrated to give a crude product which was mixed with 90 mL of a mixture of ethyl acetate and n-heptaneLiquid in ethyl acetate and n-heptane in a volume ratio of 1: 1) to give the solid product Osimertinib 7.2 g, The yield was 72percent.
57.97%
With N-ethyl-N,N-diisopropylamine In dichloromethane for 1.5 h; Cooling with ice
A solution of acryloyl chloride (41 mg, 0.45 mmol) in methylene chloride was added dropwise to the cooled in an ice water bath.N1-(2-(dimethylamino)ethyl)-5-methoxy-N1-methyl-N4-(4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl)Benzene-1,2,4-triamine (200 mg, 0.45 mmol) and DIPEA (70 mg, 0.54 mmol)Stir the mixture in dichloromethane (5 mL).After 1.5 hours of reaction,Diluted with dichloromethane,Wash with saturated sodium bicarbonate,The aqueous phase is extracted twice with dichloromethane,Combine the organic phase,Drying with anhydrous sodium sulfateconcentrate,Column chromatography afforded the title compound (130 mg, yield:57.97percent).
47%
With N-ethyl-N,N-diisopropylamine In dichloromethane at -10℃; for 1 h;
Acryloyl chloride (710 mg, 7.86 mmol) in DCM (5 mL) was added dropwise to a stirred solution of N′-(2-(dimethylamino)ethyl)-5-methoxy-N′-methyl-N-(4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl)benzene-1,2,4-triamine (33) (3.50 g, 7.86 mmol) and DIPEA (2.03 g, 15.73 mmol) in DCM (30 mL) at -10 °C. The resulting solution was maintained at -10 °C for 1 h and then washed with brine, dried with Na2SO4, and concentrated. The resulting residue was purified by silica gel chromatography (DCM: MeOH: NH3·H2O=50:1:0.1) to afford N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl)amino)phenyl)acrylamide (AZD9291) (1.80 g, 47.0percent yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ10.18 (s, 1H), 9.85 (s, 1H), 9.10 (s, 1H), 8.38 (d, J = 5.3 Hz, 1H), 8.07 (dd, J = 6.1, 2.5 Hz, 1H), 7.73 (s, 1H), 7.39 (dd, J = 6.3, 2.5 Hz, 1H), 7.27 (dt, J = 6.0, 2.4 Hz, 2H), 7.20 (d, J = 5.3 Hz, 1H), 6.80 (s, 1H), 6.46 (dd, J = 16.9, 2.2 Hz, 1H), 6.37 (dd, J = 16.9, 9.7 Hz, 1H), 5.71 (dd, J = 9.7, 2.2 Hz, 1H), 4.00 (s, 3H), 3.88 (s, 3H), 2.94 – 2.84 (m, 2H), 2.70 (s, 3H), 2.32 – 2.21 (m, 8H). 13C NMR (100 MHz, CDCl3) δ162.78, 162.10, 159.58, 157.90, 144.27, 138.25, 135.23, 134.61, 132.89, 129.64, 127.70, 125.99, 125.31, 121.72, 120.87, 120.23, 113.65, 110.03, 109.87, 107.90, 104.64, 88.28, 57.39, 56.42, 56.10, 45.42, 43.84, 33.06. MS (ESI) m/z 500.4 [M+H]+.
39%
With N-ethyl-N,N-diisopropylamine In dichloromethane for 1.5 h; Cooling with ice
A solution of acryloyl chloride (34.5 mg, 0.38 mmol) in CH2Cl2 (1 mL) was added dropwise to a stirred mixture of N1-(2-dimethylaminoethyl)-5-methoxy-N1-methyl-N4-[4-(1-methylindol-3-yl)pyrimidin-2-yl]benzene-1,2,4-triamine (Intermediate 100, 170 mg, 0.38 mmol) and DIPEA (0.073 mL, 0.42 mmol) in CH2Cl2 (5 mL), which was cooled in an ice/water bath. The mixture was stirred for 1.5h and then diluted with CH2Cl2 (25 mL) and washed with sat.NaHCO3 (50 mL). The aqueous washes were extracted with CH2Cl2 (2 x 25 mL). The combined organic solutions were dried (MgSO4) and concentrated in vacuo. Purification by FCC, eluting with 0-4percent 7N methanolic ammonia in CH2Cl2 gave the title compound (75 mg, 39percent) as a cream solid after trituration with diethyl ether; 1H NMR: 2.21 (6H, s), 2.29 (2H, t), 2.72 (3H, s), 2.89 (2H, t), 3.86 (3H, s), 3.92 (3H, s), 5.77 (1H, dd), 6.27 (1H, dd), 6.43 (1H, dd), 7.04 (1H, s), 7.15 (1H, t), 7.20-7.27 (2H, m), 7.53 (1H, d), 7.91 (1H, s), 8.24 (1H, d), 8.33 (1H, d), 8.68 (1H, s), 9.14 (1H, s), 10.22 (1H, s); m/z: ES+ MH+ 500.42.
Reference:
[1] Patent: CN109134435, 2019, A, . Location in patent: Paragraph 0056-0074
[2] Patent: CN106543060, 2017, A, . Location in patent: Paragraph 0053-0055
[3] Patent: CN108129342, 2018, A, . Location in patent: Paragraph 0057; 0058; 0059
[4] Patent: CN107216313, 2017, A, . Location in patent: Paragraph 0074; 0074
[5] Journal of Heterocyclic Chemistry, 2017, vol. 54, # 5, p. 2898 - 2901
[6] Patent: CN106366072, 2017, A, . Location in patent: Paragraph 0079; 0080; 0081; 0082; 0111-0115
[7] Patent: CN106366022, 2017, A, . Location in patent: Paragraph 0084; 0085; 0086; 0087; 0088; 0089; 0090
[8] Patent: CN107793413, 2018, A, . Location in patent: Paragraph 0191-0194
[9] European Journal of Medicinal Chemistry, 2017, vol. 135, p. 12 - 23
[10] Patent: WO2013/14448, 2013, A1, . Location in patent: Page/Page column 65
[11] Journal of Medicinal Chemistry, 2014, vol. 57, # 20, p. 8249 - 8267
3
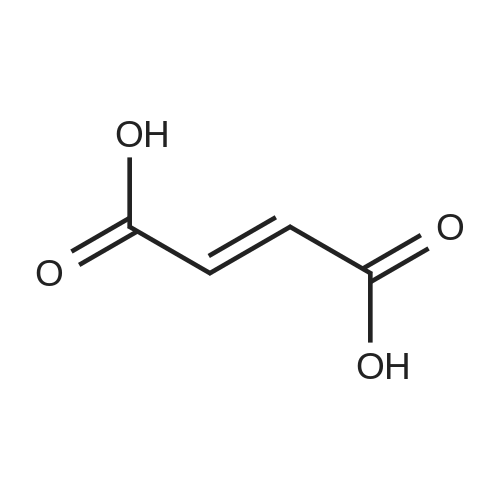
[ 1421372-66-8 ]
[ 625-36-5 ]
[ 1421373-65-0 ]
Yield
Reaction Conditions
Operation in experiment
94%
Stage #1: With N-ethyl-N,N-diisopropylamine In tetrahydrofuran; water at 0 - 20℃; for 0.25 h; Stage #2: With sodium hydroxide In tetrahydrofuran; water at 65℃; for 10 h;
To a stirred solution of N1-(2-dimethylaminoethyl)-5-methoxy-N1-methyl-N4-[4-(1-methylindol-3-yl)pyrimidin-2-yl]benzene-1,2,4-triamine (Intermediate 100, 10 g, 21.32 mmol) in THF (95 mL) and water (9.5 mL) at 0°C was added the 3-chloropropanoyl chloride (3.28 g, 25.59 mmol). The mixture was stirred at r.t. for 15 minutes then NaOH (3.48 g, 85.28 mmol) was added. The resulting mixture was heated to 65°C for 10h. The mixture was then cooled to r.t. and CH3OH (40 mL) and water (70 mL) were added. The resulting mixture was stirred overnight. The resulting solid was collected by filtration, washed with water (25 mL) and dried at 50°C for 12h to give the title compound (7.0 g, 94percent) as a solid form identified herein as polymorphic Form D. 1H NMR: 2.69 (3H, s) 2.83 (6H, d) 3.35 (4H, s) 3.84 (3H, s) 3.91 (3H, s) 5.75 (1H, d) 6.28 (1H, d) 6.67 (1H, dd) 7.05-7.23 (2H, m) 7.29 (1H, t) 7.43 (1H, d) 7.56 (1H, d) 8.21 (2H, s) 8.81 (1H, s) 9.47 (1H, s) 9.52 (1H, s) ES+ MH+ 500.26.
94%
Stage #1: at 20℃; for 0.25 h; Stage #2: With sodium hydroxide In tetrahydrofuran; water at 65℃; for 10 h;
To a solution of N1- (2-dimethylaminoethyl) -5-methoxy-N1-methyl-N4- [4- (1-methylindol-3-yl) pyrimidin-2-yl ] Benzene-1,2,4-triamine(Intermediate 100, 10 g, 21.32 mmol) in THF (95 mL) and water (9.5 mL)To the stirred solution was added 3-chloropropionyl chloride (3.28 g, 25.59 mmol). The mixture was stirred at room temperature for 15 minutes and then addedNaOH (3.48 g, 85.28 mmol). The resulting mixture was heated to 65 & lt; 0 & gt; C for 10 hours. The mixture was then cooledTo room temperature, CH3OH (40 mL) and water (70 mL) were added. The resulting mixture was stirred overnight. The resulting solid was collected by filtrationWater (25 mL) and dried at 50 ° C for 12 hours to give the title compound as a solid (7.0 g, 94percent).
79.6%
Stage #1: at 0 - 28℃; for 1 h; Stage #2: With sodium hydroxide In tetrahydrofuran; water for 8 h; Reflux
In room temperature,THF-H2O (825 ml -82.5 ml) was added to a 2 liter four-necked flask,Then, 82.5 g of the compound represented by the formula (IV)The mixture was cooled to 0 ° C.At 0 ° C to 5 ° C,23.55 g 3-chloropropionyl chloride was added dropwise.After the dropwise addition,The temperature was raised to 25 ° C to 28 ° C, stirring was continued for 1 hour,A mixture of 22.3 grams of sodium hydroxide was added in one portion.Heated to reflux for 8 hours. Cooling to 25 ~ 28 ,825 ml of ethyl acetate was added,410 ml of water and stir for 15 minutes.Standing,Dispensing. Separate organic Phase, washed with 410 ml of saturated sodium chloride once.The organic phase was separated and dried over anhydrous sodium sulfate for 1 hour.Filter, filter cake ethyl acetate 82 ml wash once,The filtrate was spin-dried to give the crude compound of formula (V).To the crude product was added 300 ml of isopropyl alcohol and 8.26 g of activated carbon,Heated to reflux for 1 hour,Hot filter.The filtrate was stirred under conditions,Down to 25 ° C,Precipitate solid.Filter, The filter cake was washed with isopropanol (2 x 40 ml)To give 74 g of an off-white solid, yield: 79.6percent.
Reference:
[1] Patent: WO2013/14448, 2013, A1, . Location in patent: Page/Page column 66
[2] Patent: CN106699736, 2017, A, . Location in patent: Paragraph 0045; 0046; 0047; 0048; 0049
[3] Patent: CN107188888, 2017, A, . Location in patent: Paragraph 0073-0075
4
[ 1032452-86-0 ]
[ 1421373-65-0 ]
Yield
Reaction Conditions
Operation in experiment
93%
With toluene-4-sulfonic acid In 2-methyl-propan-1-ol at 55℃; for 5 h;
Under nitrogen / argon,To a single-necked flask was added 100 mg (0.41 mmol)3- (2-chloropyrimidin-4-yl) -1-methylindole125.98 mg (0.43 mmol)2-methoxy-4-N, N, N'-trimethylethylenediamine-5-acrylamidoaniline,76.32 mg (0.44 mmol) of p-toluenesulfonic acid and 5 ml of isobutanol at a reaction temperature of 55 ° C for 5 h,The filter cake was dissolved in 30ml water, neutralized with saturated aqueous sodium hydrogencarbonate solution, extracted with 10ml dichloromethane twice, combined with organic layer, dried, pumping And the dichloromethane was distilled off under reduced pressure to give 190.5 mg of a pale yellow solid in a yield of 93percent
92%
With toluene-4-sulfonic acid In butan-1-ol at 40℃; for 2.5 h;
4-Methylbenzenesulfonic acid hydrate (6.5 g,34.2 mmol) was added in one portion to 3-(2-chloropyrimidin-4-yl)-1-methylindole 2 (4.2 g, 17.1 mmol) and intermediate 3 (5.0 g, 17.1 mmol)in n-butanol (80 mL). The resulting mixture was stirred at 40 °C for 2.5 h. The mixture was cooled to 0–5oC and stirred at this temperature for 1–2h. The precipitate was collected by filtration, washed withn-butanol (20 mL), and dried under vacuum to afford AZD9291 as a yellow solid. The yellow solid was triturated with MeCN to give a solid which was collected by filtration and dried under vacuum to give AZD9291 (7.8 g,15.7 mmol, 92percent) as a yellow solid. 1H NMR (CDCl3) δ 10.19 (br, 1H), 9.88 (s, 1H), 9.13(s, 1H), 8.40(d, 1H), 8.08(t, 1H),7.75(s, 1H), 7.42 (t, 1H), 7.27 (m, 1H), 6.81 (s, 1H), 6.47 (m, 1H), 5.73(m, 2H), 4.02(s, 3H), 3.91(s, 3H) , 2.92(m, 2H) , 2.73(s, 3H) , 2.31(s,8H); MS calcd for C28H33N7O2 [M+H]+ 499.27; found: 500.3.
75.5%
With potassium carbonate In N,N-dimethyl-formamide at 20 - 80℃; for 7 h;
N, N-dimethylformamide (500.0 ml) was added to a 2 L three-necked flask, the compound (8) (50.0 g, 0.17 mol, 1 eq) was then added sequentially, anhydrous potassium carbonate (26.0 g, 0.19 mol, 1.1 eq), compound (9) (41.5 g, 0.17 mol, 1.0 eq), after heating to 80 ° C, the reaction mixture was stirred for 7 hours, cooled to room temperature, stirred with water (500 ml) for 2 h, filtered, washed with a small amount of water and ethanol, dried in absolute ethanol and dried to give 64.6 g of the title compound yield 75.5percent).
Reference:
[1] Patent: CN104817541, 2017, B, . Location in patent: Paragraph 0047-0048
[2] Journal of Chemical Research, 2015, vol. 39, # 6, p. 318 - 320
[3] Patent: CN105601620, 2016, A, . Location in patent: Paragraph 0102; 0103; 0104; 0105; 0106; 0107; 0108-0114
5
[ 954421-16-0 ]
[ 1421373-65-0 ]
Yield
Reaction Conditions
Operation in experiment
91%
With sodium hydroxide In butan-1-ol at 90 - 105℃;
The reaction flask was added N- [2 - [[2- (dimethylamino) ethyl] methylamino] -4-methoxy-5-carboxamidine] phenyl-2-propenamide (II) ( 1.7g, 5mmol), (2E) -3- dimethylamino-1- (1-methyl -1H- indol-3-yl) -2-propen-1-one (III) (1.2g, 5.3 mmol), sodium hydroxide (0.8g, 10mmol) and n-butanol 35mL, gradually warming to 90-105 , the reaction was stirred for 24-30 h, TLC detection completion of the reaction. Cooling and crystallization, the resulting cake was washed with ethanol / ethyl acetate (1/1) was recrystallized, and dried in vacuo to give a yellow solid AZD9291 (I) 4.55g, yield 91.0percent
Stage #1: With lithium carbonate; methanesulfonyl chloride In acetonitrile at 10℃; for 1.5 h; Stage #2: at 0℃;
Methanesulfonyl chloride3.2 g (28 mmol) of lithium carbonate and 2.22 g (30 mmol) of lithium carbonate were added to 100 ml of acetonitrile,After stirring at 10 ° C, 2.02 g (28 mmol) of acrylic acid was added to the above solution and stirred for 1.5 h. 6.17 g (13.8 mmol) of Intermediate 3 prepared in Example 1 was added to the mixed anhydride solution at 0 ° C and monitored by TLC Raw materials disappear. The mixture was stirred for 1 h, extracted with ethyl acetate (100 ml), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under vacuum at 45 ° C to give crude AZD9291. Recrystallization from 50 ml of acetonitrile gave 6.35 g of a brown solid with a yield of 92.2percent and a purity of 99.8percent. The HPLC profile is shown in FIG. 5.
Reference:
[1] Patent: CN107216313, 2017, A, . Location in patent: Paragraph 0060; 0061; 0063; 0064
7
[ 1421372-67-9 ]
[ 79-10-7 ]
[ 1421373-65-0 ]
Yield
Reaction Conditions
Operation in experiment
61%
Stage #1: With 5%-palladium/activated carbon; hydrogen In tetrahydrofuran at 20℃; for 2 h; Stage #2: at 20℃; for 4 h; Inert atmosphere
To a 250 mL single-necked flask was added compound 5 (2.375 g, 5 mmol)And 5percent palladium-carbon (106 mg, 1 mmol)Treated with THF (60 mL) as solvent,Replace the gas three times,The reaction solution was allowed to stand in a hydrogen atmosphere (to ensure a sufficient amount of hydrogen)Stir at room temperature for 2 h,Rapid filtration of solid palladium-carbon,And acrylic acid (0.72 g, 10 mmol) was added to the filtrate,EDC · HCl (2.865 g, 15 mmol)And DIPEA (2.580 g, 20 mmol),And with nitrogen for protection, room temperature stirring 4h,TLC (CHCl3: MeOH = 8: 1, Rf = 0.8) After the reaction was complete,Concentrated under reduced pressure,Add chloroform and water extraction three times,The organic phase was taken, dried over anhydrous sodium sulfate,Filter, spin dry,A solution of AZD9291 as a yellowish solid, 1.521 g,Yield 61percent.
Reference:
[1] Patent: CN106967050, 2017, A, . Location in patent: Paragraph 0020; 0023
8
[ 1421372-67-9 ]
[ 79-10-7 ]
[ 814-68-6 ]
[ 1421373-65-0 ]
Yield
Reaction Conditions
Operation in experiment
72.1%
Stage #1: With palladium 10% on activated carbon; hydrogen In tetrahydrofuran at 20 - 50℃; Stage #2: at 20℃;
Will be 10 gramsN-(4-N,N,N'-trimethylethylenediamine-2-methoxy-5-Nitrophenyl)-4-(1-methyl-1H-indol-3-yl)Pyrimidin-2-amine is intermediate 3,Add 100ml of tetrahydrofuran solution,After heating to 50°C to dissolve it,Add 0.5 g of 10percent palladium metal on palladium carbon at room temperature.Vacuuming into the hydrogen,Response 10-12 hours (according to TLC dot board tracking)Withdrawal of hydrogen,Separately add 2.27 grams of acrylic acid,5.42 g of N,N-diisopropyl-ethylamineAnd 2.07 grams of acryloyl chloride continue stirring at room temperature for 1-2 hours (according to the TLC dot plate tracking),The reaction is completeDiluted with 100ml of water,Extract with dichloromethane,The resulting methylene chloride solution is dried over anhydrous sodium sulfate.Distilled under reduced pressure,Column purification 7.58g,Yield 72.1percent.
Reference:
[1] Patent: CN107556293, 2018, A, . Location in patent: Paragraph 0026; 0027; 0028; 0029; 0030
9
[ 199865-36-6 ]
[ 1421373-65-0 ]
Yield
Reaction Conditions
Operation in experiment
16.6 g
With diammonium sulfide In isopropyl alcohol
(7) 1-methoxy-2-nitro-5-[(N-(dimethylamino)ethyl)-N-methylamino]-4-acrylamide prepared in step (6) Benzene 15(10.46g, 7.62mol)And 2-amino-3H-4-(1-methyl-1H-3-yl)pyrimidine 7 (18.6 g, 2.53 mol) prepared in step (3)Condensation reactions occur in ammonia sulfide and isopropanol solvents,Generation of Ostinib 16 (16.6 g, 4.73 mol).
Reference:
[1] Patent: CN107915724, 2018, A, . Location in patent: Paragraph 0013; 0014; 0015
10
[ 67-56-1 ]
[ 1421373-65-0 ]
Yield
Reaction Conditions
Operation in experiment
60.08 g
at 300℃;
(4) N-{2-[[2-(dimethylamino)ethyl]methylamino]-4-hydroxy-5-[[4-(1-methyl-1H-indol-3-yl)-pyrimidin-2-yl]amino]phenyl}prop-2-enamide 14 (75.1 g, 3.2 mol) was added with methanol (100 ml) and catalyst alumina, and heated to 300C. Reacts to form N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide 15 (60.08 g, 3.0 mol), ie AZD9291.
Reference:
[1] Patent: CN107964008, 2018, A, . Location in patent: Paragraph 0020-0021; 0024-0025
11
[ 1075705-01-9 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: WO2013/14448, 2013, A1,
[2] Patent: WO2013/14448, 2013, A1,
[3] Patent: WO2013/14448, 2013, A1,
[4] Journal of Chemical Research, 2015, vol. 39, # 6, p. 318 - 320
[5] Patent: CN106366072, 2017, A,
[6] Patent: CN106366022, 2017, A,
[7] Patent: CN106543060, 2017, A,
[8] European Journal of Medicinal Chemistry, 2017, vol. 135, p. 12 - 23
[9] Patent: CN104817541, 2017, B,
[10] Journal of Heterocyclic Chemistry, 2017, vol. 54, # 5, p. 2898 - 2901
[11] Patent: CN106967050, 2017, A,
[12] Patent: CN107188888, 2017, A,
[13] Patent: CN107216313, 2017, A,
[14] Patent: CN107216313, 2017, A,
[15] Patent: CN107793413, 2018, A,
[16] Patent: CN108129342, 2018, A,
[17] Patent: CN108129342, 2018, A,
[18] Patent: CN108623567, 2018, A,
12
[ 1421372-67-9 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: WO2013/14448, 2013, A1,
[2] Patent: WO2013/14448, 2013, A1,
[3] Patent: WO2013/14448, 2013, A1,
[4] Journal of Medicinal Chemistry, 2014, vol. 57, # 20, p. 8249 - 8267
[5] European Journal of Medicinal Chemistry, 2017, vol. 135, p. 12 - 23
[6] Patent: CN107188888, 2017, A,
[7] Patent: CN107216313, 2017, A,
[8] Patent: CN107793413, 2018, A,
[9] Patent: CN108623567, 2018, A,
[10] Patent: CN109134435, 2019, A,
13
[ 1421372-94-2 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: WO2013/14448, 2013, A1,
[2] Patent: WO2013/14448, 2013, A1,
[3] Patent: WO2013/14448, 2013, A1,
[4] Journal of Medicinal Chemistry, 2014, vol. 57, # 20, p. 8249 - 8267
[5] European Journal of Medicinal Chemistry, 2017, vol. 135, p. 12 - 23
[6] Patent: CN107188888, 2017, A,
[7] Patent: CN107216313, 2017, A,
[8] Patent: CN107216313, 2017, A,
[9] Patent: CN107793413, 2018, A,
[10] Patent: CN108623567, 2018, A,
[11] Patent: CN109134435, 2019, A,
14
[ 1032452-86-0 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: WO2013/14448, 2013, A1,
[2] Patent: WO2013/14448, 2013, A1,
[3] Patent: WO2013/14448, 2013, A1,
[4] Journal of Medicinal Chemistry, 2014, vol. 57, # 20, p. 8249 - 8267
[5] European Journal of Medicinal Chemistry, 2017, vol. 135, p. 12 - 23
[6] Patent: CN106967050, 2017, A,
[7] Patent: CN107188888, 2017, A,
[8] Patent: CN107216313, 2017, A,
[9] Patent: CN107216313, 2017, A,
[10] Patent: CN108129342, 2018, A,
[11] Patent: CN108623567, 2018, A,
[12] Patent: CN109134435, 2019, A,
15
[ 450-91-9 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: WO2013/14448, 2013, A1,
[2] Patent: WO2013/14448, 2013, A1,
[3] Patent: WO2013/14448, 2013, A1,
[4] Journal of Chemical Research, 2015, vol. 39, # 6, p. 318 - 320
[5] Patent: CN106366072, 2017, A,
[6] Patent: CN106366022, 2017, A,
[7] Journal of Heterocyclic Chemistry, 2017, vol. 54, # 5, p. 2898 - 2901
16
[ 603-76-9 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: WO2013/14448, 2013, A1,
[2] Patent: WO2013/14448, 2013, A1,
[3] Patent: WO2013/14448, 2013, A1,
[4] Journal of Medicinal Chemistry, 2014, vol. 57, # 20, p. 8249 - 8267
[5] Journal of Chemical Research, 2015, vol. 39, # 6, p. 318 - 320
[6] Patent: CN109134435, 2019, A,
17
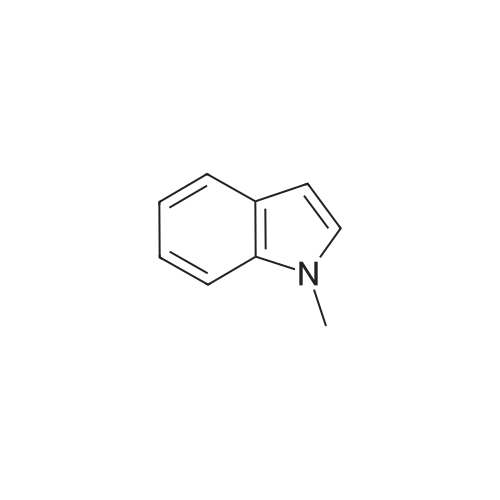
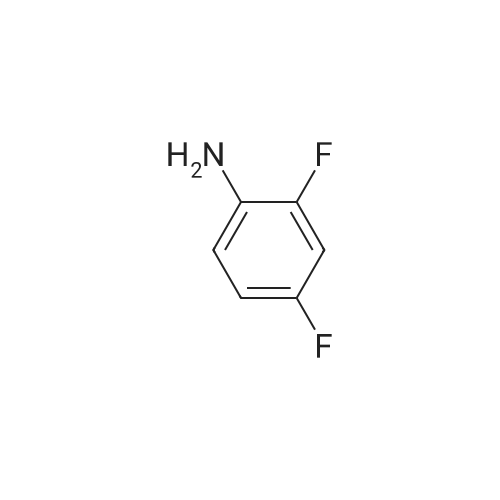
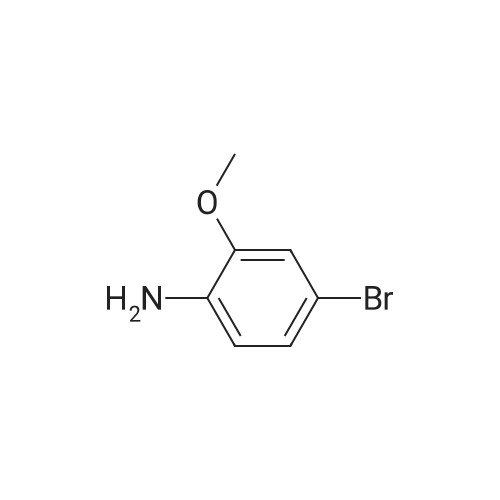
[ 1421372-66-8 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: WO2013/14448, 2013, A1,
[2] Patent: CN106674202, 2017, A,
[3] Patent: CN108623567, 2018, A,
Reference:
[1] Journal of Chemical Research, 2015, vol. 39, # 6, p. 318 - 320
[2] Patent: CN106366072, 2017, A,
[3] Patent: CN106366022, 2017, A,
21
[ 142-25-6 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: CN105601620, 2016, A,
[2] Patent: CN108129342, 2018, A,
22
[ 703-80-0 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: CN106366072, 2017, A,
[2] Journal of Heterocyclic Chemistry, 2017, vol. 54, # 5, p. 2898 - 2901
23
[ 19012-02-3 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: CN106366072, 2017, A,
[2] Journal of Heterocyclic Chemistry, 2017, vol. 54, # 5, p. 2898 - 2901
24
[ 1443291-03-9 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: CN106366072, 2017, A,
[2] Journal of Heterocyclic Chemistry, 2017, vol. 54, # 5, p. 2898 - 2901
25
[ 458-52-6 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: CN104910049, 2016, B,
26
[ 123344-02-5 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: CN107793413, 2018, A,
27
[ 367-25-9 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: CN107793413, 2018, A,
28
[ 635-22-3 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: CN107915724, 2018, A,
29
[ 610-81-1 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: CN107964008, 2018, A,
30
[ 92952-74-4 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: CN107964008, 2018, A,
31
[ 32338-02-6 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: CN108218839, 2018, A,
32
[ 59557-91-4 ]
[ 1421373-65-0 ]
Reference:
[1] Patent: CN107954989, 2018, A,
33
[ 75-75-2 ]
[ 1421373-65-0 ]
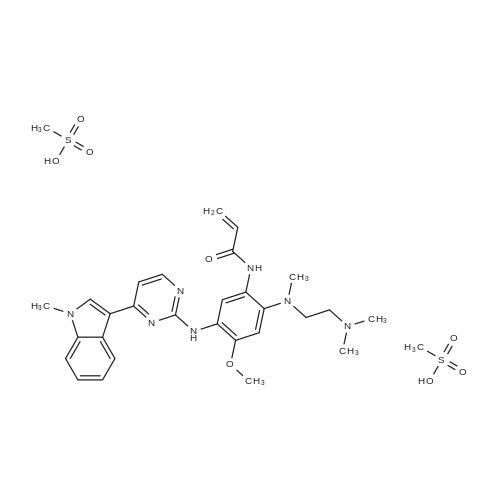
[ 1421373-66-1 ]
Yield
Reaction Conditions
Operation in experiment
94%
With N-ethyl-N,N-diisopropylamine In ethanol; ethyl acetate at 70℃; for 1.5 h;
Procedure 1: To a stirred solution of N-[2-[2-dimethylaminoethyl(methyl)amino]-4-methoxy-5-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide (Example 28, 20 g, 36.63 mmol) in ethanol (120 mL) and EtOAc (80 mL) at 70°C was added methanesulfonic acid (3.59 g, 36.63 mmol) as a solution in EtOAc (40 mL). The resulting mixture was stirred for 1.5h. The resulting solid was collected by filtration and dried at 80°C under vacuum overnight to give the title salt (20.5 g, 94percent) in a solid form defined herein as polymorphic Form B for this salt.
94%
at 70℃; for 1.5 h;
To a solution of compound A, 20 g, 36.63 mmol in ethanol (120 mL) and EtOAc (80 mL) at 70 & lt; 0 &Was added a solution of methanesulfonic acid (3.59 g, 36.63 mmol) in EtOAc (40 mL). The resulting mixture was stirred for 1.5 hours. The resulting solid was collected by filtration and dried in vacuo at 80 & lt; 0 & gt; C overnight to afford the desired solid (compound A methanesulfonic acidSalt of the polymorph of Form B) (20.5 g, 94percent).
94%
at 70℃; for 1.5 h;
A solution of methanesulfonic acid (3.59 g, 36.63 mmol) in EtOAc (40 mL) was added to a stirred solution of compound A, 20 g, 36.63 mmol in ethanol (120 mL) and Et0Ac (80 mL) at 70 ° C. The resulting mixture was stirred for 1.5 small. The resulting solid was collected by filtration and dried under vacuum at 80 ° C overnight to give the title salt (20.5 g, 94percent) in solid form (salt of polymorph B).
92.3%
at 20℃; for 4 h;
osimertinib (5.5g, 11mmol) dissolved in 1,2-dichloroethane (20 ml), stirring, adding methanesulfonic acid (1.4g, 14 . 3mmol), the reaction mixture 30 °C stirring for 3 hours, the reaction solution and concentration to dry, methanol is recrystallized, awes for the osimertinib sulfonate, a kind of white solid (6.1g), yield 92.3percent, the step of reaction with the embodiment 1.
92.2%
at 50℃; for 2 h; Large scale
2.55 kg (5.1 mol) obtained aboveN-[(4-Dimethylamino-ethylamino-2-methoxy-5-allylaminophenyl)]-4-(1-methyl-1H-indol-3-yl)-2- PyrimidinamineAdd to a mixed solvent of 23 L of acetone and 2.3 L of water, and heat to 50 degrees.490.5 g (5.1 mol) of methanesulfonic acid was diluted with 2.3 L of acetone and slowly added to the reaction system. After the addition, the reaction was continued for 2 hours.The reaction was stopped, suction filtered, and the filter cake was rinsed with 5/0.5 L of acetone/water, and dried to give 2.8 kg of bright yellow powder, yield 92.2percent (HPLC purity 99.9percent).
89.6%
at 40℃; for 0.5 h;
in room temperature,74 g of the compound represented by the formula (V) prepared in Example 5 was dissolved in 740 ml of acetone,Add 74 ml of water,Stir for five minutes.The mixture was heated to 40 ° C,A solution of 13.53 g of methanesulfonic acid in 74 ml of acetone was added dropwise,After dripping at 40 ° C for 30 minutes.Suction filter.The filter cake was rinsed with acetone (2 x 74 ml),The filter cake was dried under vacuum at 16 ° C for 16 hours,To obtain 79 g of the compound represented by the formula (I)Yield: 89.6percent.
Reference:
[1] Patent: WO2013/14448, 2013, A1, . Location in patent: Page/Page column 67
[2] Journal of Medicinal Chemistry, 2014, vol. 57, # 20, p. 8249 - 8267
[3] Patent: CN106699736, 2017, A, . Location in patent: Paragraph 0050; 0051; 0052; 0053
[4] Patent: CN106674202, 2017, A, . Location in patent: Paragraph 0076
[5] Patent: CN106543060, 2017, A, . Location in patent: Paragraph 0088; 0089
[6] Patent: CN108623567, 2018, A, . Location in patent: Paragraph 0071; 0072; 0073
[7] Patent: CN107188888, 2017, A, . Location in patent: Paragraph 0076-0078
With sodium carbonate; In N,N-dimethyl-formamide; at -5℃; for 2h;
The intermediate (VIII) (13.24 g, 29.74 mmol, 1 eq) was dissolved in 30 ml of N,N-dimethylformamide (DMF), and the mixture was stirred and cooled to -5 , and acryloyl chloride (3.23 g, 35.69 mmol, 1.2) was added dropwise. Eq), the reaction was stirred at this temperature for 2 h, TLC was followed until the reaction was completed, 100 ml of water was added dropwise, and the mixture was stirred for 1 hour, filtered, and washed with water to give intermediate (X) 14.19 g, yield 95.5%.
88%
With N-ethyl-N,N-diisopropylamine; In tetrahydrofuran; at 40℃; for 12h;
2-(4-(N-(2-(dimethylamino)ethyl)-N-methylamino)-2-methoxy-5-amino)-4-(1-methyl-1H-indole-3-yl) pyrimidine (4.0g, 9 . 0mmol) dissolved in tetrahydrofuran (10 ml), stirring, by adding acryloyl chloride (0.9g, 9 . 9mmol), dropping N,N-diisopropylethylamine (4.1g, 31.4 muM), the reaction mixture 40 C stirring for 12 hours, TLC board determine the completion of the reaction, the reaction solution and concentration to dry, adding dilute hydrochloric acid is adjusted to neutral, adding the ethyl acetate extraction, magnesium sulfate drying, and concentration to dry, methanol is recrystallized, osimertinib, white solid (4.0g), yield 88.0%, the step reaction equation is as follows:
86.37%
With triethylamine; In ethyl acetate; at 5 - 10℃;Inert atmosphere;
A 250 ml four-necked flask was taken and Compound G (2.56 g, 5.75 mmol) was added.Triethylamine (1.08 g, 10.67 mmol) was dissolved in ethyl acetate (110 ml), and the mixture was stirred and cooled to 5 C under nitrogen.A mixed solution of acryloyl chloride (0.98 g, 10.83 mmol) / ethyl acetate (20 ml) was added dropwise at 5 to 10 C.After the completion of the dropwise addition, the reaction was stirred, and water (100 ml) was added and stirred for 30 min, and then the organic phase was separated, and the aqueous phase was added to ethyl acetate (100 ml), and the organic phase was combined.The organic phase was washed once with (200 ml) saturated brine, and the organic phase was concentrated to dryness.Purification by column chromatography gave a pale yellow foamy solid 2.48 g.The molar yield was 86.37%.
83%
With N-ethyl-N,N-diisopropylamine; In dichloromethane; for 1.5h;Cooling with ice;
Intermediate 3 prepared in Comparative Example 1 in which 3.45 g (38 mmol) of acryloyl chloride in 100 ml of dichloromethane was added dropwise to an ice-cooled water bath was charged with 17 g (38 mmol) and 7.2 ml of N, N-diisopropylethylamine ) In 250 ml of dichloromethane, In, stirring 1.5h. The mixture was then diluted with 250 ml of dichloromethane, washed with 2 × 250 ml of saturated sodium bicarbonate solution and the aqueous layer extracted twice with dichloromethane. The combined organic phases were dried over anhydrous magnesium sulfate and filtered, and the filtrate was concentrated in vacuo at 40 C. The crude product was purified by column chromatography to afford 16.01 g of a brown solid in 83.0% yield with 99.3% purity
72%
With N-ethyl-N,N-diisopropylamine; In chloroform; at 5 - 15℃; for 2h;
A solution of N1- (2- (dimethylamino) ethyl) -5-methoxy-N1-methyl-N4- (4- (1-methylindol-3-yl) pyrimidin- Yl-1,2,4-triamine (8.9 g, 0.02 mol) was added to a 500 mL one-Add 250 mL of chloroform,Was added diisopropylethylamine (3.2g, 0.025mol) at 5 ,Then, acryloyl chloride (2.0 g, 0.022 mol) was added dropwise,Then reacted at 15 C for 2 h.After the reaction,The reaction solution was washed successively with 200 mL of water and 200 mL of saturated aqueous NaCl. The organic phase was dried over Na2SO4 and concentrated to give the crude product.The crude product was purified by recrystallization from 90 mL mixture of ethyl acetate and n-heptane (1: 1 by volume of ethyl acetate and n-heptane in the mixture)The resulting white solid product, Osimertinib, 7.2 g, yield 72%.
72%
With N-ethyl-N,N-diisopropylamine; In chloroform; at 5 - 15℃; for 2h;
A solution of N1- (2- (dimethylamino) ethyl) -5-methoxy-N1-methyl-N4- (4- (1-methylindol-3-yl)2-yl) phenyl-1,2,4-triamine (8.9 g, 0.02 mol) was added to 500 mLSingle mouth bottle,A mixture of 250 mL of chloroform was added to dissolve the solid, and then cooled to 5 to 10 C by adding diisopropylethylamine (3.2 g, .025 mol)Acryloyl chloride (2.0 g, 0.022 mol) was added and the reaction was carried out at 15 C for 2 h after completion of the dropwise addition.After completion of the reaction, the reaction solution was washed successively with 200 mL of water and 200 mL of saturated aqueous NaCl solution,The organic phase was dried with Na2SO4 and concentrated to give a crude product which was mixed with 90 mL of a mixture of ethyl acetate and n-heptaneLiquid in ethyl acetate and n-heptane in a volume ratio of 1: 1) to give the solid product Osimertinib 7.2 g, The yield was 72%.
57.97%
With N-ethyl-N,N-diisopropylamine; In dichloromethane; for 1.5h;Cooling with ice;
A solution of acryloyl chloride (41 mg, 0.45 mmol) in methylene chloride was added dropwise to the cooled in an ice water bath.N1-(2-(dimethylamino)ethyl)-5-methoxy-N1-methyl-N4-(4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl)Benzene-1,2,4-triamine (200 mg, 0.45 mmol) and DIPEA (70 mg, 0.54 mmol)Stir the mixture in dichloromethane (5 mL).After 1.5 hours of reaction,Diluted with dichloromethane,Wash with saturated sodium bicarbonate,The aqueous phase is extracted twice with dichloromethane,Combine the organic phase,Drying with anhydrous sodium sulfateconcentrate,Column chromatography afforded the title compound (130 mg, yield:57.97%).
47%
With N-ethyl-N,N-diisopropylamine; In dichloromethane; at -10℃; for 1h;
Acryloyl chloride (710 mg, 7.86 mmol) in DCM (5 mL) was added dropwise to a stirred solution of N?-(2-(dimethylamino)ethyl)-5-methoxy-N?-methyl-N-(4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl)benzene-1,2,4-triamine (33) (3.50 g, 7.86 mmol) and DIPEA (2.03 g, 15.73 mmol) in DCM (30 mL) at -10 C. The resulting solution was maintained at -10 C for 1 h and then washed with brine, dried with Na2SO4, and concentrated. The resulting residue was purified by silica gel chromatography (DCM: MeOH: NH3·H2O=50:1:0.1) to afford N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl)amino)phenyl)acrylamide (AZD9291) (1.80 g, 47.0% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) delta10.18 (s, 1H), 9.85 (s, 1H), 9.10 (s, 1H), 8.38 (d, J = 5.3 Hz, 1H), 8.07 (dd, J = 6.1, 2.5 Hz, 1H), 7.73 (s, 1H), 7.39 (dd, J = 6.3, 2.5 Hz, 1H), 7.27 (dt, J = 6.0, 2.4 Hz, 2H), 7.20 (d, J = 5.3 Hz, 1H), 6.80 (s, 1H), 6.46 (dd, J = 16.9, 2.2 Hz, 1H), 6.37 (dd, J = 16.9, 9.7 Hz, 1H), 5.71 (dd, J = 9.7, 2.2 Hz, 1H), 4.00 (s, 3H), 3.88 (s, 3H), 2.94 - 2.84 (m, 2H), 2.70 (s, 3H), 2.32 - 2.21 (m, 8H). 13C NMR (100 MHz, CDCl3) delta162.78, 162.10, 159.58, 157.90, 144.27, 138.25, 135.23, 134.61, 132.89, 129.64, 127.70, 125.99, 125.31, 121.72, 120.87, 120.23, 113.65, 110.03, 109.87, 107.90, 104.64, 88.28, 57.39, 56.42, 56.10, 45.42, 43.84, 33.06. MS (ESI) m/z 500.4 [M+H]+.
39%
With N-ethyl-N,N-diisopropylamine; In dichloromethane; for 1.5h;Cooling with ice;
A solution of acryloyl chloride (34.5 mg, 0.38 mmol) in CH2Cl2 (1 mL) was added dropwise to a stirred mixture of N1-(2-dimethylaminoethyl)-5-methoxy-N1-methyl-N4-[4-(1-methylindol-3-yl)pyrimidin-2-yl]benzene-1,2,4-triamine (Intermediate 100, 170 mg, 0.38 mmol) and DIPEA (0.073 mL, 0.42 mmol) in CH2Cl2 (5 mL), which was cooled in an ice/water bath. The mixture was stirred for 1.5h and then diluted with CH2Cl2 (25 mL) and washed with sat.NaHCO3 (50 mL). The aqueous washes were extracted with CH2Cl2 (2 x 25 mL). The combined organic solutions were dried (MgSO4) and concentrated in vacuo. Purification by FCC, eluting with 0-4% 7N methanolic ammonia in CH2Cl2 gave the title compound (75 mg, 39%) as a cream solid after trituration with diethyl ether; 1H NMR: 2.21 (6H, s), 2.29 (2H, t), 2.72 (3H, s), 2.89 (2H, t), 3.86 (3H, s), 3.92 (3H, s), 5.77 (1H, dd), 6.27 (1H, dd), 6.43 (1H, dd), 7.04 (1H, s), 7.15 (1H, t), 7.20-7.27 (2H, m), 7.53 (1H, d), 7.91 (1H, s), 8.24 (1H, d), 8.33 (1H, d), 8.68 (1H, s), 9.14 (1H, s), 10.22 (1H, s); m/z: ES+ MH+ 500.42.
To a stirred solution of N1-(2-dimethylaminoethyl)-5-methoxy-N1-methyl-N4-[4-(1-methylindol-3-yl)pyrimidin-2-yl]benzene-1,2,4-triamine (Intermediate 100, 10 g, 21.32 mmol) in THF (95 mL) and water (9.5 mL) at 0C was added the 3-chloropropanoyl chloride (3.28 g, 25.59 mmol). The mixture was stirred at r.t. for 15 minutes then NaOH (3.48 g, 85.28 mmol) was added. The resulting mixture was heated to 65C for 10h. The mixture was then cooled to r.t. and CH3OH (40 mL) and water (70 mL) were added. The resulting mixture was stirred overnight. The resulting solid was collected by filtration, washed with water (25 mL) and dried at 50C for 12h to give the title compound (7.0 g, 94%) as a solid form identified herein as polymorphic Form D. 1H NMR: 2.69 (3H, s) 2.83 (6H, d) 3.35 (4H, s) 3.84 (3H, s) 3.91 (3H, s) 5.75 (1H, d) 6.28 (1H, d) 6.67 (1H, dd) 7.05-7.23 (2H, m) 7.29 (1H, t) 7.43 (1H, d) 7.56 (1H, d) 8.21 (2H, s) 8.81 (1H, s) 9.47 (1H, s) 9.52 (1H, s) ES+ MH+ 500.26.
94%
To a solution of N1- (2-dimethylaminoethyl) -5-methoxy-N1-methyl-N4- [4- (1-methylindol-3-yl) pyrimidin-2-yl ] Benzene-1,2,4-triamine(Intermediate 100, 10 g, 21.32 mmol) in THF (95 mL) and water (9.5 mL)To the stirred solution was added 3-chloropropionyl chloride (3.28 g, 25.59 mmol). The mixture was stirred at room temperature for 15 minutes and then addedNaOH (3.48 g, 85.28 mmol). The resulting mixture was heated to 65 & lt; 0 & gt; C for 10 hours. The mixture was then cooledTo room temperature, CH3OH (40 mL) and water (70 mL) were added. The resulting mixture was stirred overnight. The resulting solid was collected by filtrationWater (25 mL) and dried at 50 C for 12 hours to give the title compound as a solid (7.0 g, 94%).
94%
At 0 N1- (2-dimethylaminoethyl) -5-methoxy-N1-methyl-N4- [4- (1-methylindol-3-yl) pyrimidin-2-yl] benzene-1, 2,4-triamine (10 g, 21.32 mmol)To a stirred solution in THF (95 mL) and water (9.5 mL) was added 3-chloropropionyl chloride (3.28 g, 25.59 mmol).The mixture was stirred at room temperature for 15 minutes, and then NaOH (3.48 g, 85.28 mmol) was added.The resulting mixture was heated to 65 C for 10 hours.The mixture was then cooled to room temperature, and CH3OH (40 mL) and water (70 mL) were added.The resulting mixture was stirred overnight.The resulting solid was collected by filtration, washed with water (25 mL), and dried at 50 C for 12 hours.The title compound was obtained as a solid (7.0 g, 94%).
79.6%
In room temperature,THF-H2O (825 ml -82.5 ml) was added to a 2 liter four-necked flask,Then, 82.5 g of the compound represented by the formula (IV)The mixture was cooled to 0 C.At 0 C to 5 C,23.55 g 3-chloropropionyl chloride was added dropwise.After the dropwise addition,The temperature was raised to 25 C to 28 C, stirring was continued for 1 hour,A mixture of 22.3 grams of sodium hydroxide was added in one portion.Heated to reflux for 8 hours. Cooling to 25 ~ 28 ,825 ml of ethyl acetate was added,410 ml of water and stir for 15 minutes.Standing,Dispensing. Separate organic Phase, washed with 410 ml of saturated sodium chloride once.The organic phase was separated and dried over anhydrous sodium sulfate for 1 hour.Filter, filter cake ethyl acetate 82 ml wash once,The filtrate was spin-dried to give the crude compound of formula (V).To the crude product was added 300 ml of isopropyl alcohol and 8.26 g of activated carbon,Heated to reflux for 1 hour,Hot filter.The filtrate was stirred under conditions,Down to 25 C,Precipitate solid.Filter, The filter cake was washed with isopropanol (2 x 40 ml)To give 74 g of an off-white solid, yield: 79.6%.
With triethylamine; In acetonitrile; at 80℃; for 6h;
To a stirred solution of 3-chloro-N-[2-[2-dimethylaminoethyl(methyl)amino]-4-methoxy-5-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]phenyl]propanamide (Intermediate 174, 31.5 g, 58.76 mmol) in acetonitrile (310 mL) was added triethylamine (17.84 g, 176.28 mmol) at r.t. The resulting mixture was heated to 80C for 6h then cooled to r.t.. Water (130 mL) was then added and the mixture stirred for 12h. The mixture was then filtered, washed with a mixture of water and acetonitrile (160 mL, 1:1) and dried at 50C for overnight to give the title compound (19.2 g, 94%) as a solid form identified herein as polymorphic form D. 1H NMR: 2.69 (3H, s), 2.83 (6H, d), 3.35 (4H, s), 3.84 (3H, s), 3.91 (3H, s), 5.75 (1H, d), 6.28 (1H, d), 6.67 (1H, dd), 7.05-7.23 (2H, m), 7.29 (1H, t), 7.43 (1H, d), 7.56 (1H, d), 8.21 (2H, s), 8.81 (1H, s), 9.47 (1H, s), 9.52 (1H, s), m/z: ES+ MH+ 500.26.
94%
With sodium hydroxide; In tetrahydrofuran; water; at 65℃; for 10h;
(2-dimethylaminoethyl)-5-methoxy-N1-methyl-N4-[4-(1-methylindol-3-yl)pyrimidine at 0 C Yl) benzene-1,2,4-triamine (Intermediate 100, 10 g, 21.32 mmol) in THF (95 mL) and water (9.5 mL) was added 3-chloropropionyl chloride (3.28 G, 25.59 mmol). The mixture was stirred at room temperature for 15 minutes and then NaOH (3.48 g, 85.28 mmol) was added. The resulting mixture was heated to 65 C and maintained for 10 hours. The mixture was then cooled to room temperature and CH30H (40 mL) and water (70 mL) were added. The resulting mixture was stirred overnight. The resulting solid was collected by filtration, washed with water (25 mL) and dried at 50 C for 12 hours to give compound A (7.0 g, 94%) as a solid.
94%
To a solution of 3-chloro-N- (2- [2-dimethylaminoethyl (methyl) amino] -4-methoxy-5 - [4- (1-methylindole- Yl) pyrimidin-2-yl] amino} phenyl) propanamide(31.5 g, 58.76 mmol)In acetonitrile (310 mL)In the stirred solutionTriethylamine (17.84 g, 176.28 mmol).The resulting mixture was heated to 80 & lt; 0 & gt; C for 6 hours,Then cooled to room temperature.Then add water (130 mL),The mixture was stirred for 12 hours.The mixture was then filtered,Washed with a mixture of water and acetonitrile (160 mL, 1: 1), dried at 50 C overnight,The title compound was obtained as a title compound (19.2 g, 94%).
94%
With triethylamine; In acetonitrile; at 20 - 80℃; for 6h;
At room temperature,To 3-chloro-N-(2-[2-dimethylaminoethyl(methyl)amino]-4-methoxy-5-[4-(1-methylindol-3-yl)pyrimidine 2-yl]amino}phenyl)propanamide (31.5 g, 58.76 mmol) in acetonitrile (310 mL)Triethylamine (17.84 g, 176.28 mmol) was added to the stirred solution.The resulting mixture was heated to 80 C for 6 hours and then cooled to room temperature.Water (130 mL) was then added and the mixture was stirred for 12 h.The mixture was then filtered and washed with a mixture of water and acetonitrile (160 mL, 1:1).Drying at 50 C overnight gave the title compound (19.2 g, 94%).
91.2%
With triethylamine; In acetonitrile; at 80℃;Large scale;
N-[(4-dimethylamino-ethylamino-2-methoxy-5-chloropropionylaminophenyl)]-4-(1-methyl-1H-indol-3-yl)-2-pyrimidinamine3 kg (5.6 mol) was added to 30 L of acetonitrile, and 1.7 kg (16.7 mol) of triethylamine was added thereto with stirring, and the mixture was heated at 80 C overnight.The reaction was completely monitored by TLC, then 15 L of water was added, the temperature was lowered, the mixture was stirred overnight, and the mixture was filtered, and the filter cake was rinsed with a mixture of acetonitrile and water (7.5 L / 7.5 L).Drying gave 2.55 kg of an off-white powder with a yield of 91.2% (HPLC purity: 99.6%).
70.99 g
With diethylamine; triethylamine; In acetonitrile; at 70℃; for 10.5h;
Acetonitrile (700 ml) was added to the compound B obtained in the first step. Triethylamine (79.43 g, 0.785 mol) was added to the system, and the temperature was raised to 70 C. The mixture was stirred for 10.5 h and added dropwise with water Finished, cooling and stirring.The filter cake was washed with a mixture of acetonitrile and water and dried in vacuo to give Compound C (70.99 g, 0.142 mol). HPLC purity: 99.76%. Two-step total yield of 90.5%.
To the compound 6 concentrate, 150 ml of methanol was added, stirred and dissolved, and the temperature was lowered to between 0 and 5 C, 15 g of sodium trifluoroacetate was added, and the mixture was stirred for 30 minutes, and the temperature was controlled to be 2.5 g between 0 and 5 C. The azabicyclo ring was added dropwise, stirred for 3 h, and the reaction was started until the compound 6 was completed. 9.5 g of methanesulfonic acid was added dropwise, and the mixture was heated to 35 to 40 C for 2 h, at which time the solid target was precipitated. Compound AZD9291, 41.5 g crude.
5-(N-acrylamido)-4-((N,1-(2-(N,N-dimethylamino)ethyl)-N,1-methyl)amino)-2-methoxyaniline[ No CAS ]
[ 1421373-65-0 ]
Yield
Reaction Conditions
Operation in experiment
93%
With toluene-4-sulfonic acid; In 2-methyl-propan-1-ol; at 55℃; for 5h;
Under nitrogen / argon,To a single-necked flask was added 100 mg (0.41 mmol)3- (2-chloropyrimidin-4-yl) -1-methylindole125.98 mg (0.43 mmol)2-methoxy-4-N, N, N'-trimethylethylenediamine-5-acrylamidoaniline,76.32 mg (0.44 mmol) of p-toluenesulfonic acid and 5 ml of isobutanol at a reaction temperature of 55 C for 5 h,The filter cake was dissolved in 30ml water, neutralized with saturated aqueous sodium hydrogencarbonate solution, extracted with 10ml dichloromethane twice, combined with organic layer, dried, pumping And the dichloromethane was distilled off under reduced pressure to give 190.5 mg of a pale yellow solid in a yield of 93%
92%
With toluene-4-sulfonic acid; In butan-1-ol; at 40℃; for 2.5h;
4-Methylbenzenesulfonic acid hydrate (6.5 g,34.2 mmol) was added in one portion to 3-(2-chloropyrimidin-4-yl)-1-methylindole 2 (4.2 g, 17.1 mmol) and intermediate 3 (5.0 g, 17.1 mmol)in n-butanol (80 mL). The resulting mixture was stirred at 40 C for 2.5 h. The mixture was cooled to 0-5oC and stirred at this temperature for 1-2h. The precipitate was collected by filtration, washed withn-butanol (20 mL), and dried under vacuum to afford AZD9291 as a yellow solid. The yellow solid was triturated with MeCN to give a solid which was collected by filtration and dried under vacuum to give AZD9291 (7.8 g,15.7 mmol, 92%) as a yellow solid. 1H NMR (CDCl3) delta 10.19 (br, 1H), 9.88 (s, 1H), 9.13(s, 1H), 8.40(d, 1H), 8.08(t, 1H),7.75(s, 1H), 7.42 (t, 1H), 7.27 (m, 1H), 6.81 (s, 1H), 6.47 (m, 1H), 5.73(m, 2H), 4.02(s, 3H), 3.91(s, 3H) , 2.92(m, 2H) , 2.73(s, 3H) , 2.31(s,8H); MS calcd for C28H33N7O2 [M+H]+ 499.27; found: 500.3.
75.5%
With potassium carbonate; In N,N-dimethyl-formamide; at 20 - 80℃; for 7h;
N, N-dimethylformamide (500.0 ml) was added to a 2 L three-necked flask, the compound (8) (50.0 g, 0.17 mol, 1 eq) was then added sequentially, anhydrous potassium carbonate (26.0 g, 0.19 mol, 1.1 eq), compound (9) (41.5 g, 0.17 mol, 1.0 eq), after heating to 80 C, the reaction mixture was stirred for 7 hours, cooled to room temperature, stirred with water (500 ml) for 2 h, filtered, washed with a small amount of water and ethanol, dried in absolute ethanol and dried to give 64.6 g of the title compound yield 75.5%).
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; N-ethyl-N,N-diisopropylamine; In dichloromethane;
General procedure: A solution of N1-(2-(dimethylamino)ethyl)-N4-(5-ethyl-4-(pyrazolo[1 ,5-a]pyridin-3-yl)pyrimidin- 2-yl)-5-methoxy-A/i-methylbenzene-1 ,2,4-triamine (R1) (64 mg, 0.14 mmol) in DCM (1.4 imL) was treated with EDCI (54 mg, 0.28 mmol), Hunig's base (73 uL, 0.42 mmol), and acrylic acid (19 uL, 0.28 mmol). The mixture was concentrated in vacuo and the product was purified by preparative TLC (5% MeOH/DCM) to afford A/-(2-((2-(dimethylamino)ethyl)(methyl)amino)-5-((5-ethyl-4-(pyrazolo[1 ,5- a]pyridin-3-yl)pyrimidin-2-yl)amino)-4-methoxyphenyl)acrylamide (Example 1) as an orange solid. 1H NMR (CDCIs): delta 10.01 (br. s., 1 H), 9.48 (s, 1 H), 8.50 (dt, J = 6.9, 1.1 Hz, 1 H), 8.40 (m, 2H), 8.33 (s, 1 H), 7.41 (s, 1 H), 7.21-7.25 (m, 1 H), 6.83-6.89 (m, 1 H), 6.78 (s, 1 H), 6.24-6.41 (m, 2H), 5.66 (dd, J = 9.9, 1.7 Hz, 1 H), 3.86 (s, 3H), 2.85-2.91 (m, 2H), 2.81 (q, J = 7.4 Hz, 2H), 2.70 (s, 3H), 2.27-2.31 (m, 3H), 2.25 (s, 6H), 1.28 (t, J = 7.5 Hz, 3H). ESI-MS m/z: 515.2 [M+H]+.
19
[ 1421373-65-0 ]
[ 79-03-8 ]
C31H37N7O3[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With N-ethyl-N,N-diisopropylamine; In dichloromethane; at 25℃; for 0.5h;
Under the condition of stirring in the ice bath, to AZD9291 (type II shown, 100 mg) in dichloromethane (25 ml) is added in the solution 19 mu L propionyl chloride (AZD9291 and propionyl chloride molar ratio of 1 : 1.1) and 69muLN, N-diisopropyl ethylamine. 25 C lower stirring 30 min. TLC detection after the reaction, a saturated solution of sodium bicarbonate (25 ml) washing the reaction solution, extraction three times with methylene chloride (25 ml × 3), the organic solvent layer merged together, the rotating evaporimeter remove organic solvent. Purification using silica gel (methanol/dichloromethane = 1:30), to obtain compound 2.
With N-ethyl-N,N-diisopropylamine; In dichloromethane; at 20℃; for 0.5h;Cooling with ice;
Under the condition of stirring in the ice bath to AZD9291 (type II shown, 100 mg) in dichloromethane (25 ml) is added in solution 18 mu L acryloyl chloride (AZD9291 and acrylic chloride in a molar ratio of 1 : 1.1) and 69muLN, N-diisopropyl ethylamine. Stirring the mixture at room temperature for 30 min. TLC detection after the reaction, a saturated solution of sodium bicarbonate (25 ml) washing the reaction solution, extraction three times with methylene chloride (25 ml × 3), the organic solvent layer merged together, the rotating evaporimeter remove organic solvent. Purification using silica gel (methanol/dichloromethane 1:30), to obtain compound 3.
With N-ethyl-N,N-diisopropylamine; In dichloromethane; at 20℃; for 0.5h;Cooling with ice;
Under the condition of stirring in the ice bath to AZD9291 (type II shown, 100 mg) in dichloromethane (25 ml) is added in the solution 23 mu L2-methyl propionyl chloride (AZD9291 and 2-methyl propionyl chloride molar ratio of 1 : 1.1) and 69muLN, N-diisopropyl ethylamine. Stirring the mixture at room temperature for 30 min. TLC detection after the reaction, a saturated solution of sodium bicarbonate (25 ml) washing the reaction solution, extraction three times with methylene chloride (25 ml × 3), the organic solvent layer merged together, the rotating evaporimeter remove organic solvent. Purification using silica gel (methanol/dichloromethane 1:30), get compound 4.
With N-ethyl-N,N-diisopropylamine; In dichloromethane; at 20℃; for 0.5h;Cooling with ice;
Under the condition of stirring in the ice bath to AZD9291 (type II shown, 100 mg) in dichloromethane (25 ml) is added in the solution 23 mu L butyrylchlorine (AZD9291 with butyl chloride in a molar ratio of 1 : 1.1) and 69muLN, N-diisopropyl ethylamine. Stirring the mixture at room temperature for 30 min. TLC detection after the reaction, a saturated solution of sodium bicarbonate (25 ml) washing the reaction solution, extraction three times with methylene chloride (25 ml × 3), the organic solvent layer merged together, the rotating evaporimeter remove organic solvent. Purification using silica gel (methanol/dichloromethane 1:30), get compound 5.
With N-ethyl-N,N-diisopropylamine; In dichloromethane; at 20℃; for 0.5h;Cooling with ice;
Under the condition of stirring in the ice bath to AZD9291 (type II shown, 100 mg) in dichloromethane (25 ml) is added in the solution 25 mu L benzoyl chloride (AZD9291 with benzoyl chloride in a molar ratio of 1 : 1.1) and 69muLN, N-diisopropyl ethylamine. Stirring the mixture at room temperature for 30 min. TLC detection after the reaction, a saturated solution of sodium bicarbonate (25 ml) washing the reaction solution, extraction three times with methylene chloride (25 ml × 3), the organic solvent layer merged together, the rotating evaporimeter remove organic solvent. Purification using silica gel (methanol/dichloromethane 1:30), to obtain compound 6.
With N-ethyl-N,N-diisopropylamine; In dichloromethane; at 20℃; for 0.5h;Cooling with ice;
Under the condition of stirring in the ice bath, to AZD9291 (type II shown, 100 mg) in dichloromethane (25 ml) is added in solution 16 mu L acetyl chloride (AZD9291 with acetyl chloride in a molar ratio of 1 : 1.1) and 69muLN, N-diisopropyl ethylamine. 20 C lower stirring 30 min. TLC detection after the reaction, a saturated solution of sodium bicarbonate (25 ml) washing the reaction solution, extraction three times with methylene chloride (25 ml × 3), the organic solvent layer merged together, the rotating evaporimeter remove organic solvent. Purification using silica gel (methanol/dichloromethane 1:30), to obtain compound 1
N-[2-[2-dimethylaminoethylmethylamino]-4-methoxy-5-carboxamidinephenyl]prop-2-enamide[ No CAS ]
[ 1421373-65-0 ]
Yield
Reaction Conditions
Operation in experiment
91%
With sodium hydroxide; In butan-1-ol; at 90 - 105℃;
The reaction flask was added N- [2 - [[2- (dimethylamino) ethyl] methylamino] -4-methoxy-5-carboxamidine] phenyl-2-propenamide (II) ( 1.7g, 5mmol), (2E) -3- dimethylamino-1- (1-methyl -1H- indol-3-yl) -2-propen-1-one (III) (1.2g, 5.3 mmol), sodium hydroxide (0.8g, 10mmol) and n-butanol 35mL, gradually warming to 90-105 , the reaction was stirred for 24-30 h, TLC detection completion of the reaction. Cooling and crystallization, the resulting cake was washed with ethanol / ethyl acetate (1/1) was recrystallized, and dried in vacuo to give a yellow solid AZD9291 (I) 4.55g, yield 91.0%
N-[2-[2-dimethylaminoethylmethylamino]-4-methoxy-5-[4-(1-methylindol-3-yl)pyrimidin-2-ylamino]phenyl]prop-2-enamide methanesulfonate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
94.6%
In ethanol; ethyl acetate; for 2h;Reflux;
To a 2 L three-necked flask was added anhydrous ethanol (640 ml) and ethyl acetate (320 ml), followed by addition of compound (10) (64.0 g, 0.13 mol, 1 eq), followed by refluxing and adding methanesulfonic acid (12.5 g, 0.13 mol, 1.0 eq) was refluxed for 2 h, slow cooling to 30 C, after crystallization for 3 h, the filter cake was washed with a small amount of ethanol and recrystallized from isopropanol and dried at 50 C to give 73.3 g of the title compound (1) (yield: 94.6%).
2-((2-acrylamido-5-methoxy-4-((4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl)amino)phenyl) (methyl)amino)-N,N-dimethylethan-1-amine oxide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85%
With 3-chloro-benzenecarboperoxoic acid; In dichloromethane; at 0℃; for 0.5h;
General procedure: N-oxide derivatives (compound 2, 5, 6) was prepared as,1.0 equiv. of m-CPBA was added into a solution of AZD9291 or its fluorinated derivatives in DCM (25 mL). The mixture was allowed to stir at 0 C for 30 min. Then water was added to quench the reaction.The reaction mixture was extracted with DCM for 3 times. The combined organic layer was dried over anhydrous Na2SO4 and concentratedon rotavapor under reduced pressure. The residue was purified by HPLC (MeOH:H2O = 10:1) to give desired products as yellow solid. fluorinated N-Methylindoles were used for synthesis of fluorinated derivatives of osimertinib (compound 3, 4) according to literature reported procedures.29 5.2.1. 2-((2-acrylamido-5-methoxy-4-((4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl)amino)phenyl) (methyl)amino)-N,N-dimethylethan-1-amine oxide (2) Yellow solid. Yield 85%. 1H NMR (400 MHz, DMSO-d6, d (ppm)),12.50 (s, 1H), 9.00 (s, 1H), 8.57 (s, 1H), 8.32-8.27 (m, 2H), 7.90 (s,1H), 7.51 (d, J = 8.16 Hz, 1H), 7.26-7.14 (m, 4H), 6.20-6.15 (m,1H), 5.62-5.59 (m, 1H), 3.89 (s, 3H), 3.86 (s, 3H), 3.46-3.44 (m,2H), 3.33 (s, 3H), 3.30 (s, 3H), 3.25-3.24 (m, 2H), 2.74 (s, 3H). 13CNMR (100 MHz, DMSO-d6, d (ppm)), 163.5, 161.68, 160.1, 157.5,146.2, 139.5, 137.6, 133.7, 133.6, 126.8, 125.4, 124.8, 124.6,122.0, 121.6, 120.9, 116.8, 112.5, 110.4, 107.0, 104.4, 64.3, 59.8,55.9, 53.8, 33.0; ESI-LRMS calcd. for C16H18Cl2N2O [M+H]+:516.27, found 516.35.
With 3-chloro-benzenecarboperoxoic acid; In dichloromethane; at 20℃; for 0.5h;
Under stirring condition to AZD9291 (type shown in II, 200 mg) in dichloromethane (25 ml) is added in the solution 81 mg meta-chloroperoxybenzoic acid. 20 C lower stirring 30 min. TLC detection after the reaction, a saturated solution of sodium bicarbonate (25 ml) washing the reaction solution, extraction three times with methylene chloride (25 ml × 3), the organic solvent layer merged together, the rotating evaporimeter remove organic solvent. The reverse high efficiency liquid phase purification (CH 3 OH/H 2 O10: 1), shows compound of formula I obtained.
With sulfuric acid; In water; acetone; at 20℃; for 0.5h;
To the reaction flask was added AZD9291 (5 g, 10 mmol, 1.0 eq), 50 mL of acetone was added with stirring, and an aqueous solution (20 mL) of sulfuric acid (1.18 g, 12 mmol, 1.2 eq) was added dropwise with stirring to precipitate a yellow solid Half a hour, filter, filter cake with a small amount of acetone leaching, drying, yellow granular solid 5g (8.4mmol), molar yield: 84%.After testing, purity: 99.4%
To the reaction flask was added 2.5 g (5 mmol, 1.0 eq) of AZD9291, 25 mL of acetone was added and dissolved in an aqueous solution (2.5 mL) of acetic acid (360 mg, 6 mmol, 1.2 eq) under stirring to precipitate an off-white solid , Filter, filter cake with a small amount of acetone leaching, drying, 1.8g (2.6mmol) of white granular solid, molar yield of 52%.The detection: purity 99.8%;
AZD9291 (2.5 g, 5 mmol, 1.0 eq) was added to the reaction flask at room temperature, 25 mL of acetone was added and dissolved in an aqueous solution (2.5 mL) of citric acid (1.15 g, 6 mmol, 1.2 eq) with stirring to precipitate a yellow solid, Half an hour, suction filter, filter cake with a small amount of acetone leaching, drying, like white granular solid 2.8g (4mmol), molar yield 81%.The detection: purity 99.7%;
AZD9291 (5 g, 10 mmol, 1.0 eq) was added to the reaction flask at room temperature and 50 mL of acetone was added with stirring. TsOH · H2(2.4 g, 12 mmol, 1.2 mmol) in an aqueous solution (5 ml) was added and the mixture was stirred for half an hour. The filter cake was rinsed with a small amount of acetone and dried to give 4.03 g (6.0 mmol) of a yellow granular solid, The molar yield was 60%.The test, purity: 99.7%,
To the reaction flask was added AZD9291 (5 g, 10 mmol, 1.0 eq), 50 mL of acetone was added and dissolved in an aqueous solution (5 mL) of tartaric acid (1.8 g, 12 mmol, 1.2 eq) under stirring to precipitate a pale yellow solid , Filter, filter cake with a small amount of acetone leaching, drying, 5g (7.7mmol) light yellow granular solid, molar yield of 77%.The test: purity 99.9%;
To a 250 mL single-necked flask was added compound 5 (2.375 g, 5 mmol)And 5% palladium-carbon (106 mg, 1 mmol)Treated with THF (60 mL) as solvent,Replace the gas three times,The reaction solution was allowed to stand in a hydrogen atmosphere (to ensure a sufficient amount of hydrogen)Stir at room temperature for 2 h,Rapid filtration of solid palladium-carbon,And acrylic acid (0.72 g, 10 mmol) was added to the filtrate,EDC · HCl (2.865 g, 15 mmol)And DIPEA (2.580 g, 20 mmol),And with nitrogen for protection, room temperature stirring 4h,TLC (CHCl3: MeOH = 8: 1, Rf = 0.8) After the reaction was complete,Concentrated under reduced pressure,Add chloroform and water extraction three times,The organic phase was taken, dried over anhydrous sodium sulfate,Filter, spin dry,A solution of AZD9291 as a yellowish solid, 1.521 g,Yield 61%.
In isopropyl alcohol; at 35℃; for 4h;Molecular sieve; Microwave irradiation;
Add to reaction flaskN-2-[[2- (dimethylamino) ethyl] methylamino] -4-methoxy-5-[[4- (1-methyl-1H-indol-3-yl)- 2-pyrimidinyl] amino] aniline (4.45g, 0.010mol),Add acrylic acid (0.86g, 0.012mol),HY molecular sieve (0.66g, 0.003mol),Isopropanol (32mL, 7.2ml / g), microwave heating to 35 C,Reaction for 4h, and then dried over anhydrous sodium sulfate,Finally, the solvent was removed by rotary evaporation to obtain 4.88 g of a foamy off-white solid with a yield of 97.9%.
92.2%
Methanesulfonyl chloride3.2 g (28 mmol) of lithium carbonate and 2.22 g (30 mmol) of lithium carbonate were added to 100 ml of acetonitrile,After stirring at 10 C, 2.02 g (28 mmol) of acrylic acid was added to the above solution and stirred for 1.5 h. 6.17 g (13.8 mmol) of Intermediate 3 prepared in Example 1 was added to the mixed anhydride solution at 0 C and monitored by TLC Raw materials disappear. The mixture was stirred for 1 h, extracted with ethyl acetate (100 ml), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under vacuum at 45 C to give crude AZD9291. Recrystallization from 50 ml of acetonitrile gave 6.35 g of a brown solid with a yield of 92.2% and a purity of 99.8%. The HPLC profile is shown in FIG. 5.
With water; In tetrahydrofuran; isopropyl alcohol; at 23 - 83℃; for 9h;
In a 5L reaction flask equipped with a stirrer, thermometer, and condenser, 350 g of mentetinib and 1750 ml of a mixture of water-isopropanol-tetrahydrofuran (9:0.5:0.5) were added and heated to 78-83C. Heat filtration, cooling to 50 C, 47 C -50 C, heat and stir for 4 hours; then, cooling to 25 C, 23 C -25 C, heat for 5 hours,The precipitated crystals were collected, filtered, and dried under vacuum at room temperature until constant weight, to obtain 322.3 g of high-purity melitinib hydrate crystals, with an optical purity of 99.97%. Solvent residue detection meets requirements.
3-(dimethylamino)-N-(2-[2-(dimethylamino)ethyl](methyl)amino}-4-methoxy-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl]amino}phenyl)propanamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
73.38%
In ethanol; at 20℃; for 0.5h;
The compound N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(1-methyl-1H-indole- 3-(yl)pyrimidin-2-yl)amino)phenyl)acrylamide (100 mg, 0.20 mmol)Into the reaction flask,thenAdd ethanol (5mL) to dissolve,30% dimethylamine in ethanol (1 mL) was added with stirring.Stir the reaction for 30 minutes at room temperature.After the TLC test is finished,concentrate,TLC purification gave the target compound as an off-white solid (80 mg, yield: 73.38%).
Will be 10 gramsN-(4-N,N,N'-trimethylethylenediamine-2-methoxy-5-Nitrophenyl)-4-(1-methyl-1H-indol-3-yl)Pyrimidin-2-amine is intermediate 3,Add 100ml of tetrahydrofuran solution,After heating to 50C to dissolve it,Add 0.5 g of 10% palladium metal on palladium carbon at room temperature.Vacuuming into the hydrogen,Response 10-12 hours (according to TLC dot board tracking)Withdrawal of hydrogen,Separately add 2.27 grams of acrylic acid,5.42 g of N,N-diisopropyl-ethylamineAnd 2.07 grams of acryloyl chloride continue stirring at room temperature for 1-2 hours (according to the TLC dot plate tracking),The reaction is completeDiluted with 100ml of water,Extract with dichloromethane,The resulting methylene chloride solution is dried over anhydrous sodium sulfate.Distilled under reduced pressure,Column purification 7.58g,Yield 72.1%.
At room temperature, AZD9291 (5.0 g, 10 mmol) was added to the reaction flask. Add 50 ml of acetone and stir to dissolve An aqueous solution (20 mL) of succinic acid (1.18 g, 12 mmol) was added dropwise with stirring. Yellow solids precipitated, Stir for about half an hour, Suction filtration The filter cake is rinsed with a small amount of acetone. drying, Obtained as a yellow particulate solid 4.68 g (7.6 mmol), Yield: 76%, Purity: 99.4%, Light yellow solid.
At room temperature, AZD9291 (5.0 g, 10 mmol) was added to the reaction flask. Add 50 ml of acetone and stir to dissolve Lactic acid (1.18 g, 12 mmol) was added dropwise with stirring Aqueous solution (20mL), Yellow solids precipitated, Stir for about half an hour, Suction filtration The filter cake is rinsed with a small amount of acetone. drying, Obtained as a yellow particulate solid 4.76 g (8.1 mmol), Yield: 81%, Purity: 99.5%, Light yellow solid.
At room temperature, AZD9291 (5.0 g, 10 mmol) was added to the reaction flask. Add 50 ml of acetone and stir to dissolve An aqueous solution (20 mL) of maleic acid (1.39 g, 12 mmol) was added dropwise with stirring. Yellow solids precipitated, Stir for about half an hour, Suction filtration The filter cake is rinsed with a small amount of acetone. drying, Obtained as a yellow particulate solid 4.97 g (8.1 mmol), Yield: 81%. Purity: 99.4%.
At room temperature, AZD9291 (5.0 g, 10 mmol) was added to the reaction flask. Add 50 ml of acetone and stir to dissolve Fumaric acid (1.39 g, 12 mmol) was added dropwise with stirring Aqueous solution (20mL), Yellow solids precipitated, Stir for about half an hour, Suction filtration The filter cake is rinsed with a small amount of acetone. drying, Obtained as a yellow particulate solid 5.0 g (8.2 mmol), Yield: 84%, Purity: 99.4%, pale yellow solid
At room temperature, AZD9291 (5.0 g, 10 mmol) was added to the reaction flask. Add 50 ml of acetone and stir to dissolve Gluconic acid (2.35 g, 12 mmol) was added dropwise with stirring Aqueous solution Yellow solids precipitated, Stir for about half an hour, Suction filtration The filter cake is rinsed with a small amount of acetone. drying, Obtained 5.34g (7.7 mmol) of a yellow particulate solid, Yield: 77%. Purity: 99.1%. Light yellow solid.
At room temperature, AZD9291 (5.0 g, 10 mmol) was added to the reaction flask. Add 50 ml of acetone and stir to dissolve An aqueous solution (20 mL) of malonic acid (1.25 g, 12 mmol) was added dropwise with stirring. Yellow solids precipitated, Stir for about half an hour, Suction filtration The filter cake is rinsed with a small amount of acetone. drying, Obtained 5.17 g (8.6 mmol) of a yellow particulate solid, Yield: 86%. Purity: 99.6 % Light yellow solid.
N-(2-[2-(dimethylamino)ethyl](methyl)amino}-4-methoxy-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl]amino}phenyl)-2-hydroxypropanamide[ No CAS ]
[ 1421373-65-0 ]
Yield
Reaction Conditions
Operation in experiment
24.08 g
With sulfuric acid; at 170℃;
(9) N-{2-[[2-(dimethylamino)ethyl]methylamino]-4-methoxy-5-[[4-(1-)Methyl-1H-indol-3-yl)-2-pyrimidinyl]amino]phenyl}-2-hydroxypropionamide 15 (29.36 g, 2.35 mol) at 98%In the presence of concentrated sulfuric acid (60 ml), heated to 170 C,A dehydration reaction occurs to generate N-{2-[[2-(dimethylamino)ethyl]methylamino]-4-methoxy-5-[[4-(1-methyl-1H-indole) 3-(3-yl)-2-pyrimidinyl]amino]phenyl}-2-propenamide 16(24.08g, 1.96mol),That is, a highly selective and selective EGFR mutant inhibitor AZD9291.
N-{2-[[2-(dimethylamino)ethyl]methylamino]-4-hydroxy-5-[[4-(1-methyl-1H-indol-3-yl)-pyrimidin-2-yl]amino]phenyl}prop-2-enamide[ No CAS ]
[ 1421373-65-0 ]
Yield
Reaction Conditions
Operation in experiment
60.08 g
With aluminum oxide; at 300℃;
(4) N-{2-[[2-(dimethylamino)ethyl]methylamino]-4-hydroxy-5-[[4-(1-methyl-1H-indol-3-yl)-pyrimidin-2-yl]amino]phenyl}prop-2-enamide 14 (75.1 g, 3.2 mol) was added with methanol (100 ml) and catalyst alumina, and heated to 300C. Reacts to form N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide 15 (60.08 g, 3.0 mol), ie AZD9291.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping