* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: With hydrogen In methanol at 20℃; for 3 h; Stage #2: at 20℃; for 1 h;
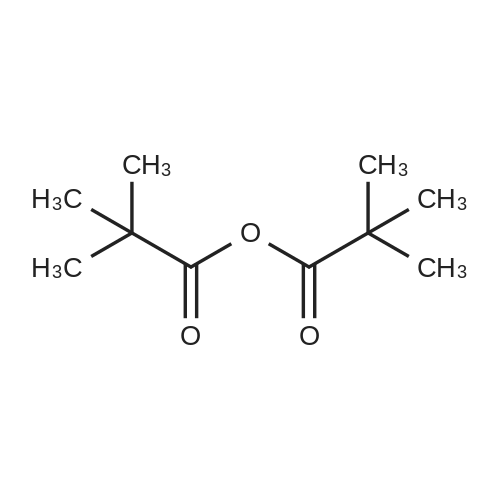
(1) 1-t-Butoxycarbonyl-3-methoxyazetidine [1493] A solution of 1-benzhydryl-3-hydroxyazetidine (10.0 g, 41.8 mmol) in methanol (300 ml) was subjected to catalytic hydrogenation in the presence of 10percent palladium (10.0 g) on charcoal at room temperature for 3 hours. After checking the completion of the reaction, the reaction mixture was filtered in order to remove the catalyst. To the filtrate was added di-t-butoxycarbonic anhydride (18.2 g, 83.6 mmol), and the reaction mixture was stirred at room temperature for 1 hour. After checking the completion of the reaction, the reaction mixture was concentrated under reduced pressure. The residue was purified by chromatography on a silica gel column using n-hexane:ethyl acetate (1:1-->1:2) as the eluant to afford 1-t-butoxycarbonyl-3-hydroxyazetidine (7.05 g, yield 97percent). [1494] Subsequently, to a solution of 1-t-butoxycarbonyl-3-hydroxyazetidine (2.5 g, 14.4 mmol) in dimethylformamide (125 ml) was added sodium hydride (55percent oil dispersion) in an ice bath. After stirring the mixture for 10 minutes in the ice bath, the resulting mixture was stirred at room temperature for 30 minutes. To the reaction mixture was added methyl iodide (1.79 ml, 28. mmol) in an ice bath. After stirring the mixture in an ice bath for 10 minutes, the reaction mixture was stirred at room temperature for 1 hour. After checking the completion of the reaction, 10percent aqueous acetic acid solution was added thereto in an ice bath and the reaction mixture was stirred in the ice bath for 30 minutes. The reaction mixture was partitioned between ethyl acetate and 10percent aqueous sodium chloride solution. The organic layer was washed successively with saturated aqueous sodium hydrogencarbonate solution and saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by chromatography on a silica gel column using n-hexane:ethyl acetate (2:1) as the eluant to afford 1-t-butoxycarbonyl-3-methoxyazetidine (2.18 g, yield 81percent) as a colorless oil. [1495] 1H-NMR (400 MHz, CDCl3): δ (ppm) 4.16-4.10 (1H, m), 4.09-4.03 (2H, m), 3.82 (2H, dd, J=10.2, 4.4 Hz), 3.28 (3H, s), 1.44 (9H, s).
96%
With ammonium formate In methanol for 1 h; Heating / reflux
To a degassed solution of 1-benzhydryl-azetidin-3-ol (4.75 g, 19.84 mmol) in methanol (methanol) (150 ml) were added ammonium formate (8.76 g, 138.91 mmol), 10percent Pd/C (450 mg) and BoC2O (di-tert-butyl dicarbonate) (13 g, 59.56 mmol). The resulting sus.not. pension was heated to reflux under N2 for 1h. It was then cooled down to room tem- perature, filtered through a short pad of celite and concentrated. The residue was dis.not. solved in CH2CI2 and washed with water. The organic layer was dried (Na2SO4) and evaporated. The raw substance was chromatographied on silica gel (heptane:ethyl acetate (ethyl acetate), 1 :1) to afford the title compound (3.30 g, 96percent) as white crys.not. tals. MS (ESI+) m/z = 118.1 [M-fBu+H]+1H NMR (400 MHz, CDCI3) : δ (ppm) 1.43 (s, 9H), 2.35 (d, J = 6.2 Hz, 1 H), 3.80 (dd, J = 10.4, 4.4 Hz, 2H), 4.15 (dd, J = 9.6, 6.7 Hz, 2H), 4.58 (m, 1 H).
96%
With ammonium formate In methanol for 1 h; Heating / reflux
To a degassed solution of 1-benzhydryl-azetidin-3-ol (4.75 g, 19.84 mmol) in methanol (MeOH) (150 ml) were added ammonium formate (8.76 g, 138.91 mmol), 10percent Pd/C (450 mg) and BOC2O (di-tert-butyl dicarbonate) (13 g, 59.56 mmol). The resulting suspension was heated to reflux under N2 for 1 h. It was then cooled down to room temperature, filtered through a short pad of celite and concentrated. The residue was dissolved in CH2CI2 and washed with water. The organic layer was dried over Na2SU4, filtered and concentrated in vacuo. Purification of the residue by flash column chromatography on silica gel (heptane:ethyl acetate (EtOAc), 1 :1 ) afforded the title compound (3.30 g, 96 percent) as white crystals.MS (ESI+) : m/z = 118.1 [M-tBu+H]+; 1H-NMR (400 MHz1 CDCI3) : δ = 1.43 (s, 9H), 2.35 (d, J = 6.2 Hz, 1 H), 3.80 (dd, J = 10.4, 4.4 Hz, 2H), 4.15 (dd, J = 9.6, 6.7 Hz, 2H), 4.58 ppm (m, 1 H).
80%
With hydrogen; triethylamine In methanol for 16 h;
Preparation Example 1-83-23-Hydroxy-azetidine-1-carboxylic acid tert-butyl ester 1.8 g (7.52 mmol) of the compound obtained from Preparation Example 1-83-1 was dissolved in 35 mL of methanol. To the solution was added 1.81 g (8.27 mmol) of di-tert-butyl dicarbonate, 0.8 g (7.9 mmol) of triethylamine, and palladium which is absorbed to active carbon (Pd/C) (10percent, 0.18 g), and stirred under hydrogen condition for 16 hours. The reaction mixture was filtered through Celite, distilled in vacuo to remove a solvent and purified by column chromatography using a mixed solution of hexane and ethyl acetate in the ratio of 1:1 to obtain the title compound 1.06 g (80percent).1H NMR (400 MHz, CDCl3); δ 4.58 (1H, m), 4.15 (2H, dd), 3.80 (2H, dd), 2.08 (1H, d), 1.44 (9H, s)
32%
Stage #1: With hydrogen In ethanol at 20℃; for 48 h; Stage #2: With N-ethyl-N,N-diisopropylamine In 1,3-dioxane; water at 0 - 20℃; for 5 h;
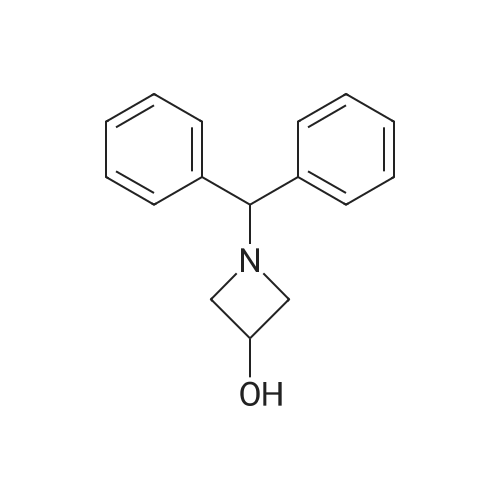
To 2.39 gm (10 mmol) of 1-(diphenylmethyl)-3-hydroxyazetidine in 50 mL of ethanol was added 239 mg of Pd/C. The reaction mixture was then hydrogenated at room temperature for 2 days. After 2 days, the suspension was filtered through celite and washed with H2O and MeOH. The combined filtrate was concentrated under reduced pressure. To the crude product were then added 50 mL of a solution containing 25 mL of H2O and 25 mL of dioxane, 2.62 gm (12 mmol) of di-t-butyl dicarbonate, and 2.1 mL (12 mmol) of DIEA at ice-bath temperature. The reaction mixture was slowly warmed to room temperature and allowed to stir at room temperature for 5 h. After 5 h, solvents were removed in vacuo. To the residue were added 100 mL of H2O and 100 mL of ethyl acetate. After removing the aqueous layer, the organic layer was washed with H2O (2 x 50 mL) and concentrated under reduced pressure. The crude product was purified by flash chromatography (2 : 1, hexane : ethyl acetate) to obtain 560 mg (32 percent) of a clear oil: 1H NMR (300 MHz, CD3OD) δ 4.48 (1H, m), 4.10 (2H, t, J = 4.5 Hz), 3.70 (2H, m), 1.43 (9H, s).
32%
With N-ethyl-N,N-diisopropylamine In 1,4-dioxane; ethanol; water; ethyl acetate
Step (a) 1-t-Butoxycarbonyl-3-hydroxyazetidine To 2.39 gm (10 mmol) of 1-(diphenylmethyl)-3-hydroxyazetidine in 50 mL of ethanol was added 239 mg of Pd/C. The reaction mixture was then hydrogenated at room temperature for 2 days. After 2 days, the suspension was filtered through celite and washed with H2O and MeOH. The combined filtrate was concentrated under reduced pressure. To the crude product were then added 50 mL of a solution containing 25 mL of H2O and 25 mL of dioxane, 2.62 gm (12 mmol) of di-t-butyl dicarbonate, and 2.1 mL (12 mmol) of DIEA at ice-bath temperature. The reaction mixture was slowly warmed to room temperature and allowed to stir at room temperature for 5 h. After 5 h, solvents were removed in vacuo. To the residue were added 100 mL of H2O and 100 mL of ethyl acetate. After removing the aqueous layer, the organic layer was washed with H2O (2*50 mL) and concentrated under reduced pressure. The crude product was purified by flash chromatography (2:1, hexane:ethyl acetate) to obtain 560 mg (32percent) of a clear oil: 1H NMR (300 MHz, CD3OD) δ 4.48 (1H, m), 4.10 (2H, t, J=4.5 Hz), 3.70 (2H, m), 1.43 (9H, s).
393.2 g
With palladium on carbon; hydrogen In tetrahydrofuran at 20℃; for 18 h;
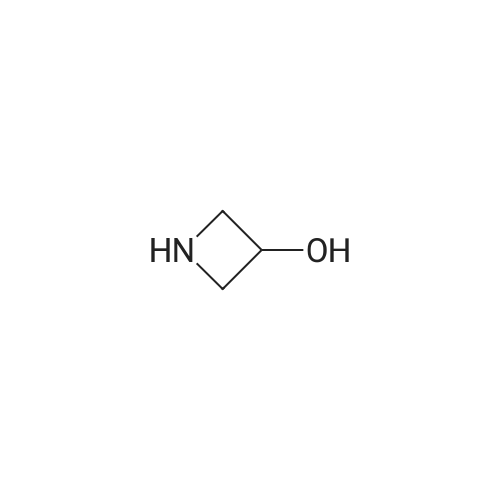
tert-Butyl 3-hydroxyazetidine-1-carboxylate (17) [0155] A suspension of 1-benzhydrylazetidin-3-ol hydrochloride (625 g, 2.27 mol) in a 10percent solution of aqueous sodium carbonate (Na2CO3, 5 L) and dichloromethane (CH2Cl2, 5 L) was stirred at room temperature until all solids were dissolved. The two layers were separated, and the aqueous layer was extracted with dichloromethane (CH2Cl2, 2 L). The combined organics extracts were dried over sodium sulfate (Na2SO4) and concentrated under reduced pressure. The resulting crude 1-benzhydrylazetidin-3-ol free base was then dissolved in THF (6 L) and the solution was placed into a large Parr bomb. Di-tert-butyl dicarbonate (BOC2O, 545 g, 2.5 mol, 1.1 equiv) and 20percent palladium (Pd) on carbon (125 g, 50percent wet) were added to the Parr bomb. The vessel was charged to 30 psi with hydrogen gas (H2) and stirred under steady hydrogen atmosphere (vessel was recharged three times to maintain the pressure at 30 psi) at room temperature for 18 h. When HPLC showed that the reaction was complete (no more hydrogen was taken up), the reaction mixture was filtered through a Celite pad and the Celite pad was washed with THF (4 L). The filtrates were concentrated under reduced pressure to remove the solvent and the residue was loaded onto a Biotage 150 column with a minimum amount of dichloromethane (CH2Cl2). The column was eluted with 20-50percent ethyl acetate in n-heptane and the fractions containing the pure desired product, tert-butyl 3-hydroxyazetidine-1-carboxylate, were collected and combined. The solvents were removed under reduced pressure to afford tert-butyl 3-hydroxyazetidine-1-carboxylate (357 g, 393.2 g theoretical, 90.8percent yield) as a colorless oil, which solidified upon standing at ambient temperature in vacuum. 1HNMR (300 MHz, CDCl3), δ 4.56 (m 1H), 4.13 (m, 2H), 3.81 (m, 2H), 1.43 (s, 9H) ppm.
With triethylamine In methanol at 0 - 20℃; for 6 h;
To a solution of 3-hydroxyazetidine hydrochloride (2.20 g) and triethyl- amine (4.0 mL) in MeOH (20 mL) at 0° C, di-tert-butyl dicarbonate (3.12 g) was added. After stirring at room temperature for 6 h, the solvent was evaporated. The residue was diluted with CH2CI2, washed with water and the organic phase was evaporated to dryness to give tert-butyl 3-hydroxy-l-azetidinecarboxylate (3.22 g, 93percent) which was used without purification in the next step. 1H-NMR (300 MHz, DMSO- de, ppm from TMS): δ 5.62 (IH, d), 4.35 (IH, m), 3.96 (2H, m), 3.55 (2H, m), 1.35 (9H, s).
88%
With triethylamine In dichloromethane at 20℃;
To a solution of azetidin-3-ol hydrochloride (2.00 g, 18.3 mmcl) in CH2CI2 (20 mL) was added TEA (5 mL) and (Boc)20 (4.80 g, 22.0 mmcl). The mixture was stirred at rt overnight. The reaction mixture was concentrated. The residue was dissolved in EtOAc (20 mL). The mixture was washed with water (20 mLx 2) and brine (20 mL), dried over Na2504 andconcentrated to give the title compound (2.80 g, yield 88percent) as yellow oil.D486 1H NMR (300 MHz, CDCI3): 6 4.58-4.56 (m, 1H), 4.17-4.11 (m, 2H), 3.82-3.77 (m, 2H), 2.51-2.49 (m, 1H), 1.43 (s, 9H).
85%
With triethylamine In ethanol at 0 - 20℃; for 16 h;
To a stirred cold (0° C.) solution of 3-hydroxyazetidine hydrochloride (75 g, 0.68 mol) in ethanol (1300 mL) was added triethylamine (208 g/280 mL, 2.05 mol) followed by Boc2O (164 g, 0.75 mol). The resultant solution was stirred at ambient temperature for 16 hours. GC/MS analysis of the reaction mixture revealed complete reaction. Volatiles were removed in vacuo and the residue was diluted with EtOAc (1300 mL) and washed with 10percent citric acid (700 mL), water (700 mL) and brine (700 mL). The organics were dried over sodium sulfate filtered, and concentrated to give the desired product (100.8 g, 85percent yield). 1H NMR (CDCl3) δ 4.6 (m, 1H), 4.2 (m, 2H), 3.8 (m, 2H), 1.4 (s, 9H).
85%
With triethylamine In ethanol at 20℃; for 16 h;
To a stirred cold (0°C) solution of 3-hydroxyazetidine hydrochloride (75 g, 0.68 mol) in ethanol (1300 mL) was added triethylamine (208g/280mL, 2.05mol) followed by B0C2O (164 g, 0.75 mol). The resultant solution was stirred at ambient temperature for 16 hours. GC/MS analysis of the reaction mixture revealed complete reaction. Volatiles were removed in vacuo and the residue was diluted with EtOAc (1300 mL) and washed with 10percent citric acid (700 mL), water (700 mL) and brine (700 mL). The organics were dried over sodium sulfate filtered, and concentrated to give the desired product (100.8 g, 85percent yield).1 H NMR (CDCI3) δ 4.6 (m, 1 H), 4.2 (m, 2 H), 3.8 (m, 2 H), 1.4 (s, 9 H).
81%
With sodium hydrogencarbonate In 1,4-dioxane; water at 20℃; for 1 h;
A mixture of 3-azetidinol hydrochloride (10 g, 91 mmol), di-tert-butyl dicarbonate (18.8 g, 86.3 mmol) and sodium bicarbonate (15.3 g, 182 mmol) in dioxane:water (400 mL, 1 :1) was stirre'd at room temperature for 15 hours. The organic portion was removed in vacuo and the aqueous portion was extracted with ethyl acetate three times. The combined organic portion was washed with 5percent aqueous HCl5 water, brine, dried over sodium sulfate, filtered and concentrated in-vacuo to afford 12.8 g, 74 mmol (81percent) of 1,1-dimethylethyl 3- hydroxyazetidine-1-carboxylate as a colorless oil without further purification. H NMR (400 MHz, DMSO): 5.62 (d, IH), 4.40-4.33 (m, IH), 4.02-3.95 (m, 2H), 3.62-3.54 (m, 2H), 1.37 (s, 9H). GC/MS for C8Hi5NO3: 173.
81%
With sodium hydrogencarbonate In 1,4-dioxane; water at 20℃; for 15 h;
A mixture of 3-azetidinol hydrochloride (10 g, 91 mmol), di-tert-buty\\ dicarbonate (18.8 g, 86.3 mmol) and sodium bicarbonate (15.3 g, 182 mmol) in <n="340"/>dioxane: water (400 mL, 1:1) was stirred at room temperature for 15 hours. The organic portion was removed in vacuo and the aqueous portion was extracted with ethyl acetate three times. The combined organic portion was washed with 5percent aqueous HCl, water, brine, dried over sodium sulfate, filtered and concentrated in- vacuo to afford 12.8 g, 74 mmol (81percent) of 1,1-dimethylethyl 3-hydroxyazetidine- 1-carboxylate as a colorless oil without further purification. 1H NMR (400 MHz, DMSO): 5.62 (d, IH), 4.40-4.33 (m, IH), 4.02-3.95 (m, 2H), 3.62-3.54 (m, 2H), 1.37 (s, 9H). GC/MS for C8Hi5NO3: 173.
81%
With sodium hydrogencarbonate In 1,4-dioxane; water at 20℃; for 15 h;
A mixture of 3-azetidinol hydrochloride (10 g, 91 mmol), di-tert-butyl dicarbonate (18.8 g, 86.3 mmol) and sodium bicarbonate (15.3 g, 182 mmol) in dioxane:water (400 mL, 1 : 1) was stirred at room temperature for 15 hours. The organic portion was removed in vacuo and the aqueous portion was extracted with ethyl acetate three times. The combined organic portion was washed with 5percent aqueous HCl, water, brine, dried over sodium sulfate, filtered and concentrated in-vacuo to afford 12.8 g, 74 mmol (81percent) of 1,1-dimethylethyl 3- hydroxyazetidine-1-carboxylate as a colorless oil without further purification. 1H NMR (400 MHz, DMSO): 5.62 (d, IH), 4.40-4.33 (m, IH), 4.02-3.95 (m, 2H), 3.62-3.54 (m, 2H), 1.37 (s, 9H). GC/MS for C8Hi5NO3: 173.
78%
With sodium hydrogencarbonate In tetrahydrofuran; water at 25 - 30℃; for 12 h; Large scale
10 kg of 3-hydroxyazetidine hydrochloride was dissolved in 100 kg of water and 84 kg of sodium bicarbonate was added.Take 52kg di-tert-butyl dicarbonate in 10L tetrahydrofuran to make a solution,And slowly adding the solution to the reaction system,The reaction was performed at 25-30[deg.] C. for 12 h.After monitoring by TLC, the reaction was completed, filtered and separated.The aqueous phase was extracted three times with ethyl acetate.Combine the organic phase,The organic phase is washed once with saturated saline solution.Dry over anhydrous sodium sulfate, filter, and concentrate under reduced pressure to give an oil.The oil was dissolved in 50 L of petroleum ether.Cool down to -50°C and stir for 12hWhite solid precipitated, filtered,It was dried to give N-Boc-3-hydroxyazetidine (white solid, 15 kg, yield: 78percent).
69%
With triethylamine In ethanol at 20℃; for 24 h;
Step A: tert-Butyl 3-hydroxy-azetidine- 1 -carboxylate To a suspension of 3-azetidinol hydrochloride (2.50 g, 22.8 mmol) in 33 mL of ethanol is added di-?-butyl dicarbonate (5.47 g, 25.10 mmol) and triethylamine (9.60 mL, 68.5 mmol) and the mixture is stirred at room temperature for 24 h. The solvents are removed in vacuo, and the residue is taken up in ethyl acetate, washed with 10percent citric acid, water, and brine. The orgainc phase is dried over magnesium sulfate, filtered and evaporated to dryness. The resulting white solid is purified on silica gel using hexanes ethyl acetate as the eluent to give the title compound (3.00 g, 69.0percent). 1H NMR (400 MHz) : 4.70 (br s, IH), 4.19 (m, 2H), 3.81 (m, 2H), 1.42 (s, 9H).
52%
With diethylamine In ethanol at 0 - 20℃;
To a cold (0 C. bath) stirred solution of compound (2) (570 mg, 5.20 mmol) in 10 mL of EtOH was added Et3N (1.8 mL, 13.0 mmol) and di-tert-butyldicarbonate (1.702 g, 7.38 mmol). The resulting mixture of clear solution was stirred at room temperature overnight. The reaction mixture was concentrated by vacuum. The residue was portioned between EtOAc (200 mL) and 0.5N citric acid solution (30 mL; brine (30 mL). The organic layer was dried (Na2SO4), then concentrated by vacuum to give 899 mg (2-) as clear oil (52percent). 1H NMR (400 MHz, chloroform-D) δ ppm 1.42 (s, 9H) 3.78 (dd, J=9.47, 4.42 Hz, 2H) 4.13 (dd, J=9.35, 6.57 Hz, 2H) 4.49-4.63 (m, 1H).
52%
With triethylamine In ethanol at 20℃;
To a cold (0°C bath) stirred solution of compound (2-2) (570 mg, 5.20 mmol) in 10mL of EtOH was added Et3N (1.8 mL, 13.0 mmol) and di-tert-butyldicarbonate (1.702 g, 7.38 mmol). The resulting mixture of clear solution was stirred at room temperature overnight. The reaction mixture was concentrated by vacuum. The residue was portioned between EtOAc (200mL) and 0.5N citric acid solution (30mL; brine (30mL). The organic layer was dried (Na2SO4), then concentrated by vacuum to give 899 mg (2-3) as clear oil (52percent). 1H NMR (400 MHz, chloroform-D) 6 ppm 1.42 (s, 9 H) 3.78 (dd, J=9.47, 4.42 Hz, 2 H) 4.13 (dd, J=9.35, 6.57 Hz, 2 H) 4.49 -4.63 (m, 1 H).
3.5 g
With sodium hydroxide In water at 0 - 35℃;
Cold aqueous NaOH (3.65 g, 91.25 mmol in 25 mL of water) was added to cold (0° C.) solution of azetidin-3-ol hydrochloride (4 g, 36.5 mmol) in water (15 mL) followed by addition of di-tert-butyl dicarbonate (8.4 mL, 38.33 mmol). The reaction mixture was stirred continuously at 20-35° C. for 12-14 h. The reaction mixture was diluted with ethyl acetate; the organic layer was separated, washed with water followed by brine solution, dried over anhydrous sodium sulphate and concentrated under reduce pressure to afford the crude compound, which was purified by column chromatography (using 60-120 silica gel and 30percent EtOAc in Hexane as eluent) to afford 3.5 g of the title compound. 1H NMR (400 MHz, DMSO) δ ppm 5.6 (1H, s), 4.4 (1H, bs), 4.0 (2H, t), 3.6-3.5 (2H, m), 1.4 (9H, s).
17 g
Stage #1: With triethylamine In methanol at -10℃; for 2.5 h; Stage #2: at -10℃;
SM2 (10.9 g, 0.1 mol) was dissolved in 200 mL of MeOH,-10 stirring reaction 30mins,After TEA (30.3 g, 0.3 mol) was added, the reaction was carried out for 2 h,(Boc) 2O (21.8 g, 0.1 mol) was added.The reaction was stirred at -10 ° C overnight.The organic phase was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated to give the crude intermediate 664602 (17 g, 98percent), which was recrystallized from water and dried over anhydrous magnesium sulfate.
Reference:
[1] Journal of Medicinal Chemistry, 2008, vol. 51, # 15, p. 4601 - 4608
[2] Patent: WO2007/118830, 2007, A1, . Location in patent: Page/Page column 91
[3] Patent: WO2017/12576, 2017, A1, . Location in patent: Page/Page column 309
[4] Patent: US2012/35122, 2012, A1, . Location in patent: Paragraph 0620; 0621
[5] Patent: WO2013/116236, 2013, A1, . Location in patent: Page/Page column 28
[6] Organic Process Research and Development, 2015, vol. 19, # 12, p. 2067 - 2074
[7] Patent: WO2007/44515, 2007, A1, . Location in patent: Page/Page column 174
[8] Patent: WO2008/76415, 2008, A1, . Location in patent: Page/Page column 338-339
[9] Patent: WO2008/124085, 2008, A2, . Location in patent: Page/Page column 189
[10] ACS Medicinal Chemistry Letters, 2012, vol. 3, # 5, p. 416 - 421
[11] Journal of Medicinal Chemistry, 2014, vol. 57, # 18, p. 7731 - 7757
[12] Patent: CN106831523, 2017, A, . Location in patent: Paragraph 0027; 0064; 0065; 0066; 0067; 0068-0073
[13] Patent: WO2007/106705, 2007, A1, . Location in patent: Page/Page column 105
[14] Patent: US2006/46991, 2006, A1, . Location in patent: Page/Page column 57-58
[15] Patent: WO2006/21881, 2006, A2, . Location in patent: Page/Page column 62; 63
[16] Patent: US2009/118287, 2009, A1, . Location in patent: Page/Page column 76
[17] Patent: WO2009/62118, 2009, A2, . Location in patent: Page/Page column 217-218
[18] Patent: WO2011/63502, 2011, A1, . Location in patent: Page/Page column 50
[19] Patent: WO2011/160020, 2011, A2, . Location in patent: Page/Page column 76
[20] Patent: WO2011/109799, 2011, A1, . Location in patent: Page/Page column 138-139; 214
[21] Patent: US2015/5280, 2015, A1, . Location in patent: Paragraph 0631 - 0633
[22] Patent: CN103709085, 2016, B, . Location in patent: Paragraph 0287-0289
[23] European Journal of Organic Chemistry, 2018, vol. 2018, # 20, p. 2587 - 2591
[24] Patent: WO2007/143823, 2007, A1, . Location in patent: Page/Page column 32-33
3
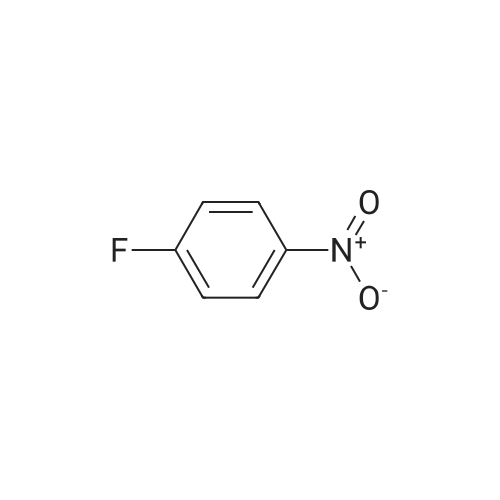
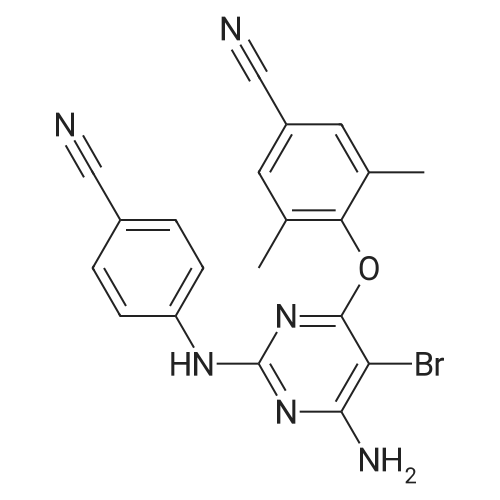
[ 45347-82-8 ]
[ 24424-99-5 ]
[ 141699-55-0 ]
Yield
Reaction Conditions
Operation in experiment
80%
With triethylamine In acetonitrile at 20℃; for 18 h;
'TO a solution of 3-hydroxyazetidine (5 g, 68.5 mmol) in 20 mL of acetonitrile at RT was added di-tert-butyl dicarbonate (11 .54 g, 52.87 mmol) and tnethylamme (7.4 mL, 53.09 mmol). The mixture was stirred for 18 hr at room temperature. The solvent was removed under reduced pressure and the residue was triturated with hexanes and the hexanes decanted. The residual oil was dried under high vacuum to give 7.78 g (80percent) of compound 3.9a as an off-white solid. :H NMR (CHLOROFORM-d) δ: 1.43 (s, 9 H) 3.55 - 3.60 (m, 1 H) 3.75 - 3.83 (m, 2 H) 4.07 - 4.18 (m, 2 H) 4.49 - 4.63 (m, 1 H).
Reference:
[1] Patent: WO2014/28800, 2014, A1, . Location in patent: Page/Page column 58; 59
[2] Journal of Medicinal Chemistry, 2001, vol. 44, # 1, p. 94 - 104
[3] Bioorganic and Medicinal Chemistry Letters, 2000, vol. 10, # 10, p. 1063 - 1066
[4] Bioorganic and Medicinal Chemistry Letters, 2009, vol. 19, # 1, p. 170 - 174
[5] Patent: US2003/229226, 2003, A1, . Location in patent: Page 21-22
4
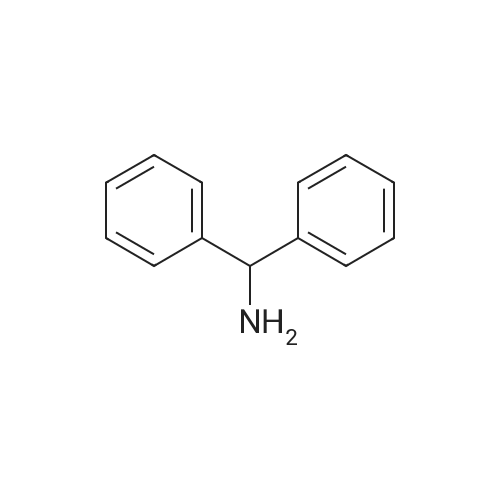
[ 18621-17-5 ]
[ 1538-75-6 ]
[ 141699-55-0 ]
Yield
Reaction Conditions
Operation in experiment
100%
With hydrogen In ethyl acetate for 24 h;
1-Benzhydryl-azetidin-3-ol 9a (4.0 g, 16.7 mmole), EtOAc (150 [ML),] [DI-TERT-BUTYL] dicarbonate (4.4 g, 20.1 [MMOLE)] and 20percent Pd (OH) 2 on carbon (0.8 g, 20 wt. percent) were sequentially added to a round bottom flask. The mixture was degassed and purged with hydrogen. The hydrogenolysis was completed after 24 hours at one atmosphere. The reaction mixture was filtered through celite and concentrated, in vacuo, to a clear oil (7.0 g). The crude product was dissolved in [CH2CI2] (10 ml) and purified over a silica gel plug (35 [G),] which was eluted with CH2CI2 (150 [ML) FOLLOWED] by EtOAc (150 [ML).] The EtOAc fractions were concentrated, in vacuo, to a clear oil (3.1 g, >100percent) : TLC (50percent ethyl acetate- cyclohexane) [R,] 0.4 [(12] stain) [; 1H-NMR (DMSO-D6,] 300 MHz) 8 5.62 (1 H, d, J = 6.4 Hz), 4.39- 4.32 (1 H, m), 3.97 (2H, t, J = 7.8 Hz), 3.57 (2H, t, J = 4.4 [HZ),] 1.35 (9H, s).
Stage #1: With formic acid In methanol for 20 h; Stage #2: With triethylamine In methanol at 20℃; for 24 h;
A stirred solution of 1-benzylazetidinol (J. Het. Chem. 1987, 24(1 ), 255-9) (0.5Og, 3.0mmole) in MeOH (15ml) and formic acid (1ml) at room temperature under argon was treated with a slurry of 10percent Pd-C catalyst (0.2Og) in MeOH (5ml) and the mixture stirred well for 2Oh, then filtered through a pad of Kieselguhr. The filtrate was treated with triethylamine (1ml) and di-tert-butyl dicarbonate (0.65g, 3.0mmole), then stirred at room temperature for 24h. The solution was concentrated under vacuum and the residue treated with 10percent Na2CO3 solution and extracted with EtOAc. The extract was dried (Na2SO4), concentrated under vacuum and the residue chromatographed on silica gel eluting with Et2O to afford the title compound as a white crystalline solid EPO <DP n="36"/>(0.32g, 60percent); 1 HNMR (400MHz, CDCI3) δ 1.43 (9H, s), 2.66 (1 H, br s), 3.79 (1 H, dd), 4.14 (1 H, dd), 4.57 (1 H, m).
With triethylamine In ethanol at 0 - 20℃; for 16 h;
Preparation 1 a: tert-butyl-3-hydroxyazetidine-1 -carboxylate; (3- hydroxyazetidine-1 -carboxylic acid tert-butyl ester) To a stirred cold (0°C) solution of 3-hydroxyazetidine hydrochloride (75 g, 0.68 mol) in ethanol (1300 mL) was added triethylamine (208 g/280 mL, 2.05 mol) followed by B0C2O (164 g, 0.75 mol). The resultant solution was stirred at ambient temperature for 16 hours. GC/MS analysis of the reaction mixture revealed complete reaction. Volatiles were removed in vacuo and the residue was diluted with EtOAc (1300 mL) and washed with 10percent citric acid (700 mL), water (700 mL) and brine (700 mL). The organics were dried over sodium sulfate filtered, and concentrated to give the desired product (100.8 g, 85percent yield).1 H NMR (CDCI3) δ 4.6 (m, 1 H), 4.2 (m, 2 H), 3.8 (m, 2 H), 1 .4 (s, 9 H).
3.5 g
With sodium hydroxide In water at 0 - 35℃;
Cold aqueous NaOH (3.65 g, 91.25 mmol in 25 mL of water) was added to cold (0° C.) solution of azetidin-3-ol hydrochloride (4 g, 36.5 mmol) in water (15 mL) followed by addition of di-tert-butyl dicarbonate (8.4 mL, 38.33 mmol). The reaction mixture was stirred continuously at 20-35° C. for 12-14 h. The reaction mixture was diluted with ethyl acetate, the organic layer was separated, washed with water followed by brine solution, dried over anhydrous sodium sulphate and concentrated under reduce pressure to afford the crude compound, which was purified by column chromatography (using 60-120 silica gel and 30percent EtOAc in Hexane as eluent) to afford 3.5 g of the title compound. 1H NMR (400 MHz, DMSO-d6) δ 5.6 (1H, s), 4.4 (1H, bs), 4.0 (2H, t), 3.6-3.5 (2H, m), 1.4 (9H, s) ppm.
This amine was dissolved in water (40 ml) then treated with sodium hydroxide (1.75 g, 44 mmol), dioxan (80 ml) and di-t-butyldicarbonate (4.79 g, 22 mmol). The reaction mixture was stirred at room temperature for 18 hours then the dioxan was evaporated. The aqueous was extracted with dichloromethane (2*50 ml), then the combined organics were dried (sodium sulphate) and evaporated to give the title compound (3.1 g, 90percent). δ (360 MHz, d6 -DMSO) 1.37 (9H, s), 3.58 (2H, dd, J1 =4, J2 =9 Hz), 3.99 (2H, dd, J1 =7, J2 =9 Hz), 4.30-4.45 (1H, m), 5.62 (1H, d, J=6 Hz).
Stage #1: With sodium carbonate In dichloromethane; water at 20℃; Stage #2: With palladium on carbon; hydrogen In tetrahydrofuran at 20℃; for 18 h;
Step 2. tert-Butyl 3-hydroxyazetidine-l -carboxylate (10) A suspension of l-benzhydrylazetidin-3-ol hydrochloride (9, 625 g, 2.27 mol) in a 10 percent solution of aqueous sodium carbonate (Na2C03, 5 L) and dichloromethane (CH2CI2, 5 L) was stirred at room temperature until all solids were dissolved. The two layers were separated, and the aqueous layer was extracted with dichloromethane (CH2CI2, 2 L). The combined organics extracts were dried over sodium sulfate (Na2SC>4) and concentrated under reduced pressure. This resulting crude free base of 9 was then dissolved in THF (6 L) and the solution was placed into a large Parr bomb. Di-teri-butyl dicarbonate (BOC20, 545 g, 2.5 mol, 1.1 equiv) and 20 percent palladium (Pd) on carbon (125 g, 50 percent wet) were added to the Parr bomb. The vessel was charged to 30 psi with hydrogen gas () and stirred under steady hydrogen atmosphere (vessel was recharged three times to maintain the pressure at 30 psi) at room temperature for 18 h. When HPLC showed that the reaction was complete (when no more hydrogen was taken up), the reaction mixture was filtered through a Celite pad and the Celite pad was washed with THF (4 L). The filtrates were concentrated under reduced pressure to remove the solvent and the residue was loaded onto a Biotage 150 column with a minimum amount of dichloromethane (CH2CI2). The column was eluted with 20 - 50 percent ethyl acetate in heptane and the fractions containing the pure desired product (10) were collected and combined. The solvents were removed under reduced pressure to afford terr-butyl 3-hydroxyazetidine-l - carboxylate (10, 357 g, 393.2 g theoretical, 90.8percent yield) as colorless oil, which solidified upon standing at room temperature in vacuum. For 10: 'iTNMR (CDCI3, 300 MHz), δ 4.56 (m 1 H), 4.13 (m, 2H), 3.81 (m, 2H), 1.43 (s, 9H) ppm.
Reference:
[1] Organic Letters, 2002, vol. 4, # 11, p. 1859 - 1862
[2] Advanced Synthesis and Catalysis, 2010, vol. 352, # 18, p. 3380 - 3390
11
[ 398489-26-4 ]
[ 141699-55-0 ]
Yield
Reaction Conditions
Operation in experiment
99%
for 2 h;
Sodium borohydride (1.01 g, 26.7 mmol) was added slowly to a solution of 1-Boc-3-azetidinone (2.28 g, 13.3 mmol) in ethanol (30 mL), stirred for 2 hours, the mixture was then concentrated under reduced pressure. The residue was treated with water (50 mL), extracted with ethyl acetate (50 mL×3). The organic layers were combined, dried over anhydrous sodium sulfate, then filtrated, the filtrate was concentrated under reduced pressure to give compound 54-d (2.28 g, yield: 99percent), which was used directly for the next step without purification. LC-MS (ESI): m/z=175 [M+H]+.
With potassium hydroxide In water at 22 - 24℃; for 0.5 h;
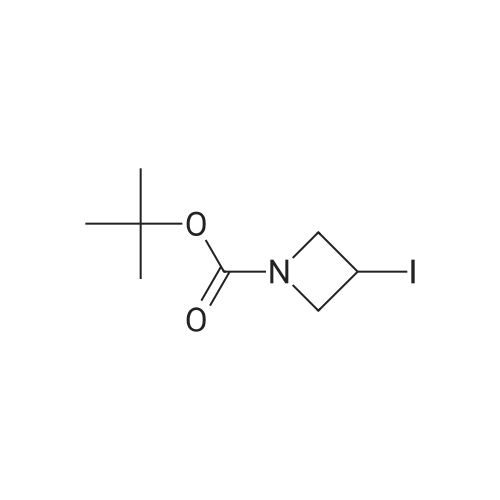
To a flame-dried reaction tube under argon was added tertbutyl3-iodoazetidine-1-carboxylate (6) (83.8 mg, 0.249 mmol, 1 equiv.), potassium acetate (36.6 mg, 0.375mmol, 1.5 equiv.) and dry DMSO (2.5 mL). The mixture was heated at 80 oC and stirred overnight. Completionof the acetoxylation step was monitored by TLC and 1H NMR of the crude reaction mixture. A solution ofpotassium hydroxide (21.0 mg, 0.374 mmol, 1.5 equiv. in 0.8 mL of H2O) was slowly added and the mixturestirred at rt for 30 min. The resulting mixture was diluted with H2O (10 mL) and extracted with ethyl acetate (3x 10 mL). The combined organic layers were washed with brine (5 mL) and dried over Na2SO4 andconcentrated under reduced pressure. The crude material was purified by flash chromatography (silica gel,10–60percent EtOAc in hexanes) to give the desired product 24 (31.2 mg, 72percent). Physical State: white solid (mp 51–52 oC); Rf = 0.13 (3:7 EtOAc/hexanes, vis. KMnO4); 1H NMR (500 MHz, CDCl3): [mixture of rotamers] δ 4.58 –4.53 (m, 1H), 4.12 (dd, J 10.6, 6.7 Hz, 2H, major), 4.12 (dd, J 8.3, 6.7 Hz, 2H, minor), 3.79 (dd, J 10.6, 4.4 Hz, 2H,major), 3.79 (dd, J 8.4, 4.4 Hz, 2H, minor), 3.12 (br s, 1H, major), 3.10 (br s, 1H, minor), 1.42 (s, 9H); 13C NMR(126 MHz, CDCl3): δ 156.6, 79.9, 61.6, 59.1 (br, 2C), 28.5 (3C). All spectral data are in accordance with thepreviously reported literature values.
With 1H-imidazole; iodine; triphenylphosphine In toluene at 100℃; for 1 h;
STAP B tert-butyl 3-iodoazetidine- 1 -carboxylate. [Chem.10][0206] To a solution of tert-butyl 3-hydroxyazetidine-l-carboxylate (3.35 g, 19.3 mmol) in toluene (200 mL) was added imidazole (3.95 g, 58.0 mmol), triphenylphosphine (10.1 g, 38.7 mmol) and iodine (7.36 g, 29.0 mmol). The mixture was heated for 1000C for Ih, cooled to room temperature, then poured into sodium bicarbonate aqueous solution (30 mL). Excess triphenylphosphine was destroyed by addition of iodine until iodine coloration persisted in organic layer. The organic layer was separated and washed with saturated sodium thiosulfate aqueous solution, dried over sodium sulfate. The crude product was purified by silica gel column chromatography (hexane - ethyl acetate = 9:1 to 1:1) to gave the title compound (5.42 g, 99percent) as clear oil.[0207] MS (ESI) m/z 284 (M+ 1)+.
95%
With 1H-imidazole; iodine; triphenylphosphine In toluene at 100℃; for 1 h;
A solution of 3-hydroxy-azetidine-i-carboxylic acid tert-butyl ester (3.35 g, 19.34 mmol) in toluene (200 ml) was treated with imidazole (3.95 g, 58.01 mmol), triphenyl- phosphine (10.14 g, 38.65 mmol) and I2 (7.36 g, 28.99 mmol). The mixture was heated at 1000C for 1 h, cooled down to room temperature and next poured into saturated aqueous NaHCO3 (30 ml). Excess triphenylphosphine was destroyed by addition of iodine until I2 coloration persisted in organic layer. The latter was washed with 5percent aqueous Na2S2O3, dried over Na2SO4 and evaporated. Purification of the crude product by flash column chromatography (heptane:ethyl acetate, 2:1) provides the title com.not. pound (5.19 g, 95percent) as a light yellow oil. MS (ESI+) m/z = 227.9 [M-ffiu+H]+1H NMR (400 MHz, CDCI3) : δ (ppm) 1.44 (s, 9H), 4.29 (dd, J = 10.4, 5.4 Hz, 2H), 4.47 (m, 1 H), 4.64 (dd, J = 9.5, 8.0 Hz, 2H).
95%
Stage #1: With 1H-imidazole; iodine; triphenylphosphine In toluene at 100℃; for 1 h; Stage #2: With sodium hydrogencarbonate In water; toluene
A solution of 3-hydroxy-azetidine-1-carboxylic acid tert-butyl ester (3.35 g, 19.34 mmol) in toluene (200 ml) was treated with imidazole (3.95 g, 58.01 mmol), triphenyl- phosphine (10.14 g, 38.65 mmol) and I2 (7.36 g, 28.99 mmol). The mixture was heated at 1000C for 1 h, cooled down to room temperature and subsequently poured into a saturated aqueous solution of NaHCO3 (30 ml). Excess triphenylphosphine was destroyed by addition of iodine until b coloration persisted in organic layer. The latter was washed with an aqueous solution of Na2S2ψ3 (5 percent strength), dried over Na2SU4, filtered and concentrated in vacuo. Purification of the residue by flash column chromatography (heptane: EtOAc, 2:1 ) provides the title compound (5.19 g, 95 percent) as a light yellow oil.MS (ESI+) : m/z = 227.9 [M-tBu+H]+ ; 1H-NMR (400 MHz, CDCI3) : δ = 1.44 (s, 9H), 4.29 (dd, J = 10.4, 5.4 Hz, 2H), 4.47 (m, 1 H), 4.64 ppm (dd, J = 9.5, 8.0 Hz, 2H).
93%
With 1H-imidazole; iodine; triphenylphosphine In toluene at 100℃; for 1 h;
A solution of ieri-butyl 3-hydroxyazetidine-l-carboxylate (25-3) (3.5 g, 0.02 mol) in toluene (200 mL) was treated with imidazole (4.08 g, 0.06 mol), PPh3 (0.6 g, 0.04 mol), and I2 (7.62 g, 0.03 mol). The mixture was heated at 100°C for 1 h and cooled down to room temperature. It was then poured into saturated NaHCC^ solution (30 mL). Excess PPh3 was destroyed by addition of iodine until I2 coloration persisted in organic layer. The mixture was washed with 5percent Na2SC>3 solution, dried over Na2S04, and evaporated in vacuo. The residue was purified on silical gel column to give 199a (5.31 g, 93percent). MS: [M+H]+ 284.
93%
With 1H-imidazole; iodine; triphenylphosphine In toluene at 100℃; for 1 h;
Example 206a tert-Butyl 3-Iodoazetidine-1-carboxylate 206a A solution of tert-butyl 3-hydroxyazetidine-1-carboxylate (3.5 g, 0.020 mol) in toluene (200 mL) was treated with imidazole (4.08 g, 0.060 mol), triphenylphosphine (0.60 g, 0.040 mol), and iodine (7.62 g, 0.030 mol). The mixture was heated at 100°C for 1 h. It was then cooled to room temperature and poured into saturated NaHCO3 solution (30 mL). Excess triphenylphosphine was destroyed by addition of iodine until iodine coloration persisted in organic layer. The mixture was washed with 5percent Na2SO3 solution, dried over Na2SO4, and evaporated in vacuo. The residue was purified by silica-gel column chromatography to afford 206a (5.31 g, 93percent). MS-ESI: [M+H]+284.
91%
With 1H-imidazole; iodine; triphenylphosphine In toluene at 110℃; for 4 h;
To a solution of tert-butyl 3-hydroxyazetidine-1-carboxylate (400 mg, 2.31 mmol) in toluene (23 ml), imidizole (472 mg, 6.93 mmol), triphenylphosphine (1.21 g, 4.62 mmol) and iodine (879 mg, 3.46 mmol) were added successively and the reaction mixture was heated at 110° C. for 4 h. The cooled reaction mixture was quenched with saturated aqueous NaHCO3 and extracted with Et2O (2*). The Et2O layers were combined and the composite was treated with iodine until a persistent brown color occurred and the mixture was stirred at RT overnight. The Et2O solution was treated with saturated aqueous Na2S2O3 until colorless, the phases were split and the organic phase was dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with 0 to 20percent EtOAc in hexane to give Compound 9a (594 mg, 91percent yield) as a clear colorless oil. MS m/z=284.0, (M+H) 1H NMR (CHLOROFORM-d) δ: 4.57 (t, J=8.4 Hz, 2H), 4.36-4.44 (m, 1H), 4.18-4.26 (m, 2H), 1.37 (s, 9H).
3.23 g
With 1H-imidazole; iodine; triphenylphosphine In toluene at 100℃; for 1.75 h;
(1) To a solution of Compound 1 (2.0 g) in toluene (115 mL) was added imidazole (2.36 g), triphenylphosphine (6.06 g), and iodine (4.4 g), and the mixture was stirred at 100°C for 1 hour and 45 minutes. The reaction mixture was cooled to room temperature, a saturated aqueous solution of sodium hydrogen carbonate was added thereto, stirred, then iodine was added thereto, stirred, and extracted with ethyl acetate. The resultant organic layer was dried, and concentrated under reduced pressure. The residue was purified with silica gel column chromatography (hexane:ethyl acetate=100:0-75:25) to give Compound 2 (3.23 g) as a colorless liquid.
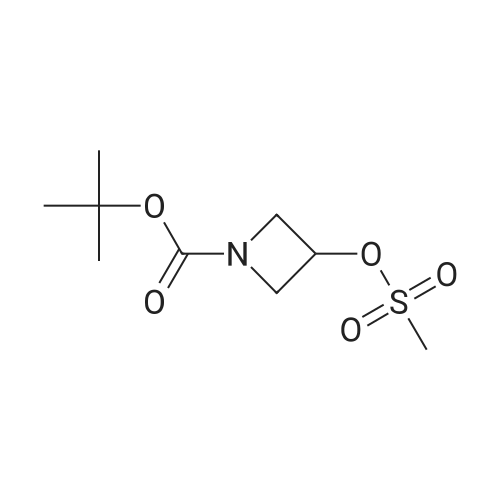
With triethylamine In tetrahydrofuran at 20℃; for 2 h; Inert atmosphere
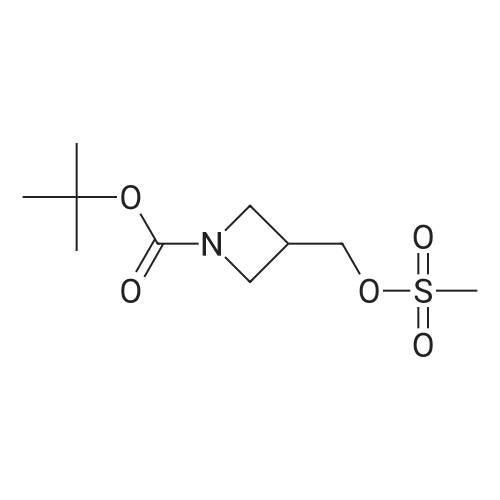
Production Example 26-2 tert-Butyl 3-((methylsulfonyl)oxy)azetidine-1-carboxylate Methanesulfonyl chloride (2.57 mL, 33.3 mmol) and triethylamine (11.6 mL, 83.1 mmol) were added to a solution of commercially available N-BOC-3-hydroxy azetidine (4.8 g, 27.7 mmol) in tetrahydrofuran (100 mL) under nitrogen atmosphere at room temperature. The reaction liquid was stirred at room temperature for 2 hours. A saturated aqueous sodium bicarbonate solution was added to the reaction liquid at room temperature, and the mixture was diluted with ethyl acetate. The organic layer was washed with a saturated saline solution and then dried over anhydrous sodium sulfate. The drying agent was separated by filtration and then the filtrate was concentrated under vacuum. The residue was purified with silica gel column chromatography (n-heptane:ethyl acetate=9:1-1:1) to quantitatively obtain the title compound. 1H-NMR Spectrum (CDCl3) δ (ppm): 1.45 (9H, s), 3.07 (3H, s), 4.03-4.18 (2H, m), 4.22-4.36 (2H, m), 5.12-5.27 (1H, m).
100%
With triethylamine In dichloromethane
(1) 1-t-Butoxycarbonyl-3-(methanesulfonyloxy)azetidine To a solution of 1-t-butoxycarbonyl-3-hydroxyazetidine (3.24 g, 18.7 mmol) (obtained as described in Reference Example 31(1)) in methylene chloride (160 ml) were added methanesulfonyl chloride (1.59 ml, 20.6 mmol) and triethylamine (2.89 ml, 20.6 mmol) in an ice bath. After stirring the mixture in the ice bath for 10 minutes, the mixture was stirred at room temperature for 6 hours. After checking the completion of the reaction, the reaction mixture was partitioned between ethyl acetate and saturated aqueous sodium hydrogencarbonate solution. The organic layer was washed with saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by chromatography on a silica gel column using n-hexane: ethyl acetate (1: 1 --> 1: 2) as the eluant to afford 1-t-butoxycarbonyl-3-(methanesulfonyloxy)azetidine (4.71 mg, yield 100percent) as a pale yellow oil. 1H-NMR (400 MHz, CDCl3): δ (ppm) 5.23 - 5.17 (1H, m), 4.28 (2H, dd, J=6.6,1.5Hz), 4.10 (2H, m), 3.07 (3H, s), 1.46 (9H, s).
99%
With triethylamine In dichloromethane at 0 - 20℃; for 6 h;
Methanesulfonyl chloride (21.4 g, 187 mmol) was added to a solution of tert-butyl3-hydroxyazetidine-1-carboxylate (25 g, 144 mmol) and triethylamine (21.8 g, 216 mmol) inDCM (500 mL) at 0 °C. After stirring at room temperature for 6 h, the reaction mixture waswashed with 1 M HC1 (50 mL) and the aqueous layer was extracted with DCM (100 mL x 2).The combined organic layers were dried over Na2SO4 and concentrated to give tert-butyl 3-((methyl sulfonyl)oxy)azetidine- 1 -carboxylate (36 g, 99percent) as a colourless oil. 1H NMR (400 MFIz, CDC13) (ppm): 5.21-5.16 (m, 1H), 4.28-4.24 (m, 2H), 4.10-4.07 (m, 2H), 3.05 (s, 3H), 1.43 (s, 9H).
98%
With triethylamine In tetrahydrofuran at 0 - 20℃;
A mixture of tert-butyl 3-hydroxyazetidine-1-carboxylate (4.98 g, 28.7 mmol) and triethylamine (4.82 mL, 62.3 mmol) in tetrahydrofuran (75 mL) was cooled to 0° C. using an ice bath. Methanesulfonyl chloride (2.46 mL, 31.8 mmol) in tetrahydrofuran (12.5 mL) was added slowly to the reaction. Once the addition was complete, the ice bath was removed and the reaction was stirred at room temperature for 4 hours. Water (100 mL) was added to the reaction, and the mixture extracted with ethyl acetate (2.x.150 mL). The combined organic phase was washed with brine (2.x.100 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to afford the title compound as a pale yellow oil (7.11 g, 98percent).1HNMR (CDCl3): δ 1.45 (s, 9H), 3.07 (s, 3H), 4.08-4.12 (m, 2H), 4.26-4.30 (m, 2H), 5.18-5.23 (m, 1H).LCMS Rt=2.53 minutes MS m/z 151.98 [M-Boc+H]+.
96%
With triethylamine In dichloromethane at 20℃;
To a solution of compound 108A (2.0 g, 11.55 mmol) in dichloromethane (10 mL) was added MsCl (1.455 g, 12.70 mmol) followed by TEA (1.519 g, 15.01 mmol). The mixture was stirred at rt overnight. The reaction was quenched with water and extracted with additional dichloromethane. The organic layer was washed, dried and concentrated to dryness to afford compound 108B (2.8 g, 96percent) as a white solid. 1H NMR (400 MHz, CDC13): δ 5.22-5.18 (m, 1H), 4.30-4.25 (m, 2H), 4.12-4.08 (m, 2H), 3.06 (s, 3H), 1.45 (s, 9H).
96%
With triethylamine In dichloromethane at 0 - 20℃;
To a stirred, cooled (0 °C) solution of terf-butyl 3-hydroxyazetidine-1 -carboxylate (500 mg, 2.89 mmol in DCM (5 mL) was added triethylamine (0.90 ml_, 6.46 mmol) followed by methanesulfonyl chloride (0.25 mL, 3.21 mmol). The mixture was allowed to warm to room temperature and stirred overnight. The mixture was poured into saturated brine and extracted twice with EtOAc. The combined organic layers were dried over Na2SC>4 and filtered. Solvent was removed under reduced pressure. The remaining material was purified on silica gel eluting with a 0percent-50percent EtOAc-hexanes gradient. The appropriate fractions (identified by TLC, silica gel, 50percent EtOAc/hexanes, ΚΜηθ4 stain) were combined, evaporated under reduced pressure and placed in vacuo to give the title compound (699 mg, 96percent) as a colorless oil that slowly solidified. 1H NMR (400 MHz, CD3SOCD3) δ 1 .39 (s, 9 H), 3.25 (s, 3 H), 3.88-3.94 (m, 2 H), 4.19-4.36 (m, 2 H), 5.22-5.28 (m, 1 H); LC-MS (LC-ES) M+H-terf-Bu = 196.
94%
With triethylamine In dichloromethane at 0℃; for 1 h;
Example 20: 3-(2'-Trifluoromethyl-biphenyl-3-yloxy)-azetidine; Step A: Preparation of 3-Methanesulfonyloxy-azetidine-1 -carboxylic acid tert-butyl ester. To 3-hydroxy-azetidine-1 -carboxylic acid tert-butyl ester (1.21 g, 6.99 mmol) and Et3N (0.85 g, 1.2 ml_, 8.4 mmol) in CH2CI2 (35 ml_) at 0 0C was added methanesulfonyl chloride (0.88 g, 0.60 ml_, 7.7 mmol). After 1 h, brine was added and the reaction extracted with CH2CI2 (2X). The combined organics were dried to give 1.65 g (94percent) of 3-methanesulfonyloxy-azetidine-1 - carboxylic acid tert-butyl ester a yellow solid that was used without further purification. 1H NMR (CDCI3): 5.20 (tt, J = 6.7, 4.2 Hz, 1 H), 4.28 (ddd, J = 10.3, 6.7, 1.2 Hz, 2H), 4.10 (ddd, J = 10.4, 4.2, 1.1 Hz, 2H), 3.07 (s, 3H), 1.44 (s, 9H).
94%
With triethylamine In dichloromethane at 20℃; for 2 h;
To a mixture of tert-butyl 3-hydroxyazetidine-1-carboxylate (1, 3.15 g. 18.3 mmol) and triethylamine (12.7 ml, 91.5 mmol) in CH2Cl2 (100 mL) was added dropwise methanesulfonyl chloride (4.17 g, 36.6 mmol) at room temperature. The reaction stirred for 2 h. The mixture was diluted with EtOAc (300 mL), washed with water (100 mL) and brine (100 mL), dried (Na2SO4), filtered and concentrated to afford the title compound (4.3 g, 94percent) as a light brown oil: 1H NMR (500 MHz, CDCl3) δ 5.16-5.23 (m, 1H), 4.25-4.30 (m, 2H), 4.07-4.12 (m, 2H), 3.06 (s, 3H), 1.44 (s, 9H)
93%
With triethylamine In dichloromethane at 0 - 20℃; for 20 h;
A solution of tert-butyl 3-hydroxyazetidine-1-carboxylate (97percent, 5.0 g, 28 mmol) in dichloromethane (50 mL) was treated with triethylamine (7.8 mL, 56 mmol) and cooled to 0° C. A solution of methanesulfonyl chloride (2.28 mL, 29.3 mmol) in dichloromethane was added drop-wise to the cold reaction, which was maintained at 0° C. for 2 h, then allowed to warm to room temp over the next 18 h. Solvents were removed in vacuo and the residue was taken up in ether and filtered: The filtrate was concentrated in vacuo, and the residue purified via silica gel chromatography (Eluant: 5:1 heptane:EtOAc, then 2:1 heptane:EtOAc) to provide C6 as a solid. Yield: 6.5 g, 26.0 mmol, 93percent. LCMS m/z 503.1 (2M+1). 1H NMR (400 MHz, CDCl3) δ 1.44 (s, 9H), 3.06 (s, 3H), 4.09 (ddd, J=10.4, 4.2, 1.2, 2H), 4.27 (ddd, J=10.4, 6.6, 1.2 Hz, 2H), 5.19 (tt, J=6.6, 4.2 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ28.23, 38.33, 56.45 (br), 67.25, 80.29, 155.80.
92%
With triethylamine In tetrahydrofuran at 20℃; for 4 h; Cooling with ice
Tert-butyl 3-hydroxyazetidin-1-carboxylate (2.0 g, 11.5 mmol) was dissolved in 50mL tetrahydrofuran, and triethylamine (2.34 g, 23.1 mmol) was added. Under the condition of ice water bath, methanesulfonyl chloride (1.58 g, 13.8 mmol) was added slowly. The resultant mixture was warmed to room temperature, and further reacted for 4 h. After the reaction, the solvent was removed by rotary evaporation, and to the residue,50mL waterwas added. After extraction with ethyl acetate (3*50mL), the organic phases were combined, dried with anhydrous sodium sulphate, and filtrated. The solvent was removed by rotary evaporation to obtain the product (2.67 g, yield: 92percent).
91%
With diethylamine In dichloromethane at 0 - 20℃;
To a solution of compound (2-3) (466 mg; 2.69 mmol) with Et3N (0.75 mL; 5.38 mmol) and 4-(dimethylamino)-pyridine (33 mg, 0.269 mmol) in 10 mL of CH2Cl2 at 0 C. was added methanesulfonyl chloride (0.25 mL 3.23 mmol). The resulting mixture of brown color solution was stirred at 0 C. to room temperature for overnight. The reaction mixture was quenched with NaHCO3, then partitioned between CH2Cl2 (200 mL) and saturated NaHCO3 solution (50 mL). The organic layer was dried (Na2SO4), then filtered through silica gel pad, eluted with hexane: EtOAc/1:1; the filtrate was concentrated by vacuum to give 614 mg (2-4) as yellow oil (91percent yield). 1H NMR (400 MHz, chloroform-D) ? ppm 1.43 (s, 9H) 3.05 (s, 3H) 4.08 (dd, J=10.36, 4.29 Hz, 2H) 4.26 (dd, J=10.36, 6.82 Hz, 2H) 5.11-5.26 (m, 1H).
91%
With dmap; triethylamine In dichloromethane at 0 - 20℃;
To a solution of compound (2-3) (466 mg; 2.69 mmol) with Et3N (0.75 mL; 5.38 mmol) and 4-(dimethylamino)-pyridine (33 mg, 0.269 mmol) in 10 mL of CH2CI2 at 0°C was added methanesulfonyl chloride (0.25 mL 3.23 mmol). The resulting mixture of brown color solution was stirred at 0°C to room temperature for overnight. The reaction mixture was quenched with NaHCO3, then partitioned between CH2CI2 (200 mL) and saturated NaHCO3 solution (50 mL). The organic layer was dried (Na2SO4), then filtered through silica gel pad, eluted with hexane: EtOAc/1:1; the filtrate was concentrated by vacuum to give 614mg (2-4) as yellow oil (91percentyield). 1H NMR (400 MHz, chloroform-D) 6 ppm 1.43 (s, 9 H) 3.05 (s, 3 H) 4.08 (dd, ^10.36, 4.29 Hz, 2 H) 4.26 (dd, J=10.36, 6.82 Hz, 2 H) 5.11 - 5.26 (m, 1 H).
83%
With triethylamine In dichloromethane at 20℃; for 1 h;
Alcohol 61 (928 mg, 5.3 mmol), Et3N (Ig, 10.7 mmol), in CH2Cl2 (20 mL), was added MsCl (733 mg, 6.4 mmol), were allowed to stirr for 1 hours at room temperature. Then the reaction mixture was diluted with diluted with CH2Cl2 (20 mL) <n="45"/>and the organic layer washed with brine (20 mL). The combined organic layer was dried over anhydrous Na2SO4 and evaporated. Purification of the resulting crude by flash silica gel chromatography provided the mesolate compound (1.11 g, 83percent) as an oil.
78%
With triethylamine In dichloromethane at 20℃; Cooling with ice
Preparation 57 tert-Butyl 3-(methylsulfonyloxy)azetidine-1-carboxylate; tert-Butyl 3-hydroxyazetidine-i-carboxylate (71g, 0.41 mol) was dissolved in dichloromethane (70OmL). Triethylamine (114mL, 0.82 mol) was added and the solution was cooled in an ice bath before the addition of methanesulfonyl chloride (33.4mL, 0.43 mol) as a solution in dichloromethane (10OmL). The reaction mixture was stirred at room temperature overnight and evaporated. The residue was dissolved in ether (50OmL), triethylamine hydrochloride was filtered off, and the filtrate was evaporated. The residue was purified by column chromatography on silica gel eluting with hexane:ethyl acetate (3:1 ) to afford the title compound (80 g, 78percent). 1H NMR (400 MHz, DMSOd6): δ = 1.38 (s, 9H), 3.23 (s, 3H), 3.90-3.94 (m, 2H), 4.19-4.24 (m, 2H), 5.22-5.28 (m, 1 H) ppm.
70%
With triethylamine In dichloromethane at 0 - 20℃; for 1 h;
To a stirred solution of tert-butyl 3-hydroxyazetidine-1-carboxylate (1.2 g, 6.92 mmol) inDCM (20 mL) was added TEA (2.0 mL, 13.8 mmol) followed by methane sulfonylchloride (0.80 mL, 10.3 mmol, spectrochem) at 0 00. The reaction mixture was stirred atRT for 1 h. Reaction completion was monitored by TLC. Reaction mixture was dilutedwith water and extracted in dichloromethane (2x50 mL). The combined extract waswashed with water, brine solution and dried over anhydrous Na2SO4 and concentrated. The crude product was isolated as pale yellow gum and was used as such for next step without further purification (1.2 g, 70percent). 1H NMR (400 MHz, DMSO-d6): 6 5.26-5.23 (m, 1 H), 4.23 (t, J = 9.6 Hz, 2H), 3.92 (d, J = 6.8 Hz, 2H), 3.24 (s, 3H), 1.37 (s, 9H). LCMS:(Method A) 152.0 (M-Boc-fH), Rt. 3.48 mm, 99.7 percent (Max).
68%
With triethylamine In dichloromethane at 0 - 20℃;
To a solution of 3-Hydroxy-azetidine-1 -carboxylic acid tert-butyl ester (2.0 g, 1 1 .2 mmol) and Et3N (3.12 ml, 22.4 mmol) in DCM (20 ml) was added MsCI (0.92 ml, 1 1 .6 mmol) dropwise at 0°C . The reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with brine (20 ml) and was extracted with ethyl acetate (20 mL χ 2). The combined organic layers was dried over anhydrous Na2S04, filtered and concentrated in vacuum. The crude product was purified by column chromatography on silica gel (eluted with petroleum ether/EtOAc = 10:1 to 2:1 ) to give compound A10-1 (2.0 g, 68
2.32 g
With triethylamine In chloroform at 0 - 20℃; for 0.5 h;
N-Boc-3-hydroxyazetidine (1.73 g) was dissolved in chloroform (20 ml). Triethylamine (2.09 ml) and methanesulfonyl chloride (856 μl) were added thereto at 0°C. After stirring at room temperature for 0.5 hours, ethyl acetate and water were added thereto to separate the organic layer. After being washed with a saturated aqueous sodium bicarbonate solution, a saturated aqueous ammonium chloride solution, and water, the organic layer was dried over anhydrous sodium sulfate. The solvent was then distilled off under reduced pressure to obtain the title compound as a colorless, oily compound (2.32 g). Physical properties: m/z[M+H]+ 252.0
2.32 g
With triethylamine In chloroform at 0 - 20℃; for 0.5 h;
N-Boc-3-hydroxyazetidine (1.73 g) was dissolved in chloroform (20 ml). Triethylamine (2.09 ml) and methanesulfonyl chloride (856 μl) were added thereto at 0° C. After stirring at room temperature for 0.5 hours, ethyl acetate and water were added thereto to separate the organic layer. After being washed with a saturated aqueous sodium bicarbonate solution, a saturated aqueous ammonium chloride solution, and water, the organic layer was dried over anhydrous sodium sulfate. The solvent was then distilled off under reduced pressure to obtain the title compound as a colorless, oily compound (2.32 g). Physical properties: m/z[M+H]+ 252.0
2.32 g
With triethylamine In chloroform at 0 - 20℃; for 0.5 h;
N-Boc-3-hydroxyazetidine (1.73 g) was dissolved in chloroform (20 ml). Triethylamine (2.09 ml) and methanesulfonyl chloride (856 μl) were added thereto at 0° C. After stirring at room temperature for 0.5 hours, ethyl acetate and water were added thereto to separate the organic layer. After being washed with a saturated aqueous sodium bicarbonate solution, a saturated aqueous ammonium chloride solution, and water, the organic layer was dried over anhydrous sodium sulfate. The solvent was then distilled off under reduced pressure to obtain the title compound as a colorless, oily compound (2.32 g). Physical properties: m/z [M+H]+ 252.0
Reference:
[1] Journal of Medicinal Chemistry, 2001, vol. 44, # 1, p. 94 - 104
[2] Patent: WO2016/206101, 2016, A1,
19
[ 141699-55-0 ]
[ 325775-44-8 ]
Reference:
[1] Journal of Medicinal Chemistry, 2001, vol. 44, # 1, p. 94 - 104
20
[ 141699-55-0 ]
[ 142253-56-3 ]
Reference:
[1] Patent: CN103709085, 2016, B,
21
[ 141699-55-0 ]
[ 193269-78-2 ]
Reference:
[1] Patent: US2011/166121, 2011, A1,
22
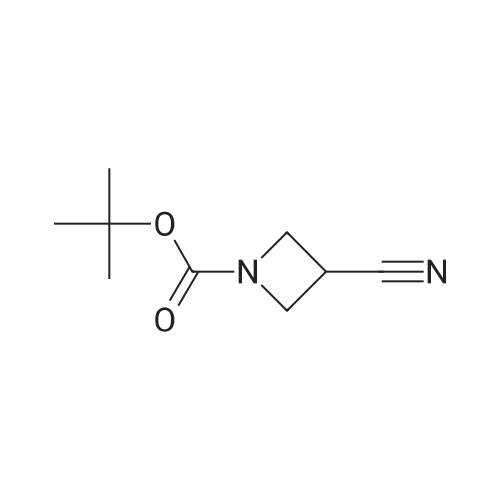
[ 141699-55-0 ]
[ 398489-26-4 ]
Yield
Reaction Conditions
Operation in experiment
95%
With pyridine-SO3 complex; triethylamine In dimethyl sulfoxide at 10 - 20℃; Inert atmosphere
A 5 L-3-neck flask equipped with mechanical stirrer, thermocouple, addition funnel and nitrogen inlet was charged with Py-SO3 (277 g, 1.74 mol) and DMSO (900 mL) and cooled to 10° C. in ice-bath. TEA (177 g/244 mL, 1.74 mol) was added. A solution of tert-butyl-3 hydroxyazetidine-1-carboxylate (Preparation 1, 100.8 g, 0.58 mol) in DMSO (500 mL) was added slowly via addition funnel at 10° C. The reaction was stirred at ambient temperature overnight. GC/MS analysis of the reaction mixture reveals that the reaction was completed. The reaction was quenched with brine (1 L). Solids were filtered and the aqueous was extracted with ethyl acetate (3×1 L). The combined organics were washed with saturated aqueous NaHCO3 (1.5 L), brine (1.5 L), dried over sodium sulfate, filtered, and concentrated to give the desired product (94 g, 95percent yield). 1H NMR (CDCl3) δ 4.6 (s, 4H), 1.4 (s, 9H).
95%
With sulfur trioxide pyridine complex; triethylamine In dimethyl sulfoxide at 10℃; for 20 h; Inert atmosphere
A 5L-3-neck flask equipped with mechanical stirrer, thermocouple, addition funnel and nitrogen inlet was charged with Py-S03 (277 g, 1.74 mol) and DMSO (900 mL) and cooled to 10°C in ice-bath. TEA (177 g/244 mL, 1 .74 mol) was added. A solution of tert-butyl-3 hydroxyazetidine-1-carboxylate (Preparation 1 a, 100.8 g, 0.58 mol) in DMSO (500 mL) was added slowly via addition funnel at 10°C. The reaction was stirred at ambient temperature overnight. GC/MS analysis of the reaction mixture reveals that the reaction was completed. The reaction was quenched with brine (1 L). Solids were filtered and the aqueous was extracted with ethyl acetate (3 x 1 L). The combined organics were washed with saturated aqueous NaHC03 (1 .5 L), brine (1.5 L), dried over sodium sulfate, filtered, and concentrated to give the desired product (94 g, 95percent yield). 1H NMR (CDCI3) δ 4.6 (s, 4 H), 1 .4 (s, 9 H).
95%
With sulfur trioxide pyridine complex; triethylamine In dimethyl sulfoxide at 10 - 20℃; Inert atmosphere; Cooling with ice
Preparation 2a: tert-butyl 3-oxoazetidine-1 -carboxylate; (3-oxoazetidine-1 - carboxylic acid tert-butyl ester) A 5L-3-neck flask equipped with mechanical stirrer, thermocouple, addition funnel and nitrogen inlet was charged with Py-S03 (277 g, 1.74 mol) and DMSO (900 ml.) and cooled to 10°C in ice-bath. TEA (177 g/244 ml_, 1 .74 mol) was added. A solution of tert-butyl-3 hydroxyazetidine-1 -carboxylate (Preparation 1 a, 100.8 g, 0.58 mol) in DMSO (500 ml.) was added slowly via addition funnel at 10°C. The reaction was stirred at ambient temperature overnight. GC/MS analysis of the reaction mixture reveals that the reaction was completed. The reaction was quenched with brine (1 L). Solids were filtered and the aqueous was extracted with ethyl acetate (3 x 1 L). The combined organics were washed with saturated aqueous NaHC03 (1 .5 L), brine (1.5 L), dried over sodium sulfate, filtered, and concentrated to give the desired product (94 g, 95percent yield). H NMR (CDCI3) δ 4.6 (s, 4 H), 1 .4 (s, 9 H).
91.1%
Stage #1: With oxalyl dichloride In dichloromethane at -78℃; Stage #2: With triethylamine In dichloromethane at 20℃;
To a -78 0C solution of 2M oxalyl chloride (31.8 mL, 63.5 mmol) in 200 mL DCM was added DMSO (9.01 mL, 127 mmol), followed by the slow addition by addition funnel of a solution of tert- butyl 3-hydroxyazetidine-l-carboxylate (10.0 g, 57.7 mmol) in 200 mL DCM. The cloudy reaction mixture was stirred at -78 0C for 15 minutes, then a solution triethylamine (32.2 mL, 231 mmol) in 40 mL DCM was added slowly by addition funnel. The reaction mixture was stirred another 15 minutes, the bath was removed, and the reaction was allowed to warm to ambient temperature and stirred for 15 hours. Water and brine were added, and the mixture was extracted with DCM. The combined extracts were dried (Na2SO4), filtered and concentrated. The crude was purified on silica gel (3-20percent ethyl acetate in hexanes gradient) to give the desired product (9.0 g, 91.1 percent yield) as a white solid.
90%
With oxalyl dichloride; dimethyl sulfoxide; triethylamine In dichloromethane at -78 - 20℃; for 15 h;
A solution of oxalyl chloride (545 μL, 6.36 mmol) in dichloromethane (25 mL) was cooled to -78 0C. While maintaining an internal temperature of -78 0C, the dropwise addition of DMSO (903 μL, 12.7 mmol) followed by 1,1-dimethylethyl 3-hydroxyazetidine- EPO <DP n="176"/>1-carboxylate (1 g, 5.78 mmol in 30 niL of dichloromethane) and finally triethylamine (3.25 mL, 23.1 mmol in 20 mL of dichloromethane) was performed. The mixture was allowed to warm to room temperature and was stirred for 15 hours. The reaction mixture was diluted with water and partitioned and the organic portion was washed twice with water. The combined aqueous portion was extracted once with dichloromethane. The combined organic portion was washed with brine, dried over sodium sulfate, filtered and concentrated in vacuo to afford a yellow oil which was purified by column chromatography. Eluting with 30percent ethyl acetate in hexanes, isolated product was concentrated in vacuo to afford 893 mg, 5.20 mmol (90percent) of 1,1-dimethylethyl 3-oxoazetidine- 1-carboxylate as a colorless oil, which solidified upon standing. 1H NMR (400 MHz, DMSO): 4.67 (s, 4H), 1.42 (s, 9H). GC/MS for C8H13NO3: 171.
90%
With oxalyl dichloride; dimethyl sulfoxide; triethylamine In dichloromethane at -78 - 20℃; for 15 h;
A solution of oxalyl chloride (545 μL, 6.36 mmol) in dichloromethane (25 mL) was cooled to -78 °C. While maintaining an internal temperature of -78 °C, the dropwise addition of DMSO (903 μL, 12.7 mmol) followed by 1,1-dimethylethyl 3 -hydroxy azetidine-1-carboxylate (1 g, 5.78 mmol in 30 mL of dichloromethane) and finally triethylamine (3.25 mL, 23.1 mmol in 20 mL of dichloromethane) was performed. The mixture was allowed to warm to room temperature and was stirred for 15 hours. The reaction mixture was diluted with water and partitioned and the organic portion was washed twice with water. The combined aqueous portion was extracted once with dichloromethane. The combined organic portion was washed with brine, dried over sodium sulfate, filtered and concentrated in vacuo to afford a yellow oil which was purified by column chromatography. Eluting with 30percent ethyl acetate in hexanes, isolated product was concentrated in vacuo to afford 893 mg, 5.20 mmol (90percent) of 1,1-dimethylethyl 3-oxoazetidine-l-carboxylate as a colorless oil, which solidified upon standing. 1H NMR (400 MHz, DMSO): 4.67 (s, 4H), 1.42 (s, 9H). GC/MS for C8Hi3NO3: 171.
90%
With oxalyl dichloride; dimethyl sulfoxide; triethylamine In dichloromethane at -78 - 20℃; for 15 h;
A solution of oxalyl chloride (545 μL, 6.36 mmol) in dichloromethane (25 mL) was cooled to -78 0C. While maintaining an internal temperature of -78 °C, the dropwise addition of DMSO (903 μL, 12.7 mmol) followed by 1,1-dimethylethyl 3 -hydroxyazetidine- 1-carboxylate (1 g, 5.78 mmol in 30 mL of dichloromethane) and finally triethylamine (3.25 mL, 23.1 mmol in 20 mL of dichloromethane) was performed. The mixture was allowed to warm to room temperature and was stirred for 15 hours. The reaction mixture was diluted with water and partitioned and the organic portion was washed twice with water. The combined aqueous portion was extracted once with dichloromethane. The combined organic portion was washed with brine, dried over sodium sulfate, filtered and concentrated in vacuo to afford a yellow oil which was purified by column chromatography. Eluting with 30percent ethyl acetate in hexanes, isolated product was concentrated in vacuo to afford 893 mg, 5.20 mmol (90percent) of 1,1-dimethylethyl 3-oxoazetidine-l-carboxylate as a colorless oil, which solidified upon standing. 1H NMR (400 MHz, DMSO): 4.67 (s, 4H), 1.42 (s, 9H). GC/MS for C8Hi3NO3: 171.
90%
With oxalyl dichloride; triethylamine In dichloromethane; dimethyl sulfoxide at -78 - 20℃; for 15 h;
A solution of oxalyl chloride (6.36 mmol) in DCM (25 ml) was cooled to -78C and DMSO (12.7 mmol), intermediate 664602 (1 g, 5.78 mmol), TEA (3.25 ml) were added sequentially. The organic phase was separated, washed twice with water, extracted with methylene chloride once, and the organic phase was concentrated.The crude product was purified by column chromatography (PE: EA = 4: 1) and the organic phase was concentrated to dryness, 1) to give intermediate 664603 (0.89 g, 90percent).
81%
With oxalyl dichloride; triethylamine In dichloromethane; dimethyl sulfoxide at -78 - 20℃;
A mixture of oxalyl chloride (2.78 ml, 31.8 mmol) in (( (80 ml) in a 500 ml round-bottomed flask was cooled to -78°C, and DMSO (4.51 ml, 63.5 mmol) was added dropwise to the mixture over 15 min. The reaction mixture was then stirred at the same temperature for 15 min. A solution of tert-butyl 3- hydroxyazetidine-l-carboxylate (5 g, 28.9 mmol) in CH2CI2 (50 ml) followed by a solution triethylamine (16.09 ml, 1 15 mmol) in CH2CI2 (70 ml) were added dropwise to the reaction mixture. The reaction mixture was warmed to room temperature, and then stirred overnight. The reaction mixture was washed with brine, and the aqueous layer back extracted with CH2CI2 (200 ml). The combined organic layers were washed with water, dried over Na2S04, filtered and concentrated in vacuum to provide a crude mixture. The crude product was then purified by column chromatography (15 percent EtOAc in Hexane) to afford tert-butyl 3-oxoazetidine-l-carboxylate Yield: 4 g (81 percent). XH NMR (400 MHz, CDC13): δ 4.69 (s, 4H), 1.49 (s, 9H).
81%
With pyridine-SO3 complex; triethylamine In dichloromethane; dimethyl sulfoxide at 10 - 20℃; for 1.5 h;
3-Oxo-azetidine- l-carboxylic acid tert-butyl ester:To a solution of l-Z?
52%
With sulfur trioxide pyridine complex; dimethyl sulfoxide; triethylamine In dichloromethane at 0℃; for 3 h;
To a mixture of tert-butyl 3-hydroxyazetidine-1-carboxylate (10.0 g, 57.7 mmol), dimethyl sulfoxide (24.0 mL, 338 mmol), triethylamine (40 mL, 300 mmol) and methylene chloride (2.0 mL) was added sulfur trioxide-pyridine complex (40 g, 200 mmol) portionwise at 0° C. The mixture was stirred for 3 hours, quenched with brine, and extracted with methylene chloride. The combined extracts were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column (0-6percent ethyl acetate (EtOAc) in hexanes) to give tert-butyl 3-oxoazetidine-1-carboxylate (5.1 g, 52percent yield).
52%
With sulfur trioxide pyridine complex; dimethyl sulfoxide; triethylamine In dichloromethane at 0℃;
Step A: tert-Butyl 3-Oxoazetidine-1-carboxylate To a mixture of tert-butyl 3-hydroxyazetidine-1-carboxylate (10.0 g, 57.7 mmol), dimethyl sulfoxide (24.0 mL, 338 mmol), triethylamine (40 mL, 300 mmol) and methylene chloride (2.0 mL) was added sulfur trioxide-pyridine complex (40 g, 200 mmol) portionwise at 0° C. The mixture was stirred for 3 hours, quenched with brine, and extracted with methylene chloride. The combined extracts were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column (0-6percent ethyl acetate (EtOAc) in hexanes) to give tert-butyl 3-oxoazetidine-1-carboxylate (5.1 g, 52percent yield).
650 mg
With pyridinium chlorochromate In dichloromethane at 20 - 35℃;
PCC (4.94 g, 22.8 mmol) was added portion wise to a stirred solution of tert-butyl-3-hydroxyazetidine-1-carboxylate (3.3 g, 19 mmol) in DCM (50 mL) at 20-35° C. and the reaction mixture was stirred continuously at the same temperature for 12-16 h. The reaction mixture was filtered and the filtrate was washed with water, dried over anhydrous sodium sulphate and concentrated under reduced pressure to afford the crude product, which was purified by column chromatography (using 60-120 silica gel and 10percent EtOAc in Hexane as eluent) to afford 650 mg of the title compound. 1H NMR (400 MHz, DMSO) δ ppm 4.66 (4H, s), 1.4 (9H, s).
650 mg
With pyridinium chlorochromate In dichloromethane at 20 - 35℃;
PCC (4.94 g, 22.8 mmol) was added portion wise to a stirred solution of tert-butyl-3-hydroxyazetidine-1-carboxylate (3.3 g, 19 mmol) in DCM (50 mL) at 20-35° C. and the reaction mixture was stirred continuously at the same temperature for 12-16 h. The reaction mixture was filtered and the filtrate was washed with water, dried over anhydrous sodium sulphate and concentrated under reduced pressure to afford the crude product, which was purified by column chromatography (using 60-120 silica gel and 10percent EtOAc in Hexane as eluent) to afford 650 mg of the title compound. 1H NMR (400 MHz, DMSO-d6) δ 4.66 (4H, s), 1.4 (9H, s) ppm.
Reference:
[1] ACS Medicinal Chemistry Letters, 2014, vol. 5, # 5, p. 550 - 555
[2] Journal of the American Chemical Society, 2013, vol. 135, # 42, p. 15742 - 15745
[3] Patent: US2012/35122, 2012, A1, . Location in patent: Paragraph 0622; 0623
[4] Patent: WO2013/116236, 2013, A1, . Location in patent: Page/Page column 28
[5] Patent: WO2013/169622, 2013, A1, . Location in patent: Page/Page column 53; 54
[6] Organic Process Research and Development, 2015, vol. 19, # 11, p. 1548 - 1553
[7] Patent: WO2010/22076, 2010, A1, . Location in patent: Page/Page column 127
[8] Patent: WO2007/44515, 2007, A1, . Location in patent: Page/Page column 174-175
[9] Patent: WO2008/76415, 2008, A1, . Location in patent: Page/Page column 339
[10] Patent: WO2008/124085, 2008, A2, . Location in patent: Page/Page column 189
[11] ACS Medicinal Chemistry Letters, 2012, vol. 3, # 5, p. 416 - 421
[12] Patent: CN103709085, 2016, B, . Location in patent: Paragraph 0290-0292
[13] Patent: WO2013/148620, 2013, A1, . Location in patent: Page/Page column 23; 24
[14] Patent: WO2009/120655, 2009, A1, . Location in patent: Page/Page column 35-36
[15] Bioorganic and Medicinal Chemistry Letters, 2009, vol. 19, # 4, p. 1084 - 1088
[16] Marine Drugs, 2016, vol. 14, # 5,
[17] Patent: US2011/224190, 2011, A1, . Location in patent: Page/Page column 36
[18] Patent: US2015/246046, 2015, A1, . Location in patent: Paragraph 0130
[19] Patent: US2009/233903, 2009, A1, . Location in patent: Page/Page column 62-63
[20] Patent: US2003/229226, 2003, A1, . Location in patent: Page 22-23
[21] Patent: WO2009/54941, 2009, A1, . Location in patent: Page/Page column 63
[22] Patent: US2010/261701, 2010, A1, . Location in patent: Page/Page column 145; 146
[23] Patent: EP2287173, 2011, A1, . Location in patent: Page/Page column 52
[24] Patent: WO2013/36611, 2013, A1, . Location in patent: Page/Page column 27; 28
[25] ACS Catalysis, 2013, vol. 3, # 11, p. 2612 - 2616
[26] Patent: US2014/256941, 2014, A1, . Location in patent: Paragraph 0156
[27] Patent: US2015/5280, 2015, A1, . Location in patent: Paragraph 0634 - 0636
[28] Patent: US2015/368238, 2015, A1, . Location in patent: Paragraph 0626; 0627
[29] Patent: WO2007/143823, 2007, A1, . Location in patent: Page/Page column 49
23
[ 141699-55-0 ]
[ 74-88-4 ]
[ 429669-07-8 ]
Yield
Reaction Conditions
Operation in experiment
93%
With sodium hydride In tetrahydrofuran; mineral oil at 0 - 20℃;
To a solution of 3-hydroxy-azetidine-1 -carboxylic acid tert-butyl ester (0.8 g, 4.62 mmol) in THF (50 mL) was added sodium hydride (60percent in mineral oil) (0.74 g, 18.5 mmol). The solution was stirred at 0 °C for 0.5 hour, followed by addition of iodomethane (2.8 mL, 46.2 mmol). The reaction mixture was stirred at room temperature overnight. The reaction was quenched with water (50 ml), and extracted with EtOAc (30 mL χ 3). The combined organic phases were dried over Na2S04, filtered and concentrated in vacuo to afford compound A9-1 (0.8 g, 93percent) as an oil. 1H NMR (400 MHz, CDCI3): δ 4.1 1 -4.13 (m, 1 H), 4.04-4.08 (m, 2 H), 3.79-3.83 (m, 2 H), 3.27 (s, 3 H), 1.43 (s, 9 H).
81%
Stage #1: With sodium hydride In DMF (N,N-dimethyl-formamide) at 0 - 20℃; for 0.666667 h; Stage #2: at 0 - 20℃; for 1.16667 h;
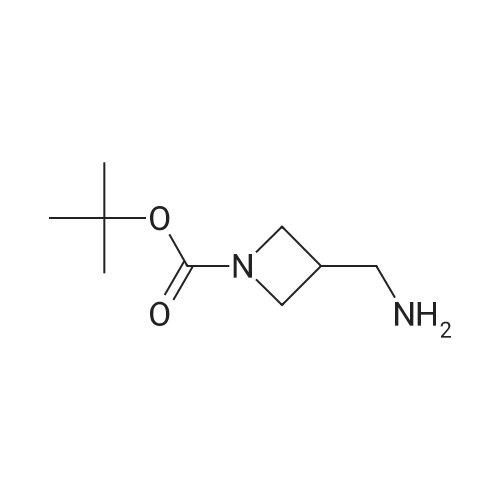
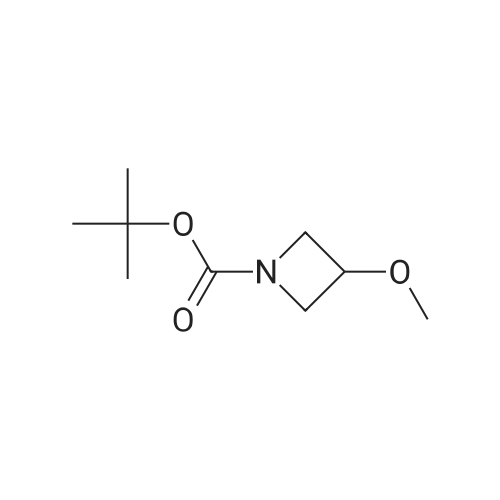
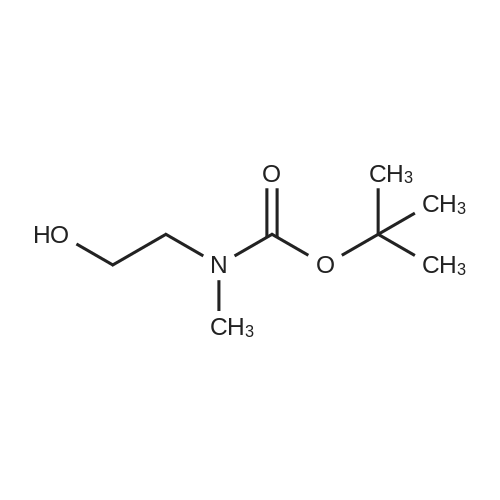
(1) 1-t-Butoxycarbonyl-3-methoxyazetidine [1493] A solution of 1-benzhydryl-3-hydroxyazetidine (10.0 g, 41.8 mmol) in methanol (300 ml) was subjected to catalytic hydrogenation in the presence of 10percent palladium (10.0 g) on charcoal at room temperature for 3 hours. After checking the completion of the reaction, the reaction mixture was filtered in order to remove the catalyst. To the filtrate was added di-t-butoxycarbonic anhydride (18.2 g, 83.6 mmol), and the reaction mixture was stirred at room temperature for 1 hour. After checking the completion of the reaction, the reaction mixture was concentrated under reduced pressure. The residue was purified by chromatography on a silica gel column using n-hexane:ethyl acetate (1:1-->1:2) as the eluant to afford 1-t-butoxycarbonyl-3-hydroxyazetidine (7.05 g, yield 97percent). [1494] Subsequently, to a solution of 1-t-butoxycarbonyl-3-hydroxyazetidine (2.5 g, 14.4 mmol) in dimethylformamide (125 ml) was added sodium hydride (55percent oil dispersion) in an ice bath. After stirring the mixture for 10 minutes in the ice bath, the resulting mixture was stirred at room temperature for 30 minutes. To the reaction mixture was added methyl iodide (1.79 ml, 28. mmol) in an ice bath. After stirring the mixture in an ice bath for 10 minutes, the reaction mixture was stirred at room temperature for 1 hour. After checking the completion of the reaction, 10percent aqueous acetic acid solution was added thereto in an ice bath and the reaction mixture was stirred in the ice bath for 30 minutes. The reaction mixture was partitioned between ethyl acetate and 10percent aqueous sodium chloride solution. The organic layer was washed successively with saturated aqueous sodium hydrogencarbonate solution and saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by chromatography on a silica gel column using n-hexane:ethyl acetate (2:1) as the eluant to afford 1-t-butoxycarbonyl-3-methoxyazetidine (2.18 g, yield 81percent) as a colorless oil. [1495] 1H-NMR (400 MHz, CDCl3): δ (ppm) 4.16-4.10 (1H, m), 4.09-4.03 (2H, m), 3.82 (2H, dd, J=10.2, 4.4 Hz), 3.28 (3H, s), 1.44 (9H, s).
33.3%
With sodium hydride In tetrahydrofuran at 0 - 20℃; for 3.25 h;
A suspension of sodium hydride (2.89 g) in tetrahydrofuran (50 ml) was stirred with cooling in an ice bath. A solution of tert-butyl 3-hydroxyazetidine-1-carboxylate (5.00 g) in tetrahydrofuran (50 ml) was gradually added, followed by stirring at the same temperature for 30 minutes. Then, the reaction mixture was stirred at room temperature for 30 minutes. The reaction mixture was stirred with cooling in an ice bath again for 15 minutes. Iodomethane (3.09 ml) was added dropwise to the reaction mixture, followed by stirring for 2 hours. Water was gradually added to the reaction mixture. When bubbling stopped, the organic layer was separated. The aqueous layer was extracted with ethyl acetate. The organic layers were combined, washed with brine, and dried over anhydrous sodium sulfate. The solvent was distilled off, and the residue was purified by silica gel column chromatography (eluent; heptane:ethyl acetate=3:1, 2:1, 1:1, then ethyl acetate). The fractions containing the target compound were concentrated to give the title compound (1.80 g, 33.3percent) as a colorless oil. The fractions containing the starting material were concentrated and collected (2.10 g, 42.0percent).1H-NMR Spectrum (CDCl3) δ (ppm): 1.44 (9H, s), 3.28 (3H, s), 3.82 (2H, m), 4.06 (2H, m), 4.14 (1H, m).
With di-isopropyl azodicarboxylate; triphenylphosphine In tetrahydrofuran at 0 - 20℃; for 16 h; Inert atmosphere
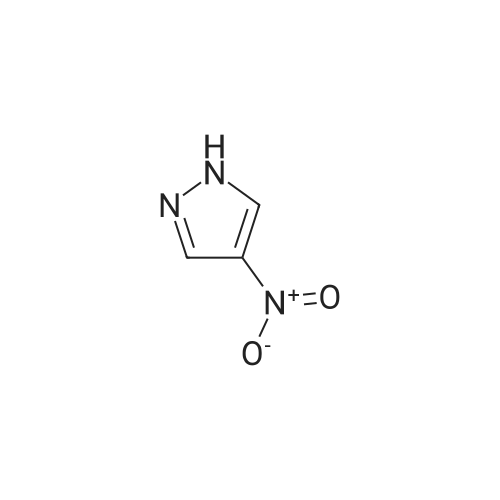
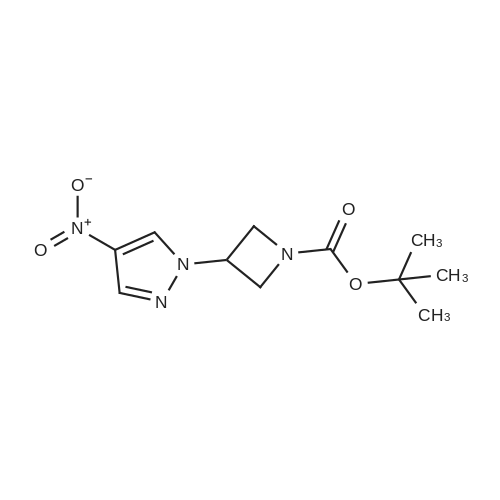
[0229j DIAD (3.92 mL, 19.9 mmmol, 1.5 equiv) was added dropwise to a stirred solution of 4-nitro-1H-pyrazole (1.5 g, 13.27 mmol), 1-Boc-3-Hydroxyazetidine (2.3 g, 13.27 mmol, 1 equiv) and triphenyiphosphine (5.22 g, 19.9 mmol, 1.5 equiv) in THF (30 mL) placed an ice-bath under N2. The mixture was stirred at 0 °C for 10 mm and allowed to warm to rt and stirred for 16 h. After diluted with EA (100 mL), the mixture was washed with water (40 mL), brine (30 mL x 2). The combined organic layer was dried, concentrated. The crude was purified through silica gel column chromatography (petroleum ether/EtOAc = 1/10) to give tert-butyl 3-(4-nitro-1H- pyrazol-1-yl)azetidine-1-carboxylate as light yellow solid (3 g, yield: 85percent). ESI-MS (M+H-56) : 213.1. ‘H NMR (400 MHz, CDC13)(5: 8.28 (s, 1H), 8.16 (s, 1H), 5.07-5.04 (m, 1H), 4.44-4.40 (m, 2H), 4.34-4.30 (m, 2H), 1.47 (s, 9H).
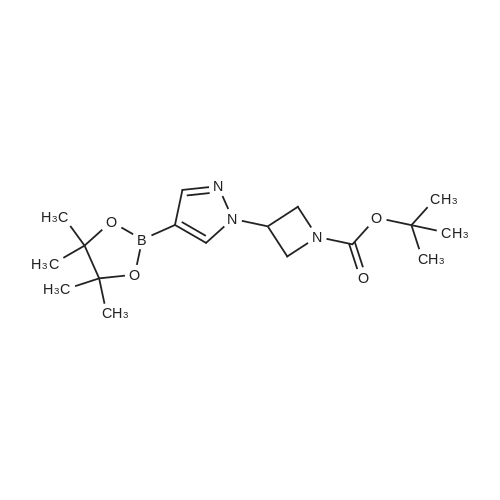
5.3 g
With di-tert-butyl (E)-azodicarboxylate; triphenylphosphine In tetrahydrofuran at 10 - 35℃;
A) tert-butyl 3-(4-nitro-1H-pyrazol-1-yl)azetidine-1-carboxylate To a solution of 4-nitro-1H-pyrazole (2.0 g), tert-butyl 3-hydroxyazetidine-1-carboxylate (3.1 g) and triphenylphosphine (5.6 g) in tetrahydrofuran (20 mL) was added di-tert-butyl (E)-diazene-1,2-dicarboxylate (5.3 g) at room temperature, and the mixture was stirred overnight at room temperature. The solvent was evaporated under reduced pressure, and the residue was purified by silica gel column chromatography (hexane/ethyl acetate) to give the title compound (5.3 g). 1H NMR(300 MHz, CDCl3)δ1.47 (9H, s), 4.28-4.37 (2H, m), 4.38-4.47 (2H, m), 5.05 (1H, tt, J = 7.9, 5.1 Hz), 8.16 (1H, s), 8.27 (1H, s).
With dmap; N-ethyl-N,N-diisopropylamine In dichloromethane at 0 - 20℃; for 16 h;
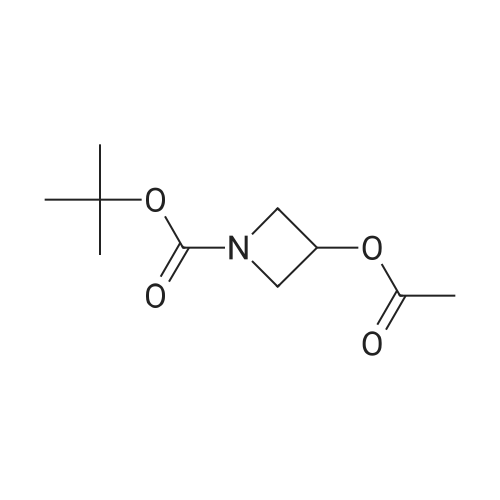
Preparation Example 1-84-13-Acetoxy-azetidine-1-carboxylic acid tert-butyl ester 0.27 g (1.559 mmol) of the compound obtained from Preparation Example 1-83-2, 0.61 g (4.677 mmol) of diisopropylethylamine and a catalytic amount of 4-dimethylaminopyridine were dissolved in 7 mL of dichloromethane and cooled to 0° C. To the solution was added 0.32 g (2 mmol) of acetic anhydride and stirred at room temperature for 16 hours. The reaction mixture was distilled in vacuo to remove a solvent and purified by column chromatography using a mixed solution of hexane and ethyl acetate in the ratio of 3:1 to obtain the title compound 0.33 g (98percent).1H NMR (400 MHz, CDCl3); δ 5.12 (1H, m), 4.23 (2H, dd), 4.12 (2H, dd), 2.09 (3H, s), 1.44 (9H, s)
Stage #1: 3-HYDROXYPYRIDINE With 1,3-bis(2,6-diisopropylphenyl)-2,2-difluoro-2,3-dihydro-1h-imidazole; 1-(Trimethylsilyl)imidazole In 1,4-dioxane at 23℃; for 0.5h; Inert atmosphere; Schlenk technique; Glovebox;
Stage #2: tert-butyl 3-hydroxyazetidine-1-carboxylate In 1,4-dioxane at 80℃; for 24h; Inert atmosphere; Schlenk technique; Glovebox;
66%
With di-isopropyl azodicarboxylate; triphenylphosphine In tetrahydrofuran at 15 - 50℃; for 16h;
8 Step 8
To a solution of pyridin-3-ol (4.94 g, 51.96 mmol, 1.5 eq) and /er/-butyl 3- hydroxyazetidine-l-carboxylate (6 g, 34.64 mmol, 1 eq) in tetrahydrofuran (100 mL) was added triphenylphosphine (10.90 g, 41.57 mmol, 1.2 eq) and diisopropyl azodicarboxylate (8.41 g, 41.57 mmol, 8.08 mL, 1.2 eq) in one portion at 15 °C, and the mixture was then stirred at 50 °C for 16 h. The reaction mixture was concentrated under reduced pressure to remove tetrahydrofuran. Water (50 mL) was poured into the mixture, and stirring continued for 1 minute. The aqueous phase was extracted with dichloromethane (60 mL x 3). The combined organic phase was washed with brine (20 mL x 2), dried with anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by flash silica gel chromatography. / -Butyl 3-(3-pyridyloxy)azetidine-l- carboxylate (6.24 g, 23.0 mmol, 66% yield) was obtained as a yellow oil.
64%
With triphenylphosphine; diethylazodicarboxylate In tetrahydrofuran
1.b Step (b)
Step (b) 3-(1-t-Butoxycarbonyl-3-azetidinyloxy)-pyridine To 315 mg (1.2 mmol) of PPh3 in 5 mL of dry THF at -20° C. was added 189 μL (1.2 mmol) of DEAD dropwise. The solution was allowed to stir 10 min. at -20° C. After 10 min, a solution containing 173 mg (1 mmol) of N-Boc-3-hydroxyazetidine and 2 mL of dry THF was added dropwise. The solution was again allowed to stir 10 min at -20° C. After 10 min, to the solution was added 95 mg (1 mmol) of 3-hydroxypyridine at once. The solution was then slowly heated to 70° C. and allowed to stir at 70° C. overnight. Next day, solvent was removed under reduced pressure. The crude product was purified by flash chromatography (2:1, Hexane: Ethyl Acetate) to obtain 160 mg (64% yield) of a yellow oil: 1H NMR (300 MHz, CD3OD) δ 8.27 (1H, d, J=4.8 Hz), 8.18 (1H, d, J=2.7 Hz), 7.23 (1H, m), 7.05 (1H, m), 4.92 (1H, m), 4.34-4.00 (2H, AB, J=6.6, 9.7 Hz), 1.45 (9H, s).
64%
Stage #1: With triphenylphosphine; diethylazodicarboxylate In tetrahydrofuran at -20℃; for 0.166667h;
Stage #2: tert-butyl 3-hydroxyazetidine-1-carboxylate In tetrahydrofuran at -20℃; for 0.166667h;
Stage #3: 3-HYDROXYPYRIDINE In tetrahydrofuran at -20 - 70℃;
1.b Step (b): 3-(1-t-Butoxycarbonyl-3-azetidinyloxy)-pyridine
To 315 mg (1.2 mmol) of PPh3 in 5 mL of dry THE at -20°C was added 189 µL (1.2 mmol) of DEAD dropwise. The solution was allowed to stir 10 min. at -20°C. After 10 min, a solution containing 173 mg (1 mmol) of N-Boc-3-hydroxyazetidine and 2 mL of dry THF was added dropwise. The solution was again allowed to stir 10 min at -20°C. After 10 min, to the solution was added 95 mg (1 mmol) of 3-hydroxypyridine at once. The solution was then slowly heated to 70°C and allowed to stir at 70°C overnight. Next day, solvent was removed under reduced pressure. The crude product was purified by flash chromatography (2 : 1, Hexane : Ethyl Acetate) to obtain 160 mg (64 % yield) of a yellow oil: 1H NMR (300 MHz, CD9OD) δ 8.27 (1H, d, J = 4.8 Hz), 8.18 (1H, d, J = 2.7 Hz), 7.23 (1H, m), 7.05 (1H, m), 4.92 (1H, m), 4.34 - 4.00 (2H, AB, J = 6.6, 9.7 Hz), 1.45 (9H, s).
With triphenylphosphine; diethylazodicarboxylate In tetrahydrofuran
Stage #1: tert-butyl 3-hydroxyazetidine-1-carboxylate With sodium hydride In N,N-dimethyl acetamide at 20℃; for 0.25h;
Stage #2: 4-Fluoronitrobenzene In N,N-dimethyl acetamide at 20℃; for 4h;
87%
With potassium hexamethylsilazane In tetrahydrofuran; toluene at 0 - 20℃; for 16.5h;
4.a a) tert-butyl 3-(4-nitrophenoxy)azetidine-l -carboxylate
To a stirred solution of tert-butyl 3-hydroxyazetidine-l-carboxylate (2.6142 g, 15.1 mmol) and l-fluoro-4-nitrobenzene (2.24 g, 15.8 mmol, CAS 350-46-9) in THF (40 mL) at 0 °C was added KHMDS in toluene (0.5 M, 31.6 mL, 15.8 mmol). After 30 min, the reaction mixture was allowed to warm to room temperature and strirred for further 16 hours. The reaction was quenched by addition of water and extracted with EtOAc. The organic phase was washed with water, brine, dried (Na2S04) and concentrated in vacuo to afford the title compound (3.85 g, 87%) as a brown oil which was used in the next step without further purification.
With tetrabutylammomium bromide; potassium hydroxide In water at 40℃;
1A Example 1A tert-butyl 3-( 4-nitrophenoxy)azetidine-1-carboxylate
tert-butyl 3-( 4-nitrophenoxy)azetidine-1-carboxylate; Tert-butyl 3-hydroxyazetidine-1-carboxylate ( 4.0 g, 23.3 mmol), 1-fluoro-4-mide nitrobenzene (6.1 g, 43.0 mmol), aqueous potassium hydroxide (35.5 ml of a 5.9 M solution,209 mmol), and tetrabutylammonium bromide (0.975 g, 3.0 mmol) were combined and stirredat 40°C overnight. The reaction was cooled, diluted with water and extracted three times withethyl acetate. The combined organics were dried (sodium sulfate), filtered and concentrated.The residue was purified by regular phase flash column chromatography to give the title compound
With tetrabutylammomium bromide; potassium hydroxide at 40℃;
1.A tert-butyl 3-(4-nitrophenoxy)azetidine-1-carboxylate
Tert-butyl 3-hydroxyazetidine-1-carboxylate (4.0 g, 23.3 mmol), 1-fluoro-4-nitrobenzene (6.1 g, 43.0 mmol), aqueous potassium hydroxide (35.5 ml of a 5.9 M solution, 209 mmol), and tetrabutylammonium bromide (0.975 g, 3.0 mmol) were combined and stirred at 40° C. overnight. The reaction was cooled, diluted with water and extracted three times with ethyl acetate. The combined organics were dried (sodium sulfate), filtered and concentrated. The residue was purified by regular phase flash column chromatography to give the title compound.
1,1-dimethylethyl 3-(4'-pyridinyloxy)-1-azetidinecarboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
57.1%
With di-isopropyl azodicarboxylate; triphenylphosphine; In toluene; at 150℃; for 3h;Sealed tube; Microwave irradiation;
To a solution of the commercially available 4-pyridinol, 17, (500 mg, 5.26mmol) in anhydrous toluene (15 mL) was added tert-butyl 3-hydroxyazetidine-1-carboxylate, R-24, (910 mg, 5.26 mmol), Ph3P (1.67 g, 6.2 mmol) and DIAD(1 .27 g, 6.3 mmol), the sealed vial was irradiated in the microwave on aBiotage Smith Synthesis at 15000 for 3 h. The resulting mixture was cooledto room temperature and concentrated under vacuo. The mixture was quenched into water, extracted with DOM, dried with anhydrous Na2504, concentrated to give the crude product which was purified by prep-TLO to give KR-30 (60 mg, 57.1%) as a pale yellow solid. ESI-MS (Mi-i): 251.1 calc.for 013H18N203: 250.1.
53%
With triphenylphosphine; diethylazodicarboxylate; In tetrahydrofuran; at 70℃;
To a solution of tert-butyl 3-hydroxyazetidine-1-carboxylate 1 (4.55 g, 26.3 mmol) inTHF (100 mL) was added <strong>[626-64-2]pyridin-4-ol</strong> (2.0 g, 21.0 mmol), PPh3 (6.89 g, 26.3 mmol)and DEAD (4.57 g, 26.3 mmol). The resulting reaction mixture was stirred at 70 Covernight. TLC indicated that the reaction was complete. The reaction mixture wasconcentrated in vacuum. The resulting oil was dissolved in 1 .0 M aqueous HCI solution C 20 mL) and extracted with DCM (50 mL x 3), The combined organic phases were washed with HCI (aq) solution (0.5 M, 150 mL). The aqueous fractions were combined and basified to pH12 using NaOH (1.0 M) and extracted with DCM(100 mL x 3) . The combined organic phases were dried over anhydrous Na2SO4,filtered and concentrated in vacuum. The residue was purified by column chromatography on silica gel to afford to afford 4 (2.81 91 53% yield) as a solid.1HNMR (400 MHz, DMSO-d6): 5 8.41 (d, J 6.0 Hz, 2 H), 6.88 (d, J = 6.0 Hz, 2 H),5.07-5.09 (m, 1 H), 4.32-4.33 (m, 2 H), 3.80-3.82 (m, 2 H)3 1.39 (s, 9 H). MS Calcd.:250; MS Found: 251 ([M+Hfl.
53%
With triphenylphosphine; diethylazodicarboxylate; In tetrahydrofuran; at 70℃;
To a solution of fert-butyl 3-hydroxyazetidine-l-carboxylate 1 (4.55 g, 26.3 mmol) in THF (100 mL) was added <strong>[626-64-2]pyridin-4-ol</strong> (2.0 g, 21.0 mmol), PPh3 (6.89 g, 26.3 mmol) and DEAD (4.57 g, 26.3 mmol). The resulting reaction mixture was stirred at 70 C overnight. TLC indicated that the reaction was complete. The reaction mixture was concentrated in vacuum. The resulting oil was dissolved in 1.0 M aqueous HCI solution (, 20 mL) and extracted with DCM (50 mL chi 3), The combined organic phases were washed with HCI (aq) solution (0.5 M, 150 mL). The aqueous fractions were combined and basified to pH12 using NaOH (1.0 M) and extracted with DCM (100 mL chi 3) . The combined organic phases were dried over anhydrous Na2S04, filtered and concentrated in vacuum. The residue was purified by column chromatography on silica gel to afford to afford 4 (2.81 g, 53% yield) as a solid. 1HNMR (400 MHz, DMSO- 6): delta 8.41 (d, J= 6.0 Hz, 2 H), 6.88 (d, J= 6.0 Hz, 2 H), 5.07-5.09 (m, 1 H), 4.32-4.33 (m, 2 H), 3.80-3.82 (m, 2 H), 1.39 (s, 9 H). MS Calcd.: 250; MS Found: 251 ([M+Hf).
With potassium tert-butylate; In tetrahydrofuran; at 0℃; for 0.5h;
Dissolve l-fluoro-2-methoxy-4-nitro-benzene (118 g, 689 mmol) and 3-hydroxy- azetidine-1-carboxylic acid tert-bupsilonXyl ester (125 g, 724 mmol) in THF (800 mL) and cool to 0 0C under nitrogen. To the above solution, add dropwise a 1 M THF solution of tert- BuOK (1 L). After the addition is complete, stir the dark brown solution for 30 min at 0 0C and then dilute with water (1 L) over a 10 min period. Stir the mixture for 5 min, then extract with tert-bvAyl methyl ether (2x ). Combine the organic solutions and wash with brine (2 x 700 mL), then dry and concentrate. Dry the solid in vacuo at 45 0C for 20 h to obtain 216 g (95percent) of the title compound as a yellow solid. 1H NMR (300 MHz, CDCl3) 6.47 (d, IH, J= 8.5), 6.30 (d, IH, J= 2.6), 6.17 (dd, IH, J= 2.4, 8.5), 4.76 (m, IH), 4.18 (m, 2H), 4.04 (m, 2H), 3.80 (s, 3H), 1.42 (s, 9H).
95%
With potassium tert-butylate; In tetrahydrofuran; at 0℃; for 0.5h;
Preparation 60; 3-(2-Methoxy-4-nitro-phenoxy)-azetidine-l-carboxylic acid tert-butyl ester; Dissolve l-fluoro-2-methoxy-4-nitro-benzene (118 g, 689 mmol) and 3 -hydroxy - azetidine-1-carboxylic acid tert-butyl ester (125 g, 724 mmol) in THF (800 mL) and cool to 0 0C. To the above solution under a nitrogen atmosphere, add dropwise a IM solution of tBuOK (I L, solution in THF). After addition is complete, stir the dark brown solution for 30 min at 0 0C, then dilute with water (1 L) over a 10 min period. Stir the mixture for5 min, then extract with MTBE twice. Combine the organic solutions and wash with brine (2 x 700 mL), then dry and concentrate. Dry the solid in vacuo at 45 0C for 20 h to <n="56"/>obtain 216 g (95percent) of the title compounds as a yellow solid. 1H NMR (300 MHz, CDCl3) .pound. 1.42 (s, 9H), 3.80 (s, 3H), 4.04 (m, 2H), 4.18 (m, 2H), 4.76 (m, IH), 6.17 (dd, IH, J = 2.4, 8.5), 6.30 (d, IH, J= 2.6), 6.47 (d, IH, J= 8.5).
With potassium tert-butylate; In tetrahydrofuran; at 20 - 70℃;
Preparation 131; 3-(4-Bromo-phenoxy)-azetidine-l-carboxylic acid tert-bvAyl ester; Add dropwise potassium tert-butoxide in THF (34.6 mL, 34.6 mmol) to a solution oip- bromofluorobenzene (6.1 g, 34.6 mmol) in THF (144 mL) and 3-hydroxy-azetidine-l- carboxylic acid tert-butyl ester (5.0 g, 28.9 mmol) at RT. Heat the mixture at 70 0C overnight. Cool the mixture to RT, quench with water, dilute with Et2O, and wash once with saturated NH4Cl. Back extract the aqueous with Et2O, dry the combined organics over Na2SO4, filter, and concentrate. Purify the crude material by flash chromatography, eluting with 25% EtOAc/hexane to give 4.4 g (46%) of the title compound. LC-MS/ES m/z (81Br) 274 [M -tertBu+H]+.
19%
With potassium tert-butylate; In tetrahydrofuran; at 20 - 70℃; for 4.0h;
Combine l-bromo-4-fluorobenzene (10.1 g, 57.7 mmol) and tert-butyl 3- hydroxyazetidine-1-carboxylate (5.0 g, 28.9 mmol) in THF (144 mL) and stir at room temperature. Add slowly potassium tert-butoxide (75.7 mL, 57.7 mmol, IM in THF). Heat the mixture at 70 0C for 4 h. Monitor the reaction completion via gas chromatography. Cool the mixture to room temperature and quench with water. Dilute the mixture with ether. Wash the organic portion with saturated NH4Cl. Back extract the aqueous with ether. Dry the combined organics with Na2SO4, filter, and concentrate. Purify the material by flash chromatography using 5-10% EtOAc/hexanes to give 1.84 g (19% yield) of the title compound as a white solid.
9b Example 9(b): 3-hydroxyazetidine-1-carboxylic acid ter-butyl ester
1-Benzhydryl-azetidin-3-ol 9a (4.0 g, 16.7 mmole), EtOAc (150 [ML),] [DI-TERT-BUTYL] dicarbonate (4.4 g, 20.1 [MMOLE)] and 20% Pd (OH) 2 on carbon (0.8 g, 20 wt. %) were sequentially added to a round bottom flask. The mixture was degassed and purged with hydrogen. The hydrogenolysis was completed after 24 hours at one atmosphere. The reaction mixture was filtered through celite and concentrated, in vacuo, to a clear oil (7.0 g). The crude product was dissolved in [CH2CI2] (10 ml) and purified over a silica gel plug (35 [G),] which was eluted with CH2CI2 (150 [ML) FOLLOWED] by EtOAc (150 [ML).] The EtOAc fractions were concentrated, in vacuo, to a clear oil (3.1 g, >100%) : TLC (50% ethyl acetate- cyclohexane) [R,] 0.4 [(12] stain) [; 1H-NMR (DMSO-D6,] 300 MHz) 8 5.62 (1 H, d, J = 6.4 Hz), 4.39- 4.32 (1 H, m), 3.97 (2H, t, J = 7.8 Hz), 3.57 (2H, t, J = 4.4 [HZ),] 1.35 (9H, s).
N-tert-Butoxycarbonyl-3-(2-methoxyethoxy)azetidine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In N,N-dimethyl-formamide;
Reference example 4 N-tert-Butoxycarbonyl-3-(2-methoxyethoxy)azetidine To a suspension of 0.25 g of 60% sodium hydride in 5 ml of dried N,N-dimethylformamide, a solution of 1.00 g of N-tert-butoxycarbonyl-3-azetidinol in 3 ml of dried N,N-dimethylformamide was added under stirring at room temperature. The mixture was stirred at room temperature for 30 minutes, then added dropwise with a solution of 0.98 g of <strong>[16427-44-4]2-methoxyethyl methanesulfonate</strong> in 2 ml of dried N,N-dimethylformamide, and stirred at the same temperature for 4 hours. The reaction solution was poured into ice water, and extracted with ethyl acetate. The extract was washed successively with water and saturated brine, and dried over sodium sulfate, and then the solvent was evaporated under reduced pressure. Theresidue was purified by column chromatography (silica gel, ethyl acetate: n-heptane = 1: 3) to obtain 0.67 g of a colorless liquid. NMR spectrum (DMSO-d6) delta ppm: 1.37(9H,s),3.25(3H,s),3.41-3.45(2H,m),3.46-3.49(2H,m),3.64(2H,dd,J=9,4Hz),3.98(2H,d d,J=9,6.5Hz),4.21-4.26(1H,m) IR spectrum nu (liq.) cm-1: 1706
With di-isopropyl azodicarboxylate; triphenylphosphine; In tetrahydrofuran; at 0 - 20℃; for 16.0h;Inert atmosphere;
[0229j DIAD (3.92 mL, 19.9 mmmol, 1.5 equiv) was added dropwise to a stirred solution of 4-nitro-1H-pyrazole (1.5 g, 13.27 mmol), 1-Boc-3-Hydroxyazetidine (2.3 g, 13.27 mmol, 1 equiv) and triphenyiphosphine (5.22 g, 19.9 mmol, 1.5 equiv) in THF (30 mL) placed an ice-bath under N2. The mixture was stirred at 0 C for 10 mm and allowed to warm to rt and stirred for 16 h. After diluted with EA (100 mL), the mixture was washed with water (40 mL), brine (30 mL x 2). The combined organic layer was dried, concentrated. The crude was purified through silica gel column chromatography (petroleum ether/EtOAc = 1/10) to give tert-butyl 3-(4-nitro-1H- pyrazol-1-yl)azetidine-1-carboxylate as light yellow solid (3 g, yield: 85%). ESI-MS (M+H-56) : 213.1. ?H NMR (400 MHz, CDC13)(5: 8.28 (s, 1H), 8.16 (s, 1H), 5.07-5.04 (m, 1H), 4.44-4.40 (m, 2H), 4.34-4.30 (m, 2H), 1.47 (s, 9H).
5.3 g
With di-tert-butyl (E)-azodicarboxylate; triphenylphosphine; In tetrahydrofuran; at 10 - 35℃;
A) tert-butyl 3-(4-nitro-1H-pyrazol-1-yl)azetidine-1-carboxylate To a solution of 4-nitro-1H-pyrazole (2.0 g), tert-butyl 3-hydroxyazetidine-1-carboxylate (3.1 g) and triphenylphosphine (5.6 g) in tetrahydrofuran (20 mL) was added di-tert-butyl (E)-diazene-1,2-dicarboxylate (5.3 g) at room temperature, and the mixture was stirred overnight at room temperature. The solvent was evaporated under reduced pressure, and the residue was purified by silica gel column chromatography (hexane/ethyl acetate) to give the title compound (5.3 g). 1H NMR(300 MHz, CDCl3)delta1.47 (9H, s), 4.28-4.37 (2H, m), 4.38-4.47 (2H, m), 5.05 (1H, tt, J = 7.9, 5.1 Hz), 8.16 (1H, s), 8.27 (1H, s).
5.6 g
With triphenylphosphine; diethylazodicarboxylate; In tetrahydrofuran; toluene; at 20℃;
A 1.9 N diisopropyl azodicarboxylate-toluene solution (8.7 mL) was added to a mixture containing 4-nitro-1H-pyrazole (1.5 g), tert-butyl 3-hydroxyazetidine-1-carboxylate (commercially available from Tokyo Chemical Industry Co., Ltd., 2.3 g), triphenyl phosphine (4.35 g)and THF (50 mL), and the mixture was stirred at room temperature overnight. Magnesium chloride (3.15 g) was added to the reaction mixture, and the mixture was then stirred at 60 C. for 2 hours, cooled, and filtered. The filtrate was concentrated, and the obtained residue was distributed in water and ethyl acetate. The organic layer was washed with a saturated saline solution, and dried over anhydrous sodium sulfate, and then concentrated under a reduced pressure. The obtained residue was purified through silica gel column chromatography (elution solvent: hexane-ethyl acetate), and thereby tert-butyl 3-(4-nitro-1H-pyrazol-1-yl)azetidine-1-carboxylate (5.6 g)as a solid was obtained.
Add sodium hydride (2.77 g, 60% w/w, 69.2 mmol) to a solution of 3-hydroxy- azetidine-1-carboxylic acid tert-butyl ester (10.9 g, 62.9 mmol) in dimethyl sulfoxide (100 mL). Stir the mixture for 30 min and then add 3-bromo-6-fluoroanisole (15.5 g, 75.5 mmol). Heat the mixture to 65 0C overnight. Cool the mixture to room temperature and then dilute the solution with saturated ammonium chloride and brine and extract with ethyl acetate. Wash the organics five times with brine, then dry over sodium sulfate and filter. Concentrate and purify via silica gel chromatography using a 0-45% gradient of ethyl acetate/hexane to give 10.4 g (46%) of the title compound as a clear oil.
With sodium hydride; In tetrahydrofuran; mineral oil; at 0 - 20℃;
To a solution of 3-hydroxy-azetidine-1 -carboxylic acid tert-butyl ester (0.8 g, 4.62 mmol) in THF (50 mL) was added sodium hydride (60% in mineral oil) (0.74 g, 18.5 mmol). The solution was stirred at 0 C for 0.5 hour, followed by addition of iodomethane (2.8 mL, 46.2 mmol). The reaction mixture was stirred at room temperature overnight. The reaction was quenched with water (50 ml), and extracted with EtOAc (30 mL chi 3). The combined organic phases were dried over Na2S04, filtered and concentrated in vacuo to afford compound A9-1 (0.8 g, 93%) as an oil. 1H NMR (400 MHz, CDCI3): delta 4.1 1 -4.13 (m, 1 H), 4.04-4.08 (m, 2 H), 3.79-3.83 (m, 2 H), 3.27 (s, 3 H), 1.43 (s, 9 H).
81%
(1) 1-t-Butoxycarbonyl-3-methoxyazetidine [1493] A solution of 1-benzhydryl-3-hydroxyazetidine (10.0 g, 41.8 mmol) in methanol (300 ml) was subjected to catalytic hydrogenation in the presence of 10% palladium (10.0 g) on charcoal at room temperature for 3 hours. After checking the completion of the reaction, the reaction mixture was filtered in order to remove the catalyst. To the filtrate was added di-t-butoxycarbonic anhydride (18.2 g, 83.6 mmol), and the reaction mixture was stirred at room temperature for 1 hour. After checking the completion of the reaction, the reaction mixture was concentrated under reduced pressure. The residue was purified by chromatography on a silica gel column using n-hexane:ethyl acetate (1:1?1:2) as the eluant to afford 1-t-butoxycarbonyl-3-hydroxyazetidine (7.05 g, yield 97%). [1494] Subsequently, to a solution of 1-t-butoxycarbonyl-3-hydroxyazetidine (2.5 g, 14.4 mmol) in dimethylformamide (125 ml) was added sodium hydride (55% oil dispersion) in an ice bath. After stirring the mixture for 10 minutes in the ice bath, the resulting mixture was stirred at room temperature for 30 minutes. To the reaction mixture was added methyl iodide (1.79 ml, 28. mmol) in an ice bath. After stirring the mixture in an ice bath for 10 minutes, the reaction mixture was stirred at room temperature for 1 hour. After checking the completion of the reaction, 10% aqueous acetic acid solution was added thereto in an ice bath and the reaction mixture was stirred in the ice bath for 30 minutes. The reaction mixture was partitioned between ethyl acetate and 10% aqueous sodium chloride solution. The organic layer was washed successively with saturated aqueous sodium hydrogencarbonate solution and saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by chromatography on a silica gel column using n-hexane:ethyl acetate (2:1) as the eluant to afford 1-t-butoxycarbonyl-3-methoxyazetidine (2.18 g, yield 81%) as a colorless oil. [1495] 1H-NMR (400 MHz, CDCl3): delta (ppm) 4.16-4.10 (1H, m), 4.09-4.03 (2H, m), 3.82 (2H, dd, J=10.2, 4.4 Hz), 3.28 (3H, s), 1.44 (9H, s).
33.3%
With sodium hydride; In tetrahydrofuran; at 0 - 20℃; for 3.25h;
A suspension of sodium hydride (2.89 g) in tetrahydrofuran (50 ml) was stirred with cooling in an ice bath. A solution of tert-butyl 3-hydroxyazetidine-1-carboxylate (5.00 g) in tetrahydrofuran (50 ml) was gradually added, followed by stirring at the same temperature for 30 minutes. Then, the reaction mixture was stirred at room temperature for 30 minutes. The reaction mixture was stirred with cooling in an ice bath again for 15 minutes. Iodomethane (3.09 ml) was added dropwise to the reaction mixture, followed by stirring for 2 hours. Water was gradually added to the reaction mixture. When bubbling stopped, the organic layer was separated. The aqueous layer was extracted with ethyl acetate. The organic layers were combined, washed with brine, and dried over anhydrous sodium sulfate. The solvent was distilled off, and the residue was purified by silica gel column chromatography (eluent; heptane:ethyl acetate=3:1, 2:1, 1:1, then ethyl acetate). The fractions containing the target compound were concentrated to give the title compound (1.80 g, 33.3%) as a colorless oil. The fractions containing the starting material were concentrated and collected (2.10 g, 42.0%).1H-NMR Spectrum (CDCl3) delta (ppm): 1.44 (9H, s), 3.28 (3H, s), 3.82 (2H, m), 4.06 (2H, m), 4.14 (1H, m).
Step 2:; 12b (372 mg, 2.15 mmol) is dissolved in THF (7 mL) and cooled to 0 C. NaH (128.9 mg of 60% suspension in oil, 3.22 mmol) is added and the mixture is stirred for 30 min. Mel (0.40 mL, 6.4 mmol) is added and the reaction mixture is capped, allowed to warm to RT and is stirred overnight. The reaction is poured into H20, extracted with EtOAc (2x), washed with brine, dried over MgS04, filtered and concentrated in vacuo. The material is purified by flash chromatography using 0 - 50% EtOAc/hexanes as the eluent to provide 12c.1H NMR (400 MHz, CDCI3): delta 4.15-4.10 (m, 1 H), 4.09-4.04 (m, 2H), 3.82 (dd, 2H, J = 10.2, 3.9 Hz), 3.28 (s, 3H), 1 .44 (s, 9H).
[0181] Following general procedure 4, to a cold (0 C.), stirred solution of the alcohol (46) (1.23 g, 1 equiv.) in EtOAc (10 mL) was added Et3N (1.28 mL, 1.3 equiv.) followed by MsCl (0.66 mL, 1.2 equiv.). The resultant suspension was stirred at 0 C. for 1 h. The undissolved material (Et3N.HCl) was filtered off and washed with EtOAc. The organic filtrate was concentrated under reduced pressure to afford an oil. This crude oil (mesylate) was dissolved in DMSO (5 mL) and NaCN (0.696 g, 2 equiv.) was added. The resultant reaction mixture was stirred at 130 C. for 2 days. The reaction mixture was then poured into H2O and extracted with Et2O (2×). The combined organic extracts were washed with H2O and brine, dried over MgSO4 and concentrated under reduced pressure to afford a residue which was purified by flash chromatography (gradient elution: 25% EtOAc in hexane to 33% EtOAc in hexane) to yield the desired nitrile (47).
With bis(1,1-dimethylethyl)-1,2-diazenedicarboxylate; triphenylphosphine In tetrahydrofuran at 20℃;
1.1.i
DIAD (0.69 mL, 3.45 mmol) was added dropwise to a suspension of 6-methoxy- [l,5]naphthyridin-4-ol (507 mg, 2.88 mmol), 3-hydroxy-azetidine-l-carboxylic acid tert-bvXy ester (commercial, 500 mg, 2.88 mmol) and PPh3 (906 mg, 3.45 mmol) in TηF (5 mL). A clear solution formed which was stirred at rt overnight. The mixture was concentrated under reduced pressure and purified by CC (ηex/EA 1 :1) to afford the title intermediate as a yellow solid (590 mg, 62% yield). MS (ESI, m/z): 332.4 [M+η+].
Example 32; Protocol for the preparation of terf-butyl 3-(3-(2-(methoxycarbonyl)-3- methylbenzo[b]thiophen-4-ylamino)pyridin-2-yloxy)azetidine-1-carboxylate; a) Preparation of terf-butyl 3-(3-nitropyridin-2-yloxy)azetidine-1-carboxylate; Sodium hydride (60 % dispersion in oil, 2.30 g, 57.64 mmol) was added portionwise into a solution of 1 -Boc-3-(hydroxy)azetidine (9.94 g, 57.64 mmol) in anhydrous THF (110 mL) at 0 C and stirred for 0.5 hour. A solution of 2-fluoro-3-nitro-pyhdine (7.80 g, 54.9 mmol) in anhydrous THF (15 mL) was added drop wise and the mixture allowed to warm to ambient temperature overnight. The mixture was cooled in an ice- bath and water (50 mL) added. The product was extracted with EtOAc, washed with water, brine and dried (Na2SO4). The product was purified by flash- column chromatography (petroleum ether (40-60 0C)/ DCM) to yield the title compound as a yellow solid (4.37 g, 27 %).1H NMR (400 MHz; CDCI3; 25 C): delta 8.36 (m, 1 H), 8.30 (d, 1 H), 7.09 (m, 1 H), 5.45 (m, 1 H), 4.35 (m, 2H), 4.06 (m, 2H), 1.45 (s, 9H). MS (ESI+): 296 (M+H). HPLC (10cm_esci_BICARB): Rt 3.47 min (HPLC purity 93 %).
With sodium hydride; In N,N-dimethyl-formamide; mineral oil; at 0℃;
Step B: Preparation of 3-(5-Chloro-3-fluoro-2-formyl-phenoxy)-azetidine- 1 -carboxylic acid tert-butyl ester.; To a solution of the title compound of Step A (270 mg, 1.5 mmol) and 3-hydroxy-azetidine-1 -carboxylic acid tert-butyl ester (270 mg, 1.5 mmol) in dry DMF (10 ml_) at 0 0C was added NaH (60% in mineral oil, 22 mg, 1.5 mmol). After 18 h, H2O and EtOAc were added and the aqueous layer was extracted with EtOAc. The combined organic extracts were dried and concentrated. The crude residue was purified by RP HPLC to provide the title compound (75 mg, 15%). MS (ESI): mass calcd. for Ci5Hi7CIFNO4, 329.1 ; m/z found, 274.0 [M+H-56]+. 1H NMR (CDCI3): 10.4 (s, 1 H), 6.84 (dd, J = 10.0, 1.6 Hz, 1 H), 6.39 (dd, J = 1.4, 1.3 Hz, 1 H), 5.00-4.92 (m, 1 H), 4.38 (dd, J = 9.9, 6.3 Hz, 2H), 4.08 (dd, J = 10.7, 4.0 Hz, 2H), 1.46 (s, 9H).
35.0 g of intermediate 11, 1.75 g of 5% wet palladium carbon catalyst and 350 ml of tetrahydrofuran were added to a 1000 ml autoclave, stirred for 10-20 minutes, replaced three times with nitrogen, and three times with hydrogen.The temperature was adjusted to 25 to 30 C, the pressure was adjusted to 45 to 50 psi, and the reaction was stirred at this temperature and pressure for 20 hours. The sample was confirmed by TLC to complete the reaction. The palladium carbon catalyst was removed by filtration, and the filtrate was concentrated to remove solvent and by-product toluene to obtain white. solid.The above white solid was dissolved in 105 ml of n-heptane, under nitrogen protection, slowly lowered to 0 to 5 C, and stirred at 0 to 5 C for 1 to 2, filtered, and the filter cake was placed in a vacuum drying oven at a temperature. Drying below 40 C gave 33.8 g of a white solid in a yield of 91%.
60%
A stirred solution of 1-benzylazetidinol (J. Het. Chem. 1987, 24(1 ), 255-9) (0.5Og, 3.0mmole) in MeOH (15ml) and formic acid (1ml) at room temperature under argon was treated with a slurry of 10% Pd-C catalyst (0.2Og) in MeOH (5ml) and the mixture stirred well for 2Oh, then filtered through a pad of Kieselguhr. The filtrate was treated with triethylamine (1ml) and di-tert-butyl dicarbonate (0.65g, 3.0mmole), then stirred at room temperature for 24h. The solution was concentrated under vacuum and the residue treated with 10% Na2CO3 solution and extracted with EtOAc. The extract was dried (Na2SO4), concentrated under vacuum and the residue chromatographed on silica gel eluting with Et2O to afford the title compound as a white crystalline solid EPO <DP n="36"/>(0.32g, 60%); 1 HNMR (400MHz, CDCI3) delta 1.43 (9H, s), 2.66 (1 H, br s), 3.79 (1 H, dd), 4.14 (1 H, dd), 4.57 (1 H, m).
Example 81A tert-butyl 3-(4-amino-1H-pyrazol-1-yl)azetidine-1-carboxylate The title compound was prepared as described in Example 41A substituting tert-butyl 3-hydroxyazetidine-1-carboxylate for 1-methylpiperidin-4-ol.
Step 3: tert- Butyl 3-(pyridin-4-ylmethoxy)azetidine-l-carboxylateA solution of A/-Boc-azetidin-3-ol (500 mg, 2.89 mmol) in THF (10 ml_) was cooled to 0 C. t-BuOK (1M in THF, 11.6 ml_) was added dropwise and the reaction mixture was stirred at room temperature for 0.25 h then cooled to 0 C. A solution of 4- bromomethylpyridine hydrobromide (2.19 g, 8.67 mmol) in dichloromethane (5 ml_) stirred with DIPEA (4 ml_, 24 mmol) for 0.5h was then added. The reaction mixture was stirred at room temperature for 24 h. The reaction mixture was diluted with H20 and extracted twice with EtOAc. The organics were separated from the aqueous layer and combined, dried over Na2S04 and concentrated to dryness. The crude mixture was purified by column chromatography (eluent: Pentane/EtOAc, 1/1 to 7/3) to give the title product (313 mg, 41%) as a yellow oil.LCMS (ESI+) m/z 265 (M)+ : 28%.*H NMR (DMSO-c/e, 300 MHz) : delta (ppm) : 8.54 (d, J = 5.9 Hz, 2H), 7.34 (d, J = 5.9 Hz, 2H), 4.49 (s, 2H), 4.39-4.25 (m, 1H), 4.06-3.90 (m, 2H), 3.80-3.65 (m, 2H), 1.37 (s, 9H).
With dmap; N-ethyl-N,N-diisopropylamine; In dichloromethane; at 0 - 20℃; for 16.0h;
Preparation Example 1-84-13-Acetoxy-azetidine-1-carboxylic acid tert-butyl ester 0.27 g (1.559 mmol) of the compound obtained from Preparation Example 1-83-2, 0.61 g (4.677 mmol) of diisopropylethylamine and a catalytic amount of 4-dimethylaminopyridine were dissolved in 7 mL of dichloromethane and cooled to 0 C. To the solution was added 0.32 g (2 mmol) of acetic anhydride and stirred at room temperature for 16 hours. The reaction mixture was distilled in vacuo to remove a solvent and purified by column chromatography using a mixed solution of hexane and ethyl acetate in the ratio of 3:1 to obtain the title compound 0.33 g (98%).1H NMR (400 MHz, CDCl3); delta 5.12 (1H, m), 4.23 (2H, dd), 4.12 (2H, dd), 2.09 (3H, s), 1.44 (9H, s)
With triphenylphosphine; diethylazodicarboxylate; In tetrahydrofuran; at 20℃; for 48h;Sealed tube;
Step 1A stock solution was made of N-Boc-3-hydroxyazetidine (532 mg) in THF (0.35M), followed by the addition of PPh3 (808 mg) (or resin bound PPh3) and DEAD (920 mg). An appropriate portion of this stock solution was added to a 2-dram round-bottom vial containing the appropriate phenol. The sealed vials were then shaken at RT for 48 h, then concentrated under reduced pressure.
3-( 6-Benzyl-5, 6, 7, 8-tetrahydro-pyrido[4, 3-d]pyrimidin-4-yloxy)-azetidine-1-carboxylic acid tert-butyl ester; 3-Hydroxy-azetidine-1-carboxylic acid tert-butyl ester (217 mg, 1.25 mmol) was dissolved under argon in THF (10 ml.) and NaH (58 mg, 1 .44 mmol) was added. The resulting suspension was stirred at rt under argon for 15 min following by the addtion of a solution of 6-benzyl-4-chloro-5,6,7,8-tetrahydro-pyrido[4,3d]pyrimidine (250 mg, 0.963 mmol). The reaction mixture was stirred at rt for 18h, quenched with water (20 ml.) and diluted withCH2CI2. The organic layer was filtered through a phase separation tube and concentrated in vacuo. Purification by flash chromatography on silica gel with heptane / CH2CI2, 50/50 to 0/100 then EtOAc to give 3-(6-benzyl-5,6,7,8-tetrahydro-pyrido[4,3-d]pyrimidin-4-yloxy)- azetidine-1 -carboxylic acid tert-butyl ester as a yellow gum (425 mg, 1 1 1 % yield) LCMS: [M+H]+= 397.4, Rt (4)= 0.98 min.
3-Hydroxy-azetidine-1 -carboxylic acid tert-butyl ester (217 mg, 1.25 mmol) was dissolved under argon in THF (10 ml_) and NaH (58 mg, 1.44 mmol) was added. The resulting suspension was stirred at rt under argon for 15 min following by the addtion of a solution of 6-benzyl-4-chloro-5,6,7,8-tetrahydro-pyrido[4,3d]pyrimidine (250 mg, 0.963 mmol). The reaction mixture was stirred at rt for 18h, quenched with water (20 ml_) and diluted withCH2CI2. The organic layer was filtered through a phase separation tube and concentrated in vacuo. Purification by flash chromatography on silica gel with heptane / CH2CI2, 50/50 to 0/100 then EtOAc to give 3-(6-benzyl-5,6,7,8-tetrahydro-pyrido[4,3-d]pyrimidin-4-yloxy)-azetidine-1 -carboxylic acid tert-butyl ester as a yellow gum (425 mg, 1 11 % yield) LCMS: [M+H]+= 397.4, Rt (4)= 0.98 min.
3-(6-Benzyl-5,6,7,8-tetrahydro-pyrido[4,3-d]pyrimidin-4-yloxy)-azetidine-1-carboxylic acid tert-butyl ester 3-Hydroxy-azetidine-1-carboxylic acid tert-butyl ester (217 mg, 1.25 mmol) was dissolved under argon in THF (10 mL) and NaH (58 mg, 1.44 mmol) was added. The resulting suspension was stirred at rt under argon for 15 min following by the addition of a solution of 6-benzyl-4-chloro-5,6,7,8-tetrahydro-pyrido[4,3d]pyrimidine (250 mg, 0.963 mmol). The reaction mixture was stirred at rt for 18 h, quenched with water (20 mL) and diluted with CH2Cl2. The organic layer was filtered through a phase separation tube and concentrated in vacuo. Purification by flash chromatography on silica gel with heptane/CH2Cl2, 50/50 to 0/100 then EtOAc to give 3-(6-benzyl-5,6,7,8-tetrahydro-pyrido[4,3-d]pyrimidin-4-yloxy)-azetidine-1-carboxylic acid tert-butyl ester as a yellow gum (425 mg, 111% yield) LCMS: [M+H]+=397.4, Rt(4)=0.98 min.
A solution of 1,1-dimethylethyl 3 -hydroxy- 1 -azetidinecarboxylate (3.8 g, 22.0 mmol) in DMF (100 mL) was treated with sodium hydride (60% in oil, 805 mg, 18.3 mmol), and the resultant mixture was stirred at ambient temperature for 10 min. 3,6-Dichloro- 4-pyridazinamine (3.0 g, 18.3 mmol) was added, and the mixture was stirred at 90 C overnight. The cooled mixture was diluted with ethyl acetate and washed with water (3x), brine and dried (MgSC^). The residue after concentration was purified by chromatography (silica gel, gradient of 20%? 50% EtOAc in hexanes) followed by trituration with ethyl ether to give the title product as a white solid (1.6 g)..H NMR (acetone-^) delta 6.71 (s, 1H), 6.18 (br s, 2H), 5.46 (m, 1H), 4.32 (br m, 2H), 3.95 (m, 2H), 1.42 (s, 9H).
With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 5h;
To a solution of 1,1 -dimethylethyl 3 -hydroxy- 1 -azetidinecarboxylate (2.61 g, 15.1 mmol) in N,N-dimethylformamide (35 mL) was added potassium carbonate (4 g, 28.9 mmol) and <strong>[117519-08-1]2-chloro-3-nitro-6-(trifluoromethyl)pyridine</strong> (i.e. the product of Step A) (3.27 g, 14.4 mmol). The mixture was then stirred for 5 h at ambient temperature. The reaction mixture was filtered, and the filtrate was poured into ice-water and extracted with ethyl acetate. The organic phase was washed with brine and dried (Na2S04). The solvent was evaporated, and the residue was purified by column chromatography (60-120 mesh silica gel, 20% ethyl acetate in petroleum ether) to give the title product (1.72 g) as off-white solid melting at 96-98 C..H NMR delta 8.43 (d, 1H), 7.5 (d, 1H), 5.5 (m, 1H), 4.4 (m, 2H), 4.1 (m, 2H), 1.5 (s, 9H). MS (ESI) 364 amu (M+l).
With dmap; acetic anhydride; triethylamine; In dichloromethane; at 0 - 20℃; for 1.33333h;
Intermediate 95: tert-Butyl 3-acetoxyazetidine-l-carboxylate [0379] A solution of tert-butyl 3-hydroxyazetidine-l-carboxylate (2.0g), DMAP(1.55g) and triethylamine (2.34g) in DCM (80mL) was cooled to 0C. A solution of acetic anhydride (2.35g) in DCM (20mL) was added dropwise over 20 minutes and the reaction mixture was stirred at room temperature for 1 hour. The mixture was washed with water and brine, dried (MgS04) and filtered. The filtrate was concentrated in vacuo and the residue was dissolved in ether and washed with 10% aqueous citric acid solution and brine, dried (MgS04), filtered. The filtrate was concentrated in vacuo to give tert-butyl 3-acetoxyazetidine-l- carboxylate (2.34g) as a clear oil.[0380] 1H NMR (CDC13) delta: 5.12 (1H, tt), 4.23 (2H, ddd), 3.89 (2H, dd), 2.09 (3H, s),1.44 (9H, s).
Stage #1: tert-butyl 3-hydroxyazetidine-1-carboxylate With sodium hydride In N,N-dimethyl-formamide; mineral oil at 20℃; Cooling with ice;
Stage #2: 2-fluoroethyl bromide In N,N-dimethyl-formamide; mineral oil at 20 - 70℃; for 5h; Cooling with ice;
16-1.1 (2S)-2-[({4-[(4-[3-(2-fluoroethoxy)azetidin-1-yl]methyl}phenyl)ethynyl]phenyl}carbonyl)(methyl)amino]-N-hydroxy-N',2-dimethylpropanediamide (Compound 559)
(1) 60% Sodium hydride (0.18 g) was added to a DMF (5.0 mL) solution of 3-hydroxyazetidine-1-carboxylic acid t-butyl ester (0.52 g) under ice cooling, and the mixture was stirred for 30 minutes at room temperature. 1-Bromo-2-fluoroethane (0.45 mL) was added under ice cooling, and the mixture was stirred for 2 hours at room temperature, then for 1.5 hours at 50°C, and further for 1.5 hours at 70°C. The reaction mixture was cooled to room temperature, and ethyl acetate and a saturated aqueous solution of ammonium chloride were added, whereafter the organic layer was isolated. The extract was washed with water and brine in this order, and dried over anhydrous sodium sulfate. Then, the desiccant was filtered out, and the solvent was distilled off under reduced pressure. The resulting residue was purified by OH type silica gel column chromatography (gradient elution with hexane/ethyl acetate = 80/20 → 70/30) to obtain 3-(2-fluoroethoxy)azetidine-1-carboxylic acid t-butyl ester (colorless oil) (0.30 g, 46%). 1 H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.44 (9 H, s), 3.58 - 3.64 (1 H, m), 3.65 - 3.72 (1 H, m), 3.82 - 3.92 (2 H, m), 4.03 - 4.14 (2 H, m), 4.24 - 4.33 (1 H, m), 4.46 - 4.53 (1 H, m), 4.59 - 4.65 (1 H, m)
Step 2. tert-Butyl 3-hydroxyazetidine-l -carboxylate (10) A suspension of l-benzhydrylazetidin-3-ol hydrochloride (9, 625 g, 2.27 mol) in a 10 % solution of aqueous sodium carbonate (Na2C03, 5 L) and dichloromethane (CH2CI2, 5 L) was stirred at room temperature until all solids were dissolved. The two layers were separated, and the aqueous layer was extracted with dichloromethane (CH2CI2, 2 L). The combined organics extracts were dried over sodium sulfate (Na2SC>4) and concentrated under reduced pressure. This resulting crude free base of 9 was then dissolved in THF (6 L) and the solution was placed into a large Parr bomb. Di-teri-butyl dicarbonate (BOC20, 545 g, 2.5 mol, 1.1 equiv) and 20 % palladium (Pd) on carbon (125 g, 50 % wet) were added to the Parr bomb. The vessel was charged to 30 psi with hydrogen gas () and stirred under steady hydrogen atmosphere (vessel was recharged three times to maintain the pressure at 30 psi) at room temperature for 18 h. When HPLC showed that the reaction was complete (when no more hydrogen was taken up), the reaction mixture was filtered through a Celite pad and the Celite pad was washed with THF (4 L). The filtrates were concentrated under reduced pressure to remove the solvent and the residue was loaded onto a Biotage 150 column with a minimum amount of dichloromethane (CH2CI2). The column was eluted with 20 - 50 % ethyl acetate in heptane and the fractions containing the pure desired product (10) were collected and combined. The solvents were removed under reduced pressure to afford terr-butyl 3-hydroxyazetidine-l - carboxylate (10, 357 g, 393.2 g theoretical, 90.8% yield) as colorless oil, which solidified upon standing at room temperature in vacuum. For 10: 'iTNMR (CDCI3, 300 MHz), delta 4.56 (m 1 H), 4.13 (m, 2H), 3.81 (m, 2H), 1.43 (s, 9H) ppm.
357 g
tert-Butyl 3-hydroxyazetidine-1-carboxylate (17) A suspension of <strong>[90604-02-7]1-benzhydrylazetidin-3-ol hydrochloride</strong> (625 g, 2.27 mol) in a 10% solution of aqueous sodium carbonate (Na2CO3, 5 L) and dichloromethane (CH2Cl2, 5 L) was stirred at room temperature until all solids were dissolved. The two layers were separated, and the aqueous layer was extracted with dichloromethane (CH2Cl2, 2 L). The combined organics extracts were dried over sodium sulfate (Na2SO4) and concentrated under reduced pressure. The resulting crude 1-benzhydrylazetidin-3-ol free base was then dissolved in THF (6 L) and the solution was placed into a large Parr bomb. Di-tert-butyl dicarbonate (BOC2O, 545 g, 2.5 mol, 1.1 equiv) and 20% palladium (Pd) on carbon (125 g, 50% wet) were added to the Parr bomb. The vessel was charged to 30 psi with hydrogen gas (H2) and stirred under steady hydrogen atmosphere (vessel was recharged three times to maintain the pressure at 30 psi) at room temperature for 18 h. When HPLC showed that the reaction was complete (no more hydrogen was taken up), the reaction mixture was filtered through a Celite pad and the Celite pad was washed with THF (4 L). The filtrates were concentrated under reduced pressure to remove the solvent and the residue was loaded onto a Biotage 150 column with a minimum amount of dichloromethane (CH2Cl2). The column was eluted with 20-50% ethyl acetate in n-heptane and the fractions containing the pure desired product, tert-butyl 3-hydroxyazetidine-1-carboxylate, were collected and combined. The solvents were removed under reduced pressure to afford tert-butyl 3-hydroxyazetidine-1-carboxylate (357 g, 393.2 g theoretical, 90.8% yield) as a colorless oil, which solidified upon standing at ambient temperature in vacuum. 1HNMR (300 MHz, CDCl3), delta 4.56 (m 1H), 4.13 (m, 2H), 3.81 (m, 2H), 1.43 (s, 9H) ppm.
With di-isopropyl azodicarboxylate; triphenylphosphine; In tetrahydrofuran; at 110℃; for 5.0h;Inert atmosphere;
A mixture of tert-butyl 3-hydroxyazetidine-1-carboxylate (200 mg, 1.15 mmol), 4-bromophenol (240 mg, 1.39 mmol), triphenylphosphine (398 mg, 1.50 mmol) and DIAD (202 mg, 1.39 mmol) in anhydrous THF (5 mL) was heated to 110 C. and stirred under nitrogen for 5 hours. The reaction mixture was concentrated in vacuo to give a brown residue, which was purified by flash chromatography (petroleum ether/ethyl acetate 10:1 to 4:1) to give tert-butyl 3-(4-bromophenoxy)azetidine-1-carboxylate (300 mg, 79.5%) as a white solid. 1H NMR (400 MHz, CDCl3) delta 7.39 (d, 2H), 6.72 (d, 2H), 4.84 (m, 1H), 4.28 (m, 2H), 3.99 (m, 2H), 1.44 (s, 9H).
72.6%
With di-isopropyl azodicarboxylate; triphenylphosphine; In toluene; at 110℃; for 1.0h;Sealed tube; Microwave irradiation;
To a solution of 4-bromophenol, 15, (130 mg, 0.76 mmol) in toluene (10 mL)was added tert-butyl 3-hydroxyazetidine-1-carboxylate, R-24, (130 mg, 0.76mmol), PPh3 (240 mg, 0.91 mmol), DIAD (186 mg, 0.91 mmol). The sealedvial was irradiated in the microwave on a Biotage Smith Synthesis at 110 00 for 1 h. The reaction mixture was quenched into water, extracted with DCM, dried with anhydrous Na2504, concentrated to give the crude product which was purified by prep-TLC to give the reagent KR-26 (180 mg, 72.6%) as apale yellow solid. ESI-MS (Mi-i): 328.1 calc. for C14H18BrNO3: 327.0.
Stage #1: tert-butyl 3-hydroxyazetidine-1-carboxylate With sodium hydride In tetrahydrofuran; mineral oil at 0℃; for 0.5h;
Stage #2: propargyl bromide In tetrahydrofuran; mineral oil at 25℃; for 2h;
Tert-butyl 3-prop-2-ynoxyazetidine-1-carboxylate (Intermediate YV)
To a mixture of tert-butyl 3-hydroxyazetidine-1-carboxylate (5.00 g, 28.8 mmol, CAS141699-55-0) in THF (5 mL) was added NaH (1.39 g, 34.6 mmol, 60% oil dispersion) at 0° C. for 0.5 hour. Then 3-bromoprop-1-yne (5.15 g, 34.6 mmol, 3.73 mL) and TBAI (1.07 g, 2.89 mmol) were added to the mixture. The reaction mixture was stirred at 25° C. for 2 hours. On completion, the reaction mixture was quenched with sat. NH4Cl (50 mL), diluted with water (100 mL) and extracted with EA (2×100 mL). The combined organic layers was dried over Na2SO4, filtered and concentrated in vacuo to give a residue. The residue was purified by column chromatography to give the title compound (5.70, 93% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 4.45-4.37 (m, 1H), 4.13 (d, J=2.4 Hz, 2H), 4.12-4.07 (m, 2H), 3.89 (dd, J=9.6 Hz, 2H), 2.45-2.43 (m, 1H), 1.43 (s, 9H).
93%
Stage #1: tert-butyl 3-hydroxyazetidine-1-carboxylate With sodium hydride In tetrahydrofuran; mineral oil at 0℃; for 0.5h; Inert atmosphere;
Stage #2: propargyl bromide With tetra-(n-butyl)ammonium iodide In tetrahydrofuran; mineral oil at 25℃; for 2h; Inert atmosphere;
Tert-butyl 3-prop-2-ynoxyazetidine-1-carboxylate (Intermediate YV)
To a mixture of tert-butyl 3-hydroxyazetidine-1-carboxylate (5.00 g, 28.8 mmol, CAS 141699-55-0) in THF (5 mL) was added NaH (1.39 g, 34.6 mmol, 60% oil dispersion) at 0 °C for 0.5 hour. Then 3-bromoprop-1- yne (5.15 g, 34.6 mmol, 3.73 mL) and TBAI (1.07 g, 2.89 mmol) were added to the mixture. The reaction mixture was stirred at 25 °C for 2 hours. On completion, the reaction mixture was quenched with sat. NH4Cl (50 mL), diluted with water (100 mL) and extracted with EA (2 X 100 mL). The combined organic layers was dried over Na2SO4, filtered and concentrated in vacuo to give a residue. The residue was purified by column chromatography to give the title compound (5.70, 93 % yield) as a yellow solid.1H NMR (400 MHz, CDCl3) d 4.45 - 4.37 (m, 1H), 4.13 (d, J = 2.4 Hz, 2H), 4.12 - 4.07 (m, 2H), 3.89 (dd, J = 9.6 Hz, 2H), 2.45 - 2.43 (m, 1H), 1.43 (s, 9H).
84%
Stage #1: tert-butyl 3-hydroxyazetidine-1-carboxylate With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0 - 20℃; for 0.5h;
Stage #2: propargyl bromide In N,N-dimethyl-formamide; mineral oil at 50℃;
1 Step 1: tert-butyl 3-(prop-2-yn-1-yloxy)azetidine-1-carboxylate
To a stirred solution of tert-butyl 3-hydroxyazetidine-6): -carboxylate 1.72-g, 12.2 mmol) in N,N-dimethylformamide (10 ml) was added sodium hydride (60% in mineral oil) (255 mg, 6.36 mmol) at 0° C., and the resulting mixture was stirred at 0° C. for 30 minutes. The reaction mixture was allowed to warm up to room temperature and stirred for additional 30 min, then 3-bromoprop-1-yne (818 mg, 6.94 mmol) was added, and the resulting reaction mixture was stirred at 50° C. overnight. LCMS showed the reaction was complete. The reaction mixture was quenched with water (10 ml) at 0° C. and extracted with ethyl acetate (30 ml*2). The combined organic layers were washed with brine (30 ml*2), dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give a residue which was purified by silica gel flash chromatography (eluted with 10% ethyl acetate in hexane) to afford tert-butyl 3-(prop-2-yn-1-yloxy)azetidine-1-carboxylate (1.03 g, yield 84%) as colorless oil.
67.5%
Stage #1: tert-butyl 3-hydroxyazetidine-1-carboxylate With sodium hydride In N,N-dimethyl-formamide at 0 - 20℃; for 0.5h; Inert atmosphere;
Stage #2: propargyl bromide In N,N-dimethyl-formamide at 50℃; for 20h; Inert atmosphere;
4.1 The first step: Synthesis of 3-(prop-2-ynyloxy) tert-butyl azetidine-1-carboxylate (004B)Under nitrogen protection, tert-butyl 3-hydroxyazetidine-1-carboxylate (004A)
(680 mg, 3.93 mmol) was dissolved in N,N-dimethylformamide (10 mL) and cooled to 0-5 °C, sodium hydride (157 mg, 3.93 mmol, 60%) was added, and the mixture was stirred at room temperature for 30 min. 3-Bromo-1-propyne (700 mg, 5.89 mmol) was added, and the temperature was raised to 50° C. to react for 20 h. The reaction solution was cooled to room temperature, quenched by adding saturated ammonium chloride solution (50 mL), extracted with ethyl acetate (40 mL×3), the organic phases were combined, dried over anhydrous sodium sulfate, filtered, concentrated, and the residue was separated and purified by silica gel column ( Petroleum ether:ethyl acetate (V/V)=10:1) to give yellow liquid tert-butyl 3-(prop-2-ynyloxy)azetidine-1-carboxylate (004B) (560 mg, yield 67.5%).
67.5%
Stage #1: tert-butyl 3-hydroxyazetidine-1-carboxylate With sodium hydride In N,N-dimethyl-formamide at 0 - 20℃; for 0.5h; Inert atmosphere;
Stage #2: propargyl bromide In N,N-dimethyl-formamide at 50℃; for 20h; Inert atmosphere;
4.1 The first step: Synthesis of 3-(prop-2-ynyloxy) tert-butyl azetidine-1-carboxylate (004B)Under nitrogen protection, tert-butyl 3-hydroxyazetidine-1-carboxylate (004A)
(680 mg, 3.93 mmol) was dissolved in N,N-dimethylformamide (10 mL) and cooled to 0-5 °C, sodium hydride (157 mg, 3.93 mmol, 60%) was added, and the mixture was stirred at room temperature for 30 min. 3-Bromo-1-propyne (700 mg, 5.89 mmol) was added, and the temperature was raised to 50° C. to react for 20 h. The reaction solution was cooled to room temperature, quenched by adding saturated ammonium chloride solution (50 mL), extracted with ethyl acetate (40 mL×3), the organic phases were combined, dried over anhydrous sodium sulfate, filtered, concentrated, and the residue was separated and purified by silica gel column ( Petroleum ether:ethyl acetate (V/V)=10:1) to give yellow liquid tert-butyl 3-(prop-2-ynyloxy)azetidine-1-carboxylate (004B) (560 mg, yield 67.5%).
65%
With sodium hydride In N,N-dimethyl-formamide at 0 - 70℃;
Stage #1: tert-butyl 3-hydroxyazetidine-1-carboxylate With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0 - 20℃; for 1h;
Stage #2: propargyl bromide In N,N-dimethyl-formamide; mineral oil at 50℃;
1 Step 1 : tert-butyl 3-(prop-2-yn-l-yloxy)azetidine-l-carboxylate
To a stirred solution of tert-butyl 3-hydroxyazetidine-l-carboxylate (1.0 g, 12.2 mmol) in N,N-dimethylformamide (10 ml) was added sodium hydride (60% in mineral oil) (255 mg, 6.36 mmol) at 0°C, and the resulting mixture was stirred at 0°C for 30 minutes. The reaction mixture was allowed to warm up to room temperature and stirred for additional 30 min, then 3-bromoprop- 1-yne (818 mg, 6.94 mmol) was added, and the resulting reaction mixture was stirred at 50°C overnight. LCMS showed the reaction was complete. The reaction mixture was quenched with water (10 ml) at 0°C and extracted with ethyl acetate (30 ml x 2). The combined organic layers were washed with brine (30 ml x 2), dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give a residue which was purified by silica gel flash chromatography (eluted with 10% ethyl acetate in hexane) to afford tert-butyl 3-(prop-2-yn-l-yloxy)azetidine-l- carboxylate (1.03 g, yield 84%) as colorless oil.
With triphenylphosphine; diethylazodicarboxylate; In tetrahydrofuran; at 0 - 35℃; for 16h;
Dissolve <strong>[349-57-5]3-nitro-5-(trifluoromethyl)phenol</strong> (500 mg, 2.4 mmol), /ert-butyl 3-hydroxyazetidine-l-carboxylate (500 mg, 2.9 mmol), PPh3 (9.5 g, 3.6 mmol) in THF (8 mL) and cool to 0C on ice bath. Add slowly diethyl azodicarboxylate (630 mg, 3.6 mmol). Stir the mixture at 35C for 16 hrs. Pour the mixture into water (50 mL), adjust pH to neutral with saturated NaHC03 solution. Extract with EtOAc (15 mLx3), combine the organic layers; wash with brine (100 mL), dry over anhydrous Na2S04. Concentrate under reduced pressure to give the crude product. Purification by chromatography (silica gel, EtOAc:PE=15:85) affords the target compound (760 mg, 87%).
87%
With triphenylphosphine; diethylazodicarboxylate; In tetrahydrofuran; at 0 - 35℃; for 16h;
Dissolve <strong>[349-57-5]3-nitro-5-(trifluoromethyl)phenol</strong> (500 mg, 2.4 mmol), tert-butyl 3-hydroxyazetidine-1-carboxylate (500 mg, 2.9 mmol), PPh3 (9.5 g, 3.6 mmol) in THF (8 mL) and cool to 0 C. on ice bath. Add slowly diethyl azodicarboxylate (630 mg, 3.6 mmol). Stir the mixture at 35 C. for 16 hrs. Pour the mixture into water (50 mL), adjust pH to neutral with saturated NaHCO3 solution. Extract with EtOAc (15 mL*3), combine the organic layers; wash with brine (100 mL), dry over anhydrous Na2SO4. Concentrate under reduced pressure to give the crude product. Purification by chromatography (silica gel, EtOAc:PE=15:85) affords the target compound (760 mg, 87%).
1.52 g
With di-isopropyl azodicarboxylate; triphenylphosphine; In tetrahydrofuran; at 20℃;
General procedure: To a mixture of 4,6-dichloropyrimidin-5-ol (step 1 intermediate) (4.0 g, 24.2 mmol), ethyl glycolate (3.02 g, 29.1 mmol) and triphenylphosphine (12.7 g, 48.5 mmol) in THF (40 mL) was slowly added diethyl azodicarboxylate (DEAD) (9.8 g, 48.5 mmol) at RT. The mixture was stirred overnight at RT. The mixture was diluted with diethyl ether and filtered off theprecipitated solid. The filtrate was concentrated under reduced pressure and the residue was purified by silica gel column chromatography to yield 450 mg of the desired compound. ‘H NMR (400 MHz, DMSO-d6) ö 1.34 (t, J= 7.2 Hz, 3H), 4.31 (q, J= 7.2 Hz, 2H), 4.80 (s, 2H), 8.51 (s, 1H).
tert-butyl 3-[4-(4-methoxycarbonylphenyl)phenoxy]azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
69.6%
With di-isopropyl azodicarboxylate; triphenylphosphine; In toluene; at 110℃; for 1h;Sealed tube; Microwave irradiation;
To a solution of KR-24 (400 mg, 1 .76 mmol) in anhydrous toluene (15 mL) was added tert-butyl 3-hydroxyazetidine-1 -carboxylate, R-24, (304 mg, 1 .76 mmol), Ph3P (694 mg, 2.64mmol) and DIAD (538 mg, 2.64 mmol). The sealedvial was irradiated in the microwave on a Biotage Smith Synthesis at 110 00 for 1 h. The resulting mixture was cooled to room temperature and concentrated under vacuo. The mixture was quenched into water, extracted with DOM, dried with anhydrous Na2504, concentrated to give the crude product which was purified by prep-TLO (PE:EA=i :0 to 3:1) to give KR-25(470 mg, 69.6%) as a pale yellow solid. ESI-MS (Mi-i): 384.2 calc. for022H25N05: 383.2.
tert-butyl 3-[4-[(E)-3-methoxy-3-oxo-prop-1-enyl]phenoxy]azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
69.9%
With di-isopropyl azodicarboxylate; triphenylphosphine In toluene at 110℃; for 1h; Sealed tube; Microwave irradiation;
Preparation of reagent KR-29: tert-Butyl 3-[4-[(E)-3-methoxy-3-oxo-ro-1 - enyllrhenoxylazetidine-1 -carboxylate
To a solution of KR-28 (200 mg, 1.12 mmol) in anhydrous toluene (15 mL) was added tert-butyl 3-hydroxyazetidine-i-carboxylate, R-24, (194 mg, 1.12 mmol), Ph3P (353 mg, 1.13 mmol) and DIAD (230 mg, 1.13 mmol), the sealed vial was irradiated in the microwave on a Biotage Smith Synthesis at 11000 for 1 h. The resulting mixture was cooled to room temperature and concentrated under vacuo. The mixture was quenched into water, extracted with DCM, dried with anhydrous Na2504, concentrated to give the crudeproduct which was purified by prep-TLC (PE:EA= 1:0 to 3:1) to give KR-29 (260 mg, 69.9%) as a pale yellow solid. ESI-MS (Mi-i ):334.2 calc. for 018H23N05: 333.1.
1-Boc-3-hydroxyazetidine (Compound 43-26) (0.6 g, 3.46 mmol) was dissolved in anhydrous DMF (6 mL) and NaH (60% in mineral oil, 152 mg, 3.8 mmol) was added. After stirring at RT for 30 min a solution of <strong>[69045-82-5]2-fluoro-5-(trifluoromethyl)pyridine</strong> (Compound 43-25) (0.57 g, 3.46 mmol) in DMF (5 mL) was added and the resulting mixture was stirred at 80C for 2 h. The mixture was cooled to RT, quenched with water and extracted with EtOAc. The EtOAc was dried over MgSO4 and evaporated on a rotary evaporator. The residue was dissolved in DCM (5 mL) and TFA (3 mL) was added. The mixture was stirred at RT for 2 h and evaporated to give crude Compound 43-27, which was used in the next step without purification.
tert-butyl 3-[(5-carbamoyl-6-chloro-3-ethylpyrazin-2-yl)oxy]azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
318 mg
Preparation Example 352 To a mixture of tert-butyl 3-hydroxyazetidine-1-carboxylate (420 mg) and N,N-dimethylformamide (12.5 mL) was added potassium tert-butoxide (260 mg) under ice-cooling, followed by stirring for 1 hour, and then <strong>[313340-08-8]3,5-dichloro-6-ethylpyrazine-2-carboxamide</strong> (500 mg) was added thereto, followed by stirring for 1 hour. The reaction mixture was poured into ice water, followed by extraction with ethyl acetate. The organic phase was washed with saturated brine and then dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel column chromatography (eluent; hexane:ethyl acetate), and then washed with a mixed solvent of hexane:diisopropyl ether to obtain tert-butyl 3-[(5-carbamoyl-6-chloro-3-ethylpyrazin-2-yl)oxy]azetidine-1-carboxylate (318 mg) as a white solid.
tert-butyl 3-(2-(methoxycarbonyl)phenoxy)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
17%
With caesium carbonate In N,N-dimethyl-formamide at 75℃; for 18h;
33 tert-butyl 3-(2-(methoxycarbonyl)phenoxy)azetidine-1 -carboxylate
tert-butyl 3-(2-(methoxycarbonyl)phenoxy)azetidine-1 -carboxylate To a solution of methyl 2-fluorobenzoate (0.83 ml, 6.49 mmol) in DMF (16.2 ml) was added t-butyl 3-hydroxyazetidine-1-carboxylate (1.124 g, 6.49 mmol) and cesium carbonate (10.6 g, 32.4 mmol) and the reaction mixture was stirred at 75°C for 18h. The reaction was extracted with water and DCM. The organic layers were combined, dried over Na2S04, filtered and concentrated to dryness. Purification of the crude product by chromatography on silica eluting with 0 - 100% EtOAc in heptane afforded the title compound as a colorless oil in 17% yield; UPLC-MS: Rt = 2.10 mins; MS m/z [M+Na]+ ; Method E.
tert-butyl-3-(2-hydroxypropoxy)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Stage #1: tert-butyl 3-hydroxyazetidine-1-carboxylate With sodium hydride In N,N-dimethyl-formamide at 0℃; for 0.5h;
Stage #2: 1,2-propylene cyclic carbonate In N,N-dimethyl-formamide at 120℃;
22.1
step 1: To a solution of tert-butyl 3-hydroxyazetidine (2.5 g, 14.5 mmol) in DMF (100 mL) was added NaH (1.45 g, 36.3 mmol) at 0° C. and stirred for 30 min. After addition of 4-methyl-1,3-dioxolane-2-one (3 g, 29 mmol) the reaction was stirred at 120° C. overnight. The reaction mixture was cooled to RT and quenched by adding a saturated aqueous NH4HCO3 solution (5 mL). The mixture was concentrated under reduced pressure to afford tert-butyl 3-(2-hydroxypropoxy)azetidine-1-carboxylate (4 g) as yellow solid, which was used in the next step without further purification. LCMS (ESI): m/z=232.1 [M+1]+
tert-butyl 3-(4-carbamoylphenoxy)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
50%
Stage #1: p-hydroxybenzamide With 1,3-bis(2,6-diisopropylphenyl)-2,2-difluoro-2,3-dihydro-1h-imidazole; 1-(Trimethylsilyl)imidazole In 1,4-dioxane at 23℃; for 0.5h; Inert atmosphere; Schlenk technique; Glovebox;
Stage #2: tert-butyl 3-hydroxyazetidine-1-carboxylate In 1,4-dioxane at 80℃; for 26h; Inert atmosphere; Schlenk technique; Glovebox;
tert-butyl 3-(3-methoxy-4-nitro-1H-pyrazol-1-yl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
72.9%
With diethylazodicarboxylate; In tetrahydrofuran; at 0 - 20℃; for 15h;
To a cooled (0 C.) suspension of <strong>[400755-41-1]3-methoxy-4-nitro-1H-pyrazole</strong> (1.00 g, 6.99 mmol, 1.00 eq), tert-butyl-3-hydroxypyrrolidine-1-carboxylate (2.12 g, 12.2 mmol, 1.75 eq), and polystyrene bound triphenylphosphine (4.06 g, 12.2 mmol, 1.75 eq, 3 mmol/gram) in THF (45 mL) was added diethyl azodicarboxylate (2.42 mL, 13.0 mmol, 1.90 eq) in a drop-wise manner over 3 min. The reaction mixture was allowed to warm to ambient temperature and stirred for 15 hr. The reaction mixture was then diluted with EtOAc (60 mL), filtered and the filtrate concentrated. The crude reaction mixture was purified via flash chromatography on silica gel eluting with a gradient of 0-60% EtOAc in heptane to give the title compound (1.52 g, 72.9% yield) as a white solid. 1H NMR (400 MHz, CDCl3) delta ppm 8.12 (s, 1H) 4.87 (tt, J=5.6, 7.5 Hz, 1H) 4.40-4.28 (m, 4H) 4.09 (s, 3H) 1.48 (s, 9H). m/z (APCI+) for C7H11N4O3 198.9 (M-Boc+H)+
tert-butyl 3-(2-bromo-4-(methoxycarbonyl) phenoxy)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85%
With di-isopropyl azodicarboxylate; triphenylphosphine; In tetrahydrofuran; at 0 - 20℃; for 1h;
To a solution of <strong>[29415-97-2]methyl <strong>[29415-97-2]3-bromo-4-hydroxybenzoate</strong></strong> (0.50 g, 2.16 mmol), tert- butyl 3-hydroxyazetidine-1-carboxylate (0.40 g, 2.16 mmol) and triphenylphosphine (0.68 g, 2.59 mmol) in dry THF (30 mL) was added diisopropyl azodicarboxylate (0.52 g, 2.59 mmol) at 0 C. Then, the temperature was allowed to rise to room temperature and stir for another 1 h. Upon completion, the reaction was diluted with CH2Cl2 (150 mL), washed with NaOH (1M) (50 mL × 2), brine (50 mL × 2), dried over Na2SO4, and concentrated to give the crude product. The crude product was then purified by column chromatography (silica gel, hexanes:acetone = 10:1 to 8:1) to yield 59b (0.71 g, 85% yield) as a white solid.1H NMR (500 MHz, CDCl3) delta ppm 8.23 (s, 1H), 7.92 (dd, J = 2.0, 8.5 Hz, 1H), 6.53 (d, J = 8.5 Hz, 1H), 4.97-4.93 (m, 1H), 4.33 (dd, J = 6.5, 10.0 Hz, 2H), 4.06 (dd, J = 5.0, 10.0 Hz, 2H), 3.88 (s, 3H), 1.44 (s, 9H).13C NMR (125 MHz, CDCl3): delta ppm 165.84, 156.96, 156.16, 135.51, 130.71, 124.90, 112.09, 112.04, 80.30, 67.25, 52.45, 28.55. MS (ESI) m/z = 385.9 [M + H]+ .
Stage #1: tert-butyl 3-hydroxyazetidine-1-carboxylate With triphenylphosphine; diethylazodicarboxylate In tetrahydrofuran at 20℃; for 0.333333h;
Stage #2: With thiobenzoic acid In tetrahydrofuran at 20℃; for 1h;
Stage #3: With sodium methylate In methanol for 1h;
tert-butyl 3-mercaptoazetidine-1-carboxylate (4)
To a solution of 3 (2.48 g) in tetrahydrofuran (35 ml) were added triphenylphosphine(5.64 g), diethyl azodicarboxylate (3.92 ml) and the mixture wasstirred at room temperature for 20 min. To the mixture was added thiobenzoicacid (2.53 ml) and the mixture was stirred at room temperature for 1 h. Themixture was concentrated in vacuo and the residue was purified by silica gelcolumn chromatography (hexane-AcOEt) to give a colorless amorphous(6.0 g). To a stirred solution of the compound obtained above (6.0 g) inMeOH (50 ml) was added NaOMe (772 mg) and the reaction mixture wasstirred for 1 h. Saturated aqueous NaHCO3 was added to the mixture andextracted with AcOEt and the organic phase was dried over Na2SO4, filteredand concentrated in vacuo. The residue was purified by silica gel columnchromatography (hexane-AcOEt) to afford 4 (1.81 g, 67%) as a colorless solid.1H NMR (400 MHz, CDCl3) δ 4.32-4.37 (m, 2H), 3.77-3.82 (m, 2H),3.61-3.71 (m, 1H), 2.00 (d, J=8.5 Hz, 1H), 1.39-1.50 (m, 9H).
Multi-step reaction with 3 steps
1: triethylamine / tetrahydrofuran / 12 h / 0 - 25 °C
2: N,N-dimethyl-formamide / 12 h / 80 °C
3: lithium hydroxide monohydrate; water / tetrahydrofuran / 2 h / 70 °C
Multi-step reaction with 3 steps
1: triethylamine / dichloromethane / 5.75 h / 0 - 20 °C / Inert atmosphere
2: 30 h / 60 °C
3: potassium carbonate; methanol / 2 h / 50 °C / Inert atmosphere
3-(4-fluorobenzyloxy)azetidine-1-carboxylic acid tert-butyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
94%
Stage #1: tert-butyl 3-hydroxyazetidine-1-carboxylate With sodium hydride In N,N-dimethyl-formamide for 1h; Cooling with ice;
Stage #2: 1-chloromethyl-4-fluorobenzene In N,N-dimethyl-formamide at 20℃;
synthesis of 3-(4-fluorobenzyloxy)azetidine-1-carboxylic acid tert-butyl ester(2)
To a solution of compound 3-hydroxy-azetidine-1-carboxylic acid tert-butyl ester I (5.30 g, 30 mmoi) in DMF (60 mL) was added NaH (1 .80 g, 45 mmol) whilst coolingon an ice-water bath. The suspension was then stirred at this temperature for one hour, followed by the addition of ichlorom:ethyl4fluorobenzene (8.94 g, 60 mmol). The resulting mixture was stirred at ambient temperature overnight. The reaction mixture was poured into water (200 mL) and extracted with EtOAc (150 mL x 3). The organic combined phases were dried over Na2SO4, filtered and concentrated in vacuoto get the crude product. The residue was purified by chromatography on a silica gel column eluted with PE/EtOAc = 10:1 to 2:1) to afford compound 2 (7.90 g, 94% yield) as an oil. 1H NMR (300 MHz, DMSO-d6): 5 7.417.37 (m, 2H), T21-7.14 (m, 2H), 4.40 (s, 2H),4.33-429 (m, IH), 4.02-3.97 (m, 2H), 3.68-3.66 (rn, 2H), 1.37 (s, 9H). MS Calcd.:281; MS Found: 282 ([M÷Hfl.
94%
Stage #1: tert-butyl 3-hydroxyazetidine-1-carboxylate With sodium hydride In N,N-dimethyl-formamide for 1h; Cooling with ice;
Stage #2: 1-chloromethyl-4-fluorobenzene In N,N-dimethyl-formamide at 20℃;
1 Synthesis of 3-(4-fluoro-ben lox -azetidine-l-carboxylic acid tert-butyl ester (2)
To a solution of compound 3-hydroxy-azetidine-l-carboxylic acid tert- butyl ester 1 (5.30 g, 30 mmol) in DMF (60 mL) was added NaH (1.80 g, 45 mmol) whilst cooling on an ice-water bath. The suspension was then stirred at this temperature for one hour, followed by the addition of l-chloromethyl-4-fluoro- benzene (8.94 g, 60 mmol). The resulting mixture was stirred at ambient temperature overnight. The reaction mixture was poured into water (200 mL) and extracted with EtOAc ( 150 mL χ 3). The organic combined phases were dried over azSCU, filtered and concentrated in vacuo to get the crude product. The residue was purified by chromatography on a silica gel column (eiirted with PE/'EtOAc ::: 10: 1 to 2: 1) to afford compound 2 (7.90 g, 94% yield) as an oil. ΝΜΡν (300 MHz, DMSO- 6): S 7.41-7.37 (m, 2H), 7.21-7.14 (m, 2H), 4.40 (s, 2H), 4.33-4.29 (m, 1H), 4.02-3.97 (m, 2H), 3.68-3.66 (m, 2H), 1.37 (s, 9H). MS Calcd.: 281 ; MS Found: 282 ([M+H]+).
With potassium hydroxide; In water; at 22 - 24℃; for 0.5h;
To a flame-dried reaction tube under argon was added tertbutyl3-iodoazetidine-1-carboxylate (6) (83.8 mg, 0.249 mmol, 1 equiv.), potassium acetate (36.6 mg, 0.375mmol, 1.5 equiv.) and dry DMSO (2.5 mL). The mixture was heated at 80 oC and stirred overnight. Completionof the acetoxylation step was monitored by TLC and 1H NMR of the crude reaction mixture. A solution ofpotassium hydroxide (21.0 mg, 0.374 mmol, 1.5 equiv. in 0.8 mL of H2O) was slowly added and the mixturestirred at rt for 30 min. The resulting mixture was diluted with H2O (10 mL) and extracted with ethyl acetate (3x 10 mL). The combined organic layers were washed with brine (5 mL) and dried over Na2SO4 andconcentrated under reduced pressure. The crude material was purified by flash chromatography (silica gel,10-60% EtOAc in hexanes) to give the desired product 24 (31.2 mg, 72%). Physical State: white solid (mp 51-52 oC); Rf = 0.13 (3:7 EtOAc/hexanes, vis. KMnO4); 1H NMR (500 MHz, CDCl3): [mixture of rotamers] delta 4.58 -4.53 (m, 1H), 4.12 (dd, J 10.6, 6.7 Hz, 2H, major), 4.12 (dd, J 8.3, 6.7 Hz, 2H, minor), 3.79 (dd, J 10.6, 4.4 Hz, 2H,major), 3.79 (dd, J 8.4, 4.4 Hz, 2H, minor), 3.12 (br s, 1H, major), 3.10 (br s, 1H, minor), 1.42 (s, 9H); 13C NMR(126 MHz, CDCl3): delta 156.6, 79.9, 61.6, 59.1 (br, 2C), 28.5 (3C). All spectral data are in accordance with thepreviously reported literature values.
Stage #1: tert-butyl 3-hydroxyazetidine-1-carboxylate With sodium hydride In N,N-dimethyl-formamide; mineral oil at 20℃;
Stage #2: 2-fluoroethyl tosylate In N,N-dimethyl-formamide; mineral oil
4.1.2 General synthetic procedure for compounds 7a-f
NaH 60% dispersion in mineral oil (3.3mmol) was slowly added to a stirring solution of hydroxyl amine (5a-f) (3.0mmol) in 6mL of DMF at room temp, and stirred for 30min. Compound 6 was then slowly added to reaction mixture, followed by an additional hour of stirring. A saturated NaHCO3 (aq) solution (40mL) was then added to the crude reaction mixture and stirred at room temp for 1h. The reaction mixture was extracted with CH2Cl2 (3×20mL) to afford the crude product. The subsequent residue was loaded onto a Biotage SNAP flash purification cartridge, eluting with a 2:5 EtOAc/hexanes gradient, to afford the desired Boc-protected O-alkylated fluoroethoxy compounds, identified by LCMS. The intermediate was then dissolved in CH2Cl2 (2mL), followed by dropwise addition of CF3COOH (2mL), and stirred at room temperature for 3h. Volatiles were then removed under reduced pressure and the crude product was neutralized with a saturated NaHCO3 (aq) solution (10mL). The reaction mixture was extracted with CH2Cl2 (3×20mL), and the organic layers were combined, dried, and concentrated to afford the free-amine intermediates (7a-f) as oils used in the next step without any further purification.
tert-butyl 3-(2,3-difluorophenoxy)azetidin-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
91%
With triphenylphosphine; In tetrahydrofuran; at 70℃; for 20h;
Triphenylphosphine (7.11 g, 27.1 mmol) was dissolved in dry THF (100 mL).DEAD (10.8 g, 24.9 mmol) (5 min) was added dropwise, then 3-hydroxyazetidine-1-carboxylic acid tert-butyl ester (3.99 g, 22.6 mmol) was added in small portions in the order given.And <strong>[6418-38-8]2,3-difluorophenol</strong> (3.00 g, 22.6 mmol). The reaction mixture was stirred at 70 C for 20 hours and then separated between water and MTBE. Extract the aqueous solution with another portion of MTBE and dissolve the combined organic solutionThe solution was washed with aqueous KOH (10%), water and brine.After drying (Na2SO4) and filtering, the volatiles were evaporated.Purified by silica gel chromatography,Where a mixture of isooctane and EtOAc (gradient 30%-100% EtOAc) eluted5.9 g (91%) of the title compound are obtained.
tert-butyl 3-(2,3,6-trifluorophenoxy)azetidin-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
72%
With di-isopropyl azodicarboxylate; triphenylphosphine; In dichloromethane; at 0 - 20℃; for 72h;
2,3,6-Trifluorophenol (0.95 g, 6.4 mmol) with 3-hydroxyazetidin-1-carboxylic acid tert-butyl ester (1.45 g, 8.4 mmol) and triphenylphosphine (2.2) g, 8.4 mmol) was dissolved together in DCM (25 mL).Diisopropyl azodicarboxylate (1.7 mL, 8.6 mmol) was then added dropwise to the resulting solution at 0 C. The mixture was stirred under cooling for 1 hour and then at room temperature for 3 days.After diluting with DCM, the solution was washed with brine, dried over Na.The residue obtained was purified by silica gel column chromatography.Where isooctane / EtOAc (0-17% EtOAc) was usedGradient gave 1.40 g (72%) of title compound.It is an oily substance.
3-(2,4,6-trifluorophenoxy)azetidin-1-carboxylic acid tert-butyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
With di-isopropyl azodicarboxylate; triphenylphosphine In dichloromethane at 0 - 20℃; for 21h;
9 3-(2,4,6-trifluorophenoxy)azetidin-1-carboxylic acid tert-butyl ester
2,4,6-Trifluorophenol (0.91 g, 6.1 mmol) with 3-hydroxyazetidine-1-carboxylic acid tert-butyl ester (1.40 g, 8.1 mmol) and triphenylphosphine (2.1) g, 8.0 mmol) was dissolved together in DCM (20 mL).Diisopropyl azodicarboxylate (1.6 mL, 7.9 mmol) was then added dropwise to the resulting solution at 0 °C.The mixture was stirred under cooling for 1 hour and then at room temperature for 20 hours.After diluting with DCM, the solution was washed with brine, dried over Na. The residue obtained was purified by silica gel column chromatography.Where a gradient of isooctane / EtOAc (0-18% EtOAc) was used.1.5 g (80%) of the title compound was obtained as an oil.
tert-butyl 3-(2,4,5-trifluorophenoxy)azetidin-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
49%
With di-isopropyl azodicarboxylate; triphenylphosphine; In dichloromethane; at 0 - 20℃; for 21h;
2,4,5-Trifluorophenol (0.53 g, 3.6 mmol) and tert-butyl 3-hydroxyazetidin-1-carboxylate (0.80 g, 4.6 mmol)Dissolved in DCM (20 mL) with triphenylphosphine (1.25 g, 4.8 mmol).Diisopropyl azodicarboxylate (0.9 mL, 4.6 mmol) was then added dropwise to the resulting solution at 0 C.The mixture was stirred under cooling for 1 hour and then at room temperature for 20 hours.After dilution with DCM, the solution was washed with brine.It was dried over Na 2 SO 4 and then concentrated. The residue obtained was purified by silica gel column chromatography.Using isooctane/EtOAc (0-18% Et0Ac) gradient,0.53 g (49%) of title compound was obtained as an oil.
4-[1-(tert-butoxycarbonyl)azetidin-3-yl]oxy-3-chloro-2,5-difluorobenzoic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
0.189 g
Sodium hydride (55% in oil) was suspended to 1 mL of N,N-dimethylformamide and 1 mL of N,N-dimethylformamide solution of 0.520 g of tert-butyl 3-hydroxyazetidin-1-ylcarboxylate was'added under ice cooling in a nitrogen atmosphere. The mixture was stirred at room temperature for 10 minutes. 1 mL of a N,N-dimethylformamide solution of 0.210 g of <strong>[101513-77-3]3-chloro-2,4,5-trifluorobenzoic acid</strong> was added under ice cooling and the mixture was stirred at room temperature for 30 minutes. 30 mL of a 10% aqueous solution of citric acid was added and then the mixture was extracted with ethyl acetate. The extract was washed with water and dried over anhydrous sodium sulfate and then the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (chloroform:methanol=3 to 30% gradient and hexane:ethyl acetate 40% isocratic) to obtain 0.189 g of the title compound. 1H-NMR (DMSO-d6): delta 1.45 (9H, s), 4.17 (2H, dd, J=4.0 Hz, 10.0 Hz), 4.32 (2H, dd, J=6.5 Hz, 10.0 Hz), 5.08-5.13 (1H, m), 7.65-7.69 (1H, m), 10.47 (1H, brs)
3-[[2,4-bis(trifluoromethyl)phenyl]methoxy]azetidine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With trifluoroacetic acid; In dichloromethane; at 20℃; for 3h;Inert atmosphere;
General procedure: To a solution of tert-butyl 3 -((2 -fluoro-4-(trifluoromethoxy)benzyl)oxy)azetidine- 1 -carboxylate(415 mg, 1.14 mmol) in DCM (5 mL) was added TFA (1.3 g, 875 tL, 11.4 mmol) and thereaction mixture was stirred at RT for 3 h. Volatiles were removed in vacuo to yield 455 mg oflight yellow oil that was used in the next step without further purification. MS (ESI): mlz =266.1 [M+H].
3-[[2-methyl-3-(trifluoromethyl)phenyl]methoxy]azetidine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With trifluoroacetic acid; In dichloromethane; at 20℃; for 3h;Inert atmosphere;
General procedure: To a solution of tert-butyl 3 -((2 -fluoro-4-(trifluoromethoxy)benzyl)oxy)azetidine- 1 -carboxylate(415 mg, 1.14 mmol) in DCM (5 mL) was added TFA (1.3 g, 875 tL, 11.4 mmol) and thereaction mixture was stirred at RT for 3 h. Volatiles were removed in vacuo to yield 455 mg oflight yellow oil that was used in the next step without further purification. MS (ESI): mlz =266.1 [M+H].
tert-butyl 3-[[2-fluoro-6-(trifluoromethyl)phenyl]methoxy]azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80.9%
With potassium tert-butylate; In tetrahydrofuran; at 25℃; for 12h;Inert atmosphere;
To a solution of <strong>[239087-08-2]2-fluoro-6-(trifluoromethyl)benzyl bromide</strong> (1000.0 mg, 3.89 mmol, CAS RN239087-08-2) and tert-butyl 3-hydroxyazetidine-1-carboxylate (673.92 mg, 3.89 mmol, CAS RN141699-55-0 ) in dry THF (10 mL) at 25 C, was added t-BuOK (5.84 mL, 5.84 mmol; 1.0 M in dryTHF) and the mixture was stirred at 25 C for 12 h. The mixture was poured into H20 (10 mL) andextracted three times with EA (20 mL each). The combined organic layers were combined, dried overanhydrous Na2SO4 and filtered. The filtrate was concentrated under reduced pressure, purified by flash chromatography on silica gel (gradient PE : EA = 10: 1 to 2: 8) to give the title compound as colorless oil (1100 mg, 80.9%). MS (ESI): m/z = 294.0 [M-56+H].
80.9%
With potassium tert-butylate; In tetrahydrofuran; at 25℃; for 12h;
To a solution of <strong>[239087-08-2]2-fluoro-6-(trifluoromethyl)benzyl bromide</strong> (1000.0 mg, 3.89 mmol, CAS RN 239087-08-2) and tert-butyl 3-hydroxyazetidine-l-carboxylate (673.92 mg, 3.89 mmol, CAS RN 141699-55-0 ) in dry THF (10 mL) at 25 C, was added t-BuOK (5.84 mL, 5.84 mmol; 1.0 M in dry THF) and the mixture was stirred at 25 C for 12 h. The mixture was poured into H20 (10 mL) and extracted three times with EA (20 mL each). The combined organic layers were combined, dried over anhydrous Na2S04 and filtered. The filtrate was concentrated under reduced pressure, purified by flash chromatography on silica gel (gradient PE : EA = 10 : 1 to 2 : 8) to give the title compound as colorless oil (1100 mg, 80.9%). MS (ESI): m/z = 294.0 [M-56+H]+.
tert-butyl 3-(3-bromo-4-chlorophenoxy)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
53%
With cyanomethylenetributyl-phosphorane; In toluene; at 100℃; for 0.5h;Sealed tube; Inert atmosphere;
In a sealed tube, <strong>[13659-24-0]3-bromo-4-chlorophenol</strong> (0.1 mg, 0.482 mmol) and tert-butyl 3- hydroxyazetidine-1-carboxylate (0.083 g, 0.482 mmol) were dissolved in toluene (1.5 mL). Thevial was degassed with argon, then (tributylphosphoranylidene)acetonitrile (CAS RN 157141-27-0, 0.195 mL, 0.723 mmol) was added and the reaction mixture heated to 100C for 30 minutes. The mixture was diluted with EtOAc, poured into sat. aq. NaHCO3 solution and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2504, filtered and concentrated in vacuo. The residue was purified by silica gelflash chromatography eluting with a 0 to 20% EtOAc/heptane gradient to give the title compound (0.116 g, 53%) as a yellow oil. MS (ESI): mlz = 308.1 [M-56+H]t
53%
With cyanomethylenetributyl-phosphorane; In toluene; at 100℃; for 0.5h;Inert atmosphere; Sealed tube;
In a sealed tube, <strong>[13659-24-0]3-bromo-4-chlorophenol</strong> (0.1 mg, 0.482 mmol) and tert- butyl 3- hydroxyazetidine- 1 -carboxylate (0.083 g, 0.482 mmol) were dissolved in toluene (1.5 mL). The vial was degassed with argon, then (tributylphosphoranylidene)acetonitrile (CAS RN 157141- 27-0, 0.195 mL, 0.723 mmol) was added and the reaction mixture heated to l00C for 30 minutes. The mixture was diluted with EtOAc, poured into sat. aq. NaHCCb solution and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with brine, dried over Na?S04, filtered and concentrated in vacuo. The residue was purified by silica gel flash chromatography eluting with a 0 to 20% EtO Ac/heptane gradient to give the title compound (0.116 g, 53%) as a yellow oil. MS (ESI): m/z = 308.1 [M-56+H]+.
tert-butyl 3-[[4-bromo-2-(trifluoromethyl)phenyl]methoxy]azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
69.4%
With potassium tert-butylate; In tetrahydrofuran; at 20℃; for 12h;
To a solution of tert-butyl 3 -hydro xyazeti dine- 1 -carboxylate (337.5 mg, 1.95 mmol, CAS RN 22214-30-8) in THF (9 mL) was added t-BuOK (1.95 mL, 1.95 mmol), then 4-bromo-l-(bromomethyl)- 2-(trifluoromethyl) benzene (590.0 mg, 1.86 mmol) was added and the mixture was stirred at 20 C for 12 h. The mixture was poured into aq. NFLCl solution (200 mL) and extracted three times with EtOAc (50 mL). The combined organic layers were washed with brine, dried over Na2SC>4 and concentrated. The residue was purified by prep-HPLC (ACN and water containing 0.225% v/v FA) to give the desired product as light brown oil (300 mg, 39.4%). MS (ESI): m/z = 356.3 [M-56+H]+.
39.4%
With potassium tert-butylate; In tetrahydrofuran; at 20℃; for 12h;Inert atmosphere;
To a solution of tert-butyl 3-hydroxyazetidine-1-carboxylate (337.5 mg, 1.95 mmol, CAS RN22214-30-8) in THF (9 mL) was added t-BuOK (1.95 mL, 1.95 mmol), then 4-bromo-1-(bromomethyl)-2-(trifluoromethyl) benzene (590.0 mg, 1.86 mmol) was added and the mixture was stirred at 20 C for12 h. The mixture was poured into aq. NH4C1 solution (200 mL) and extracted three times with EtOAc(50 mL). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated. The residue was purified by prep-HPLC (ACN and water containing 0.225% v/v FA) to give the desired product as light brown oil (300 mg, 39.4%). MS (ESI): m/z = 356.3 [M-56+H].
tert-butyl 3-[[2-fluoro-4-(trifluoromethyl)phenyl]methoxy]azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
37.1%
With cyanomethyl tributylphosphorane at 100℃; for 1h; Microwave irradiation; Inert atmosphere;
9.a Step a) tert-Butyl 3-[[2-fluoro-4-(trifluoromethyl)phenyl]methoxy]azetidine-l-carboxylate
To a solution of [2-fluoro-4-(trifluoromethyl)phenyl]methanol (1500.0 mg, 7.73 mmol CAS RN 197239-49-9) and tert-butyl 3-hydroxyazetidine-l-carboxylate (1405.3 mg, 8.11 mmol, CAS RN 141699-55-0) in toluene (15 mL) was added cyanomethyltributylphosphorane (2797.3 mg, 11.6 mmol) and the mixture was stirred at 100 °C under microwave heating for 1 h. After cooling to RT the mixture was concentrated, and the residue purified by reverse column chromatography (0.1% v/v FA in water and MeCN) to give the title compound (1000 mg, 2.86 mmol, 37.1%) as a light yellow oil. MS (ESI): m/z = 294.1 [M-C4Hs+H]+.
With sodium hydride In N,N-dimethyl-formamide at 0 - 25℃; for 1h;
1.1 Step 1:
To a solution of tert-butyl 3-hydroxyazetidine-1-carboxylate (1.13 g, 6.53 mmol) in DMF (20 mL) was added NaH (203.65 mg, 8.49 mmol) at 0 °C in portions, the reaction mixture was stirred at 25 °C for 1 h. Then 2-(benzyloxy)ethyl 4-methylbenzenesulfonate (2 g, 6.53 mmol) was added and stirred at 25 °C for 16 h. The resulting solution was quenched by saturated NH4Cl (50 mL), extracted with EA (50 mLx3), washed with water (50 mL) and brine (50 mL), dried over Na2SO4 and concentrated. The residue was purified by column chromatography to afford tert-butyl 3-(2-(benzyloxy)ethoxy)azetidine-1-carboxylate (1.3 g, 64.79% yield) as a yellow oil. MS: m/z = 330.2 (M+23, ESI+).
64.79%
Stage #1: 3-hydroxyazetidine-1-carboxylic acid tert-butyl ester With sodium hydride In N,N-dimethyl-formamide at 0 - 25℃; for 1h;
Stage #2: 2-(benzyloxy)ethyl 4-methylbenzenesulfonate In N,N-dimethyl-formamide at 25℃; for 16h;
1.1 Synthesis of Compound 25
Step 1: To a solution of tert-butyl 3 -hydroxyazetidine- 1 -carboxylate (1.13 g, 6.53 mmol) in DMF (20 mL) was added NaH (203.65 mg, 8.49 mmol) atO °C in portions, the reaction mixture was stirred at 25 °C for 1 h. Then 2-(benzyloxy)ethyl 4-methylbenzenesulfonate (2 g, 6.53 mmol) was added and stirred at 25 °C for 16 h. The resulting solution was quenched by saturated NH4C1 (50 mL), extracted with EA (50 mLx3), washed with water (50 mL) and brine (50 mL), dried over Na2SO4 and concentrated. The residue was purified by column chromatography to afford tert-butyl 3-(2-(benzyloxy)ethoxy)azetidine-1-carboxylate (1.3 g, 64.79% yield) as a yellow oil. MS: m/z = 330.2 (M+23, ESI+).
64.79%
Stage #1: 3-hydroxyazetidine-1-carboxylic acid tert-butyl ester With sodium hydride In N,N-dimethyl-formamide at 0 - 25℃; for 1h;
Stage #2: 2-(benzyloxy)ethyl 4-methylbenzenesulfonate In N,N-dimethyl-formamide at 25℃; for 16h;
1.1 Synthesis of Compound 25
Step 1: To a solution of tert-butyl 3 -hydroxyazetidine- 1 -carboxylate (1.13 g, 6.53 mmol) in DMF (20 mL) was added NaH (203.65 mg, 8.49 mmol) atO °C in portions, the reaction mixture was stirred at 25 °C for 1 h. Then 2-(benzyloxy)ethyl 4-methylbenzenesulfonate (2 g, 6.53 mmol) was added and stirred at 25 °C for 16 h. The resulting solution was quenched by saturated NH4C1 (50 mL), extracted with EA (50 mLx3), washed with water (50 mL) and brine (50 mL), dried over Na2SO4 and concentrated. The residue was purified by column chromatography to afford tert-butyl 3-(2-(benzyloxy)ethoxy)azetidine-1-carboxylate (1.3 g, 64.79% yield) as a yellow oil. MS: m/z = 330.2 (M+23, ESI+).
tert-butyl 3-((4-chloro-3-(trifluoromethyl)benzyl)oxy)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
89%
Stage #1: tert-butyl 3-hydroxyazetidine-1-carboxylate With sodium hydride In N,N-dimethyl-formamide at 0℃; for 0.5h;
Stage #2: 4-(bromomethyl)-1-chloro-2-(trifluoromethyl)benzene In N,N-dimethyl-formamide at 0 - 20℃;
Stage #1: 3-chloro-4-fluorophenol With caesium carbonate In N,N-dimethyl-formamide at 20℃; for 0.166667h;
Stage #2: tert-butyl 3-hydroxyazetidine-1-carboxylate In N,N-dimethyl-formamide at 90℃; for 16h;
Intermediate A-5:
A mixture of 3-chloro-4-fluoro-phenol (1.5 g, 10.2 mmol) and cesium carbonate (6.7 g, 20.5 mmol) in DMA (10 mL) is stirred for 10 min at RT, then tert-butyl-3-methanesulfonyloxy)azetidine-l- carboxylate (2.6 g, 10.2 mmol) is added. After stirring at 100°C for 16 h the mixture is cooled to RT. Water and DCM are added. Phases are separated. The organic phase is dried over Na2SO4 and concentrated under reduced pressure. The remainder is purified by preparative HPLC to provide the product A-5.ESI-MS: 302/304 [M+H]+; HPLC (Rt): 0.78 min (method A)
With caesium carbonate In N,N-dimethyl-formamide at 90℃; for 0.533333h;
Intermediate A-7:
A mixture of 3-fluoro-5-(trifluoromethyl)-phenol (0.7 g, 4.0 mmol), cesium carbonate (2.6 g, 8.0 mmol) and tert-butyl-3-methanesulfonyloxy)azetidine-l-carboxylate (1.0 g, 4.0 mmol) in DMF (30 mL) is stirred for 16h at 90°C for 16 h. Then the mixture is cooled to RT and dilute with ethyl acetate. The organic phase is consecutively washed with saturated aqueous solution of NaHCO3, brine and saturated aqueous solution of NH4CI and is concentrated under reduced pressure to provide the product A-7.ESI-MS: 336 [M+H]+, 280 [M +H-isobuten]+; HPLC (Rt): 1.44 min (method J)
tert-butyl 3-[6-(trifluoromethyl)pyrazin-2-yl]oxyazetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
71%
Stage #1: tert-butyl 3-hydroxyazetidine-1-carboxylate With sodium hydride In 1,4-dioxane; mineral oil at 0℃; for 0.25h;
Stage #2: 2-chloro-6-(trifluoromethyl)pyrazine In 1,4-dioxane; mineral oil at 50℃; for 0.75h;
a) tert-butyl 3-[6-(trifluoromethyl)pyrazin-2-yl]oxyazetidine-l -carboxylate
To a solution of 3 -hydroxy- azetidine-1 -carboxylic acid tert-butyl ester (500 mg, 2.89 mmol) in 1,4-dioxane (25 mL) was added sodium hydride (60% dispersion in mineral oil, 174 mg, 4.33 mmol) at 0 °C. After stirring for 15 minutes, a solution of 2-chloro-6-trifluoro methyl -pyrazine (527 mg, 2.89 mmol) in 1,4-dioxane (5 mL) was added and the reaction mixture was stirred at 50 °C for 45 min. After cooling to room temperature the reaction mixture was quenched by addition of saturated aqueous NEECl and extracted with ethyl acetate (3 x 20 mL). The combined organic layers were washed with brine, dried (Na2S04), filtered and concentrated in vacuo. Purification by flash chromatography (silica, 40% ethyl acetate in hexane) afforded the title compound (660 mg, 71%) as a colorless solid.
Stage #1: 2-chloro-5-fluoropyridin-4-ol With di-isopropyl azodicarboxylate; triphenylphosphine In tetrahydrofuran at 20℃; for 0.5h; Inert atmosphere;
Stage #2: tert-butyl 3-hydroxyazetidine-1-carboxylate In tetrahydrofuran for 3h; Inert atmosphere; Reflux;
tert-butyl 3-(4-(ethoxycarbonyl)phenoxy)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
94%
With di-isopropyl azodicarboxylate; triphenylphosphine In toluene at 105℃; for 16h;
8.1 Step 1. tert-Butyl 3-(4-(ethoxycarbonyl)phenoxy)azetidine-1-carboxylate
To a mixture of ethyl 4-hydroxybenzoate (0.35 g, 2.106 mmol), tert-butyl 3- hydroxyazetidine-1-carboxylate (0.456 g, 2.63 mmol), and triphenylphosphine (0.829 g, 3.16 mmol) in Toluene (12 mL) at rt was added diisopropyl azodicarboxylate (DIAD) (0.622 mL, 3.16 mmol) over 1 min. The resulting solution was heated at 105C for 16 h. Upon cooling to rt, the reaction solution was diluted with ethyl acetate (50 mL) and silica gel (5 g) was added. The volatiles were removed under vacuum. The residue was subjected to ISCO chromatography (80 g silica gel, 0-35% ethyl acetate/hexane) to provide tert-butyl 3-(4-(ethoxycarbonyl)phenoxy)azetidine-1-carboxylate (637 mg, 1.982 mmol, 94 % yield) as viscous oil.1H NMR (400 MHz, chloroform-d) δ 8.15 - 7.74 (m, 2H), 6.95 - 6.58 (m, 2H), 4.93 (tt, J = 6.4, 4.1 Hz, 1H), 4.40 - 4.28 (m, 4H), 4.01 (ddd, J = 9.7, 4.1, 1.1 Hz, 2H), 1.45 (s, 9H), 1.38 (t, J = 7.1 Hz, 3H).
94%
With di-isopropyl azodicarboxylate; triphenylphosphine In toluene at 105℃; for 16h;
8.1 Step 1. tert-Butyl 3-(4-(ethoxycarbonyl)phenoxy)azetidine-1-carboxylate
To a mixture of ethyl 4-hydroxybenzoate (0.35 g, 2.106 mmol), tert-butyl 3- hydroxyazetidine-1-carboxylate (0.456 g, 2.63 mmol), and triphenylphosphine (0.829 g, 3.16 mmol) in Toluene (12 mL) at rt was added diisopropyl azodicarboxylate (DIAD) (0.622 mL, 3.16 mmol) over 1 min. The resulting solution was heated at 105C for 16 h. Upon cooling to rt, the reaction solution was diluted with ethyl acetate (50 mL) and silica gel (5 g) was added. The volatiles were removed under vacuum. The residue was subjected to ISCO chromatography (80 g silica gel, 0-35% ethyl acetate/hexane) to provide tert-butyl 3-(4-(ethoxycarbonyl)phenoxy)azetidine-1-carboxylate (637 mg, 1.982 mmol, 94 % yield) as viscous oil.1H NMR (400 MHz, chloroform-d) δ 8.15 - 7.74 (m, 2H), 6.95 - 6.58 (m, 2H), 4.93 (tt, J = 6.4, 4.1 Hz, 1H), 4.40 - 4.28 (m, 4H), 4.01 (ddd, J = 9.7, 4.1, 1.1 Hz, 2H), 1.45 (s, 9H), 1.38 (t, J = 7.1 Hz, 3H).
94%
With di-isopropyl azodicarboxylate; triphenylphosphine In toluene at 105℃; for 16h;
8.1 Step 1. tert-Butyl 3-(4-(ethoxycarbonyl)phenoxy)azetidine-1-carboxylate
To a mixture of ethyl 4-hydroxybenzoate (0.35 g, 2.106 mmol), tert-butyl 3- hydroxyazetidine-1-carboxylate (0.456 g, 2.63 mmol), and triphenylphosphine (0.829 g, 3.16 mmol) in Toluene (12 mL) at rt was added diisopropyl azodicarboxylate (DIAD) (0.622 mL, 3.16 mmol) over 1 min. The resulting solution was heated at 105C for 16 h. Upon cooling to rt, the reaction solution was diluted with ethyl acetate (50 mL) and silica gel (5 g) was added. The volatiles were removed under vacuum. The residue was subjected to ISCO chromatography (80 g silica gel, 0-35% ethyl acetate/hexane) to provide tert-butyl 3-(4-(ethoxycarbonyl)phenoxy)azetidine-1-carboxylate (637 mg, 1.982 mmol, 94 % yield) as viscous oil.1H NMR (400 MHz, chloroform-d) δ 8.15 - 7.74 (m, 2H), 6.95 - 6.58 (m, 2H), 4.93 (tt, J = 6.4, 4.1 Hz, 1H), 4.40 - 4.28 (m, 4H), 4.01 (ddd, J = 9.7, 4.1, 1.1 Hz, 2H), 1.45 (s, 9H), 1.38 (t, J = 7.1 Hz, 3H).
tert-butyl 3-[[2-[5-[(3-methyloxetan-3-yl)methoxy]benzimidazol-1-yl]-8-quinolyl]oxy]azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
34.98%
With (tributylphosphoranylidene)acetonitrile In toluene at 100℃; for 12h; Inert atmosphere;
Intermediate 8. tert-Butyl 3-[[2-[5-[(3-methyloxetan-3-yl) methoxy]benzimidazol-1-yl]-8- quinolyl]oxy]azetidine-1-carboxylate
A mixture of 2-[5-[(3-methyloxetan-3-yl)methoxy]benzimidazol-1-yl]quinolin-8-ol (200 mg, 553.41 µmol, 1 eq), tert-butyl 3-hydroxyazetidine-1-carboxylate (124.61 mg, 719.44 µmol, 1.3 eq), and 2-(tributyl-phosphanylidene)acetonitrile (200.35 mg, 830.12 µmol, 1.5 eq) in toluene (3 mL) was degassed and purged with N2 (3x), and then the mixture was stirred at 100°C for 12 h under N2. The reaction mixture was then concentrated to afford the title compound, which was purified by prep-TLC (SiO2, ethyl acetate:methanol, 10:1) as a yellow solid (100 mg, 193.58 µmol, 34.98% yield).
34.98%
With (tributylphosphoranylidene)acetonitrile In toluene at 100℃; for 12h; Inert atmosphere;
Intermediate 8. tert-Butyl 3-[[2-[5-[(3-methyloxetan-3-yl) methoxy]benzimidazol-1-yl]-8- quinolyl]oxy]azetidine-1-carboxylate
A mixture of 2-[5-[(3-methyloxetan-3-yl)methoxy]benzimidazol-1-yl]quinolin-8-ol (200 mg, 553.41 µmol, 1 eq), tert-butyl 3-hydroxyazetidine-1-carboxylate (124.61 mg, 719.44 µmol, 1.3 eq), and 2-(tributyl-phosphanylidene)acetonitrile (200.35 mg, 830.12 µmol, 1.5 eq) in toluene (3 mL) was degassed and purged with N2 (3x), and then the mixture was stirred at 100°C for 12 h under N2. The reaction mixture was then concentrated to afford the title compound, which was purified by prep-TLC (SiO2, ethyl acetate:methanol, 10:1) as a yellow solid (100 mg, 193.58 µmol, 34.98% yield).
tert-butyl 3-((3-(trifluoromethyl)benzyl)oxy)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
74.7%
Stage #1: 3-hydroxyazetidine-1-carboxylic acid tert-butyl ester With sodium hydride In N,N-dimethyl-formamide at 0 - 20℃; for 0.5h;
Stage #2: 1-bromomethyl-3-trifluoromethylbenzene In N,N-dimethyl-formamide at 20℃; for 1.5h;
55 Preparation of tert-butyl 3-((3-(trifluoromethyl)benzyl)oxy)azetidine-1-carboxylate (57-1).
To a solution of tert-butyl 3-hydroxyazetidine-1-carboxylate (500 mg, 2.887 mmol) in DMF (10 mL) at 0 °C was added 60% NaH (173.20 mg, 4.330 mmol). The reaction mixture was stirred at the room temperature for 0.5 h, Then added 1(b-romomethyl)-3- (trifluoromethyl)benzene (762.22 mg, 3.175 mmol) and stirred at the room temperature for 1.5 h. LCMS showed the reaction was completed. The reaction solution was quenched with sat.NH4Cl (20 mL) and extracted with EA (20 mL*3), the organic phase was dried over Na2SO4, filtered, and concentrated to afford the crude product. The residue was purified by Flash Chromatography (PE /EtOAc=5:2) to give the product tert-butyl 3-((3- (trifluoromethyl)benzyl)oxy)azetidine-1-carboxylate (717 mg, 2.16 mmol , 74.7%) as light- yellow oil. Mass Spectrum (ESI) m/z = 354.0 (M+23).
74.7%
Stage #1: 3-hydroxyazetidine-1-carboxylic acid tert-butyl ester With sodium hydride In N,N-dimethyl-formamide at 0 - 20℃; for 0.5h;
Stage #2: 1-bromomethyl-3-trifluoromethylbenzene In N,N-dimethyl-formamide at 20℃; for 1.5h;
55 Preparation of tert-butyl 3-((3-(trifluoromethyl)benzyl)oxy)azetidine-1-carboxylate (57-1).
To a solution of tert-butyl 3-hydroxyazetidine-1-carboxylate (500 mg, 2.887 mmol) in DMF (10 mL) at 0 °C was added 60% NaH (173.20 mg, 4.330 mmol). The reaction mixture was stirred at the room temperature for 0.5 h, Then added 1(b-romomethyl)-3- (trifluoromethyl)benzene (762.22 mg, 3.175 mmol) and stirred at the room temperature for 1.5 h. LCMS showed the reaction was completed. The reaction solution was quenched with sat.NH4Cl (20 mL) and extracted with EA (20 mL*3), the organic phase was dried over Na2SO4, filtered, and concentrated to afford the crude product. The residue was purified by Flash Chromatography (PE /EtOAc=5:2) to give the product tert-butyl 3-((3- (trifluoromethyl)benzyl)oxy)azetidine-1-carboxylate (717 mg, 2.16 mmol , 74.7%) as light- yellow oil. Mass Spectrum (ESI) m/z = 354.0 (M+23).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping