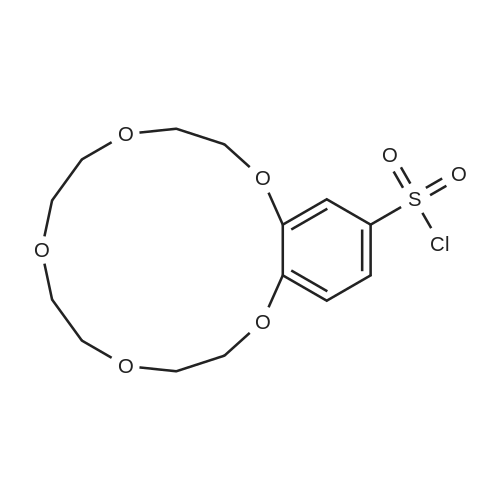
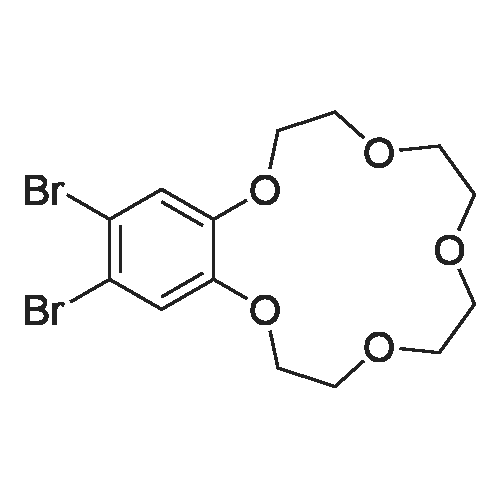
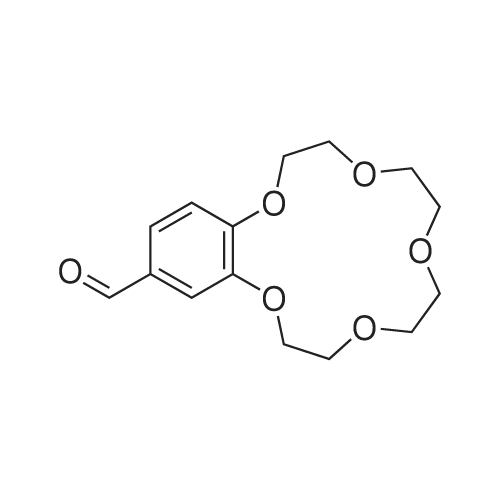
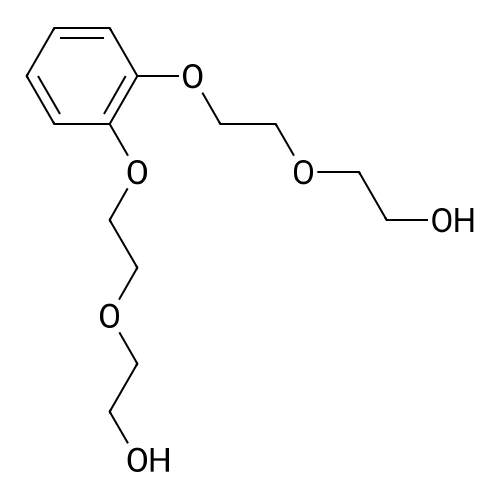
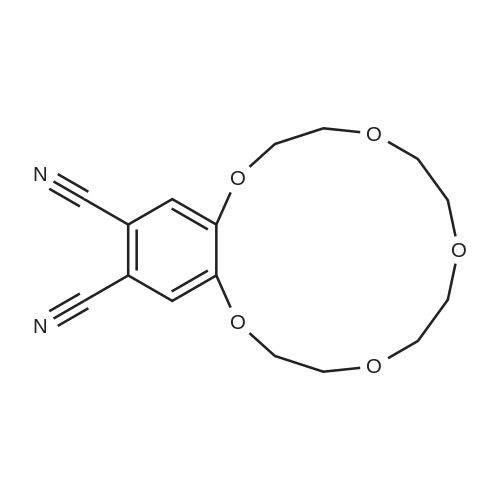
| 99.5% |
With nitric acid; In acetonitrile; |
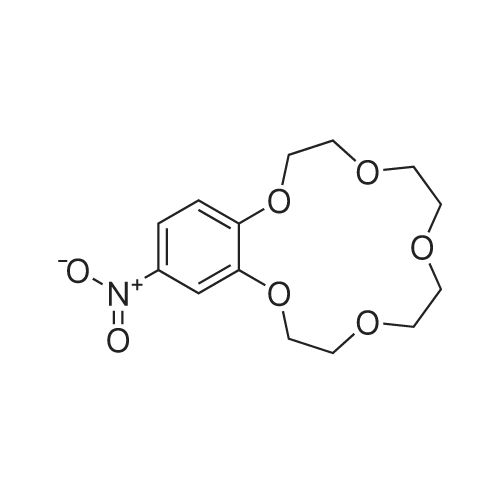
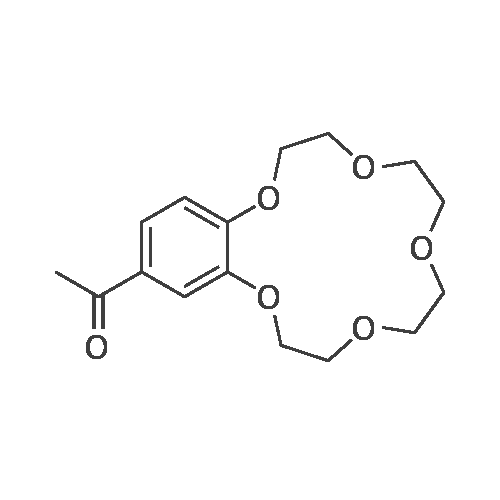
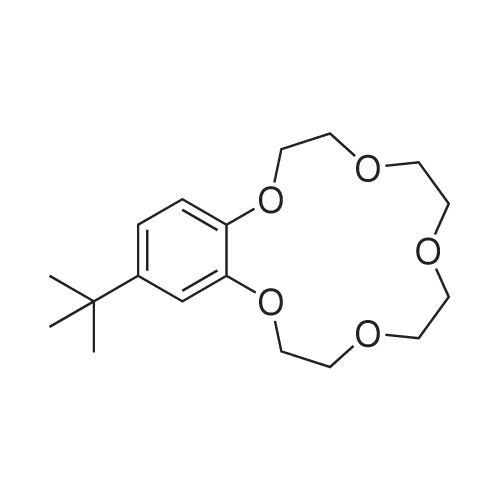
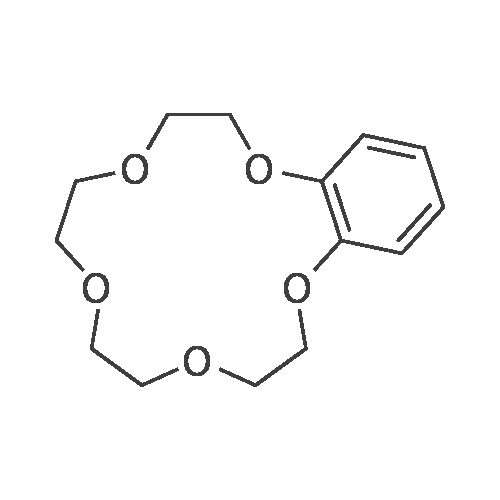
Benzo-15-crown-5 nitration was carried outby nitric acid, 58%, over boiling reaction mass in theacetonitrile media, using relevant method17.Nitric acid of various concentrations can beused in nitration reaction. In some cases, diluted nitric acid (which is poor nitration agent and quite strongoxidizing agent) is also used due to the constantpresence of nitrous acid that hinders nitration.Another drawback of diluted nitric acid, which limitsits use, is its ability to express nitrating effect only athigh temperatures, which are featured by prevailingside oxidation reaction. Concentrated nitric acidshows nitrating effect at lower temperatures and lesslikely to cause undesirable oxidation, which is why wecarried out a nitration of resulted <strong>[14098-44-3]benzo-15-crown-5</strong>with nitric acid, 56%, in the acetonitrile media. Theyield of target product was 99.5%, while the meltingpoint of resulted compound was 94-95 0C.Infrared spectrum (KBr, nu, cm-1): 497(weak), 538 (weak), 618 (weak), 654 (medium)(aromatic C-H), 723 (weak), 745 (medium) (NO2),787 (weak), 806 (medium), 868 (medium) (N-O), 913(weak), 934 (medium), 980 (medium), 1010(weak),1047 (medium), 1093 (strong) (C-N), 1138 (strong)(C-O-C), 1240 (strong) (NO2), 1276 (strong) (NO2),1335 (strong) (NO2), 1417 (strong), 1428 (strong),1448 (medium) (N=O), 1517 (strong) (CH2 + NO2),1587 (medium) (aromatic C-C), 2868 (medium)(C-H), 2904 (medium) (C-H), 2929 (medium) (C-H),3083 (medium) (aromatic C-H).1H N u c l e a r m a g n e t i c r e s o n a n c e(DMSO-d6, 300 MHz): 3.62 (broad singlet, 8H,CH2+CH2+CH2+CH2), 3.74-3.85 (multiplet, 4H,CH2+CH2), 4.13-4.24 (multiplet, 4H, CH2+CH2), 7.15(doublet, 1H, J=8.9, CH), 7.73 (doublet, 1H, J=2.7,CH), 7.89 (double doublet, 1H, J=8.9, J=2.7, CH).13C Nuclear magnetic resonance (DMSO-d6,75 MHz): 67.77, 68.84, 69.27, 69.71, 70.09, 70.16,70.30, 100.96, 105.53, 117.49, 139.35, 143.95,149.92. |
| 72% |
With nitric acid; In acetonitrile; at 70℃; for 0.333333h; |
A solution of 26.8 g(0.10 mol) of <strong>[14098-44-3]benzo-15-crown-5</strong> in 14 mL ofacetonitrile was heated to 70C, 12 mL (0.15 mol) of58% nitric acid was added dropwise, and the mixturewas stirred for 20 min, cooled to room temperature,and diluted with 120 mL of cold water. The lightyellow crystalline solid was filtered off, recrystallizedfrom propan-2-ol, and dried in air. Yield 72%, mp 95-96C, purity 97% (GC/MS). IR spectrum, nu, cm-1: 497w, 538 w, 618 w, 654 m, 723 w, 745 m, 787 w, 806 m,868 m, 913 w, 934 m, 980 m, 1010 w, 1047 m, 1093 s,1138 s, 1240 s, 1276 s, 1335 s, 1417 s, 1428 s, 1448 m,1517 s, 1587 m, 2868 m, 2904 m, 2929 m, 3083 m. 1HNMR spectrum, delta, ppm: 3.62 br.s (8H), 3.74-3.85 m(4H), 4.13-4.24 m (4H), 7.15 d (1H, J = 8.9), 7.73 d(1H, J = 2.7 Hz), 7.89 d (1H, J = 8.9 Hz). 13C NMRspectrum, deltaC, ppm: 70.09, 70.16, 70.30, 100.96,105.53, 117.49, 139.35, 143.95, 149.92. Found, %: C54.01; H 6.23 N 4.27. C14H19NO7. Calculated, %: C53.62; H 6.06; N 4.47. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping