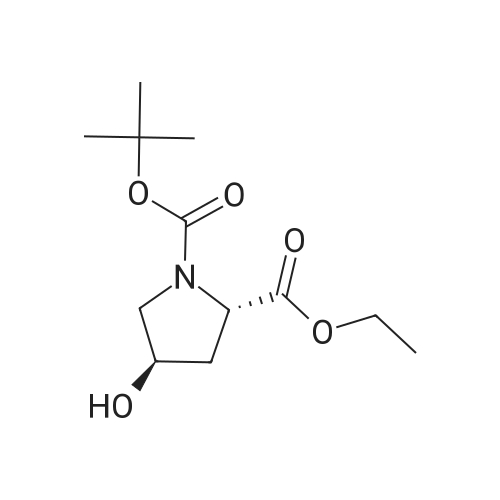
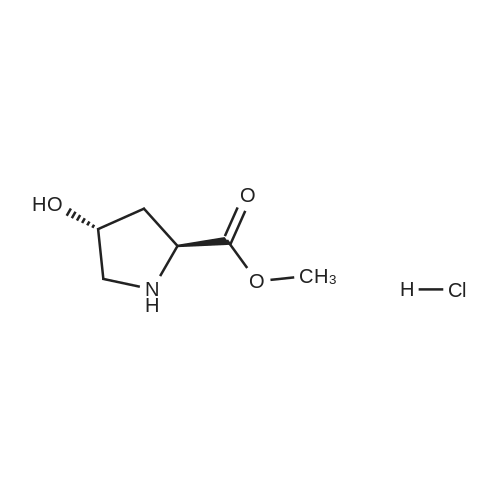
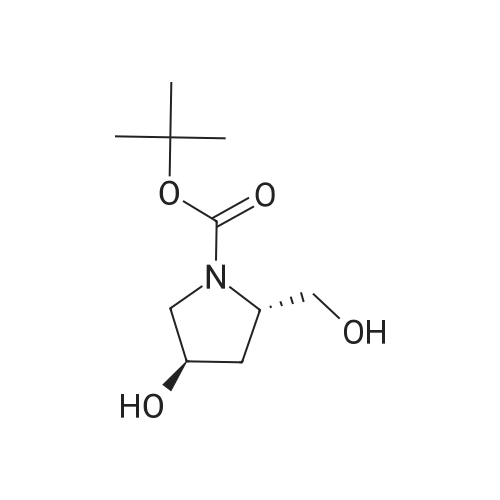
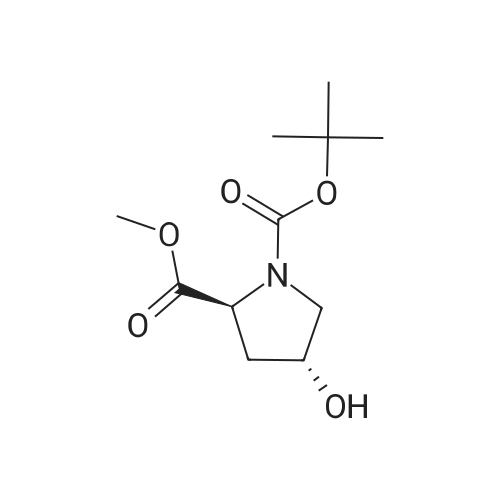
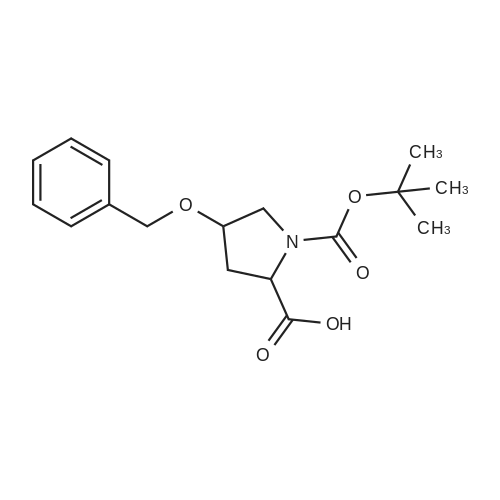
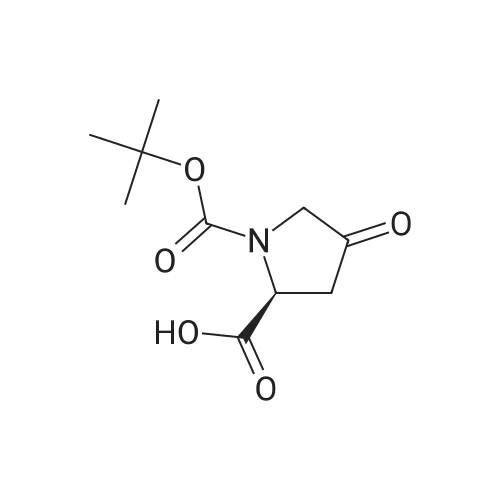
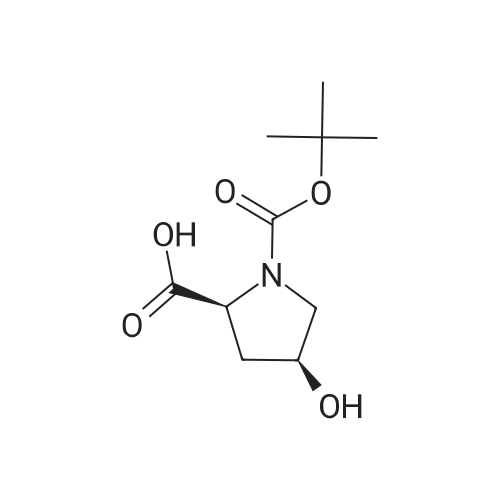
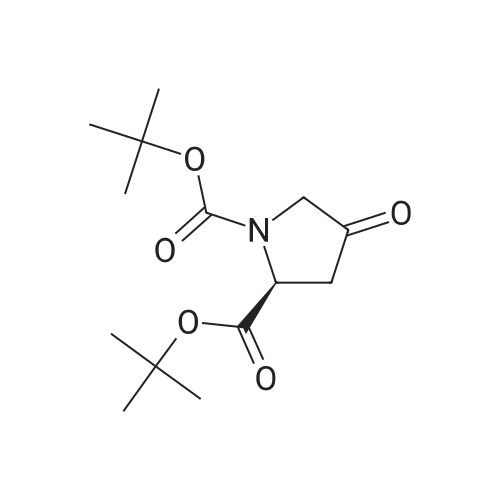
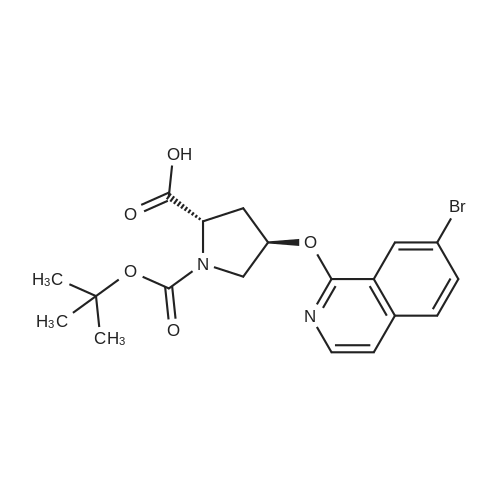
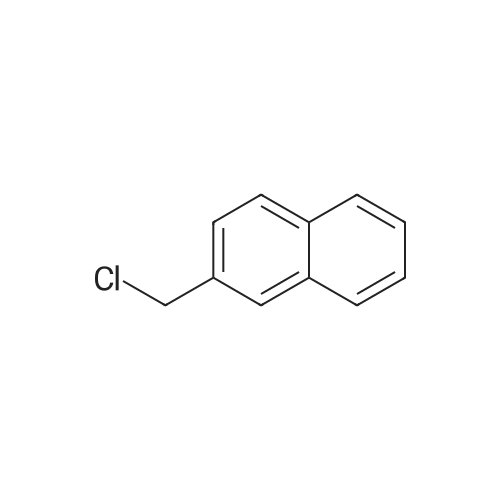
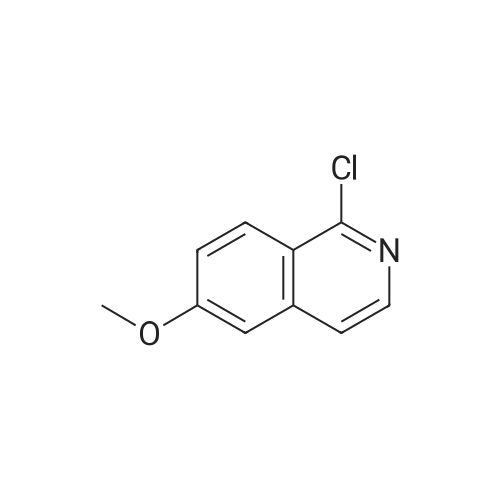
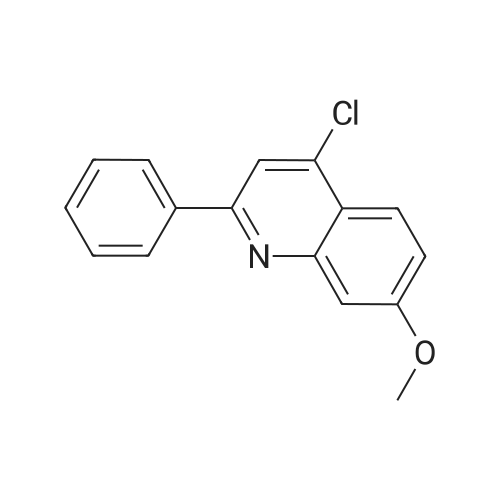
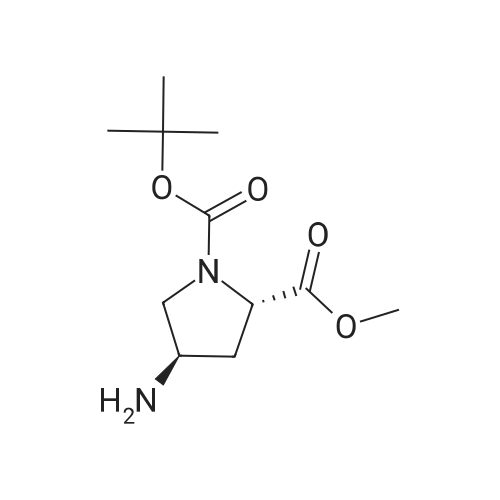
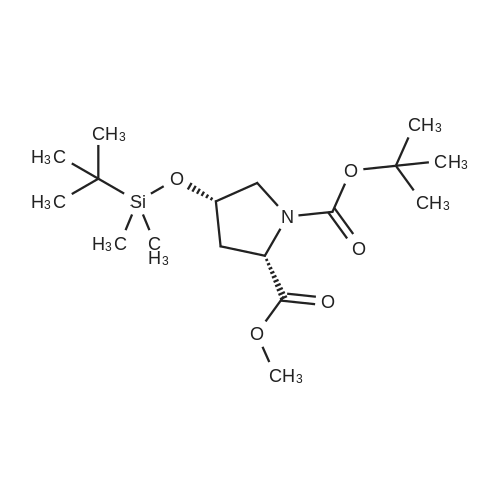
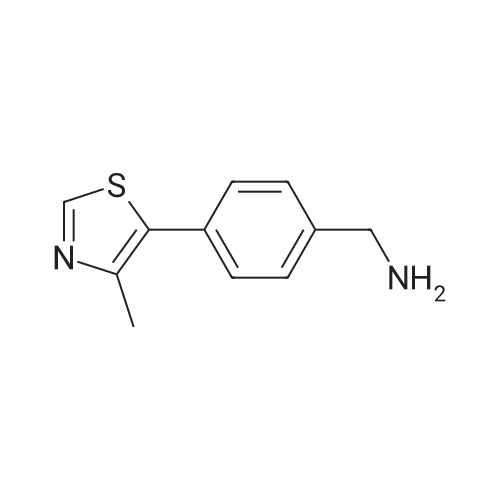
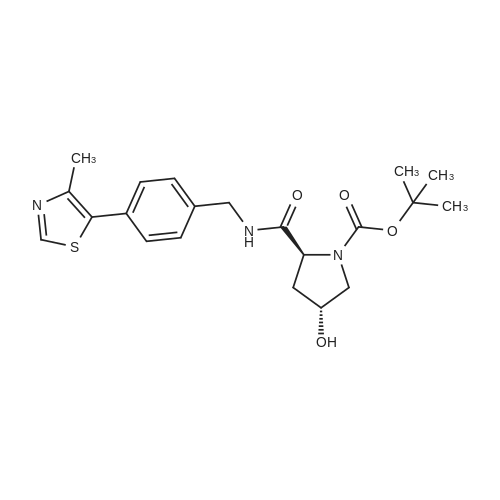
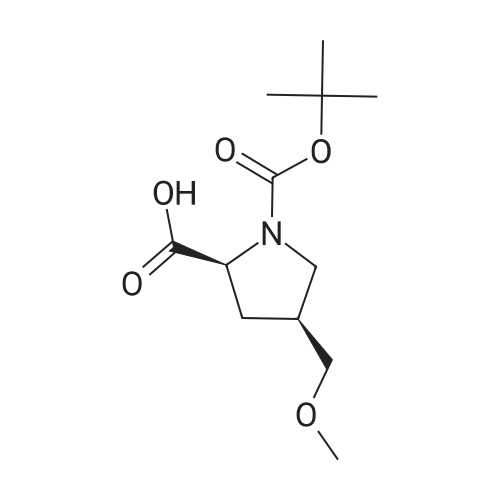
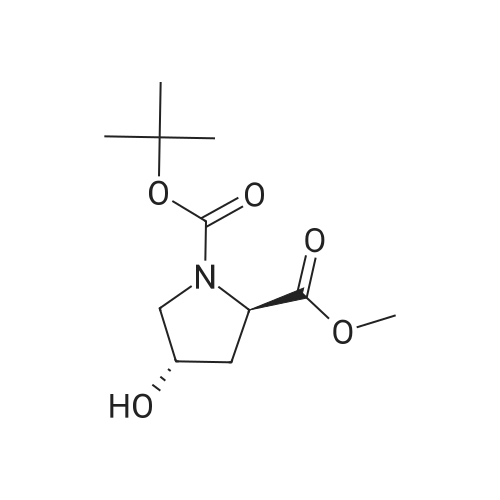
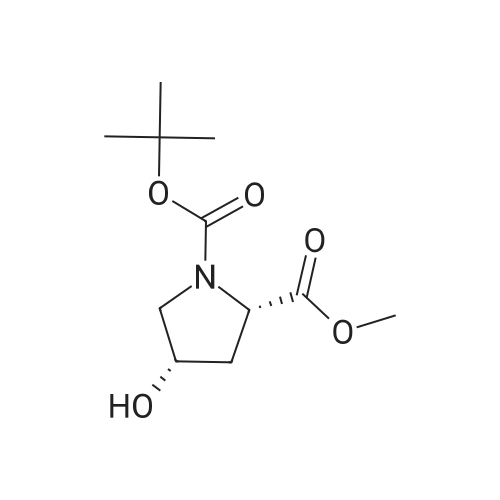
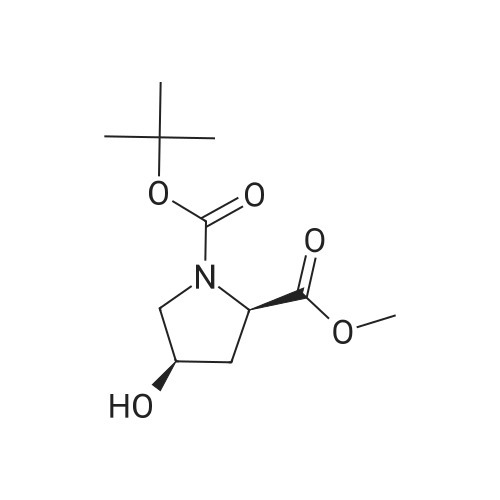
| 99% |
With sodium hydroxide In tetrahydrofuran; lithium hydroxide monohydrate Ambient temperature; |
|
| 99% |
Stage #1: di-<i>tert</i>-butyl dicarbonate; (2S,4R)-4-hydroxypyrrolidine-2-carboxylic acid With sodium hydroxide In 1,4-dioxane; lithium hydroxide monohydrate at 0 - 20℃; for 8h;
Stage #2: With hydrogenchloride; lithium hydroxide monohydrate |
A1-1.A
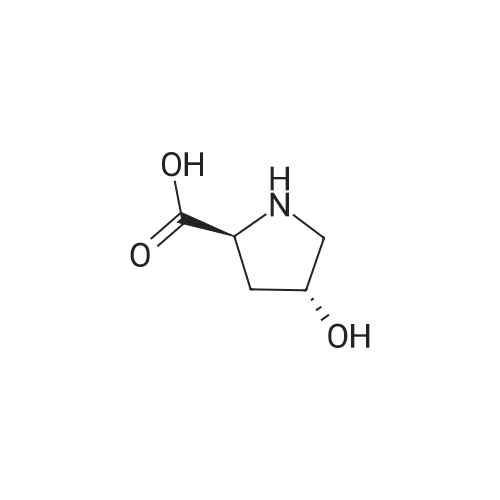
Step A: (4RVl-Boc-4-hydroxy-L-proline; [431] To a solution of (4R)-hydroxy-L-proline (5.08 g, 38.77 mmol) in IN NaOH (40 ml) and 1,4-dioxane (40 ml) was added dropwise di-t-butyl dicarbonate (9.3 g, 42.6 mmol) at 0°C. The reaction mixture was stirred at rt for 8 h, concentrated in vacuo, acidified with IN HCl, and extracted with EtOAc. The organic extracts were washed with brine, dried over MgSO , filtered, and concentrated in vacuo to give the title compound (8.8 g, 99 %).[432] MS[M+H] = 232(M+1) [433] |
| 99% |
Stage #1: di-<i>tert</i>-butyl dicarbonate; (2S,4R)-4-hydroxypyrrolidine-2-carboxylic acid With sodium hydroxide In 1,4-dioxane at 0 - 20℃;
Stage #2: With hydrogenchloride In lithium hydroxide monohydrate |
1.A
(4R)-hydroxy-L-proline (5.08 g, 38.77 mmol) was dissolved in 1 N NaOH (40 ml) and 1,4-dioxane (40 ml), and to the resulting solution, di-t-butyl dicarbonate (9.3 g, 42.6 mmol) was added dropwise at 0° C. The reaction mixture was stirred at room temperature for 8 hours, concentrated in vacuo, acidified with 1 N HCl, and extracted with EtOAc. The organic extracts were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo to give the title compound (8.84 g, 99%).MS[M+H]=232 (M+1) |
| 99% |
With potassium carbonate In tetrahydrofuran; lithium hydroxide monohydrate at 23℃; for 18h; |
1 Example 1
In a 2 L reaction bottle, 131.13 g (1 mol) of L-hydroxyproline and 500 mL of water were added,Stirring to dissolve; then add about 240g saturated potassium carbonate solution to adjust PH = 8 ~ 9.The reaction solution was heated to 23 ° C and 218.25 g (1 mol) of (Boc) 2O in 500 mL of THF was added dropwise. After the addition, the reaction was incubated at 23 ° C for 18 h. After completion of the reaction, the THF in the system was distilled off under reduced pressure. The aqueous phase was extracted with 2 x 250 mL of methyl t-butyl ether and then cooled to 3 ° C and adjusted to pH = 2 to 3 with 4N HCl. 1.1 L ethyl acetate. The organic phase was washed with 300 mL of saturated brine, and the organic phase was dried over anhydrous sodium sulfate for 2 h. After filtration, the filtrate was concentrated to give an oily substance which was placed in a white solid with a weight of 230.0 g and a yield of 99%. |
| 98% |
With triethylamine In methanol at 40℃; |
109.1; 39.O-2
To a suspension of trans-D-hydroxyproline (3 g, 22.88 mmol) in Et3N (6 mL) and MeOH (30 mL) was added Boc anhydride (5.49 g, 25.2 mmol). The mixture was stirred at 40 0C until a clear solution was obtained. The mixture was then concentrated, diluted with IN NaOH (20 mL), washed with hexanes, acidified with 3N HCl, salted and extracted with copious amounts of ethyl acetate (four times). Organics were dried over Na2SO4 and concentrated to afford title compound 235 (5.2 g, 98%) as a white foam. LRMS(ESI): (calc.) 231.11 (found) 230.2 (MH)-. |
| 98% |
With triethylamine In methanol at 40℃; |
109.1
To a suspension of trans-D-hydroxyproline (3 g, 22.88 mmol) in Et3N (6 mL) and MeOH (30 niL) was added Boc anhydride (5.49 g, 25.2 mmol). The mixture was stirred at 40 0C until a clear solution was obtained. The mixture was then concentrated, diluted with IN NaOH (20 mL), washed with hexanes, acidified with 3N HCl, salted and extracted with copious amounts of ethyl acetate (four times). Organics were dried over Na2SO4 and concentrated to afford title compound 235 (5.2 g, 98%) as a white foam. LRMS(ESI): (calc.) 231.11 (found) 230.2 (MH)-. |
| 98% |
With triethylamine In methanol at 40℃; |
109.1 Step 1: (2S,4R)-1-(tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid (235)
To a suspension of trans-D-hydroxyproline (3 g, 22.88 mmol) in Et3N (6 mL) and MeOH (30 mL) was added Boc anhydride (5.49 g, 25.2 mmol). The mixture was stirred at 40° C. until a clear solution was obtained. The mixture was then concentrated, diluted with 1N NaOH (20 mL), washed with hexanes, acidified with 3N HCl, salted and extracted with copious amounts of ethyl acetate (four times). Organics were dried over Na2SO4 and concentrated to afford title compound 235 (5.2 g, 98%) as a white foam. LRMS(ESI): (calc.) 231.11 (found) 230.2 (MH)-. |
| 97% |
Stage #1: di-<i>tert</i>-butyl dicarbonate; (2S,4R)-4-hydroxypyrrolidine-2-carboxylic acid With Sodium hydrogenocarbonate In 1,4-dioxane; lithium hydroxide monohydrate at 0 - 20℃;
Stage #2: With hydrogenchloride In 1,4-dioxane; lithium hydroxide monohydrate |
|
| 97% |
With Sodium hydrogenocarbonate In 1,4-dioxane; lithium hydroxide monohydrate Inert atmosphere; |
|
| 96% |
With sodium hydroxide In methanol for 3h; Ambient temperature; |
|
| 96% |
Stage #1: (2S,4R)-4-hydroxypyrrolidine-2-carboxylic acid With Sodium hydrogenocarbonate In 1,4-dioxane; lithium hydroxide monohydrate at 0℃; for 0.166667h;
Stage #2: di-<i>tert</i>-butyl dicarbonate In 1,4-dioxane; lithium hydroxide monohydrate at 20℃; for 18h; |
1 Step 1: Synthesis of ( 2S,4R)-1 -( tert-butox carbonyl)-4-hydroxypyrrolidine-2-carboxylic acid:
Step 1: Synthesis of ( 2S,4R)-1 -( tert-butox carbonyl)-4-hydroxypyrrolidine-2-carboxylic acid:To a stirred solution of (2S,4R)-4-hydroxypyrrolidine-2-carboxylic acid (50.0 g, 381.3 mmol) in 1,4-dioxane (250 ml) at 0 °C was added aqueous NaHC03 (96.10 g dissolved in 500 ml of H20, 1144 mmol). After stirring about 10 minutes at 0 °C, the reaction mixture was treated with di-tert-butyl dicarbonate (99.86 g, 457.5 mmol) and the resulting solution was stirred at room temperature for about 18 hours. TLC indicated starting material was consumed and the desired product was observed. The reaction mixture was poured into ice cold water, the solution pH was adjusted to 2 to 3 by addition of 4N HC1 {Note: the temperature of reaction mixture should be 5-10 °C). The aqueous layer was extracted with ethyl acetate (2x500 ml), combined organic layer was washed with brine, dried over Na2S04i filtered and solvent was evaporated under reduced pressure to provide the title compound (85.0 g, yield: 96%). |
| 95% |
With Sodium hydrogenocarbonate In 1,4-dioxane; lithium hydroxide monohydrate |
|
| 95% |
With sodium hydroxide In 1,4-dioxane at 20℃; for 7h; |
|
| 95% |
With sodium hydroxide In tetrahydrofuran at 0℃; |
|
| 95% |
Stage #1: di-<i>tert</i>-butyl dicarbonate; (2S,4R)-4-hydroxypyrrolidine-2-carboxylic acid With sodium hydroxide In lithium hydroxide monohydrate; <i>tert</i>-butyl alcohol at 20 - 35℃; for 19h;
Stage #2: With sulfuric acid potassium salt In lithium hydroxide monohydrate |
5C.A
To a 3-necked 5 L RB flask equipped with a mechanical stirrer was added 1N NaOH (aq) solution (1.68 L, 1.68 mol, 1.1 eq), followed by trans-L-4-hydroxyproline 5cl (200 g, 1.525 mol, 1.0 eq) and tert-butanol (1000 mL). The Boc2O (400 g, 1.83 mol, 1.2 eq) was added portionwise over ca. 60 minutes, keeping the internal temperature below 35° C. Upon completion of the addition, the reaction was allowed to stir 18 hours at RT. The mixture was extracted with pentane (2×300 mL) and the aqueous phase acidified to pH 1.0-1.5 with KHSO4 (aq) [prepared by dissolving 315 g of KHSO4 in 2L H2O]. The turbid aqueous mixture was then extracted with EtOAc (4×500 mL), and washed with sat. brine (1×1 L), dried over MgSO4, filtered and concentrated under vacuum. The residue was co-evaporated with methylene chloride/hexane to give white solid 5c2, 333.3 g (95% yield). MS (electrospray): 132: (MH-Boc)+, and 230 (M-H)-. 1H NMR (DMSO-d6), δ12.47 (bs, 1H), 5.04 (s, 1H), 4.11 (t, 1H), 3.44-3.20 (m, 1H), 3.24 (d, J=11.2 Hz, 1H), 2.16-2.04 (m, 1H), 1.94-1.83 (m, 1H), 1.39 and 1.34 rotamers (2 x s, 9H). |
| 95.4% |
With sodium hydroxide In tetrahydrofuran; lithium hydroxide monohydrate at 5 - 25℃; |
1 Example 1 : Preparation of (4R)-1-(tert-butoxycarbonyl)-4-hydroxy-L-proline (Formula IV)
Di-tert-butyl dicarbonate (183 g) was added to a solution of trans-4-hydroxy-L-proline (100 g) in tetrahydrofuran (500 mL). The reaction mixture was cooled to a temperature of 5°C to 10°C. A solution of sodium hydroxide (prepared by adding 30.5 g sodium hydroxide to 305 mL deionized water) was added slowly to the reaction mixture over 10 minutes to 20 minutes. The reaction mixture was allowed to warm to a temperature of 20°C to 25°C, and stirred at the same temperature for 20 hours to 24 hours. The reaction mixture was cooled to a temperature of 5°C to 10°C. The pH of the reaction mixture was adjusted to 2.0 to 2.5 using aqueous hydrochloric acid (290 mL; 3M). The reaction mixture was stirred for 10 minutes, and then allowed to settle for 15 minutes. The layers were separated. The aqueous layer was extracted with ethyl acetate (3 x 200 mL). The combined organic layers were washed with an aqueous solution of sodium chloride (200 mL; prepared by adding 40 g sodium hydroxide to 200 mL deionized water). The organic layer was concentrated at a temperature of about 50°C to 55°C under reduced pressure to obtain a residue. The residue was dissolved in ethyl acetate (500 mL), and again concentrated at 50°C to 55°C under reduced pressure. The residue obtained was again dissolved in ethyl acetate (200 mL) to obtain a solution. Hexanes (100 mL) were slowly added to this solution over a period of 20 minutes to 30 minutes. The reaction mixture was stirred at 20°C to 25°C for 60 minutes. Hexanes (700 mL) were again slowly added to the reaction mixture over a period of 60 minutes. The reaction mixture was stirred at 20°C to 25°C for 2 hours. The reaction mixture was cooled to a temperature of about 0°C to 5°C, and stirred at the same temperature for 2 hours to obtain a solid. The solid was filtered, then washed with a pre-cooled mixture of ethyl acetate and hexanes (0°C to 5°C; 2 x 100 mL, prepared by mixing 40 mL ethyl acetate and 160 mL hexanes). The solid was dried at 40°C to 45 °C under reduced pressure to obtain (4R)-1-(tert- butoxycarbonyl)-4-hydroxy-L-proline. Yield: 95.4% |
| 95.8% |
With sodium hydroxide In lithium hydroxide monohydrate at 40 - 45℃; for 3 - 4h; |
1 Example 1: Preparation of (4R)- 1 -(tert-butoxycarbonyl)-4-hydroxy-L-proline (Formula IV)
Di-tert-butyl dicarbonate (183 g) was added to a solution of trans-4-hydroxy-L- proline (100 g) in water (200 mL). A solution of sodium hydroxide (33.6 g sodium hydroxide in 100 mL deionized water) was added slowly to the reaction mixture over a period of 10 minutes to 20 minutes. The reaction mixture was allowed to warm to 40°C to 45°C, then was stirred at the same temperature for 3 hours to 4 hours. The reactionmixture was cooled to 25°C to 3 0°C. Ethyl acetate (1000 mL) was added to the reaction mixture, and then the mixture was cooled to 0°C to 5°C. The pH of the reaction mixture was adjusted to 2.0 to 2.5 using aqueous hydrochloric acid (275 mL; 3M). The reaction mixture was stirred for 10 minutes, and then allowed to settle for 15 minutes. The layers were separated. The aqueous layer was extracted with ethyl acetate (4 x 200 mL) at 15°C to 20°C. The combined organic layers were washed with an aqueous solution of sodium chloride (100 mL; 20 g sodium chloride in 100 mL deionized water). The organic layerwas concentrated at 50°C to 55°C under reduced pressure to obtain a residue. The residuewas dissolved in ethyl acetate (200 mL), then stirred at 40°C to 45°C for 20 minutes.Hexanes (100 mL) were slowly added to the solution over a period of 20 minutes to 30minutes at 25°C to 3 0°C, and then the mixture was stirred at the same temperature for 60minutes to obtain a solid. Hexanes (700 mL) were slowly added to the solid to obtain aslurry, and then the mixture was stirred over a period of 60 minutes. The slurry wascooled to 0°C to 5°C, then stirred at the same temperature for 2 hours. The solid wasfiltered, then washed with a precooled mixture of ethyl acetate and hexanes (0°C to 5°C; 2x 100 mL, a mixture of 40 mL ethyl acetate and 160 mL hexanes). The solid was dried at40°C to 45°C under reduced pressure to obtain (4R)-1-(tert-butoxycarbonyl)-4-hydroxy-L-proline.Yield: 95.8 %HPLC Purity: 99.93 % |
| 94% |
With triethylamine In methanol at 50 - 60℃; |
|
| 93.2% |
With sodium hydroxide In tetrahydrofuran Ambient temperature; |
|
| 93% |
With sodium hydroxide In tetrahydrofuran; lithium hydroxide monohydrate for 16h; Inert atmosphere; |
|
| 92% |
With sodium hydroxide In tetrahydrofuran; lithium hydroxide monohydrate at 20℃; |
|
| 92% |
With triethylamine In methanol at 20℃; for 23.5h; Reflux; |
(2S,4R)-1-(Tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic Acid (Boc-trans-L-4-hydroxyproline, v)
To a stirred solution of trans-L-4-hydroxyproline i (2.0 g,15.3 mmol) in MeOH (36.0 mL) was added Et3N (4.0 mL,28.7 mmol) and Boc anhydride (6.7 g, 30.5 mmol) and the reaction was refluxed for 3.5 h, cooled to room temperature, and stirred for 20 h. Solvent was removed under vacuum and the residue cooled to 0oC. Following the addition of NaH2PO4 (150 mg), the solution was acidified to pH 2 with 0.5 M HCl.The mixture was stirred at 0oC for 30 min before extractingthe product with EtOAc (420 mL). The combined organic layers were dried with MgSO4 and filtered. The solvent was removed under vacuum yielding v as a white foam (3.23 g,14 mmol, 92 %) |
| 92% |
With N,N-dimethyl-4-aminopyridine In dichloromethane at 20℃; for 6h; |
3.C.1 Synthesis of (2S,4R)-1-(tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid (2)
To a stirred solution of compound 1 (1.2 g, 7.35 mmol) in dichloromethane (50 mL), N,N-dimethylaminopyridine (0.898 g, 7.35 mmol) followed by di-tertiary butyl dicarbonate (3.2 g, 14.70 mmol) were added and the reaction mixture was allowed to stir at room temperature for 6 h. The reaction mixture was quenched with water and the compound was extracted in ethyl acetate. The organic layers were washed with 0.5 M HCl and brine, separated, dried over sodium sulphate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using 1-2% methanol in dichloromethane as eluent to afford compound 2 (1.75 g, 92%). MS (ESI) m/z 232 [M+1]+. |
| 90% |
With anhydrous sodium carbonate |
|
| 90% |
With Sodium hydrogenocarbonate In 1,4-dioxane; lithium hydroxide monohydrate at 20℃; for 16h; |
Boc-(2S,4R)-Hyp-OH(2)
H-Hyp-OH1(21.5g,163.96mmol,1equiv.)wasdissolvedin1MNaHCO3aq.(378mL,378mmol,2.3equiv.).AsolutionofBoc2O(42.94g,196.75mmol,1.2equiv.)in1,4-dioxane(150mL wasaddeddropwiseover1h. Thereactionwasstirredatroomtemperaturefor16h.ThereactionmixturewaswashedwithEt2O(2x100mL),acidifiedwithconc.HCltopH2,extractedwithEtOAc(3x100mL),washedwithbrine,driedoverMgSO4,filteredandthesolventevaporatedprovidingthepureproduct2(34.14g,90%)asawhitepowder. |
| 89% |
With sodium hydroxide In lithium hydroxide monohydrate at 20℃; for 10h; |
1 Intermediate 1; tert-butyl (2S,4R)-4-[tert-butyl(dimethyl)silyl]oxy}-2-formyl pyrrolidine- 1-carboxylate (cf Scheme 6, compound XLL)
To a mixture of commercial (4R)-4-hydroxy-L-proline (75g, 0. 57MOL) in 10% NAOH (11) was added (Boc) 20 (186g, 0. 855mol) at 0 C with stirring. The reaction was allowed to stir at RT for 10h and then washed with petroleum ether. The aqueous layer was acidified with citric acid to pH=4 and extracted with ethyl acetate. The combined organic layer was washed with brine and dried over NA2S04. The solvent was removed under vacuum to give crude (4R)-I-(TERT-BUTOXYCARBONYL)-4-HYDROXY-L-PROLINE (118G) as viscous liquid. Yield: 89% To a solution of (4R)-1-(tert-butoxycarbonylo)-4-hydroxy-L-proline (100g, 0. 432MOL) in dry DMF (600ML), at 0 C was added K2CO3 (179g, 1. 3mol) followed by iodoethane (LOLG, 0. 65MOL). After stirring at RT for 12h, K2CO3 was filtered off and DMF was distilled off under reduce pressure. The residue was diluted with dichloromethane (11), washed with brine and dried. The solvent was removed under vacuum to give crude 1-1-TERT-BUTYL 2- ethyl (2S, 4R)-4-hydroxypyrrolidine-1, 2-dicarboxylate (103g) as yellow liquid. Yield: 91% H NMR (300MHZ, CDC ) : 1.25 (t, 3H), 1.4 (d, 9H), 1.9-2. 2 (M, 2H), 2.5 (M, IH), 3.5 (M, 2H), 4.2 (M, 2H), 4.4-4. 6 (M, 2H). To a solution of 1-tert-butyl 2-ethyl (2S, 4R)-4-HYDROXYPYRROLIDINE-1, 2-dicarboxylate (100g, 0. 38MOL) in dry dichloromethane (1.51) at 25 C was added DMAP (47g, 0. 38MOL), followed by triethylamine (39g, 0. 38mol). To the above reaction mixture was added TBDMSCl (64g, 0. 42mol) dissolved in dry dichloromethane (200ML) drop-wise over a period OF 45MIN. After stirring at RT for 20H, the reaction mixture was diluted with water and separated the organic layer. The organic layer was washed with 5% aq. citric acid, brine and dried over NA2SO4. The solvent was removed under vacuum to give I-TERT-BUTYL 2-ethyl (2S, 4R)-4- [TERT-BUTYL (DIMETHYL) silyl] oxy} pyrrolidine-1, 2-DICARBOXYLATE (140g) as a colorless liquid. Yield: 97% 'H NMR (300MHz, CDC13) : 0.0 (s, 6H), 0. 8 (s, 9H), 1.2 (t, 3H), 1. 45 (d, 9H), 1. 7-1. 9 (M, 2H), 2.2 (M, 1H), 3.3-3. 5 (M, 2H), 4.2 (M, 2H), 4.5 (M, 1H). To a suspension of lithium aluminium hydride (lOg) in dry tetrahydrofuran (750ml) at- 40 C was added 1-tert-butyl 2-ethyl 1-TERT-BUTYL 2-ethyl (2S,4R)-4-[(tert- butyl (dimethyl) silyl] oxy} pyrrolidine-1, 2-dicarboxylate (100g, 0. 289mol) drop-wise in tetrahydrofuran (250ML). After stirring AT-TO C for 5H, the reaction mixture was quenched with 10% NAOH (40ML). Filtered off the solid residue, washed with tetrahydrofuran and the filtrate was evaporated under reduce pressure to give tert-butyl (2S, 4R)-4-[TERT- butyl (dimethyl) silyl] OXY}-2- (HYDROXYMETHYL) PYRROLIDINE-1-CARBOXYLATE (85g) as a colorless liquid. Yield: 94% LCMS: ESI+: 232 (M-Boc+H)+, 276 (M-tBu+H) +, 354 (M+Na) + 'H NMR (300MHZ, DMSO-d6) : 0.05 (s, 6H), 0.78 (s, 9H), 1.33 (s, 9H), 1.65-2. 00 (m, 2H), 3.20 (m, 2H), 3. 38 (m, 2H), 3.70 (m, 1H), 4.30 (m, 1H), 4. 60 (t, 1H). To a mixture of DMSO (53g, 0. 68MOL) and oxalylchloride (43G, 0. 34MOL) in dry dichloromethane (1. 51) at-78oC was added tert-butyl (2S, 4R)-4-[tert-butyl (dimethyl) silyl] OXY}-2-(HYDROXYMETHYL) PYLROLIDINE-L-CARBOXYLATE (75g, 0. 226MOL) drop-wise. After stirring at-78oC for IH, was added triethylamine (158ml, 1. 13MOL) drop-wise and warmed the reaction mixture slowly to RT. The reaction mixture was diluted with water and separated the organic layer. The organic layer was washed with brine and dried. The solvent was removed under vacuum to give tert-butyl (2S,4R)-4-[tert-butyl(dimethyl)silyl] OXY}-2-FORMYLPYRROLIDINE-L-CARBOXYLATE (70g) as pale yellow liquid. Yield: 94 % |
| 89% |
Stage #1: di-<i>tert</i>-butyl dicarbonate; (2S,4R)-4-hydroxypyrrolidine-2-carboxylic acid With sodium hydroxide In tetrahydrofuran; lithium hydroxide monohydrate at 20℃;
Stage #2: With sulfuric acid potassium salt In lithium hydroxide monohydrate |
H.1
To a suspension of trans-L-4-hydroxy proline (28.17 g, 215 mmol) in 2:1 THF:H2O (700 mL) was added 2.5 M NaOH (118 mL), then Boc2O (67.1 mL, 292 mmol). The reaction was stirred overnight at room temperature. The THF was then removed in vacuo, and 10% KHSO4 solution (423 ml) was added. The mixture was extracted three times with ethyl acetate. The combined organic layers were partitioned with brine, dried over MgSO4, and concentrated to afford trans-L-N-Boc-4-hydroxy proline (44.41 g, 89% yield). 1H NMR (500 MHz, DMSO-d6) δ ppm 12.49 (s, 1H), 4.86-5.23 (m, 1H), 4.20-4.30 (m, 1H), 4.07-4.16 (m, 1H), 3.31-3.46 (m, 1H), 3.19-3.29 (m, 1H), 2.03-2.18 (m, 1H), 1.81-1.95 (m, 1H), 1.39 (s, 3H), 1.34 (s, 6H). |
| 88% |
With anhydrous sodium carbonate In 1,4-dioxane; lithium hydroxide monohydrate |
|
| 88% |
With sodium hydroxide In 1,4-dioxane at 20℃; for 12h; |
1.A; S1
Example 1 . Amino Acid Analogue Synthetic Approaches A. Synthesis of Fmoc-2S,4/R-perfluoro-tert-butyl-hydoxyproline (7) |
| 86% |
With triethylamine In 1,4-dioxane; methanol; lithium hydroxide monohydrate at 20℃; for 4h; |
|
| 86% |
With sodium hydroxide In tetrahydrofuran; lithium hydroxide monohydrate at 20℃; for 16h; |
|
| 86% |
With Sodium hydrogenocarbonate In 1,4-dioxane; lithium hydroxide monohydrate at 0 - 20℃; for 24h; Inert atmosphere; |
Synthesis of compounds P-1 to P-6
To a solution of 4-OH-L-Pro (10 g, 76.3 mmol) in Dioxane : H2O = 1 : 1 (200 mL) at 0 °C was added NaHCO3 (19.2 g, 228.9 mmol) and Boc2O (25 g, 11.5 mmol), and the reaction mixture was stirred at rt for 24 h. Then the reaction was treated with 3 M aq. HCl, and extracted with EtOAc (three times). Then the combined organic extracts were washed with brine. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. The resulting residue was purified by silica gel flash chromatography (DCM : MeOH = 10:1) to give 15.17 g compound P-1 in 86% yield. 1H NMR (300 MHz, CD3OD, mixture of two amide rotamers) δ: 4.34-4.29 (1 H, m), 3.61-3.32 (3 H, overlap), 2.32-2.25 (1 H, m), 2.11-2.03 (1 H, m), 1.48 and 1.44 (9 H, s); 13C NMR (75 MHz, CD3OD, mixture of two amide rotamers) δ: 177.2, 156.5 and 156.1, 81.7 and 81.4, 70.7 and 70.1, 59.6 and 59.3, 55.9 and 55.5, 40.1 and 39.5, 28.7 and 28.6; HRMS(ESI) calcd for C10H17NO5 [M+Na]+: 254.0999; found: 254.0996. |
| 86.49% |
With sodium hydroxide In tetrahydrofuran; lithium hydroxide monohydrate at 15 - 30℃; for 16h; |
(2S,4R)-1-tert-butoxycarbonyl-4-hydroxy-pyrrolidine-2-carboxylic acid
To a solution of L-hydroxyprobne (2.49 g, 19 mmol, 1 eq) in THF (26 mL) / water (13 mL) was added sodium hydroxide (13.67 mL, 38 mmol, 2 eq) followed the addtion of di-t- butyl dicarbonate (6.22 g, 28.5 mmol, 1.5 eq) at 15 °C and stirred at 30 °C for 16 h. The reaction mixture was extracted with EtOAc while pH was about 9~10. The water layer was adjusted pH to 1~2 with 2.0 N HCl aqueous and then lyophilized. 100 mL CH3CN and 50 mL EtOAc was added and stirred at room temperature. After 2 hr, the mixture was filtered and concentrated to give the title compound (3.8 g, 86.49% yield) as white solid. |
| 86.49% |
With sodium hydroxide In tetrahydrofuran; lithium hydroxide monohydrate at 15 - 30℃; for 16h; |
(2S,4R)-1-tert-butoxycarbonyl-4-hydroxy-pyrrolidine-2-carboxylic acid
To a solution of L-hydroxyprobne (2.49 g, 19 mmol, 1 eq) in THF (26 mL) / water (13 mL) was added sodium hydroxide (13.67 mL, 38 mmol, 2 eq) followed the addtion of di-t- butyl dicarbonate (6.22 g, 28.5 mmol, 1.5 eq) at 15 °C and stirred at 30 °C for 16 h. The reaction mixture was extracted with EtOAc while pH was about 9~10. The water layer was adjusted pH to 1~2 with 2.0 N HCl aqueous and then lyophilized. 100 mL CH3CN and 50 mL EtOAc was added and stirred at room temperature. After 2 hr, the mixture was filtered and concentrated to give the title compound (3.8 g, 86.49% yield) as white solid. |
| 86.49% |
With sodium hydroxide In tetrahydrofuran; lithium hydroxide monohydrate at 15 - 30℃; for 16h; Inert atmosphere; |
(2S,4R)-1-tert-butoxycarbonyl-4-hydroxy-pyrrolidine-2-carboxylic acid
To a solution of L-hydroxyprobne (2.49 g, 19 mmol, 1 eq) in THF (26 mL) / water (13 mL) was added sodium hydroxide (13.67 mL, 38 mmol, 2 eq) followed the addtion of di-t- butyl dicarbonate (6.22 g, 28.5 mmol, 1.5 eq) at 15 °C and stirred at 30 °C for 16 h. The reaction mixture was extracted with EtOAc while pH was about 9~10. The water layer was adjusted pH to 1~2 with 2.0 N HCl aqueous and then lyophilized. 100 mL CH3CN and 50 mL EtOAc was added and stirred at room temperature. After 2 hr, the mixture was filtered and concentrated to give the title compound (3.8 g, 86.49% yield) as white solid. |
| 85% |
With sodium hydroxide In <i>tert</i>-butyl alcohol for 18h; |
|
| 85% |
With triethylamine In methanol for 2h; Heating; |
|
| 82% |
Stage #1: di-<i>tert</i>-butyl dicarbonate; (2S,4R)-4-hydroxypyrrolidine-2-carboxylic acid With Sodium hydrogenocarbonate In 1,4-dioxane; lithium hydroxide monohydrate
Stage #2: With hydrogenchloride In 1,4-dioxane; lithium hydroxide monohydrate |
Synthesis of N-Boc trans-4-hydroxyproline (71)
Synthesis of N-Boc trans-4-hydroxyproline (71) trans-4-hydroxyproline (70) (5 g, 38 mmol) was dissolved in dioxane/water (1:1) (50 mL), and to the solution was added NaHCO3 (80 mmol) and Boc anhydride (30 mmol, 6.5 gram). The reaction was stirred for 4 hours. NaHCO3 was added to keep the pH above 7. The crude reaction mixture was acidified using 0.5 N HCl. Dioxane was evaporated. Boc-trans-4-hydroxyproline was recovered by extraction using EtOAc/water. The organic phase was dried using MgSO4 and subsequently evaporated to yield N-Boc-4-hydroxyproline (71) as a clear oil (5.6 g, 82%). |
| 82% |
With Sodium hydrogenocarbonate In 1,4-dioxane; lithium hydroxide monohydrate for 4h; |
trans-4-hydroxyproline (70) (5 g, 38 mmol) was dissolved in dioxane/water (1:1) (50 mL), and to the solution was added NaHCO3 (80 mmol) and Boc anhydride (30 mmol, 6.5 gram). The reaction was stirred for 4 hours. NaHCO3 was added to keep the pH above 7. The crude reaction mixture was acidified using 0.5 N HCl. Dioxane was evaporated. Boc-trans-4-hydroxyproline was recovered by extraction using EtOActwater. The organic phase was dried using MgSO4 and subsequently evaporated to yield N-Boc-4-hydroxyproline (71) as a clear oil (5.6 g, 82%). |
| 81% |
With sodium hydroxide In tetrahydrofuran; lithium hydroxide monohydrate at 20℃; |
|
| 80% |
With sodium hydroxide In lithium hydroxide monohydrate; <i>tert</i>-butyl alcohol for 12h; Ambient temperature; |
|
| 80% |
With sodium hydroxide In lithium hydroxide monohydrate at 20 - 30℃; for 1 - 2h; |
Preparation of N-Boc-L-hydroxyproline; HO, HO, ON H 0 H O No.ONo.O Batch: 132 g (1.00 mol) of L-hydroxyproline 700 ml of deionized water 230 g (1.05 mol) of Boc20 300 ml of acetone L-Hydroxyproline is dissolved in 700 ml of water at 20-25°C. When the hydroxyproline has dissolved, the pH is adjusted from about 5.5 to 10.5 (10-lL) with NaOH (50%). Then, at a temperature of 25-28°C (max. 30°C), the Boc2O solution in acetone and the sodium hydroxide solution (50%) are simultaneously metered in so that the temperature is kept between 25-28°C (ma=. 30°C) and the pH at 10.5 (limits 10-11). Duration of addition about 1-2 h. After the metering is complete, the pH is kept further at 10.5 until constant. An easily stirrable suspension results at the end of the reaction. The total consumption of NaOH (50%) was 174 g (2.18 mol). The pH is subsequently adjusted to pH 2.6 with HC1 (37%). The temperature is always kept at 25°C ; keep it at 25°C by cooling if it rises a little. C02 is liberated during the acidification. The evolution of gas starts at a pH of about 7.5 (CO2). Further addition of HCl (37%) should take place sufficiently slowly for the resulting C02 to be driven out to the same extent. Duration about 30 min in the laboratory. The total amount of HCl (37%) used was: 206 g (2.1 mol). When the pH of 2.6 (2.5-2. 8) is reached, stirring is continued for 15 min in order to stir out residues of dissolved CO2. Subsequently, 600 ml of MIBK are added, and the mixture is heated to 35-40°C while stirring (15-20 min). It is subsequently allowed to settle, and the phases are separated. 1st Org. phase: 920 g The aqueous phase (about 1 1) is again-extracted with 400 ml of MIBK at 35-40°C. 2nd Org. phase: 360 g The first organic phase (contains most of the acetone from the reaction) is then distilled under a vacuum down to 200 mbar at a bottom temperature not exceeding 50°C until almost no further distillate is produced. The 2nd org. phase is added to the distillation bottom, and distillation is continued at 200 mbar. The vacuum is then slowly lowered further, and distillate is collected. When about 600 ml have distilled out, the solution is filtered to remove entrained salt (a few g). It is distilled until an approx. 50% strength solution is produced (approx. 400-450 g). Subsequently, 200 ml of MIBK are added and distilled out until the water content in the bottom reaches < 0.5 (preferably 0. 3%). (Possibly renewed addition of MIBK and distillation). When the water content of 0.5 is reached, 100-200 ml of MIBK are added and the mixture is cooled to 40°C. 400 ml of hexane are slowly added to the solution at 40°C. During the addition or thereafter, seed crystals are added to the mixture and, after crystallization has started, a further 200 ml of hexane are added. The suspension is then cooled over the course of 2 h to 15-20 and then with ice/salt further to 0-5°C. And subsequently stirred for 1-2 h. The suspension is filtered and the solid is washed with hexane (2x) 1 l/kg. 258 g of moist product are obtained (heavy crystals). The solid is then dried in vacuo at a max. temperature of 50°C under 15 mbar. 186 g (80% yield) are obtained. The final product is checked via HPLC purity (laboratory > 98 area%) and NMR (identity), and loss on drying (< 0.2%). The product should no longer have an ocLor of MIBK (prior sample from dryer). |
| 80% |
Stage #1: di-<i>tert</i>-butyl dicarbonate; (2S,4R)-4-hydroxypyrrolidine-2-carboxylic acid With sodium hydroxide In tetrahydrofuran; lithium hydroxide monohydrate at 20℃;
Stage #2: With hydrogenchloride In lithium hydroxide monohydrate |
13.1
Step 1 Preparation of (2S,4R)-1-(tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid L-Hydroxyproline 13a (60 g, 0.458 mol) was added to a solution of 750 mL of solvent mixture of tetrahydrofuran/water (2:1) followed by 252 mL of aqueous sodium hydroxide (10%) and a solution of di-tert-butyl dicarbonate (136 g, 0.624 mol) in 750 mL of solvent mixture of tetrahydrofuran/water (2:1). The reaction mixture was reacted overnight at room temperature and diluted with 500 mL of ethyl acetate. The layers were separated and then the organic layer was discarded, and the aqueous layer was adjusted to pH 2 with concentrated hydrochloric acid. The mixture was extracted with 1.5 L of ethyl acetate. The combined organic extracts were washed with saturated brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain the title compound (2S,4R)-1-(tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid 13b (86.4 g, yield 80%) as a colorless oil. |
| 70% |
With sodium hydroxide In tetrahydrofuran; lithium hydroxide monohydrate for 24h; Inert atmosphere; |
|
| 68% |
Stage #1: di-<i>tert</i>-butyl dicarbonate; (2S,4R)-4-hydroxypyrrolidine-2-carboxylic acid With anhydrous sodium carbonate In tetrahydrofuran; 1,4-dioxane; lithium hydroxide monohydrate
Stage #2: With hydrogenchloride In 1,4-dioxane; lithium hydroxide monohydrate at 0℃; |
a; 31.a
A solution of L-hydroxyproline (10.0 g, 76.2 mmol) in 80 ml of 10% aq Na2CO3 was added dropwise to a solution of Boc-anhydride (15.8 g, 72.4 mmol) in 44 ml of 10:1 THF/dioxane, and the mixture was stirred overnight. After removal of THF, the mixture was then washed with Et2O, cooled to 0 degree, and acidified carefully to PH=2 with 3N HCl. The aqueous solution was extracted with EA and the organic layer was dried and concentrated to afford 4-hydroxy-pyrrolidine-1,2-dicarboxylic acid 1-tert-butyl ester, 12 g (68%) as yellow oil. MS m/e=322.1 [M+H]+. |
| 62% |
With sodium hydroxide In <i>tert</i>-butyl alcohol at 20℃; |
|
| 60% |
With Sodium hydrogenocarbonate In 1,4-dioxane; lithium hydroxide monohydrate at 0 - 20℃; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping