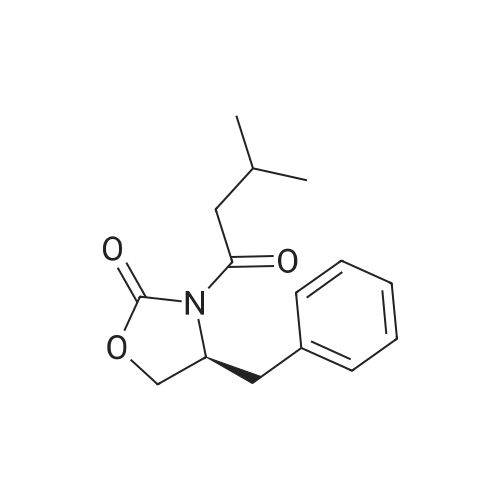
| 99% |
Stage #1: (R)-4-(phenylmethyl)-2-oxazolidinone With n-butyllithium In tetrahydrofuran at -78℃; for 0.333333h; Inert atmosphere;
Stage #2: propionyl chloride In tetrahydrofuran at -78 - 20℃; Inert atmosphere; |
|
| 99% |
Stage #1: (R)-4-(phenylmethyl)-2-oxazolidinone With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 1h;
Stage #2: propionyl chloride In tetrahydrofuran; hexane at -78℃; for 3h; |
|
| 99% |
Stage #1: (R)-4-(phenylmethyl)-2-oxazolidinone With n-butyllithium In tetrahydrofuran at -78℃; for 0.333333h; Inert atmosphere;
Stage #2: propionyl chloride In tetrahydrofuran at -78 - 25℃; for 1h; Inert atmosphere; |
|
| 99% |
Stage #1: (R)-4-(phenylmethyl)-2-oxazolidinone With n-butyllithium In tetrahydrofuran at -78℃; for 0.5h;
Stage #2: propionyl chloride In tetrahydrofuran at -78℃; for 0.5h; |
|
| 98% |
With n-butyllithium In tetrahydrofuran; hexane at -78 - 20℃; Inert atmosphere; |
|
| 97% |
With n-butyllithium In tetrahydrofuran at -78 - 0℃; |
|
| 97% |
Stage #1: (R)-4-(phenylmethyl)-2-oxazolidinone With n-butyllithium In tetrahydrofuran; hexane at -78℃;
Stage #2: propionyl chloride In tetrahydrofuran; hexane at -78℃; |
|
| 97% |
Stage #1: (R)-4-(phenylmethyl)-2-oxazolidinone With n-butyllithium In tetrahydrofuran at -78℃; for 0.0833333h;
Stage #2: propionyl chloride In tetrahydrofuran at -78 - 20℃; for 0.5h; |
|
| 94% |
Stage #1: (R)-4-(phenylmethyl)-2-oxazolidinone With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 0.333333h;
Stage #2: propionyl chloride In tetrahydrofuran; hexane at 20℃; |
55.A
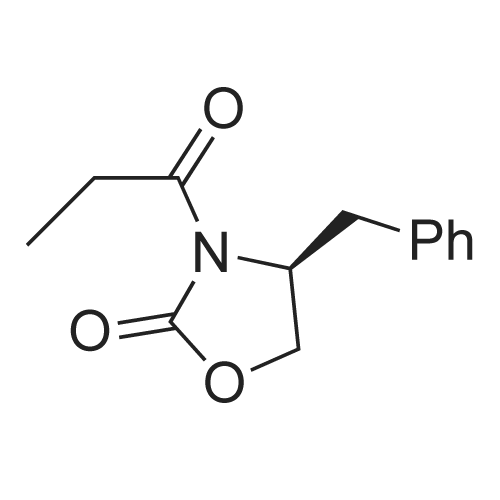
A. (R)-4-Benzyl-3-propionyl-oxazolidin-2-one. ; To a solution of (R)-4-benzyl-oxazolidin-2-one (3.0 g, 16.9 mmol) in THF (100 mL) at -78° C. was added n-BuLi (2.5 M hexanes, 7.1 mL, 17.8 mmol) in rapid drops via syringe. The solution was stirred for 20 min at -78° C. Propionyl chloride (1.6 mL, 18.4 mmol) was added rapidly via syringe. The reaction solution was allowed to warm slowly to rt overnight then was quenched by the addition of satd. aq. NH4Cl. The mixture was concentrated, diluted with water, and extracted with DCM (3×). The combined organic layers were dried (Na2SO4) and concentrated to give the crude product, which was purified by flash chromatography (EtOAc/hexanes) to afford the title compound as a white solid (3.70 g, 94%). 1H NMR (500 MHz, CDCl3): 7.36-7.31 (m, 2H), 7.30-7.25 (m, 1H), 7.23-7.19 (m, 2H), 4.71-4.64 (m, 1H), 4.20 (dd, J=9.1, 7.5, 1H), 4.17 (dd, J=9.1, 3.1, 1H), 3.31 (dd, J=13.4, 3.3, 1H), 2.99 (dq, J=17.9, 7.4, 1H), 2.93 (dq, J=17.9, 7.3, 1H), 2.77 (dd, J=13.4, 9.6, 1H), 1.21 (t, J=7.3, 3H). |
| 93% |
With dmap; triethylamine In dichloromethane at 0℃; for 1h; |
2 Example 2Synthesis of compound (III-b)
(R) -4-benzyl-2-oxazolidinone (20 g, 1 eq.) Was dissolved in 200 ml of dichloromethane, triethylamine (17.1 g, 1.5 eq) was added with stirring at 0 ° C with stirring,4-dimethylaminopyridine (414 mg, 0.03 equiv)Followed by the addition of propionyl chloride (10.4 g, 1 equiv)The mixture was stirred at 0 ° C for 1 hour, diluted with dichloromethane,Washed with water, saturated with sodium bicarbonate, and the organic phase was dried over anhydrous sodium sulfate.The solvent was removed by evaporation under reduced pressure to give the crude product. The crude product was purified by column chromatography,To give the title compound (III-b) (24.3 g, yield 93%). |
| 91% |
Stage #1: (R)-4-(phenylmethyl)-2-oxazolidinone With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 0.583333h;
Stage #2: propionyl chloride In tetrahydrofuran; hexane at -78℃; for 2.33333h; |
(R)-4-Benzyl-3-propionyloxazolidin-2-one (14)
To a solution of the commerciallyavailable oxazolidinone S4 (5.00 g, 28.2 mmol, 1.00 eq) in THF (120 mL) was added n-BuLi (19.4 mL 1.6 M in Hexanes, 31.0 mmol, 1.10 eq) dropwise at -78 °C. The resultingdark orange solution was aged at -78 °C for 35 min. Then propionyl chloride (4.93 mL,56.4 mmol, 2.00 eq) was added at -78 °C. The dark colour discharged rapidly. The mixturewas stirred for further 2 h 20 min at -78 °C. Then Na2CO3 (sat. aq., 30 mL) was added toquench the remaining propionyl chloride and the mixture was diluted with NaCl (sat. aq.,15 mL) and EtOAc (20 mL). The layers were separated and the aqueous phase wasextracted with EtOAc (3×20 mL). The combined organic layers were washed with brine,dried over MgSO4 and concentrated in vacuo. The crude was purified by columnchromatography (CH2Cl2:acetone 15:1) to afford the title compound as a yellow oil, whichcrystallised in a freezer (-18 °C). The material was recrystallysed from hex:EtOAc (ca.10:1, some hexane added). The yellow mother liquour was discarded and the crystalswere washed with cold hexane to afford the title compound 14 (5.97 g, 91%) in colourlessneedles.TLC (CH2Cl2:acetone 15:1): Rf = 0.701H-NMR (CDCl3, 400.1 MHz): δ = 7.38-7.27 (m, 3H), 7.24-7.16 (m, 2H), 4.68 (mc, 1H), 4.25-4.12 (m, 2H), 3.31 (dd, J = 3.3 Hz, J = 13.4 Hz, 1H), 2.96 (dd, J = 7.3 Hz, J = 9.3 Hz, 1H), 2.96(mc, 1H), 2.77 (dd, J = 9.6 Hz, J = 13.4 Hz, 1H), 1.21 (t, J = 7.3 Hz, 3H). |
| 91% |
Stage #1: (R)-4-(phenylmethyl)-2-oxazolidinone With n-butyllithium In tetrahydrofuran; hexane at -78℃;
Stage #2: propionyl chloride In tetrahydrofuran; hexane at -78 - 20℃; |
General procedure for the synthesis of propionylated oxazolidinones [1]
General procedure: To a suspension of oxazolidinone (1.0 eq.) in anhydrous tetrahydrofuran a 2.5 M solution of n-butyllithium in hexane (1.0 eq.) was added dropwise at -78 °C. After stirring for 30 min at -78 °C propionylchloride (1.0 eq) was added dropwise at -78 °C. After stirring for 35 minutes at -78 °C and for 135 min at room temperature the reaction mixture was poured on an ice-water mixture and was extracted with diethylether (3 x 150 mL). The combined organic phases were washed with sat. sodiumhydrogencarbonate solution (1 x 50 mL) and sat. NaCl-solution (1 x 50 mL), were dried over sodium sulfate and concentrated under reduced pressure. Column chromatography (10-15 % EtOAc in cyclohexane) of the crude product gave the desired propionylated oxazolidinone. |
| 90% |
Stage #1: (R)-4-(phenylmethyl)-2-oxazolidinone With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 0.166667h;
Stage #2: propionyl chloride In tetrahydrofuran; hexane at -78 - 20℃; for 1h; |
|
|
With n-butyllithium In tetrahydrofuran at -78℃; |
|
|
With n-butyllithium In tetrahydrofuran |
8
Example 8; Propionyl Oxazolidinone 28; Compound 28 was prepared by reaction of (i?)-(+)-4-benzyl-2-oxazolidinone with BuLi and propionyl chloride in THF according to standard literature procedures (See, Evans, D. A. Aldrichimica Acta 1982, 15, 23). |
|
With n-butyllithium In tetrahydrofuran |
8 Example 8
Propionyl OXAZOLIDINONE 28 : Compound 28 was prepared by reaction of (R)- (+)-4-BENZYL-2-OXAZOLIDINONE with BuLi and propionyl chloride in THF according to standard literature procedures (See, Evans, D. A. ALDRICHIMICA ACTA 1982, 15, 23). |
|
Stage #1: (R)-4-(phenylmethyl)-2-oxazolidinone With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 0.5h;
Stage #2: propionyl chloride In tetrahydrofuran; hexane for 0.5h; |
16
Eaxmple 16 (4R)-4-benzyl-3-propionyl-1,3-oxazolidin-2-one: To a solution of (4R)-4-benzyl-1,3-oxazolidin-2-one (60 g) in tetrahydrofuran (600 mL), n-butyllithium-hexane solution (1.54M, 230 mL) was slowly dropped at -78°C, followed by stirring for 30 minutes. Then, propionyl chloride (30.9 mL) was added in portions thereto, followed by stirring for 30 minutes. The reaction mixture was added with water and was raised to room temperature. Then, the mixture was extracted with ethyl acetate. The organic layer was successively washed with water and brine, dried and concentrated. Thus, the title compound (79.4 g) having the following physical properties was obtained. TLC: Rf 0.52 (hexane:ethyl acetate = 3 :1); NMR (CDCl3): δ 7.13-7.42 (m, 5H), 4.60-4.76 (m, 1H), 4.08-4.30 (m, 2H), 3.31 (dd, J = 13.20, 3.20 Hz, 1H), 2.86-3.09 (m, 2H), 2.77 (dd, J = 13.20, 9.70 Hz, 1H), 1.21 (t, J = 7.30 Hz, 3H). |
|
With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 0.5h; |
12.1
[Step 12]; Synthesis of (4R)-4-benzyl-3-[(2R,3S)-3-hydroxy-2-methylpentanoyl]-1,3-oxazolidin-2-one; (1) Synthesis of (4R)-4-benzyl-3-propionyl-1,3-oxazolidin-2-one (P26); This reaction was performed with reference to the literature (Gage, J. R.; Evans, D. A., Organic Synthesis, 1989, 68, 83-91. Chan, P. C.-M.; Chong. J. M.; Kousha, K., Tetrahedron, 1994, 150(9), 2703-2714.). 1.57M n-butyllithium hexane solution (96 ml, 150 mmol) was slowly added dropwise to a THF (400 ml) solution of (R)-4-benzyl-2-oxazolidinone (25.36 g, 143 mmol) at -78° C. under stirring. Subsequently propionyl chloride (13.7 ml, 157 mmol) was added at once and the reaction solution was stirred at the same temperature for 30 minutes. After the reaction solution was warmed to room temperature for 30 minutes, it was diluted with ethyl acetate and washed with water and brine. The organic layer was dried over anhydrous sodium sulfate. After the drying agent was filtered off, the organic layer was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (Merck, commercial name Silica Gel 60, 230-400 mesh; heptane:ethyl acetate=5:1) to obtain the title compound (36.58 g) as a white solid. 400 MHz 1H-NMR (CDCl3) δ (ppm) 1.21 (t, J=7.2 Hz, 3H), 2.77 (dd, J=9.6, 13.2 Hz, 1H), 2.88-3.05 (m, 2H), 3.31 (dd, J=3.2, 13.2 Hz, 1H), 4.11-4.23 (m, 2H), 4.65-4.70 (m, 1H), 7.20-7.36 (m, 5H); 100 MHz 13C-NMR (CDCl3) δ (ppm) 8.52, 29.42, 38.15, 55.39, 66.44, 127.56, 129.17, 129.64, 135.55, 164.52, 174.30; IR (KBr) 3082, 3026, 2984, 2942, 2872, 2360, 2338, 1787, 1702, 1496 cm-1; HRMS C13H15NNaO3 (M+Na+) Calcd: 256.0950, Found: 256.0928; [α]D27-101.20 (c 1.11, CH3CH2OH), [α]D25-63.9 (c 1.00, CHCl3) |
|
With n-butyllithium In tetrahydrofuran |
|
|
With dmap; triethylamine In dichloromethane at 0 - 20℃; Inert atmosphere; |
|
| 9.52 g |
Stage #1: (R)-4-(phenylmethyl)-2-oxazolidinone With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 0.166667h; Inert atmosphere;
Stage #2: propionyl chloride In tetrahydrofuran; hexane at -78 - 0℃; for 0.666667h; Inert atmosphere; |
To a solution of (R)-4-benzyl-2-oxazolidinone (10 g, 56.4 mmol) in anhydrousTHF (200 mL) was added n-BuLi (2.5 M in hexanes, 25 mL) dropwise at -78 °C. Thereaction was stirred for 10 min followed by the addition of propionyl chloride (5.9 g,63 mmol) with stirring for additional 10 min. The mixture was allowed to warm to0 °C over 30 min before quenching with sat aq NH4Cl (50 mL). After the volatilematerials were removed by concentrating on a rotary evaporator, the resulted residueswere diluted with CH2Cl2 (100 mL). The organic layers were washed with 10% aqNaOH (2 × 50 mL), and the aqueous layer was extracted with CH2Cl2 (2 × 100 mL).The combined organic layers were dried over Na2SO4, filtered and concentrated underreduced pressure. Purification of the crude product by silica gel flash columnchromatography (gradient eluent: 0-1% of EtOAc/petroleum ether) afforded S9 (9.52g, 73%) as a white solid.8 |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping