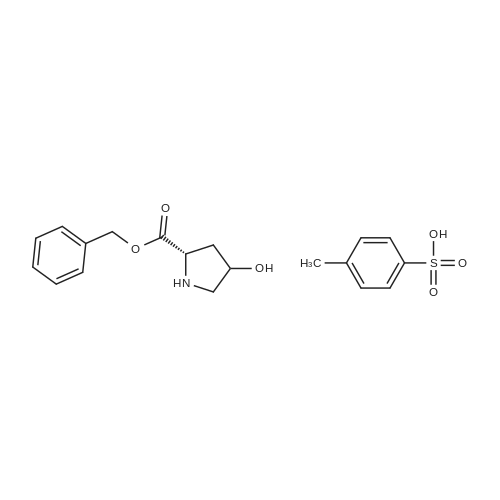
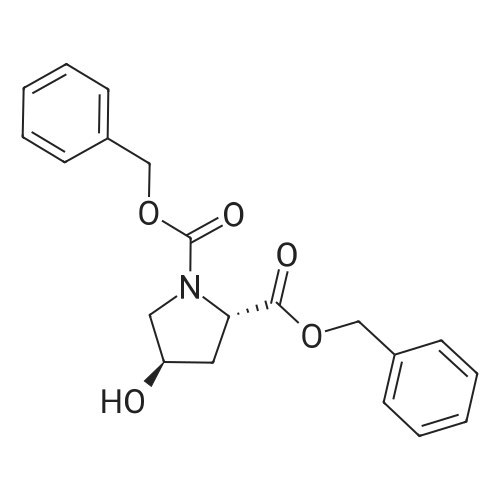
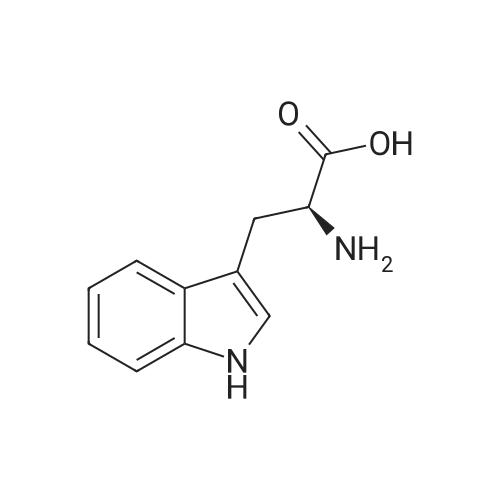
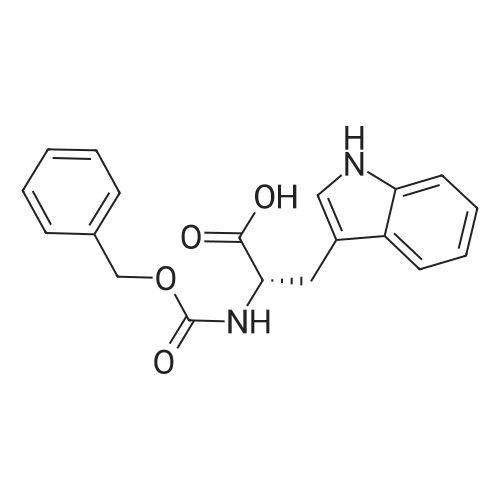
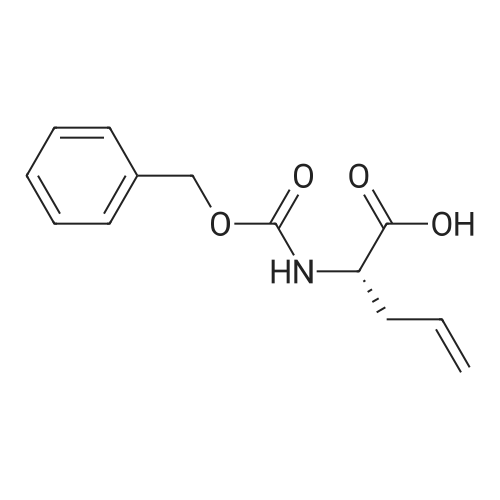
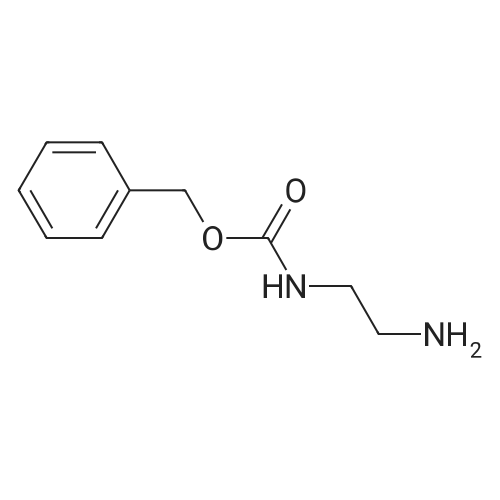
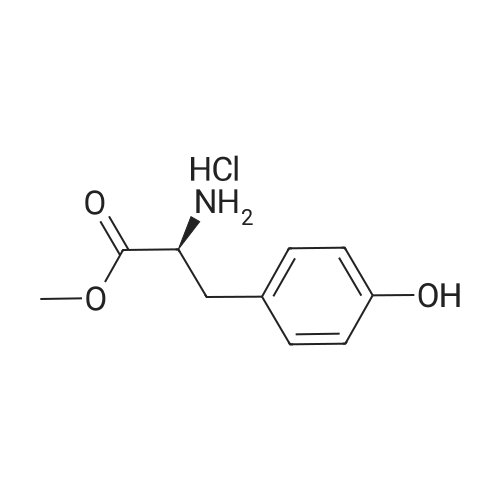
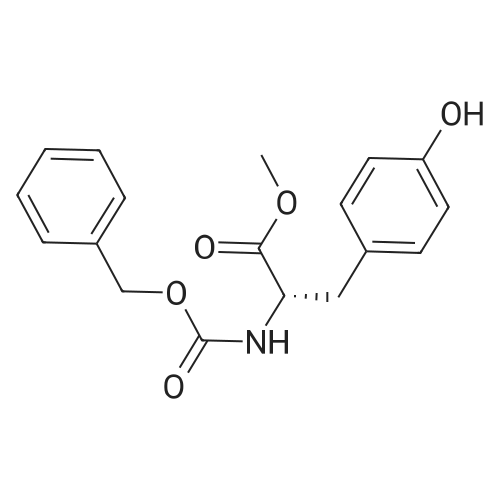
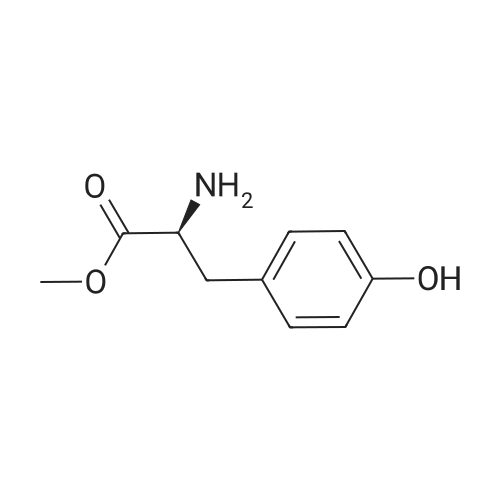
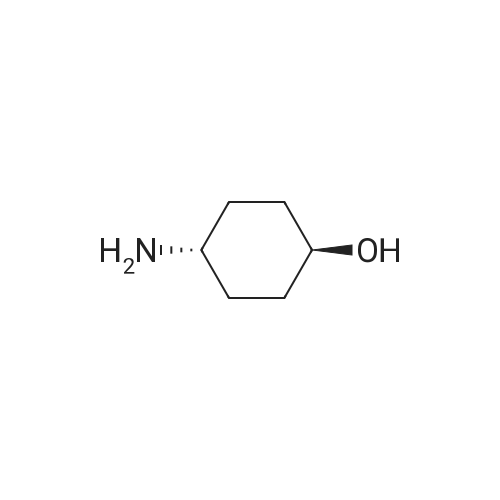
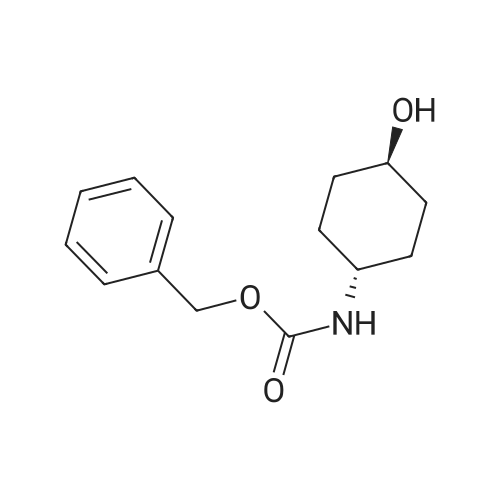
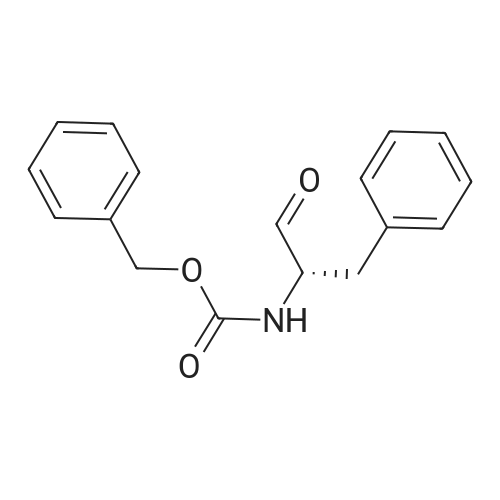
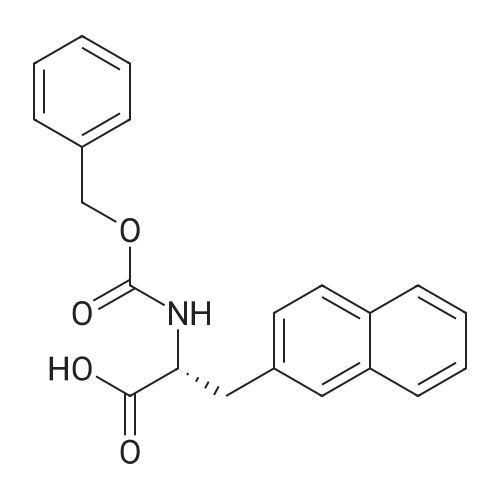
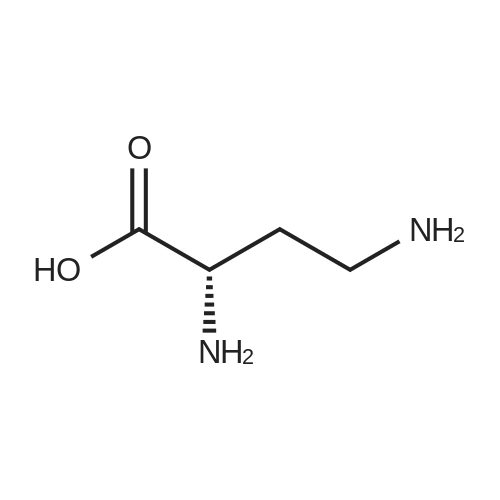
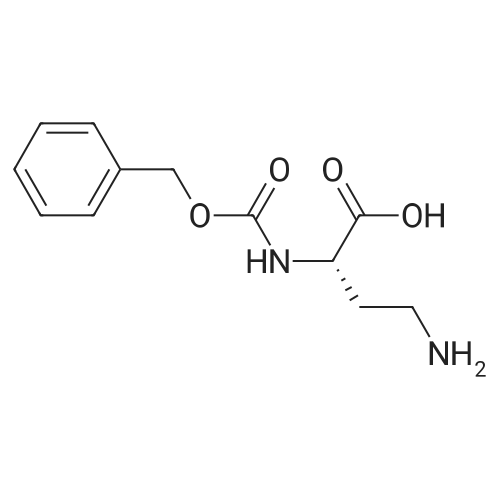
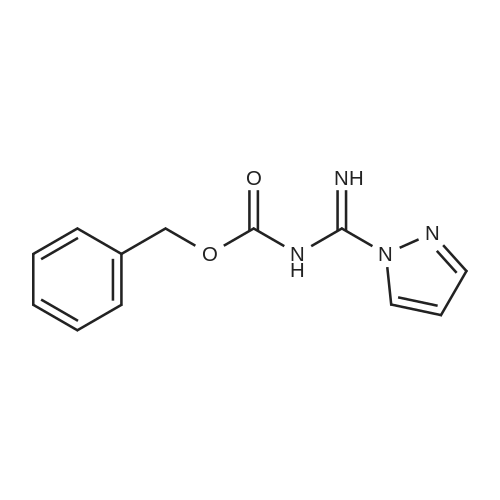
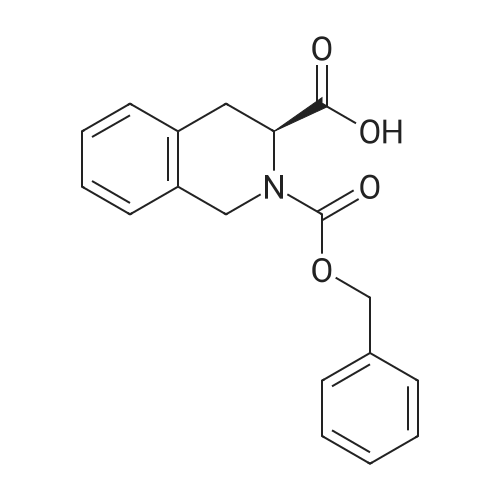
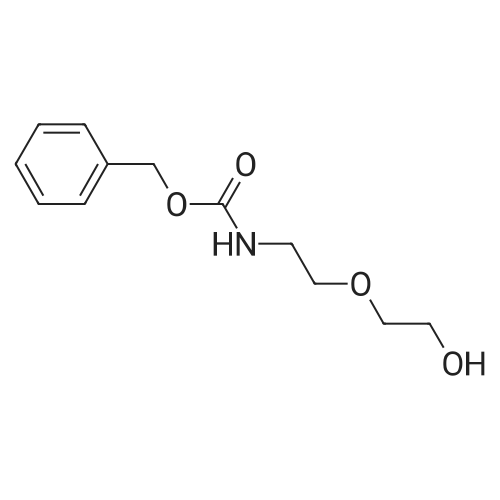
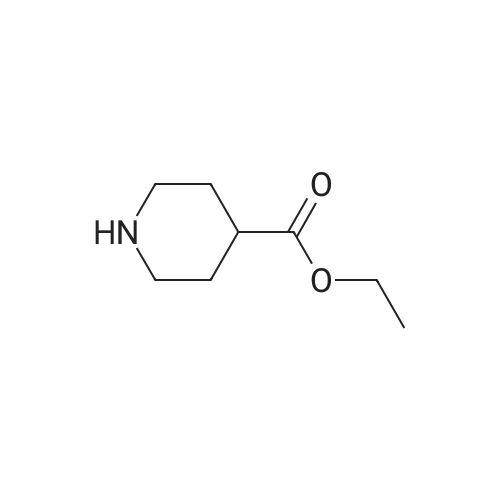
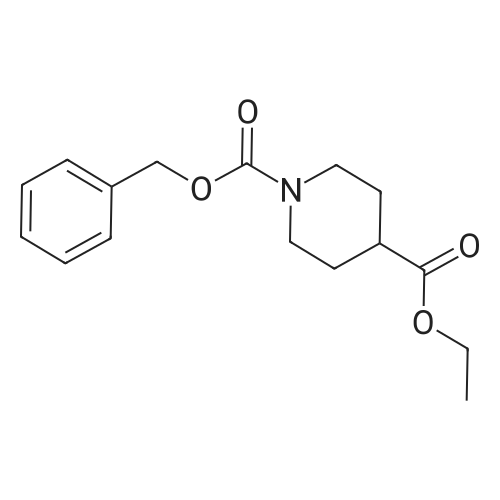
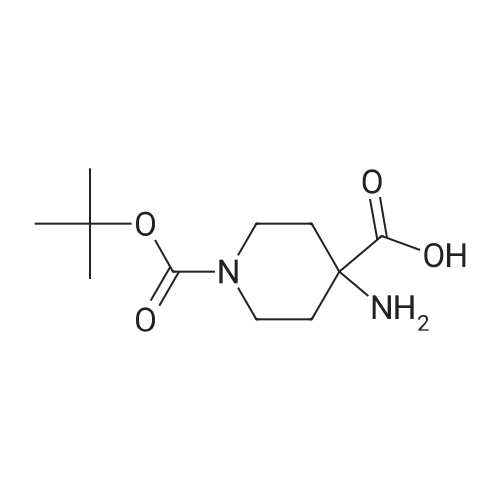
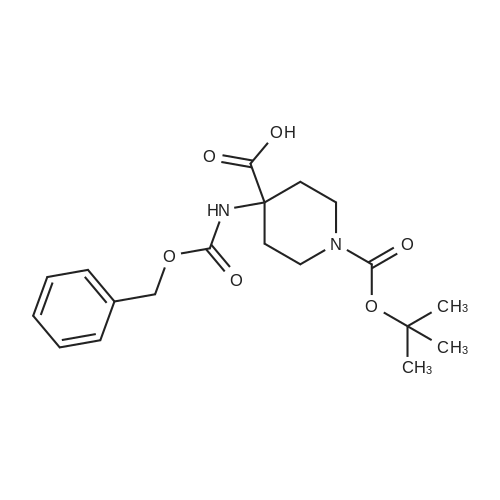
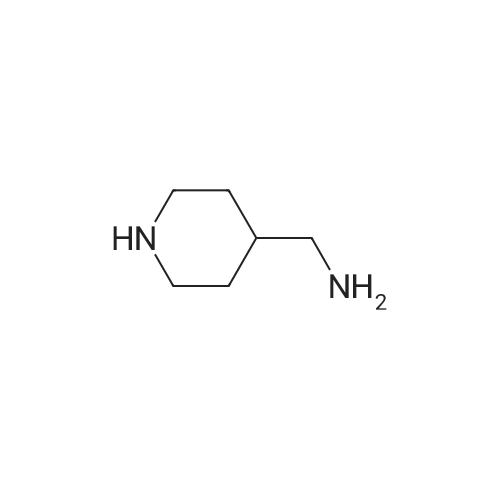
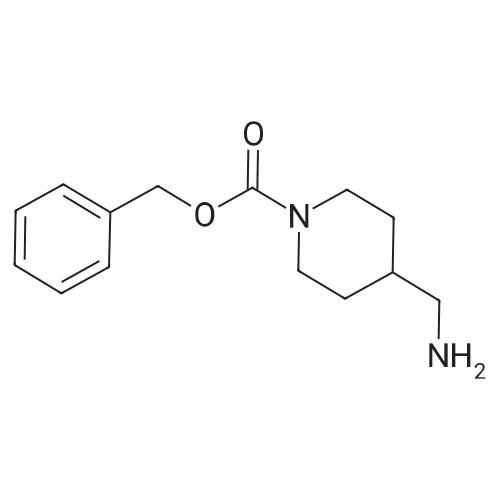
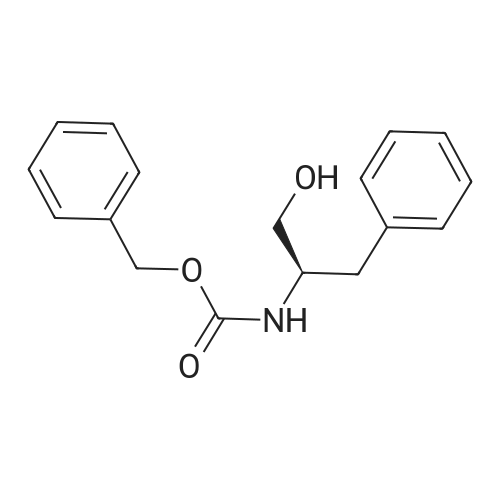
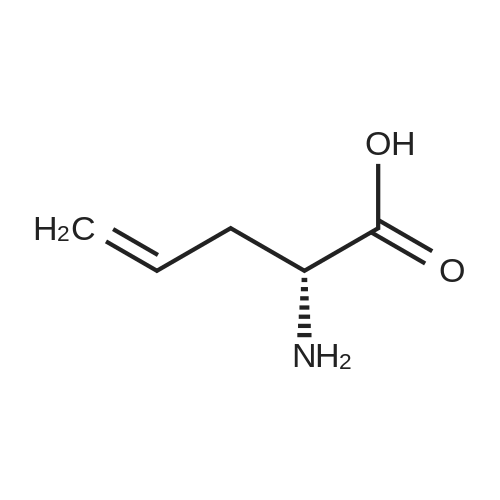
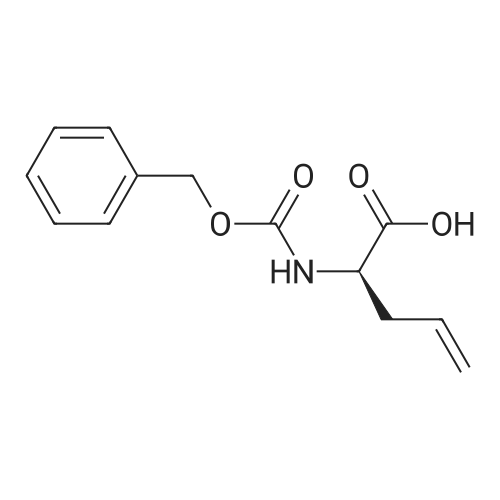
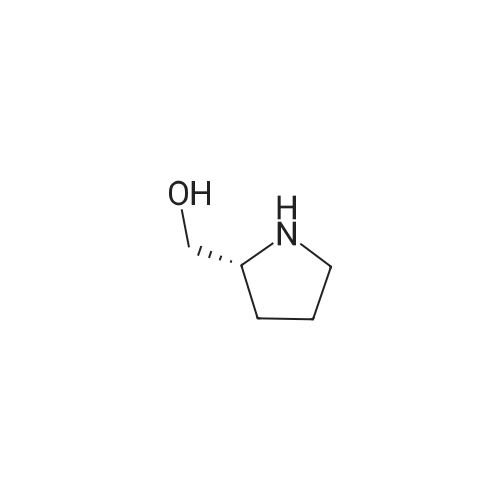
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
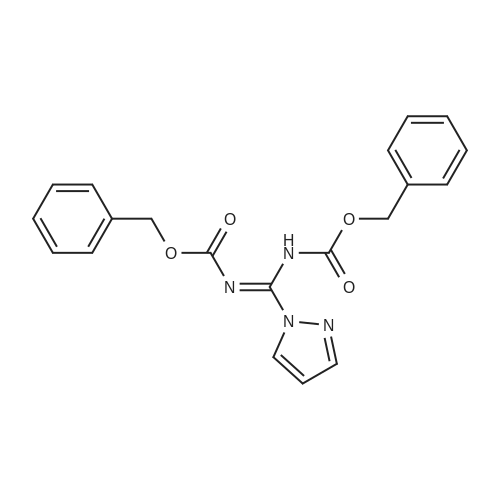
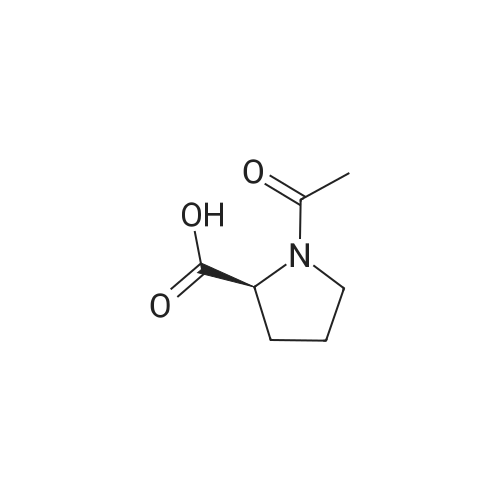
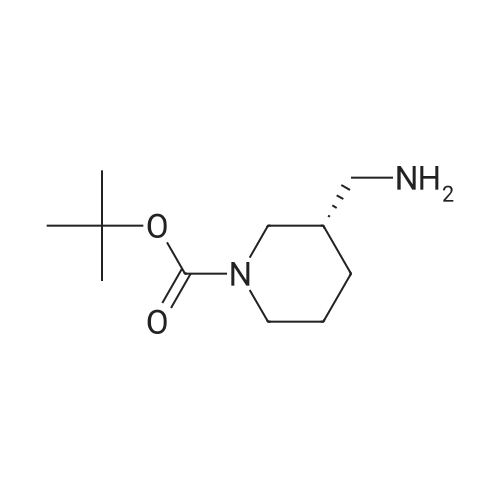
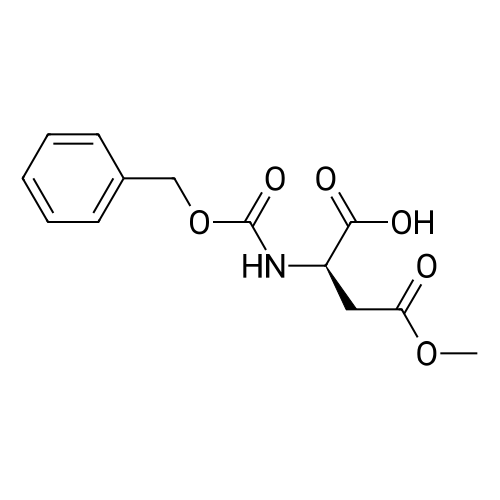
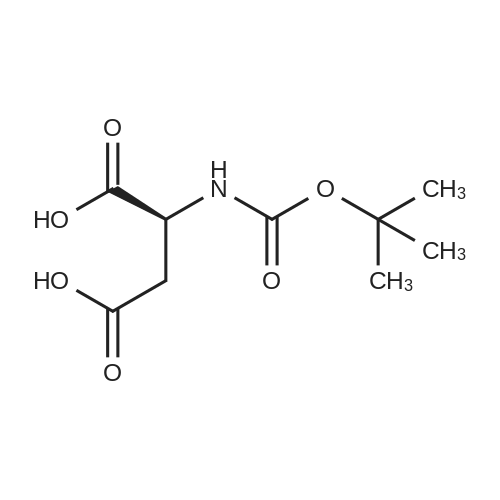
[1] Journal of Organic Chemistry, 2004, vol. 69, # 23, p. 8077 - 8085
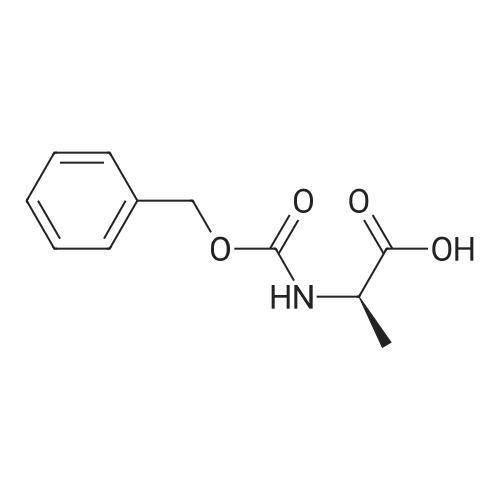
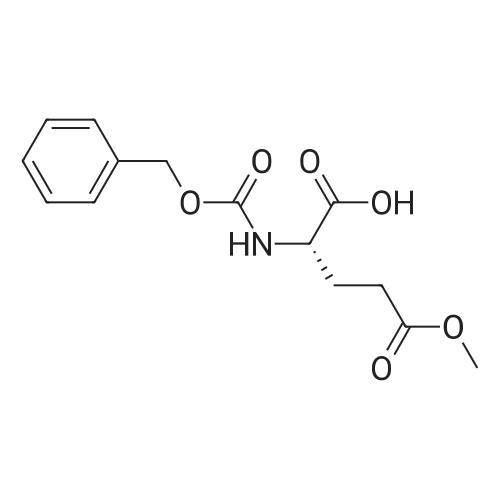
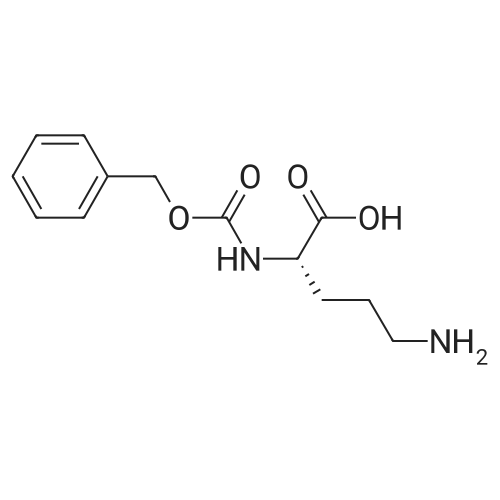
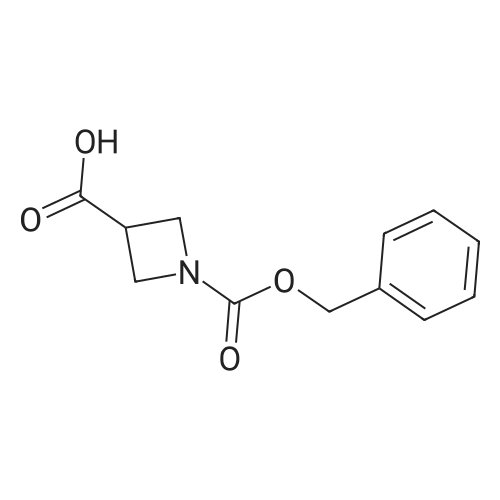
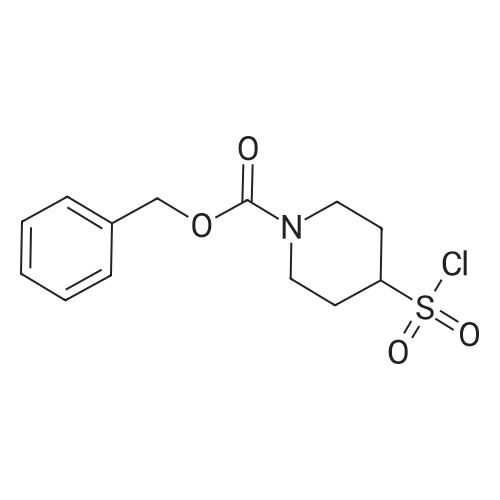
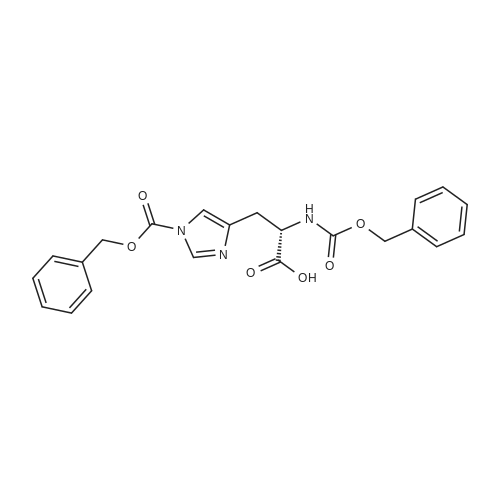
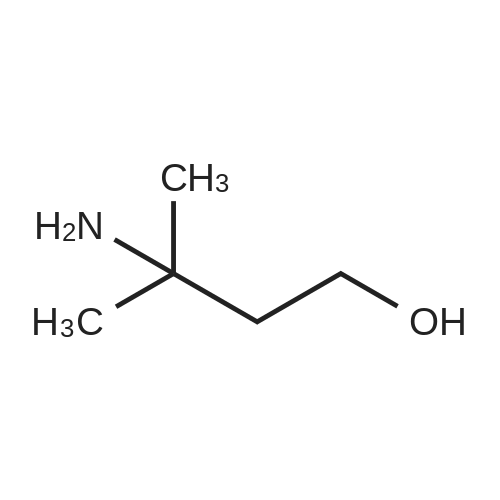
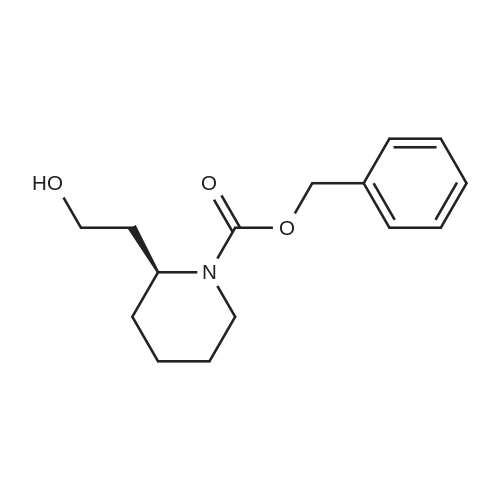
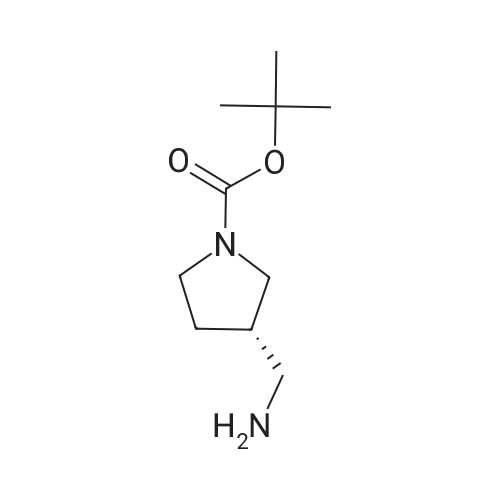
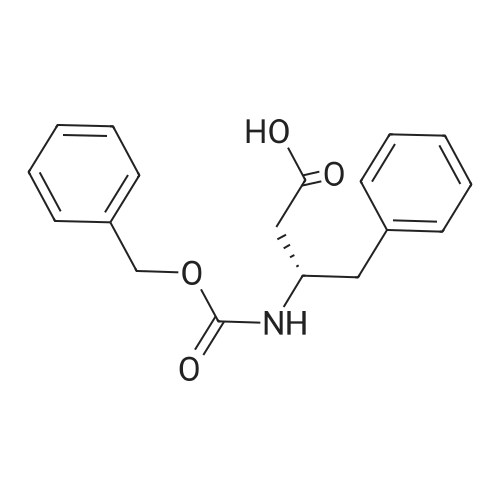
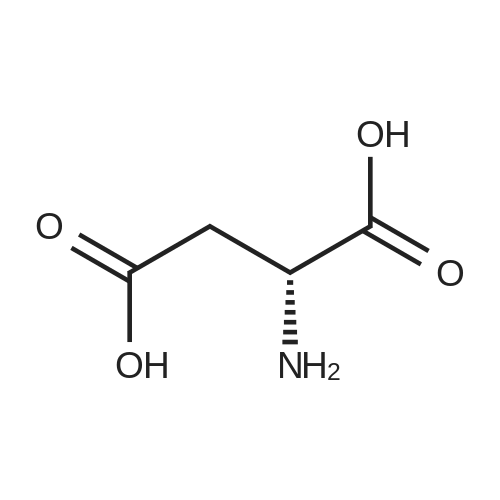
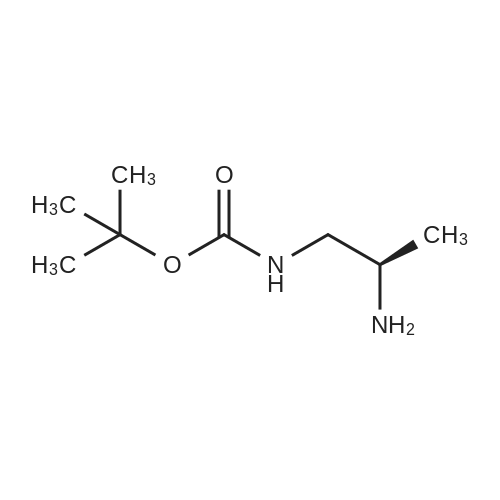
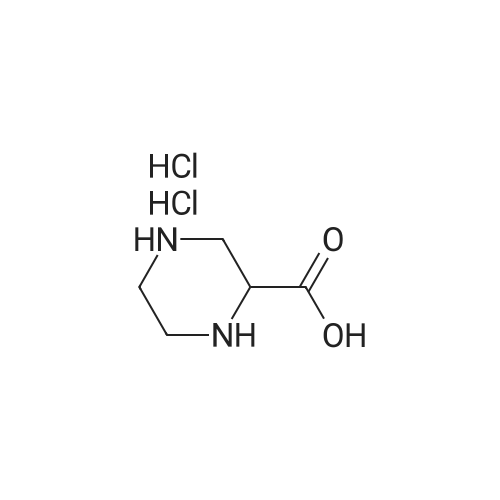
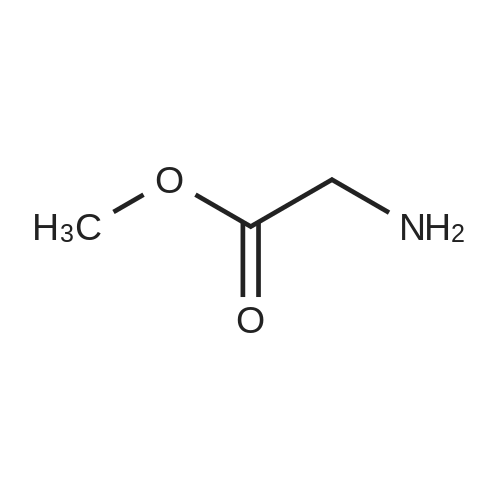
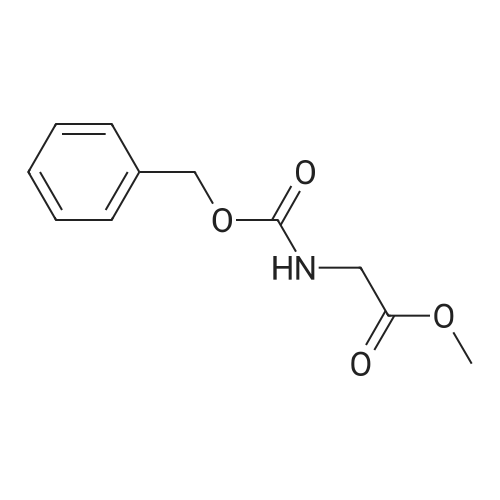
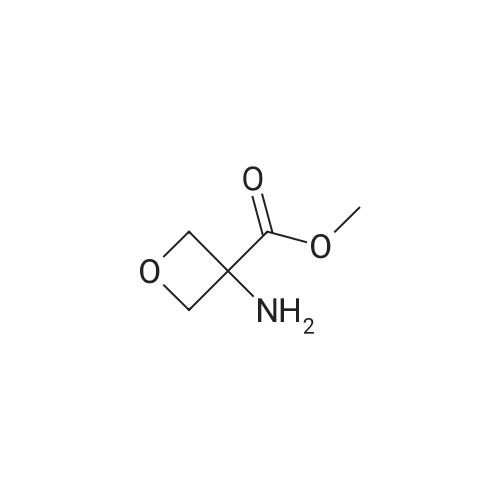
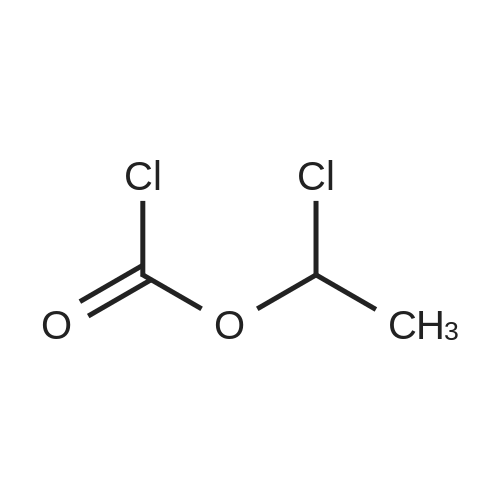
2
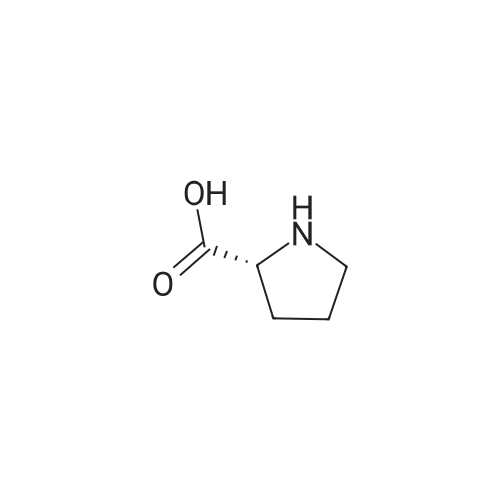
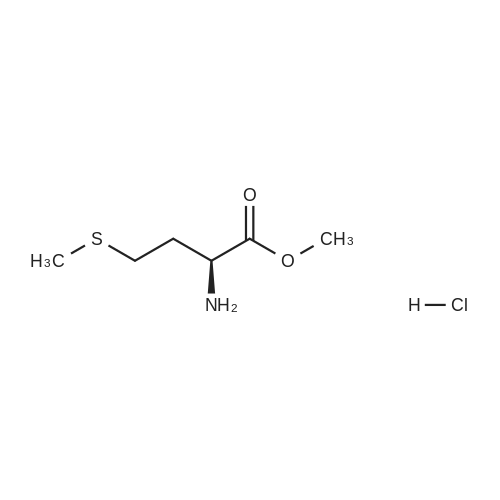
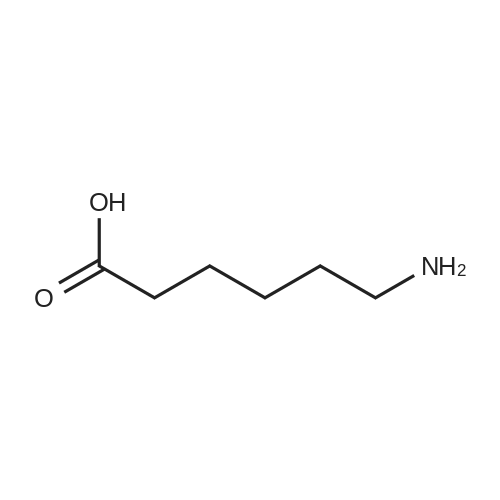
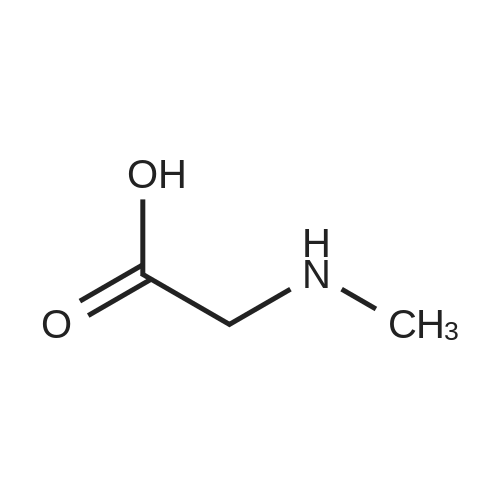
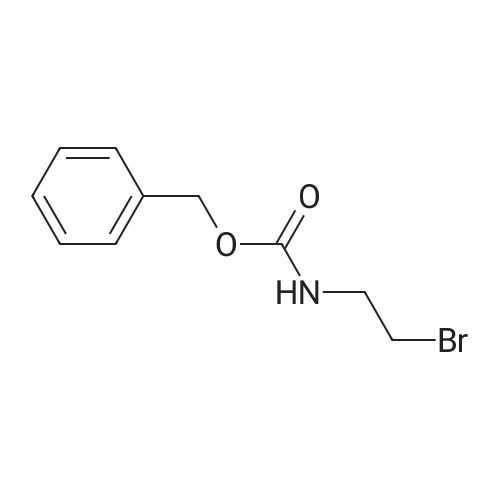
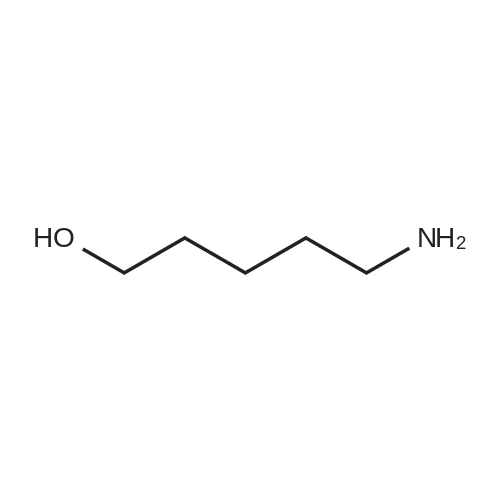
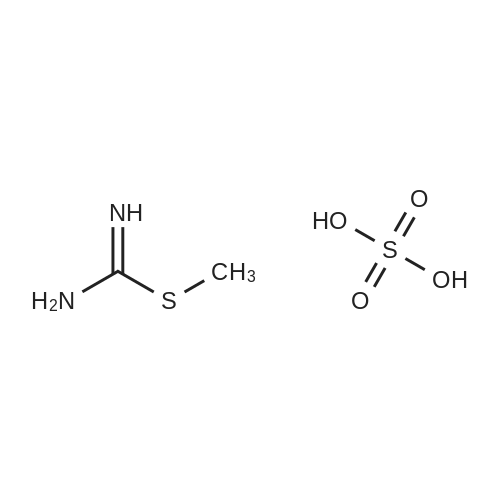
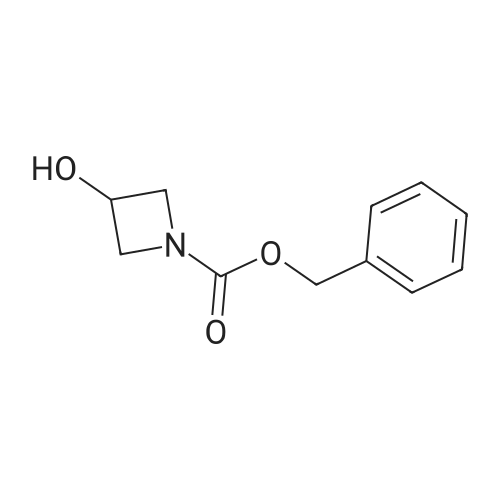
[ 13139-17-8 ]
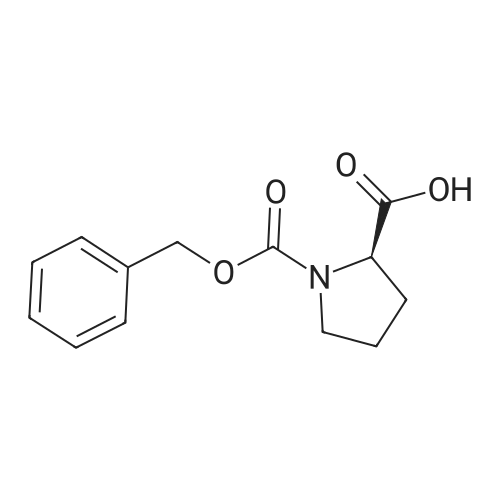
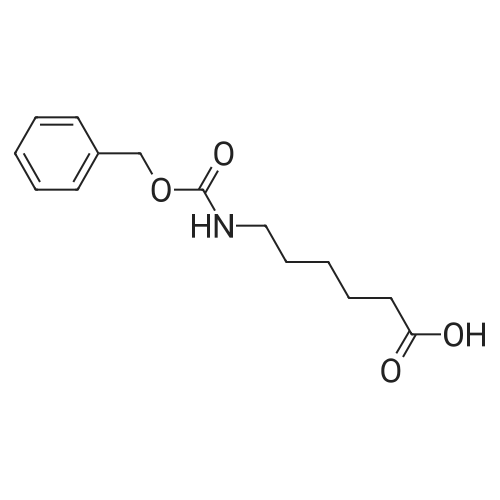
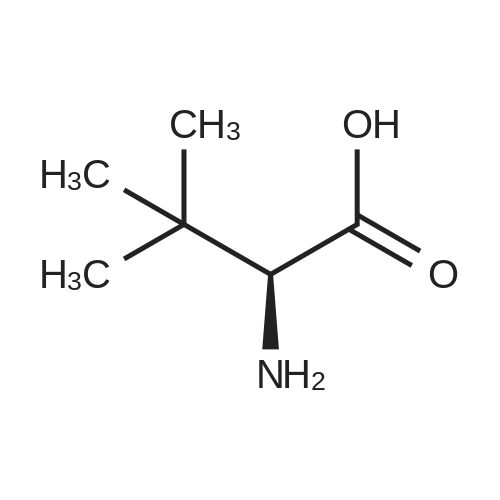
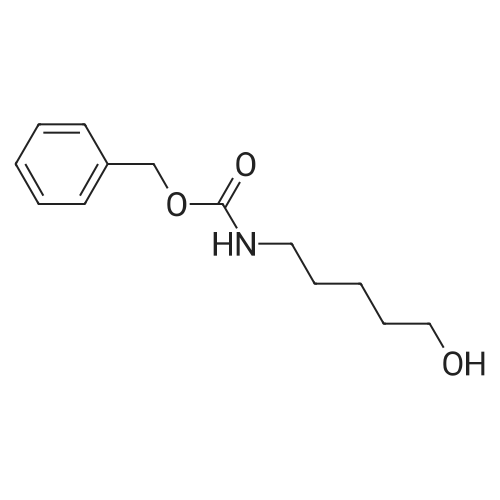
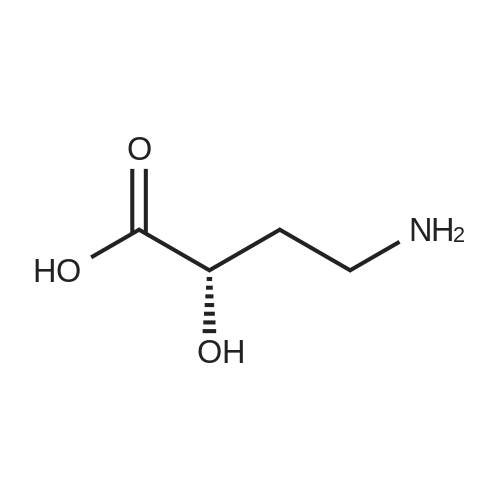
[ 73-22-3 ]
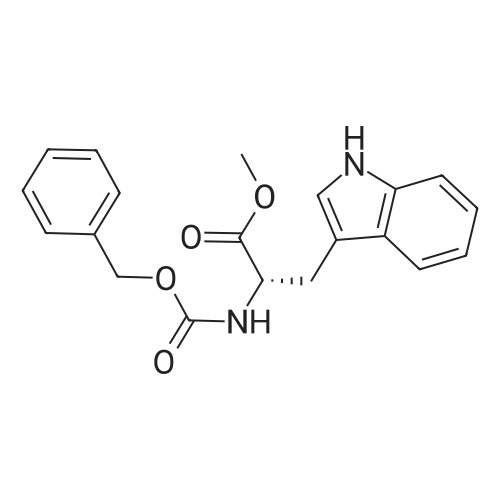
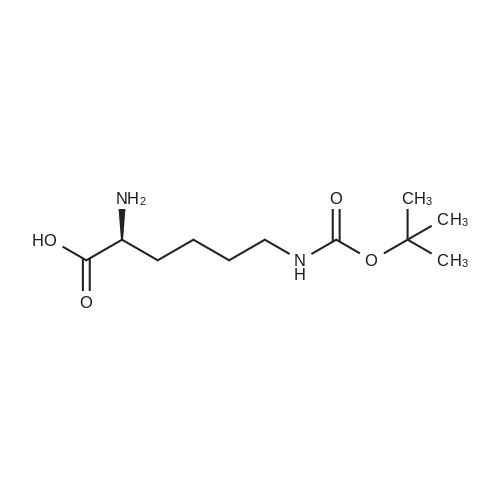
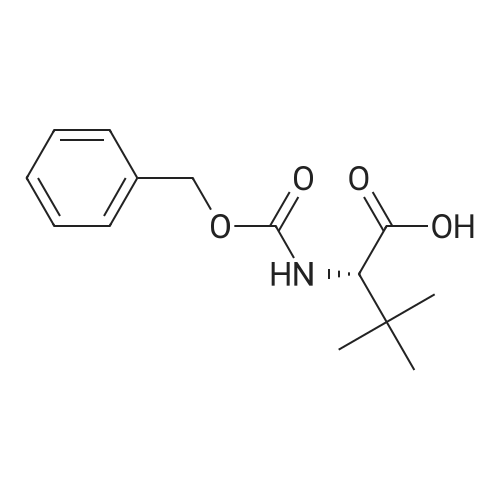
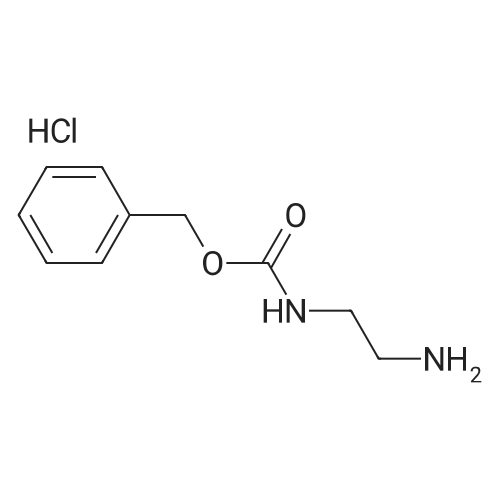
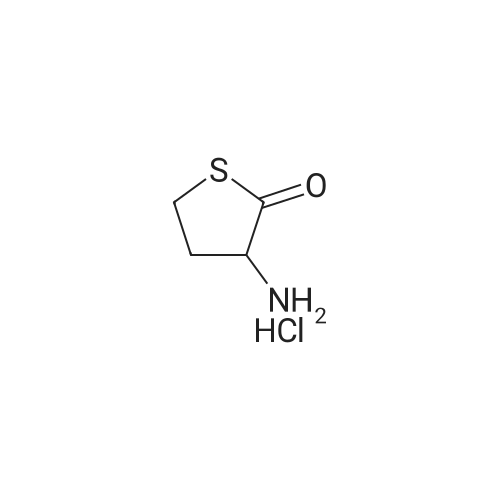
[ 7432-21-5 ]
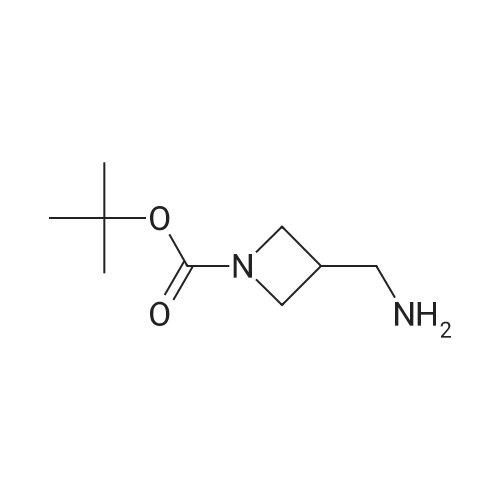
Reference:
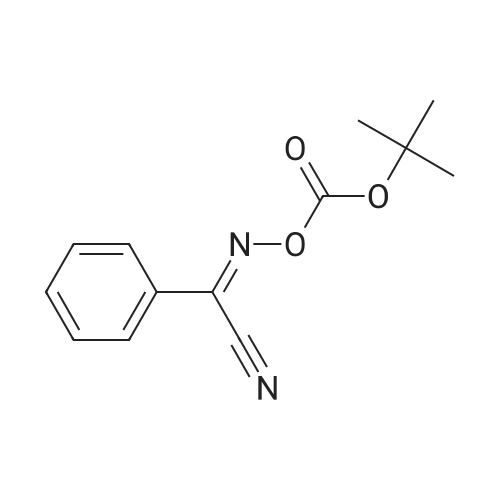
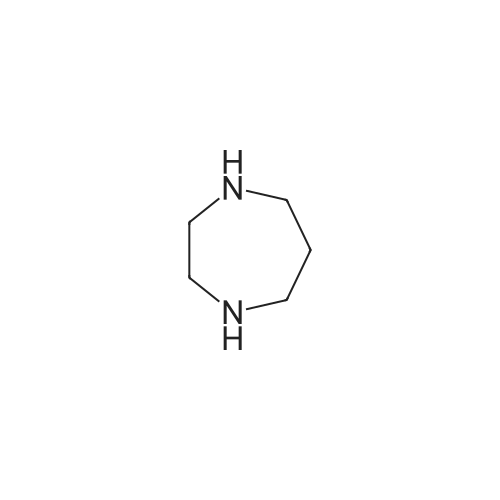
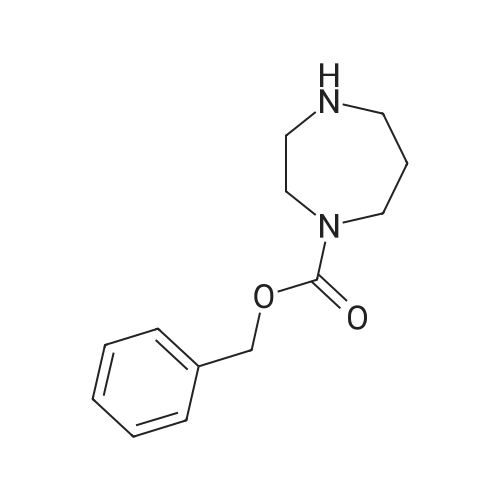
[1] Russian Journal of Bioorganic Chemistry, 1999, vol. 25, # 5, p. 283 - 287[2] Bioorganicheskaya Khimiya, 1999, vol. 25, # 5, p. 323 - 328
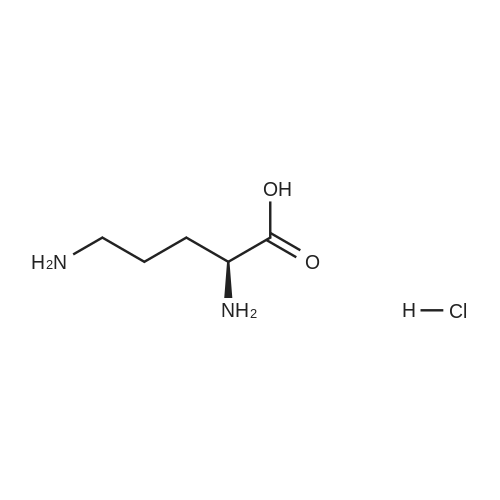
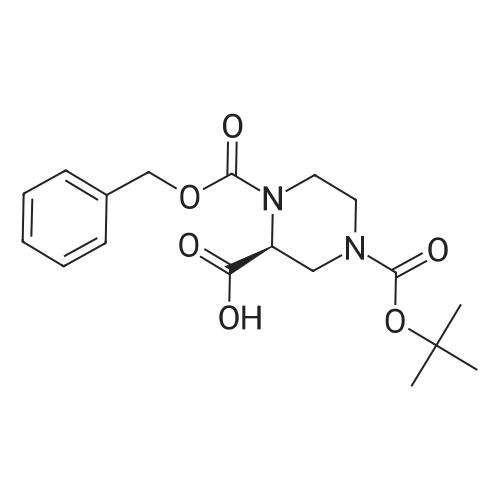
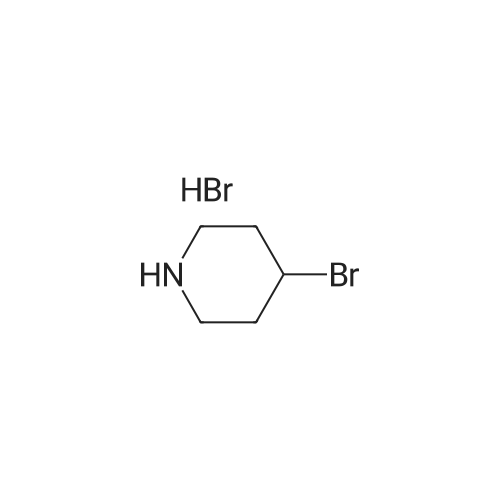
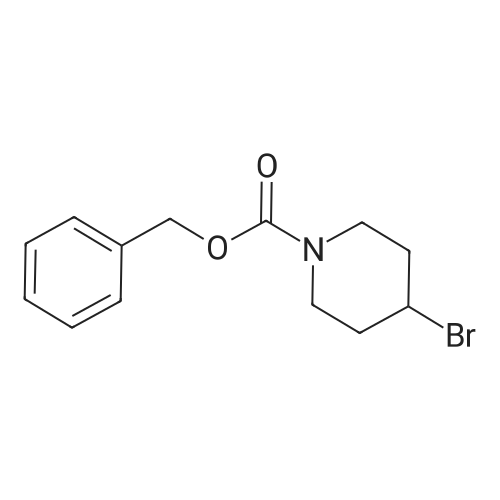
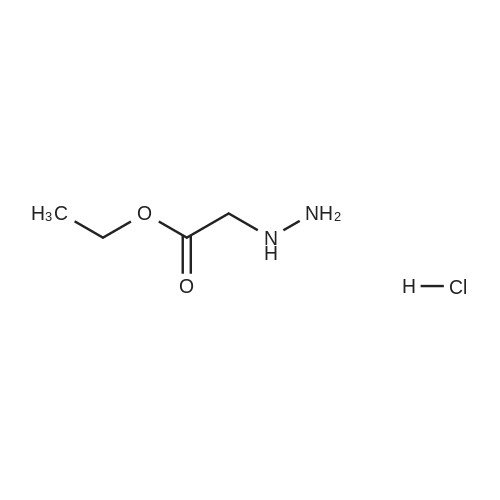
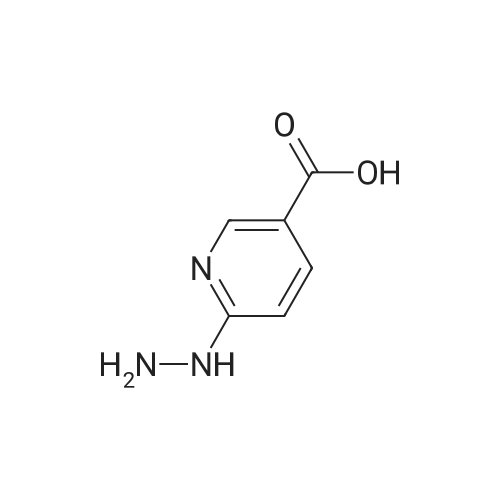
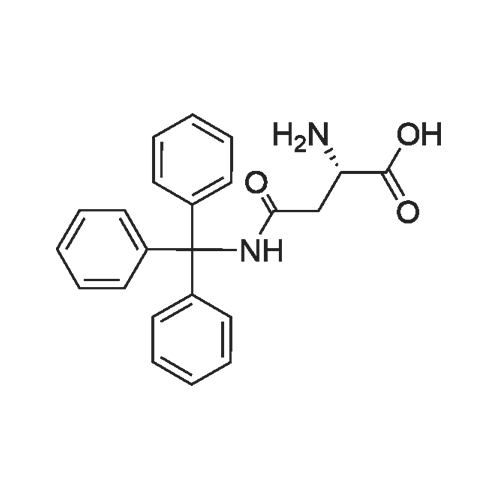
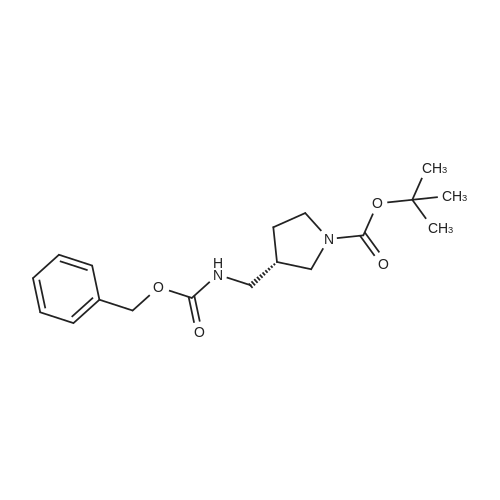
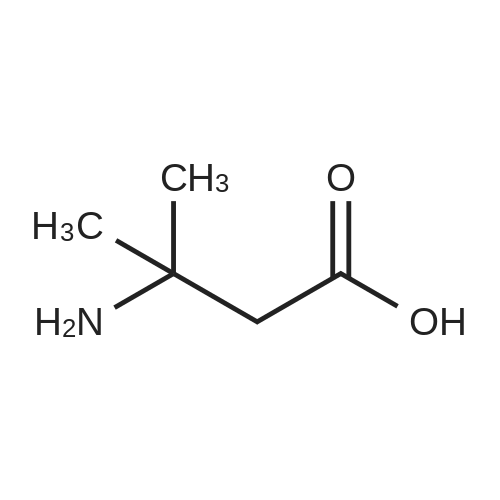
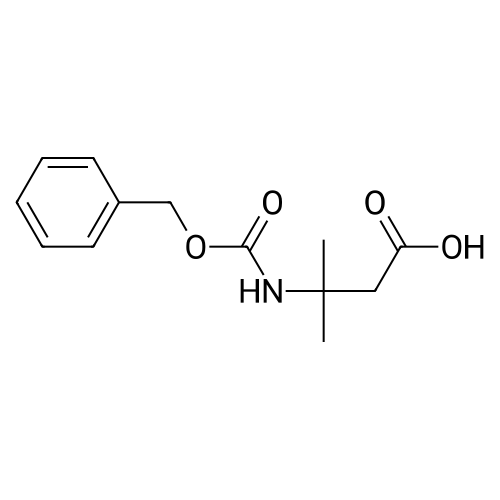
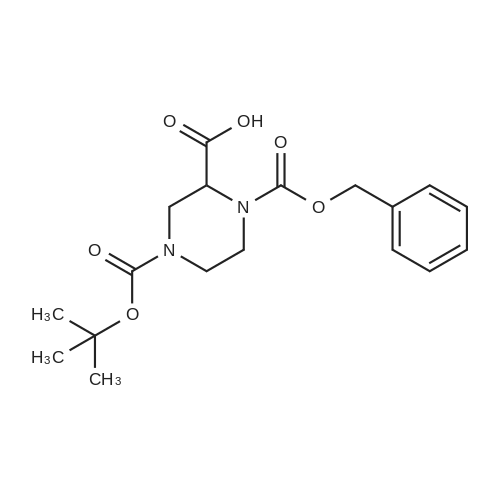
3
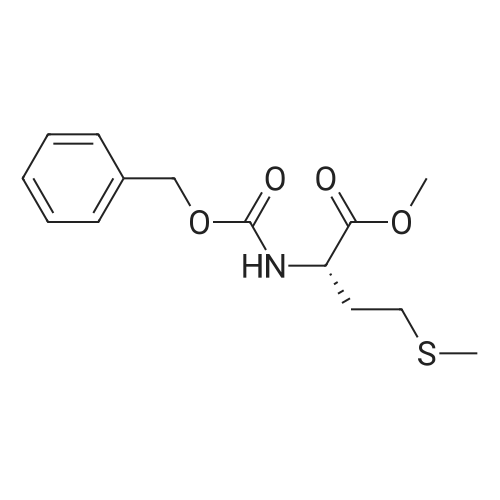
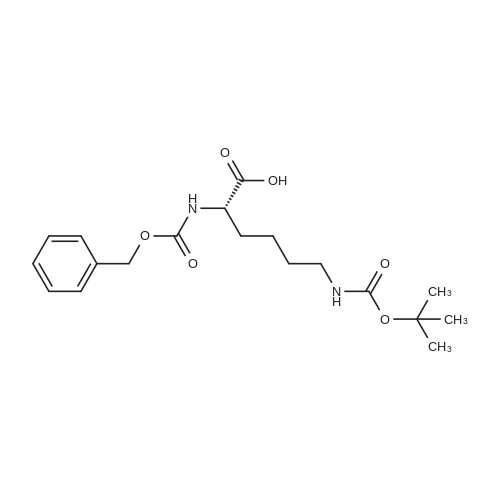
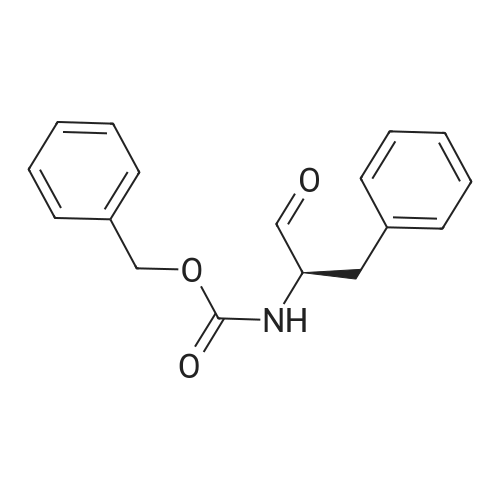
[ 498-94-2 ]
[ 13139-17-8 ]
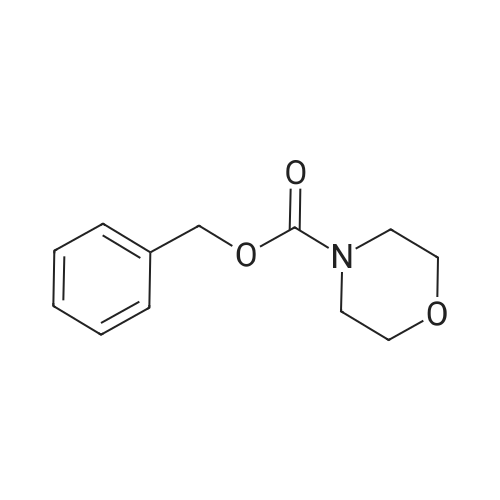
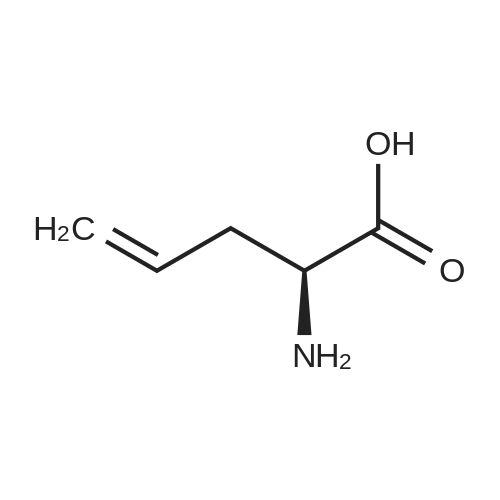
[ 10314-98-4 ]
Yield
Reaction Conditions
Operation in experiment
22%
With sodium carbonate In water; acetonitrile at 20℃; for 16 h;
To a solution of piperidine-4-carboxylic acid (10.0 g, 77.4 mmol) and Na2CO3 (8.21 g, 77.4 mmol) in water (100 mL) was added a solution of benzyl 2,5-dioxopyrrolidin-1-yl carbonate (19.3 g, 77.4 mmol) in MeCN (100 mL). The reaction was stirred at ambient temperature for about 16 h and then concentrated under reduced pressure. The resulting aqueous solution was quenched with NH4Cl and was then extracted with EtOAc (2.x.100 mL). The combined organic extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure to give 1-(benzyloxycarbonyl)piperidine-4-carboxylic acid as a white solid (4.56 g, 22percent): LC/MS (Table 2, Method a) Rt=1.93 min; MS m/z: 262 (M-H)-.
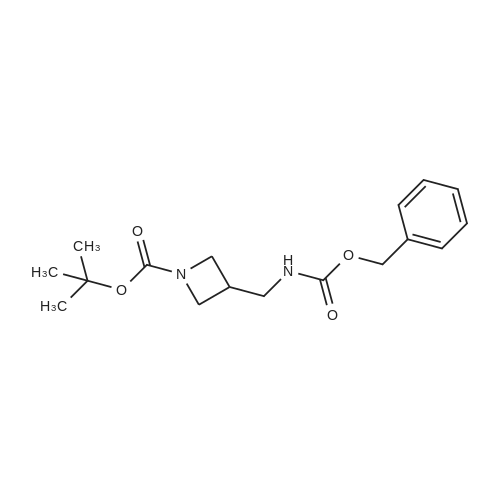
Reference:
[1] Bulletin des Societes Chimiques Belges, 1987, vol. 96, # 10, p. 775 - 782
[2] Bulletin of the Chemical Society of Japan, 1987, vol. 60, # 7, p. 2409 - 2418
[3] Chemical and Pharmaceutical Bulletin, 1999, vol. 47, # 10, p. 1489 - 1490
[4] Organic Preparations and Procedures International, 2002, vol. 34, # 5, p. 531 - 537
[5] Patent: US3974137, 1976, A,
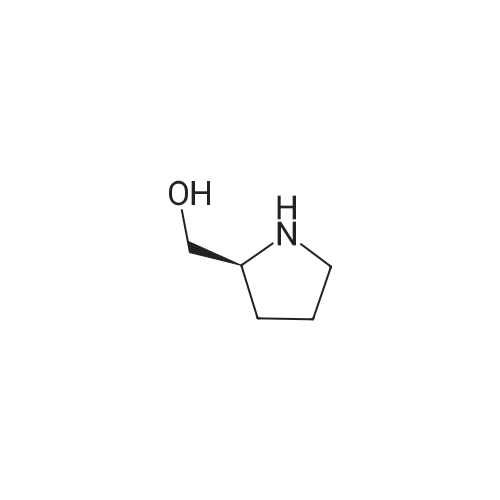
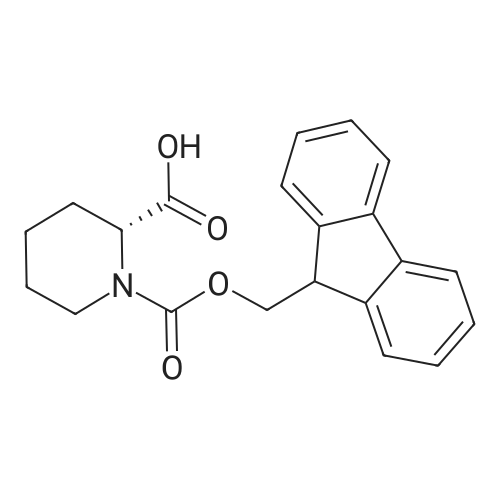
10
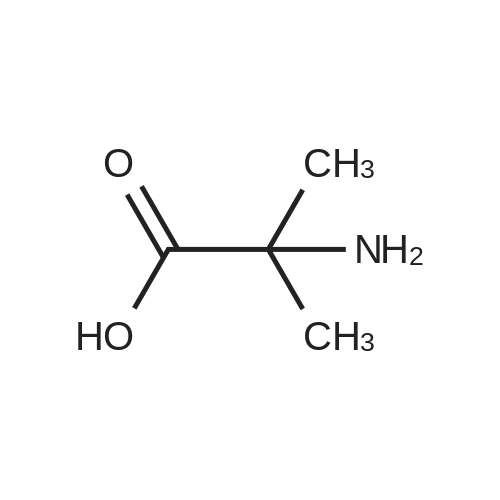
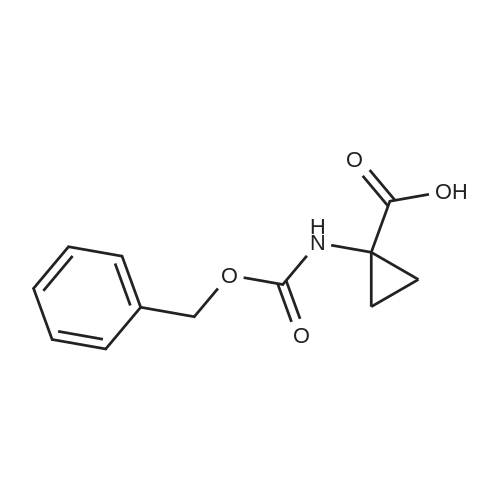
[ 74124-79-1 ]
[ 100-51-6 ]
[ 13139-17-8 ]
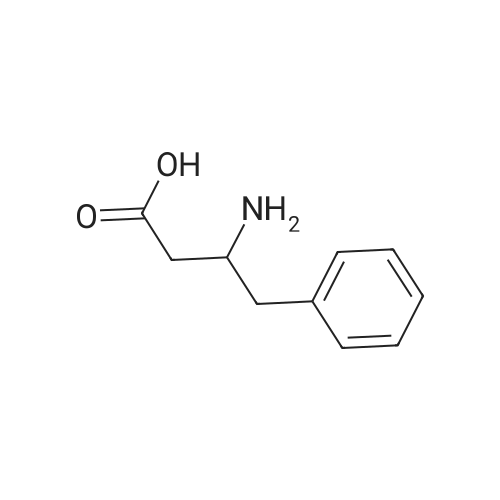
Reference:
[1] European Journal of Medicinal Chemistry, 1999, vol. 34, # 7-8, p. 625 - 638
[2] Patent: WO2008/64218, 2008, A2, . Location in patent: Page/Page column 72; 83-85; 86-87; 98-99; 125
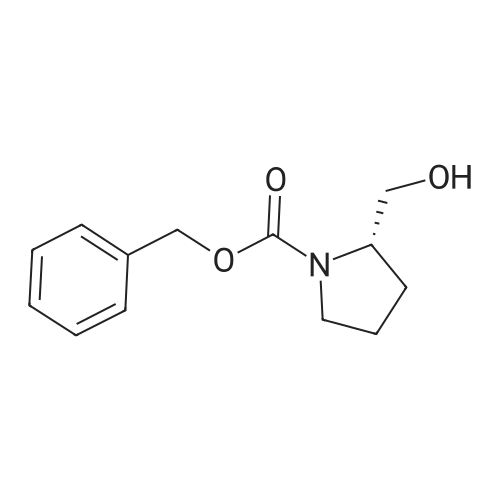
11
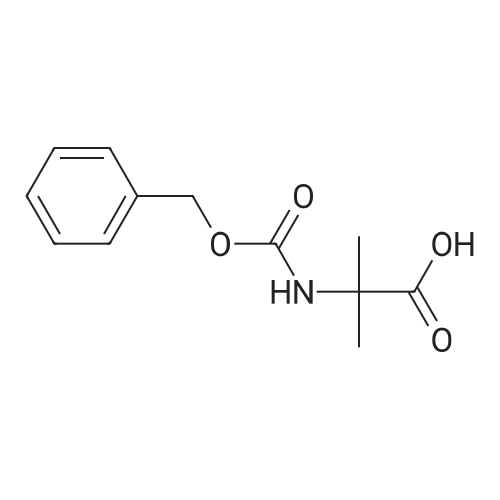
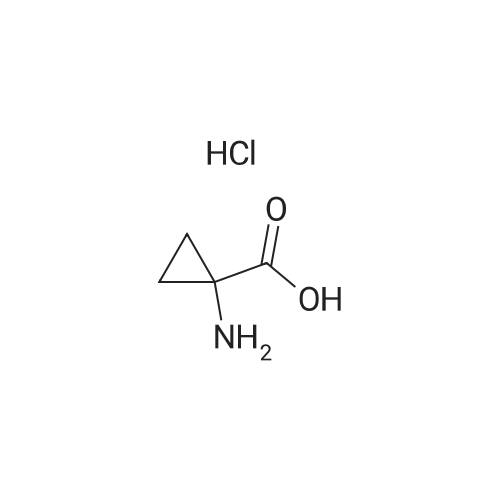
[ 82911-72-6 ]
[ 501-53-1 ]
[ 13139-17-8 ]
Reference:
[1] Canadian Journal of Chemistry, 1982, vol. 60, p. 976 - 980
12
[ 5382-16-1 ]
[ 13139-17-8 ]
[ 95798-23-5 ]
Reference:
[1] Tetrahedron, 1998, vol. 54, # 46, p. 13981 - 13996
[2] Organic and Biomolecular Chemistry, 2012, vol. 10, # 2, p. 293 - 304
[3] Tetrahedron, 2000, vol. 56, # 43, p. 8433 - 8441
13
[ 694-05-3 ]
[ 13139-17-8 ]
[ 66207-23-6 ]
Yield
Reaction Conditions
Operation in experiment
100%
With triethylamine In dichloromethane at 20℃;
To a solution of 1,2,3,6-tetrabydropyridine (1.66 g, 20.0 mmol) in dry CH2Cl2 (10 mL) was added triethyl amine (3.35 mL, 24.0 mmol), followed by a solution of N-(benzyloxycarbonyloxy)succinimide (5.23 g, 21.0 mmol) in dry CH2Cl2 (10 mL). The reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with CH2Cl2 (50 mL) and washed with 10percent citric acid, sat'd NaHCO3, brine and dried over anhydrous Na2SO4. Concentration under reduced pressure afforded 4.34 g of Compound 135A: (100percent) as an oil. Analytical HPLC retention time = 2.996 min. (Chromolith SpeedROD column 4.6 x 50 mm, 10-90percent aqueous methanol containing 0.1percent TFA over 4 minutes, 4 mL/min, monitoring at 254 nm). 1H-NNM (CDCl3): 7.20-7.35 (m, 5H), 5.88 (bs, 1H), 5.60-5.78 (m, 1H), 5.18 (s, 2H), 3.99 (t, J = 2.64, 2H), 3.59 (t, J = 5.69, 2H), 2.18 (m, 2H).
With sodium hydrogencarbonate In tetrahydrofuran; water at 0 - 30℃; for 16 h;
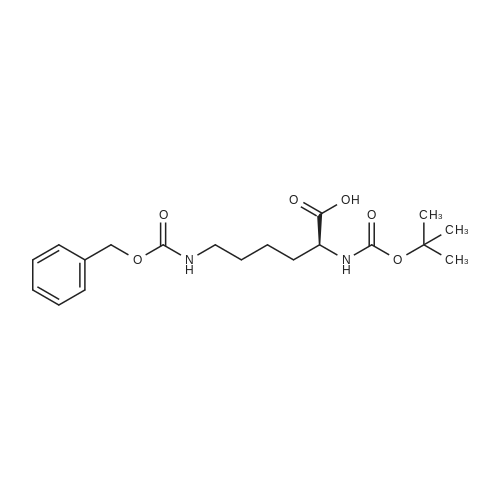
(2S)-6-(benzyloxycarbonylamino)-2-(tert-butoxycarbonylamino)hexanoic acid (ii) To a solution of (2S)-6-amino-2-(tert-butoxycarbonylamino)hexanoic acid (100.00 g, 406.01 mmol, 1.00 eq), NaHCO3 (102.33 g, 1.22 mol, 3.00 eq) in THF (1000 mL) and H2O (1000 mL) was added CbzOSu (101.19 g, 406.01 mmol, 1.00 eq) at 0° C. Then the mixture was stirred at 30° C. for 16 hr. The mixture was adjusted to pH=5-6 with 1N HCl, extracted with EA (600 mL*3), dried over Na2SO4, concentrated to give (2S)-6-(benzyloxycarbonylamino)-2-(tert-butoxycarbonylamino)hexanoic acid (142.00 g, 373.26 mmol, 91.9percent yield) as a yellow oil.
With triethylamine In dichloromethane at 20℃; for 1 h; Inert atmosphere
00568] Compound 1001 - Starting compound 1000 (10 g, 85.38 mmol) was added to the reaction flask, evacuated and purged with argon. The starting material was dissolved in dichloromethane and triethylamine (23.79 rriL, 170.7 mmol) was added via syringe. N- (Benzyloxycarbonyloxy)succinimide (31.9 g, 128 mmol) was dissolved in anhydrous dichloromethane and then added to the reaction mixture via syringe. The reaction was stirred at room temperature for 1 hour. The reaction was checked by TLC (50percent EtOAc/hexanes) and the reaction was concentrated under reduced pressure. The residue was dissolved in dichloromethane, added to separation funnel and organic layer was washed with 10percent citric acid solution. The organic layer was separated and washed with a brine solution. The organic layer was separated and dried with sodium sulfate. The solid was filtered off and the mother liquor was concentrated. The residue was purified by flash chromatography on silica gel (10percent to 100percent EtOAc/hexanes) and the product fractions combined and concentrated on reduced pressure to yield 17.9g, (83percent) of 1001. NMR (400 MHz, DMSO-de): δ 7.40 - 7.25 (m, 4H), 7.21 (t, J = 5.7 Hz, 1H), 4.99 (s, 2H), 4.31 (t, J = 5.1 Hz, 1H), 3.36 (m, 2H), 2.96 (q, J = 6.7 Hz, 2H), 1.38 (m, 4H), 1.32 - 1.17 (m, J = 5.2, 4.6 Hz, 4H).
Reference:
[1] Patent: WO2018/136620, 2018, A2, . Location in patent: Paragraph 00568
[2] Patent: US4550163, 1985, A,
22
[ 13139-17-8 ]
[ 2491-18-1 ]
[ 56762-93-7 ]
Reference:
[1] Organic and Biomolecular Chemistry, 2017, vol. 15, # 16, p. 3507 - 3518
23
[ 13139-17-8 ]
[ 62-57-7 ]
[ 15030-72-5 ]
Yield
Reaction Conditions
Operation in experiment
97.8%
With sodium carbonate In tetrahydrofuran; water at 20℃; for 16 h;
Step 1. Cbz-OSu (57 g, 0.23 mol) in THF (150 mL) was added a mixture of aq Na2C03 (82 g, 0.78 mol) and 80-1 (20 g, 0.19 mol). After it was stirred at r.t. for 16 h, it was adjusted to pH > 10 and washed with EtOAc (400 mL x 2). The aqueous layer was pooled and acidified to pH < 1 with cone. HC1. It was extracted with EtOAc (500 mL x 2). The organic layer was dried over Na2S04 and evaporated to give 80-2 (45 g, 97.8percent).
72%
Stage #1: With sodium hydrogencarbonate In tetrahydrofuran; water at 0 - 20℃; for 12 h; Stage #2: at 0℃;
Synthesis of Compound S.2. To S.1 (10 g, 0.0969 mol) in THF (60 ml) and water (60 mL) at 0° C. was added sodium bicarbonate (16.27 g, 0.193 mole) followed by N-(benzyloxy carbonyloxy) succinimide (60.37 g, 0.242 mol). The reaction mixture was stirred at RT for 12 hr. The THF was removed under vacuum and the aqueous phase was washed with ether (2.x.100 mL). The aqueous phase was cooled to 0° C. and acidified to pH=2 with 5N HCL (50 mL). The reaction mixture was extracted with ethyl acetate (2.x.100 mL); the combined organic layer was dried over sodium sulfate and concentrated under reduced pressure. The crude material was purified by column chromatography (1percent MeOH in dichloromethane) to give S.2 (16 g, 72percent). 1H NMR (CDCl3, 200 MHz) δ 7.45-7.32 (m, 5H), 5.40 (bs, 1H,) 5.12 (s, 2H), 1.82 (s, 6H); MS: m/z 238 [M+1]+.
72%
Stage #1: With sodium hydrogencarbonate In tetrahydrofuran; water at 0 - 20℃; for 12 h; Stage #2: With hydrogenchloride In water
To S.1 (10 g, 0.0969 mol) in THF (60 ml) and water (60 mL) at 0° C. was added sodium bicarbonate (16.27 g, 0.193 mole) followed by N-(benzyloxy carbonyloxy) succinimide (60.37 g, 0.242 mol). The reaction mixture was stirred at RT for 12 hr. The THF was removed under vacuum and the aqueous phase was washed with ether (2.x.100 mL). The aqueous phase was cooled to 0° C. and acidified to pH=2 with 5N HCL (50 mL). The reaction mixture was extracted with ethyl acetate (2.x.100 mL); the combined organic layer was dried over sodium sulfate and concentrated under reduced pressure. The crude material was purified by column chromatography (1percent MeOH in dichloromethane) to give S.2 (16 g, 72percent). 1H NMR (CDCl3, 200 MHz) δ 7.45-7.32 (m, 5H), 5.40 (bs, 1H,) 5.12 (s, 2H), 1.82 (s, 6H); LCMS: m/z 238 [M+1]+.
Reference:
[1] Patent: WO2015/95227, 2015, A2, . Location in patent: Page/Page column 215
[2] Patent: US2009/5359, 2009, A1, . Location in patent: Page/Page column 48-49
[3] Patent: US2009/36419, 2009, A1, . Location in patent: Page/Page column 93
[4] Chemical Communications, 2013, vol. 49, # 86, p. 10133 - 10135
[5] Journal of Organic Chemistry, 2004, vol. 69, # 11, p. 3849 - 3856
24
[ 13139-17-8 ]
[ 60-32-2 ]
[ 1947-00-8 ]
Reference:
[1] Patent: US4550163, 1985, A,
25
[ 13139-17-8 ]
[ 2418-95-3 ]
[ 2389-60-8 ]
Reference:
[1] Chemical and Pharmaceutical Bulletin, 1999, vol. 47, # 10, p. 1489 - 1490
26
[ 13139-17-8 ]
[ 107-97-1 ]
[ 39608-31-6 ]
Reference:
[1] Journal of Medicinal Chemistry, 2012, vol. 55, # 5, p. 2212 - 2226
[2] Journal of Inorganic Biochemistry, 2012, vol. 117, p. 248 - 260
27
[ 13139-17-8 ]
[ 20859-02-3 ]
[ 62965-10-0 ]
Yield
Reaction Conditions
Operation in experiment
100%
With triethylamine In methanol at 20℃; for 14 h;
Step A: 2S-Benzyloxycarbonylamino-3,3-dimethyl-butyric acid; To a suspension of L-tert-leucine (11.88 g, 90.7 mmol) in methanol (200 ml) were added triethylamine (26.56 ml, 190 mmol) and N-(benzyloxycarbonyl- oxy)-succinimide (24.88 g, 99.8 mmol). The reaction mixture was stirred at room temperature for 14 h. Methanol was removed in vacuo to afford a viscous pale yellow oil, which was dissolved in ethyl acetate (100 ml). The organic layer was washed with 1M hydrochloric acid (15 ml) and brine, dried over anhydrous magnesium sulfate and filtered. The solvent was removed in vacuo to fumish the title compound as an oil (24 g, quant.). 1H-NMR; δ(CDCl3), 7.43-7.36 (5H, m), 5.36 (1H, d, J = 9.4 Hz), 5.12 (2H, br s), 4.20 (1H, d, J = 9.6 Hz) and 1.02 (9H, s). LRMS: +ve ion 266 [M+H], -ve ion 264 [M-H], 529 [2M-H].
To a suspension of L-2-amino-4-pentenoic acid (2.0 g) in Dioxane (80 mL) at RT was added triethylamine (4. 3 g), water (2.0 mL), and N- (BENZYLOXYCARBONYLOXY) succinimide (9.1 g). The reaction mixture was stirred overnight at RT, concentrated in vacuo, diluted with saturated NaHCO3 and extracted with ether (3x). The basic aqueous layer was acidified to pH 2 with 2N HCl and extracted with EtOAc (3x). The combined EtOAc layer was washed with brine, dried (NA2S04), filtered and evaporated, then concentrated from Toluene (2x) to give a viscous oil (4. 51 g, QUANTITATIVE). LH NMR (300 MHz, DMSO-d6) 6 2.31-2. 50 (M, 2H), 4. 00-4. 06 (m, 1H), 5.03 (s, 2H), 5.05-5. 12 (m, 2H), 5.71-5. 84 (m, 1H), 7.35 (M, 5H), 7.53 (d, 1H), 12.57 (bs, 1H). MS APCI, m/z = 250 (M+1). LC/MS 2.30 min. , Method A.
95%
Stage #1: With sodium hydroxide In water; acetone Stage #2: With sodium hydrogencarbonate In water; acetone for 20 h;
To a stirred solution of NaOH (700mg, 17.4mmol) in water (40mL) was added l-allyl (glycine)–OH (2.00g, 17.4mmol). N-(Benzyloxycarbonyloxy)-succinimide (4.30g, 17.4mmol) in acetone (25mL) was added in one portion with vigorous stirring. After the addition was complete, NaHCO3 (1.50g, 17.4mmol) was added and stirring was continued for 20h. The reaction mixture was extracted once with Et2O (50mL) to remove any excess neutral organic compounds and the aqueous layer was acidified to pH 2 with 5M aq HCl. The aqueous layer was extracted with EtOAc (3×25mL) and dried (MgSO4) and evaporated in vacuo to give 14 as a brown oil (4.10g, 95percent yield), with spectra identical to those of an authentic sample.
Reference:
[1] Patent: WO2004/80983, 2004, A1, . Location in patent: Page 47
[2] Tetrahedron, 2013, vol. 69, # 30, p. 6275 - 6284
[3] Organic Letters, 2006, vol. 8, # 2, p. 239 - 242
[4] Patent: WO2011/15241, 2011, A1, . Location in patent: Page/Page column 187; 398
[5] Patent: US2012/270881, 2012, A1, . Location in patent: Page/Page column 116
[6] Patent: CN105294732, 2016, A, . Location in patent: Paragraph 0782; 1408
[7] Patent: JP2015/155430, 2015, A, . Location in patent: Paragraph 0106; 0111
29
[ 13139-17-8 ]
[ 107-15-3 ]
[ 72080-83-2 ]
Yield
Reaction Conditions
Operation in experiment
80%
for 1 h;
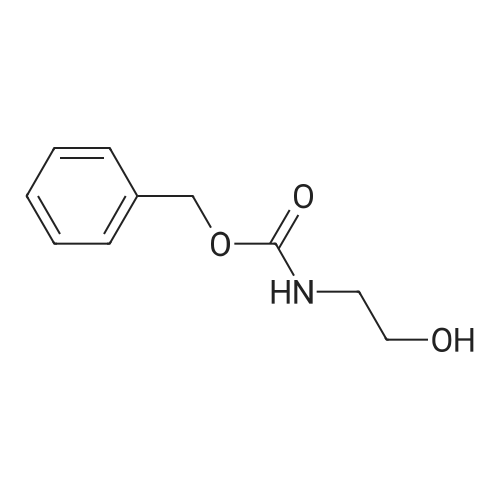
Step 1 To a solution of 2-aminoethylamine LVIII (150 mL; 2.25 mol) in chloroform (1.5 L), cooled to 0° C. was added a solution of carbobenzoxy N-hydroxysuccinimide (112.14 g; 0.45 mol) in water (0.5 L) with vigorously stirring for one hour. The solid was filtered and the solution was washed with brine (3*), once with water and dried over anhydrous MgSO4. The solvent was evaporated under reduced pressure and the residue was purified on a silica gel column (100:1-->30:1 DCM:MeOH) to yield benzyl 2-aminoethylcarbamate LIX as a colorless viscous oil (350 g; 1.80 mol; 80percent yield). 1H NMR (CDCl3) 2.79-2.83 (m, 2H), 3.21-3.28 (m, 2H), 5.10 (s, 2H), 5.19 (brs, 1H), 7.29-7.34 (m, 1H), 7.35-7.38 (m, 4H); ESIMS found for C10H14N2O2 m/z 195.2 (M+H).
Reference:
[1] Patent: US2008/318957, 2008, A1, . Location in patent: Page/Page column 57; 58
[2] Chemical Communications, 2011, vol. 47, # 33, p. 9330 - 9332
[3] Organic Letters, 2012, vol. 14, # 17, p. 4450 - 4453
[4] Patent: WO2016/102562, 2016, A1, . Location in patent: Page/Page column 52
30
[ 13139-17-8 ]
[ 3417-91-2 ]
[ 13512-31-7 ]
Reference:
[1] Journal of Medicinal Chemistry, 2004, vol. 47, # 16, p. 4041 - 4053
[2] Bioorganic and Medicinal Chemistry, 2003, vol. 11, # 24, p. 5449 - 5460
31
[ 13139-17-8 ]
[ 1080-06-4 ]
[ 13512-31-7 ]
Reference:
[1] Synthesis, 2009, # 22, p. 3765 - 3768
32
[ 13139-17-8 ]
[ 27489-62-9 ]
[ 27489-63-0 ]
Yield
Reaction Conditions
Operation in experiment
90%
Stage #1: at 0 - 20℃; for 24 h; Stage #2: With hydrogenchloride In water
To a stirred solution of trans-4-aminocyclohexanol (35.5 g, 306.6 mmol) in methanol (621 mL) at 0° C. under nitrogen was added N-(benzyloxycarbonyloxy)succinimide (72.8 g, 291.9 mmol). The reaction mixture was slowly warmed to room temperature and after 1 d, poured into 0.25 M aqueous hydrochloric acid solution (2.13 L). The resulting slurry was stirred vigorously for 1 h and then filtered. The filtered solid was washed with water to obtain the title compound (65.65 g, 90percent) as a white solid. 13C NMR (400 MHz, (CD3)2SO) δ 155.3, 137.2, 128.3, 127.7, 127.6, 68.1, 65.0, 49.1, 33.9, 30.3. 1H NMR (400 MHz, (CD3)2SO) δ 7.29 (m, 5H), 7.10 (d, J=7.9 Hz, 1H), 4.96 (d, J=2.3 Hz, 2H), 4.76 (d, J=4.0 Hz, 1H), 3.29 (s, 1H), 3.17 (br s, 1H), 1.71 (m, 4H), 1.22 (t, J=9.7 Hz, 4H).
Reference:
[1] Bioorganic and Medicinal Chemistry, 2003, vol. 11, # 24, p. 5449 - 5460
38
[ 13139-17-8 ]
[ 22059-21-8 ]
[ 84677-06-5 ]
Yield
Reaction Conditions
Operation in experiment
53 g
With sodium hydrogencarbonate In tetrahydrofuran; water at 20℃; for 16 h; Cooling with ice
Step A: intermediate VI A solution of benzyl (2,5-dioxopyrrolidin-1-yl) carbonate (83 g, 334 mmol) in tetrahydrofuran (THF) (500 ml) was added to a solution of 1-aminocyclopropanecarboxylic acid (25 g, 247 mmol) in water (500 ml). At ice temperature, a solution of sodium bicarbonate (91 g, 1088 mmol) in water (500 ml) was poured into the mixture. After the addition, the reaction was allowed to warm to room temperature and was stirred for 16 h. The solvent was removedunder reduced pressure. The residue was suspended in200 mL of methylene chloride and extracted with water (500 mL). The water solution was acidified with acetic acid and the precipitate was filtered yielding 1- (((benzyloxy)carbonyl)amino)cyclopropanecarboxyiic acid (53 g, 171 mmol, 69.2 percent yield) as a white solid. Used crude in next reaction. LC/MS: m/z calculated 235.1 , found 258.0 ( +Na)+.
Stage #1: With ethylenediamine In dichloromethane for 2.5 h; Stage #2: With hydrogenchloride In diethyl ether; hexane
Solid Phase Synthetic protocol: A solution of N-(benzyloxycarbonyloxy)succinimide (ZOSu, 100 g, 401 mmol) in dichloromethane (500 ml) was added dropwise over 2 hours to a solution of 5 ethylenediamine (1, 189 ml, 2.81 mol) in dichloromethane (750 ml). After 30 minutes the suspension was filtered and solids washed with dichloromethane. The filtrate was evaporated to dryness and the residue diluted with toluene (1.00 L) and water (0.50 L). The resulting mixture was filtered and the filtrate was separated to afford two phases. The aqueous phase contained the product; therefore it was extracted with 10 dichloromethane (2 x 250 ml). All organic phases were combined, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was diluted with toluene (750 ml) and extracted with 2 M aqueous hydrochloric acid (500 ml) and 1 M aqueous hydrochloric acid (100 ml). Acidic aqueous phases were combined and basified with a solution of sodium hydroxide (60.0 g, 1.50 mol) in water (90 ml). The resulting mixture 15 was extracted with dichloromethane (4 x 200 ml), dried over anhydrous sodium sulfate, filtered, concentrated in vacuo and diluted with hexanes (200 ml). 4 M Solution of hydrogen chloride in ether (100 ml, 400 mmol) was added to the solution, the resulting suspension was concentrated in vacuo and diluted with hexanes (1.00 L). The precipitated solid was filtered, washed with hexanes and dried in vacuo to give (2-amino20 ethyl)-carbamic acid benzyl ester hydrochloride as white powder. 25 Yield: 62.62 g (68percent). RF (Si02, dichloromethane/methanol 4: 1): 0.25 (free base). 1H NMR spectrum (300 MHz, Ac0D-d4, 80 °C, dH): 7.42-7.26 (m, 5 H); 5.16 (s, 2 H); 3.60 (t, J=5.7 Hz, 2 H); 3.32 (t, J=5.7 Hz, 2 H).
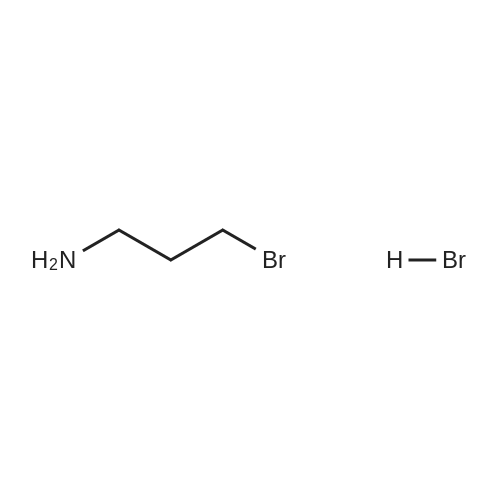
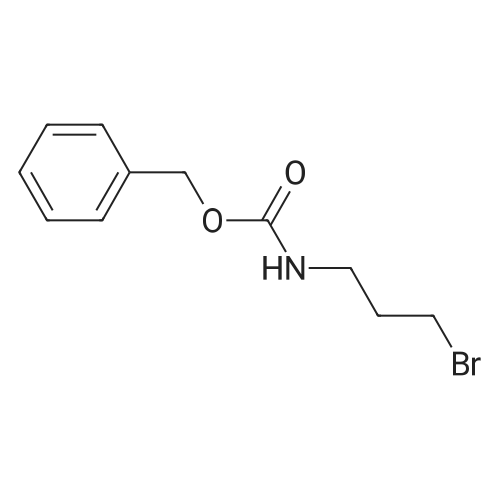
TEA ( 6.91 g, 68.5 mmol) was added dropwise to an ice-cooled mixture of 3- bromopropylamine.hydrobromide (10.Og, 45.6 mmol) and N-(Benzyloxycarbonyloxy)- succinimide (11.22g, 47.9 mmol) in DCM (200 mL). The stirred mixture was allowed to warm to room temperature overnight, then washed with water (3x), brine, dried (MgSO4), filtered and concentrated, providing 11.43 g (92percent) of N-(Benzyloxycarbonyl)- 3-bromopropylamine, as a pale yellow oil .
Reference:
[1] Journal of Medicinal Chemistry, 1997, vol. 40, # 19, p. 3100 - 3108
49
[ 13139-17-8 ]
[ 929-06-6 ]
[ 145881-74-9 ]
Yield
Reaction Conditions
Operation in experiment
40 g
With triethylamine In dichloromethane at 20℃;
To 2-(2-aminoethoxyl)ethanol (20 g) dissolved in dichloromethane (200 ml) was added stepwise triethylamine (26.5 ml) and benzyloxy(carbonyloxy)succinimide (47.47 g). The reaction mixture was stirred overnight at room temperature. The solvent was distilled off under reduced pressure and the residue was dissolved in deionized water (500 ml). The insoluble layer was separated, and the aqueous layer was mixed with 50 g of NaCl and the pH adjusted to 5.0 with 5percent H3PO4. The product was extracted with dichloromethane (100 ml, 50 ml, and 50 ml) and the extract was dried with MgSO4, filtered, and evaporated to dryness under reduced pressure. The residue was dried under vacuum overnight, then combined with the previously separated insoluble layer and dissolved in dichloromethane (300 ml). The solution was washed with 5-percent aqueous NaCl solution (3×50 ml) and dried with MgSO4. The solvent was distilled under reduced pressure, and the residue was dried overnight under vacuum, giving 40 g of the desired product.
With sodium hydrogencarbonate In tetrahydrofuran; water at 20℃; for 1 h;
Piperidin-4-ylmethanol (5.2 g, 45.1 mmol) was dissolved in 60 mE of THF and 60 mE of saturated NaHCO3 aq and then benzyl (2,5-dioxopyrrolidin- 1 -yl) carbonate (11.25 g, 45.1 mmol) added portionwise. The mixture was stirred for 1 h at RT, partitioned between AcOEt and water, washed twice with 1M HClaq and saturated NaClaq. Organic layer was dried over Na2504, evaporated under reduced pressure to afford the title compound (10.77 g, 43.2 mmol, 96percent yield). The product obtained was used in the following steps without thrther purifications. UPLC-MS: 0.80 mm, 250.2 [M+H]+, method 1.
Stage #1: With sodium hydroxide In water at 5℃; Stage #2: at 5 - 20℃; for 1 h; Stage #3: With hydrogenchloride In water
R (-) [ETHYINIPECONATE] tartaric acid salt (117 g, 382 [MMOL)] was dissolved in 2N [NAOH] (1200 ml, 2.40 mol) and cooled to 5 [°C.] [Z-OSU] (100 g, 401 [MMOL)] dissolved in 100 [ML] THF was added to the chilled reaction. The mixture was allowed to stir at 5 [°C] for 1 h. and at RT overnight. Approx. 100 ml of solvent was removed by rotary evaporation in vacuo, and the remaining solution was acidified to pH 2-3 with conc. HCI (app. 75 [ML).] The resulting crystal were isolated by filtration and dried in vacuo overnight. Yield 86 g (85 percent) 1H-NMR (dmso-d6,400 MHz) [D :] 12.5 (s broad, 1H), (7.42-7. 30 (m, 5H), 5.1 (s, 2H), 4.0 (s, [1H),] 3. [80] (s, 1H), 3.12 (m, [1H),] 2.92 (t, 1H), 2.36 (m, 1H), 1.92 (m, 1H), 1.7-1. 48 (m, 2H), 1.48-1. 30 (m, [1 H).] HPLC-MS (Method B): [MILZ =] 264 (M+1), Rt = 3.30 min.
Stage #1: With sodium hydrogencarbonate In tetrahydrofuran; water at 20℃; Stage #2: With hydrogenchloride In water
Preparation of Intermediate 1 (Int. l); (Z)-tert-Butyl 1 -(4-(N'-hydroxycarbamimidoyl)-benzyl)azetidine-3 -carboxylate; Int.1-A. l-(Benzyloxycarbonyl)azetidine-3-carboxylic acid [00103] To a solution of azetidine-3-carboxylic acid (88 g, 0.871 mol) and sodium bicarbonate (161 g, 1.92 mol) in water (1.75 L) at room temperature was added a solution of benzyl 2,5-dioxopyrrolidin-l-ylcarbonate (239 g, 0.959 mol) in tetrahydrofuran (3.5 L). The reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the aqueous layer was washed with ethyl acetate (2 x 500 mL). The aqueous layer was acidified with a 1.0 N aqueous solution of hydrochloric acid and was then extracted with ethyl acetate (3 x 750 mL). The organic layer was washed with water, followed by brine, and dried over anhydrous sodium sulfate. Concentration under reduced pressure afforded 1- (benzyloxycarbonyl) azetidine-3-carboxylic acid as colorless oil (202 g, 99percent yield). The compound had an HPLC retention time = 2.27 min. - Column: YMC COMBISCREEN.(R). ODS-A 4.6 x 50 mm (4 min.); Solvent A = 10percent MeOH, 90percent H2O, 0.1percent TFA; Solvent B = 90percent MeOH, 10percent H2O, 0.1percent TFA. LC/MS M+1 = 236.15. 1H NMR (400 MHz, CDCl3) δ ppm 3.39 - 3.49 (m, IH), 4.22 (d, J=7.28 Hz, 4H), 5.11 (s, 2H), and 7.29 - 7.39 (m, 5H).
99%
Stage #1: With sodium hydrogencarbonate In tetrahydrofuran at 20℃; Stage #2: With hydrogenchloride In water
Preparation of Intermediate 1 (Int. l); tert-Butyl 1 -(4-(N'-hydroxycarbamimidoyl)-benzyl)azetidine-3 -carboxylate; Int.1-A. l-(Benzyloxycarbonyl)azetidine-3-carboxylic acid; [00109] To a solution of azetidine-3-carboxylic acid (88 g, 0.871 mol) and sodium bicarbonate (161 g, 1.92 mol) in water (1.75 L) at room temperature was added a solution of benzyl 2,5-dioxopyrrolidin-l-ylcarbonate (239 g, 0.959 mol) in tetrahydrofuran (3.5 L). The reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the aqueous layer was washed with ethyl acetate (2 x 500 mL). The aqueous layer was acidified with a 1.0 N aqueous hydrochloric acid solution and was then extracted with ethyl acetate (3 x 750 mL). The organic layer was washed with water, followed by brine, and dried over anhydrous sodium sulfate. Concentration under reduced pressure afforded 1- (benzyloxycarbonyl) azetidine-3-carboxylic acid as colorless oil (202 g, 99percent yield). The compound had an HPLC retention time = 2.27 min. - Column: YMC COMBISCREEN.(R). ODS-A 4.6 x 50 mm (4 min.); Solvent A = 10percent MeOH, 90percent H2O, 0.1percent TFA; Solvent B = 90percent MeOH, 10percent H2O, 0.1percent TFA. LC/MS M+1 = 236.15. 1H NMR (400 MHz, CDCl3) δ ppm 3.39 - 3.49 (m, IH), 4.22 (d, J=7.28 Hz, 4H), 5.11 (s, 2H), and 7.29 - 7.39 (m, 5H).
99%
Stage #1: With sodium hydrogencarbonate In tetrahydrofuran; water at 20℃; Stage #2: With hydrogenchloride In water; ethyl acetate
Preparation 25A: l-(Benzyloxycarbonyl)azetidine-3 -carboxylic acidCbZlM^-CO2Hv (25A) [00262] To a solution of azetidine-3-carboxylic acid (88 g, 0.871 mol) and sodium bicarbonate (161 g, 1.92 mol) in water (1.75 L) at room temperature was added a solution of benzyl 2,5-dioxopyrrolidin-l-ylcarbonate (239 g, 0.959 mol) in tetrahydrofuran (3.5 L). The reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the aqueous layer was washed with ethyl acetate (2 x 500 mL). The aqueous layer was acidified with a 1.0 N aqueous hydrochloric acid solution and was then extracted with ethyl acetate (3 x 750 mL). The organic layer was washed with water, followed by brine, and dried over anhydrous sodium sulfate.Concentration under reduced pressure afforded l-(benzyloxycarbonyl) azetidine-3- carboxylic acid as colorless oil (202 g, 99percent yield). The compound had an HPLC retention time = 2.27 min. - Column: YMC COMBISCREEN.(R). ODS-A 4.6 x 50 mm (4 min.); Solvent A = 10percent MeOH, 90percent H2O, 0.1percent TFA; Solvent B = 90percent MeOH, 10percent H2O, 0.1percent TFA. LC/MS M+1 = 236.15. 1H NMR (400 MHz, CDCl3) δ ppm 3.39 - 3.49 (m, IH), 4.22 (d, J=7.28 Hz, 4H), 5.11 (s, 2H), and 7.29 - 7.39 (m, 5H).
99%
With sodium hydrogencarbonate In tetrahydrofuran; water at 20℃;
INTERMEDIATE 6tert-Butyl azetidine-3 -carboxylate acetic acid saltStep A: l-(Benzyloxycarbonyl)azetidine-3-carboxylic acidCbzN-C°2H (I-6A)[00202] To a solution of azetidine-3 -carboxylic acid (88 g, 0.871 mol) and sodium bicarbonate (161 g, 1.92 mol) in water (1.75 L) at room temperature was added a solution of benzyl 2,5-dioxopyrrolidin-l-ylcarbonate (239 g, 0.959 mol) in THF (3.5 L). The reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the aqueous layer was washed with ethyl acetate (2 x 500 mL). The aqueous layer was acidified with a 1.0 N aqueous hydrochloric acid solution and was then extracted with ethyl acetate (3 x 750 mL). The organic layer was washed with water, followed by brine, and dried over anhydrous sodium sulfate. Concentration under reduced pressure afforded l-(benzyloxycarbonyl) azetidine-3 -carboxylic acid as colorless oil (202 g, 99percent yield). The compound had an HPLC retention time = 2.27 min (condition B); LC/MS M+1 = 236.15. XH NMR (400 MHz, CDC13) δ ppm 3.39 - 3.49 (m, 1H), 4.22 (d, J=7.28 Hz, 4H), 5.1 1 (s, 2H), and 7.29-7.39 (m, 5H).
99%
With sodium hydrogencarbonate In tetrahydrofuran; water at 20℃;
1-A: l-(Benzyloxycarbonyl)azetidine-3 -carboxylic acid[0119] To a solution of azetidine-3 -carboxylic acid (88 g, 0.871 mol) and sodium bicarbonate (161 g, 1.92 mol) in water (1.75 L) at room temperature was added a solution of benzyl 2,5-dioxopyrrolidin-l-ylcarbonate (239 g, 0.959 mol) in tetrahydrofuran (3.5 L). The reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the aqueous layer was washed with ethyl acetate (2 x 500 mL). The aqueous layer was acidified with a 1.0 N aqueous solution of hydrochloric acid and extracted with ethyl acetate (3 x 750 mL). The organic layer was washed with water, washed with brine, and dried over anhydrous sodium sulfate. Concentration under reduced pressure afforded l-(benzyloxycarbonyl) azetidine-3 -carboxylic acid as colorless oil (202 g, 99percent yield). The compound had an HPLC retention time = 2.27 min. - Column: Column: YMC Combiscreen ODS-A 4.6 x 50 mm (4 min.); Solvent A = 10percent MeOH, 90percent H20, 0.1percent TFA; Solvent B = 90percent MeOH, 10percent H20, 0.1percent TFA. LC/MS M+1 = 236.15. XH-NMR (400 MHz, CDC13) δ ppm 3.39-3.49 (m, 1H), 4.22 (d, J=7.28 Hz, 4H), 5.11 (s, 2H), and 7.29-7.39 (m, 5H).
99%
Stage #1: With sodium hydrogencarbonate In tetrahydrofuran; water at 20℃; Stage #2: With hydrogenchloride In water
Preparation of Intermediate 1 (Int. l); tert-Butyl 1 -(4-(N'-hydroxycarbamimidoyl)-benzyl)azetidine-3 -carboxylate; Int. l-A. l-(Benzyloxycarbonyl)azetidine-3-carboxylic acid; [00101] To a solution of azetidine-3-carboxylic acid (88 g, 0.871 mol) and sodium bicarbonate (161 g, 1.92 mol) in water (1.75 L) at room temperature was added a solution of benzyl 2,5-dioxopyrrolidin-l-ylcarbonate (239 g, 0.959 mol) in tetrahydrofuran (3.5 L). The reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the aqueous layer was washed with ethyl acetate (2 x 500 mL). The aqueous layer was acidified with a 1.0 N aqueous solution of hydrochloric acid and extracted with ethyl acetate (3 x 750 mL). The organic layer was washed with water, washed with brine, and dried over anhydrous sodium sulfate. Concentration under reduced pressure afforded 1- (benzyloxycarbonyl) azetidine-3-carboxylic acid as colorless oil (202 g, 99percent yield). The compound had an HPLC retention time = 2.27 min. - Column: YMC COMBISCREEN.(R). ODS-A 4.6 x 50 mm (4 min.); Solvent A = 10percent MeOH, 90percent H2O, 0.1percent TFA; Solvent B = 90percent MeOH, 10percent H2O, 0.1percent TFA. LC/MS M+1 =236.15. 1H NMR (400 MHz, CDCl3) δ ppm 3.39 - 3.49 (m, IH), 4.22 (d, J=7.28 Hz, 4H), 5.11 (s, 2H), and 7.29 - 7.39 (m, 5H).
To a solution of 4-bromopiperidine hydrobromide (3.0 g, 12.2 mmol) in tetrahydrofuran (30 ml) were added 1-[(benzyloxy)carbonyl]oxy}-2,5-pyrrolidinedione (3.20 g, 12.9 mmol), N-methylmorpholine (1.62 ml, 14.7 mmol) and 4-N,N-dimethylaminopyridine (30 mg) at room temperature, and stirred at room temperature for 16 hours. The reaction solution was poured into water and extracted with ethyl acetate. The organic layer was washed with a 1N-aqueous hydrochloric acid solution, dried over anhydrous magnesium sulfate and distilled under reduced pressure to remove the solvent, and the resulting residue was purified by a silica gel column chromatography (eluent: hexane/ethyl acetate = 1/1) to obtain benzyl 4-bromo-1-piperidinecarboxylate (3.58 g, 98percent).
Stage #1: With sodium hydroxide In 1,4-dioxane; water at 20℃; for 0.75 h; Stage #2: at 20℃; for 17 h; Stage #3: With hydrogenchloride In 1,4-dioxane; water
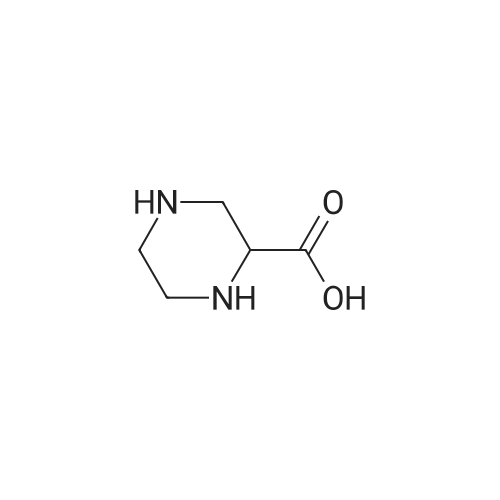
54a. 4-((Benzyloxy)carbonyl)-1 -(tert-butoxycarbonyl)piperazine-2- carboxylic acidThe tile compound was prepared according to the procedure of [Kempf, D. J.; Norbeck, D. W.; Sham, H. L U.S. Patent 5,455,351 , Oct 3, 1995]. Piperazine-2-carboxylic acid (10.0 g, 77.0 mmol) was dissolved in a 1 :1 solution of 1 ,4-dioxane:water (100 mL) at room temperature with vigorous stirring. The clear solution was adjusted to pH 11 by the addition of an aqueous solution of sodium hydroxide (80 mL of a 1Λ/ solution). The pH was monitored in situ with a pH meter throughout the reaction. The reaction flask was fitted with an addition funnel that contained a solution of Λ/-α- (benzyloxycarbonyloxy) succinamide (13.6 g, 55 mmol) in 1 ,4-dioxane (50 mL). The Λ/-α-(benzyloxycarbonyloxy) succinamide solution was added over 45 minutes at room temperature and the pH was kept above 10 by the periodic addition of 1 Λ/ sodium hydroxide. The pH of the solution was adjusted to 9.5 and 2-(teAf-butoxycarbonyloxyimino)-2-phenylacetonitrile (13.4 g, 55 mmol) was added as a solution in 1 ,4-dioxane (50 mL) over 10 minutes. The pH was maintained at 9.5 and the solution was stirred at room temperature for 17 hours. The solution was then acidified to pH 2 and the aqueous solution was washed with diethyl ether (3 x 150 mL). The aqueous solution was cooled to O0C and acidified by adding of concentrated hydrochloric acid. The acidic solution was extracted with ethyl acetate (5 x 150 mL). The combined organic phases were dried over sodium sulfate, filtered and concentrated in vacuo. The residue was triturated with a 1 :1 solution of dichloromethane: hexanes (150 mL) and the solvent was removed in vacuo to provide the product as a viscous yellow oil (15.7 g, 43 mmol, 80percent). Rf = 0.60 (66:34 dichloromethane: ethyl acetate + 0.1percent (v/v acetic acid); 1H-NMR (400 MHz, DMSO) δ 13.0 (br s, 1 H), 7.37-7.36 (m, 5H), 5.05 (s, 2H), 4.54-4.33 (m, 2H), 3.90-3.66 (m, 2H), 3.07-2.81 (m, 4H), 1.38 (s, 9H); Mass spectrum (ESI +ve) m/z 365.1 (MH+).
With sodium hydrogencarbonate; In tetrahydrofuran; water; at 0 - 20℃;
To a solution of N-(Benzyloxycarbonyloxy)succinimide (1.80g, 7.23 mmol) in THF (9 mL) was added 1-aminocyclopropanecarboxylic acid, HC1 (722 mg, 5.24 mmol) in water(1.4mL). The mixture was cooled to 0C and shaken for 5 minutes. To this mixture was added at 0C an aqueous solution of sodium bicarbonate (2.46 g, 29.3 mmol)) in 5mL of water. The mixture was allowed to warm to room temperature and shaken overnight. The product mixture was filtered and the solids set aside. Volatiles were evaporated to near dryness then the crude mixture was diluted with lOmL of water and 40mL of DCM. The mixture was extracted and the DCM layer was set aside. The resulting aqueous layer was cooled in an ice bath then acidified with lOmL of acetic acid. The solid was filtered, washed with lmL of water x2 then dried under vaccuum for 2 hours giving 856mgs of l-(benzyloxycarbonylamino)cyclopropanecarboxylic acid as a fluffy white solid (63% yield). XH NMR (400 MHz, DMSO-i/6) delta ppm 0.93 - 1.10 (m, 2 H), 1.26 - 1.37 (m, 2 H), 5.02 (s, 2 H), 7.25 - 7.44 (m, 5 H), 7.89 (s, 1 H), 12.44 (d, 1 H). LCMS rt = 2.1 12min., m/z 236.1(M + H), 90% purity. The LC/MS data was obtained on a Shimadzu analytical LC /Micromass Platform LC (ESI+) at 220nm using the following set of conditions: Phenomenex Luna 3muiotaeta C18, 2 x 50mm column, with a gradient of 0-100%B (B = 90% HPLC grade acetonitrile/ 0.1% trifluoroacetic acid/ 10% HPLC grade water), (A = 90% HPLC grade water / 0.1% trifluoroacetic acid/ 10% HPLC grade acetonitrile), in 4 minutes with a 1 minute hold at a rate of 0.8 mL/minute.
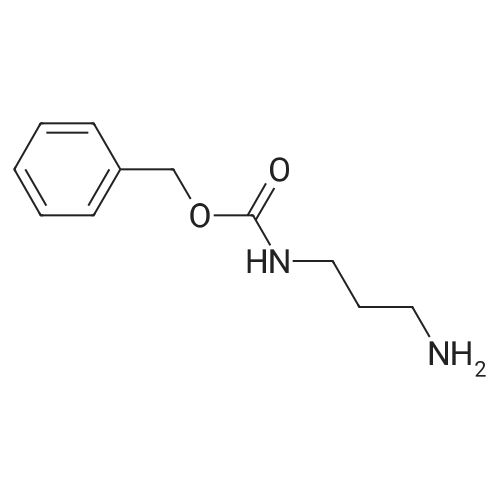
A. 3-(Benzyloxycarbonylamino)propylamine. 1,3-Diaminopropane (35.5 g, 0.48 mol) was dissolved in 300 mL CHCl3, and the solution was cooled to 0° C. A solution of N-(benzyloxycarbonyloxy) succinimide (4.5 g, 0.018 mol) in 150 mL CHCl3 was added dropwise over 6 h, with the internal temperature maintained below 10° C. After addition was complete, the reaction solution was stirred at room temperature overnight. The solution was washed with water, dried (Na2 S04), filtered and concentrated in vacuo to provide 3.0 g (80percent) of the title compound as a low melting solid: 1 H NMR (CDCl3) delta1.25 (bs, 2H), 1.63 (quintet, 2H), 2.77 (t, 2H), 3.2-3.34 (bm, 2H), 5.09 (s, 2H), 5.37 (bm, 1H), 7.28-7.40 (m, 5H); MS, m/e 209 ((M+H)+).
With sodium carbonate; In tetrahydrofuran; water; at 20℃; for 16h;
Step 1. To a mixture of aq Na2C03 (82 g, 0.78 mol) and compound 73-1 (20 g, 0.19 mol) was added Cbz-OSu (57 g, 0.23 mol) in THF (150 mL). After the mixture was stirred at r.t. for 16 h, it was adjusted to pH > 10 and the solution was extracted with EtOAc (400 mL x 2). The aqueous layer was acidified to pH < 1 with conc.HCl and the solution was extracted with EtOAc (500 mL x 2). The organic layers were dried over Na2S04 and concentrated to give compound 73-2 (45 g, 0.19 mol), which was used for next step without further purification.
With sodium hydrogencarbonate; In tetrahydrofuran; water; at 20℃; for 16h;Cooling with ice;
Step A: intermediate VI A solution of benzyl (2,5-dioxopyrrolidin-1-yl) carbonate (83 g, 334 mmol) in tetrahydrofuran (THF) (500 ml) was added to a solution of 1-aminocyclopropanecarboxylic acid (25 g, 247 mmol) in water (500 ml). At ice temperature, a solution of sodium bicarbonate (91 g, 1088 mmol) in water (500 ml) was poured into the mixture. After the addition, the reaction was allowed to warm to room temperature and was stirred for 16 h. The solvent was removedunder reduced pressure. The residue was suspended in200 mL of methylene chloride and extracted with water (500 mL). The water solution was acidified with acetic acid and the precipitate was filtered yielding 1- (((benzyloxy)carbonyl)amino)cyclopropanecarboxyiic acid (53 g, 171 mmol, 69.2 % yield) as a white solid. Used crude in next reaction. LC/MS: m/z calculated 235.1 , found 258.0 ( +Na)+.
With sodium carbonate; In tetrahydrofuran; water; at 20℃; for 16h;
Step 1. To a mixture of 82-1 (10 g, 98.9 mmol) in aq. solution Na2C03 (41.9 g, 395.6 mmol) and was added Cbz-OSu (29.6 g, 118.7 mmol) in THF (150 mL). After the mixture was stirred at r.t. for 16 h, it was adjusted to pH > 10 and the solution was washed with EtOAc (200 mL x 2). The aqueous layer was acidified to pH < 1 with cone. HC1 and the solution was extracted with EtOAc (250 mL x 2). The organic layer was dried over Na2S04 and concentrated to give 82-2 (23 g, crude), which was used for next step without further purification.
With water; triethylamine; In 1,4-dioxane; at 20℃;
To a suspension of D-2-amino-4-pentenoic acid (500 mg) in Dioxane (20 mL) at RT was added triethylamine (1.09 g), water (0. 5 mL), and N- (BENZYLOXYCARBONYLOXY) SUCCINIMIDE (2.27 g). The reaction mixture was stirred overnight at RT, concentrated in vacuo, diluted with saturated NAHCO3 and extracted with ether (3x). The basic aqueous layer was acidified to pH 2 with 2N HCI and extracted with EtOAc (3x). The combined EtOAc layer was washed with brine, dried (NA2S04), filtered and concentrated to give a clear viscous oil (1.17 g, quantitative). H NMR (300 MHz, DMSO-d6) 8 2.29-2. 50 (M, 2H), 3.98-4. 06 (M, 1H), 5.03 (s, 2H), 5.05-5. 12 (M, 2H), 5. 71-5. 84 (m, 1H), 7.35 (M, 5H), 7.54 (d, 1H), 12.57 (bs, 1H). MS APCI, m/z = 250 (M+1). LC/MS 1.90 min. , Method A.
76%
With triethylamine; In tetrahydrofuran; water; at 20℃;
D-allylglycine (2.07 g, 18.0 mmol) and N-(benzyloxycarbonyloxy)succinimide (Cbz-OSu) (4.49 g, 18.0 mmol) are added to a round bottomed flask containing THF (60 mL) and water (20 mL). The mixture is stirred at room temperature and Et3N (10.1 mL, 72.0 mmol) is added, and the reaction is stirred overnight at room temperature. The clear solution is diluted with EtOAc (200 mL), washed with IN HCl (3 x 100 mL) and brine (1 x 100 mL), and dried with MgSO4. Solvent is evaporated in vacuo to afford 10-B as a white solid, which is used without further purification.
To a solution of <strong>[54288-70-9]4-bromopiperidine hydrobromide</strong> (3.0 g, 12.2 mmol) in tetrahydrofuran (30 ml) were added 1-[(benzyloxy)carbonyl]oxy}-2,5-pyrrolidinedione (3.20 g, 12.9 mmol), N-methylmorpholine (1.62 ml, 14.7 mmol) and 4-N,N-dimethylaminopyridine (30 mg) at room temperature, and stirred at room temperature for 16 hours. The reaction solution was poured into water and extracted with ethyl acetate. The organic layer was washed with a 1N-aqueous hydrochloric acid solution, dried over anhydrous magnesium sulfate and distilled under reduced pressure to remove the solvent, and the resulting residue was purified by a silica gel column chromatography (eluent: hexane/ethyl acetate = 1/1) to obtain benzyl 4-bromo-1-piperidinecarboxylate (3.58 g, 98percent).
Step A: 2S-Benzyloxycarbonylamino-3,3-dimethyl-butyric acid; To a suspension of L-tert-leucine (11.88 g, 90.7 mmol) in methanol (200 ml) were added triethylamine (26.56 ml, 190 mmol) and N-(benzyloxycarbonyl- oxy)-succinimide (24.88 g, 99.8 mmol). The reaction mixture was stirred at room temperature for 14 h. Methanol was removed in vacuo to afford a viscous pale yellow oil, which was dissolved in ethyl acetate (100 ml). The organic layer was washed with 1M hydrochloric acid (15 ml) and brine, dried over anhydrous magnesium sulfate and filtered. The solvent was removed in vacuo to fumish the title compound as an oil (24 g, quant.). 1H-NMR; delta(CDCl3), 7.43-7.36 (5H, m), 5.36 (1H, d, J = 9.4 Hz), 5.12 (2H, br s), 4.20 (1H, d, J = 9.6 Hz) and 1.02 (9H, s). LRMS: +ve ion 266 [M+H], -ve ion 264 [M-H], 529 [2M-H].
With triethylamine; In dichloromethane; at 0℃; for 1h;
2-BROMOETHYLAMINE hydrobromide (15 g, 73 MMOL) AND N- (BENZYLOXYCARBONYLOXY)- succinimide (15.5 g, 62 MMOL) are suspended in CH2CI2 (150 mL) at 0C. TEA (9 mL, 65 MMOL) is added slowly keeping the temperature at 0C. After 1h the mixture is washed with 0.5M aq. KHS04 (50 mL) and sat. aq. NaCl (50 mL), the organic phase is dried (NA2SO4), filtered and evaporated to provide the title compound
To N-Benzyloxycarbonyl-D-1,2, 3, 4-tetrahydro-isoquinoline-1-carboxylic acid (3.11 g, 10 mmol, made from commercially available D-1,2, 3, 4-tetrahydro- ISOQUINOLINE-1-CARBOXYLIC acid and O-BENZYLOXYCARBONYL-N-HYDROXYSUCCINAMIDE) in THF (10 mL) was added BH3 (1M solution in THF; 30 mL, 30 mmol) over 5 min, and the reaction mixture was stirred for 3 hours. Acetic acid (9 mL) in MEOH (90 mL) was added and the mixture was stirred for 30 min. Solvents were evaporated, the residue was taken up in EtOAc and was washed with saturated aqueous NAHC03 (90 mL, aq. phase pH 7-8) and brine. The organic layer was dried over NA2S04 and concentrated to give compound 13a (2.97 g, 100%). MS (CI) M/Z 253.9 (MH+-CO2), 297. 9 (MH+) ; TR = 2.695 min (method 4).
To a solution of 28 (1.07 g, 6.28 mmol) in CH2Cl2 (60 mL), was added N-(benzyloxycarbonyloxy)succinimide (1.72 g, 6.91 mmol). The reaction mixture stirred at room temperature for approximately one hour before most of the solvent was removed (55 mL) in vacuo. The crude product was purified by a silica gel chromatography (8percent-12percent MeOH/CH2Cl2) to give benzyl 1,4-dioxaspiro[4.5]dec-8-ylmethylcarbamate. LCMS (ES) m/z 306.2 (M+H)+.
To a stirred solution of trans-4-aminocyclohexanol (35.5 g, 306.6 mmol) in methanol (621 mL) at 0 C. under nitrogen was added N-(benzyloxycarbonyloxy)succinimide (72.8 g, 291.9 mmol). The reaction mixture was slowly warmed to room temperature and after 1 d, poured into 0.25 M aqueous hydrochloric acid solution (2.13 L). The resulting slurry was stirred vigorously for 1 h and then filtered. The filtered solid was washed with water to obtain the title compound (65.65 g, 90%) as a white solid. 13C NMR (400 MHz, (CD3)2SO) delta 155.3, 137.2, 128.3, 127.7, 127.6, 68.1, 65.0, 49.1, 33.9, 30.3. 1H NMR (400 MHz, (CD3)2SO) delta 7.29 (m, 5H), 7.10 (d, J=7.9 Hz, 1H), 4.96 (d, J=2.3 Hz, 2H), 4.76 (d, J=4.0 Hz, 1H), 3.29 (s, 1H), 3.17 (br s, 1H), 1.71 (m, 4H), 1.22 (t, J=9.7 Hz, 4H).
With triethylamine; In toluene; at 5 - 30℃; for 1.25h;
N-(Benzyloxycarbonyloxy)succinimide (74.5 Kg) was charged to a 200-gallon reactor, followed by toluene (235.1 Kg). The mixture was stirred for 15 minutes and cooled to 5 C. A solution of aminoacetaldehyde dimethylacetal (30 Kg) and triethylamine (28.9 Kg) in toluene (26.1 Kg) was charged slowly to the reactor over a 1 hour period while maintaining the internal temperature below 30 C. The mixture was stirred at 20 C. until there remained no more than 1.0% CbzOSu, as determined by HPLC. Water (150 Kg) was charged to the reactor and the contents of the reactor were stirred for 15 minutes. The layers were separated and the organic layer was washed with 5% ammonium chloride (2×151 Kg) followed by a water wash (150 Kg). The product was taken onto the next step as a solution, without isolation.
With triethylamine; In N,N-dimethyl-formamide; for 18h;
Preparation 23; Preparation of phenylmethyl f(3f?)-3-pyrrolidinylmethvncarbamate; a) 1 ,1-dimethylethyl (3S)-3-[([(phenylmethyl)oxy]carbonyl}amino)methyl]-1- pyrrolidinecarboxylate; To a solution of (S)-3-(aminomethyl)-1-Lambda/-Boc-pyrrolidine (3.0 g, 14.98 mmole) in DMF (40 mL) was added triethylamine (2.5 ml_, 17.94 mmole) and N- (benzyloxycarbonyloxy)succinimide (4.46 g, 17.90 mmole). After 18 hours, the reaction solution was concentrated under vacuum and redissolved in diethyl ether (250 mL). The organic solution was washed with H2O (2 x 150 mL), brine (100 mL) and dried over Na2SO4 and concentrated. The remaining residue was purified on silica (CHCl3/MeOH, 9:1 containing 5% NH4OH) to give the title compound (4.46 g, 91%) as a light yellow oil:LC-MS (ES) m/e 335 (M + H)+.
With hydrogenchloride; benzaldehyde; In tetrahydrofuran; dichloromethane; toluene;
Step 1 Preparation of Benzyl 4-(aminomethyl)piperidine-1-carboxylate 4-Aminomethylpiperidine (40 g, 350 mmol) and benzaldehyde (37.3 mL, 368 mmol) in toluene (600 mL) were heated to reflux under dean stark conditions for 2 h. The resulting reaction mixture was cooled to room temperature and 500 mL dichloromethane was added. The resulting solution was cooled to 5 C. and treated with N-(benzyloxycarbonyloxy)succinimide (91.7 g, 368 mmol). After 10 min, the cooling bath was removed and the reaction mixture stirred for 1 h. The solvents were evaporated and the resulting residue was stirred with 400 mL THF and 400 mL 2 M HCl for 1 h. The mixture was concentrated to remove organics and was then extracted with ether (3*300 mL). The aqueous phase was adjusted to pH14 with 50% NaOH and extracted with ethyl acetate. The organic layer was washed with water and brine, dried over anhydrous sodium sulfate, and the solvent evaporated to give benzyl 4-(aminomethyl)piperidine-1-carboxylate as an oil. 1H-NMR 500 MHz (delta, CDCl3) delta7.4-7.2 (m, 5H); 5.12 (s, 2H); 4.20 (brs, 2H); 2.77 (brs, 2H); 2.58 (d, J=6.6 Hz, 2H) 1.9-1.7 (m, 2H); 1.0-1.5 (m, 5H).
With hydrogenchloride; benzaldehyde; In tetrahydrofuran; dichloromethane; toluene;
Step 1 Benzyl 4-(aminomethyl)piperidine-1-carboxylate 4-Aminomethylpiperidine (40 g, 350 mmol) and benzaldehyde (37.3 mL, 368 mmol) in toluene (600 mL) were heated to reflux under Dean Stark conditions for 2 h. The resulting reaction mixture was cooled to room temperature and 500 mL dichloromethane was added. The resulting solution was cooled to 5 C. and treated with N-(benzyloxycarbonyloxy)succinimide (91.7 g, 368 mmol). After 10 min, the cooling bath was removed and the resulting reaction mixture stirred for 1 h. The solvents were evaporated and the residue stirred with 400 mL THF and 400 mL 2M HCl for 1 h. The mixture was concentrated to remove organics and extracted with ether (3*300 mL). The aqueous phase was adjusted to pH14 with 50% NaOH and extracted with ethyl acetate. The organic layer was washed with water and brine, dried over anhydrous sodium sulfate, and the solvent evaporated to give benzyl 4-(aminomethyl)piperidine-1-carboxylate as an oil. 1H NMR (500 MHz CDCl3) delta: 7.4-7.2 (m, 5H); 5.12 (s, 2H); 4.20 (brs, 2H); 2.77 (brs, 2H); 2.58 (d, J=6.6 Hz, 2H) 1.9-1.7 (m, 2H); 1.0-1.5 (m, 5H).
With hydrogenchloride; benzaldehyde; In tetrahydrofuran; dichloromethane; toluene;
Step 1 Benzyl 4-(aminomethyl)piperidine-1-carboxylate 4-Aminomethylpiperidine (40 g, 350 mmol) and benzaldehyde (37.3 mL, 368 mmol) in toluene (600 mL) were heated to reflux under dean stark conditions for 2 h. The reaction mixture was cooled to room temperature and 500 mL dichloromethane added. The solution was cooled to 5 C. and treated with N-(benzyloxycarbonyloxy)succinimide (91.7 g, 368 mmol). After 10 min., the cooling bath was removed and the reaction mixture stirred for 1 h. The solvents were evaporated and the residue stirred with 400 mL THF and 400 mL 2M HCl for 1 h. The mixture was concentrated to remove organics and extracted with ether (3*300 mL). The aqueous phase was adjusted to pH14 with 50% NaOH and extracted with ethyl acetate. The organic layer was washed with water and brine, dried over anhydrous sodium sulfate, and the solvent evaporated to give benzyl 4-(aminomethyl)piperidine-1-carboxylate compound.
5-Benzyloxycarboxyaminoresorcinol To a suspension of <strong>[6318-56-5]5-aminoresorcinol hydrochloride</strong> (808 mg, 5.0 mmole) in dry dimethylformamide (10 ml) under argon was added N-ethyl,di-isopropylamine (1.5 g) then N-(benzyloxycarbonyloxy)succinimide (1.24 g, 5 mmole) and the mixture stirred for 24 hours at 20 C. to give a clear brown solution. The solution was cooled in ice and acidified to pH2 by the addition of 2M hydrochloric acid. Solvent was removed in vacuo and the residue partitioned between ethyl acetate and water. The organic layer was washed twice with icewater, dried over magnesium sulphate and filtered through a layer of silica gel. The solution was evaporated to give a light tan solid. 1.05 g 86% 1H nmr [(CD3)2SO]: 5.15, s, 2H; 5.90, m, 1H; 6.50, m, 2H; 7.45, m, 5H; 9.15, s, 2H
Method A (S)-Piperidine-1,2-dicarboxylic acid 1-benzyl ester 2-methyl ester To a stirred suspension of <strong>[18650-39-0](S)-piperidine-2-carboxylic acid methyl ester hydrochloride</strong> salt (5.0 g, 27.8 mmol) in dichloromethane (DCM) (35 mL) at 0° C. was added diisopropylethylamine (DIPEA) (10.1 mL, 58.4 mmol) followed by N-(benzyloxycarbonyloxy) succinimide (7.63 g, 30.6 mmol). The reaction mixture was allowed to warm to room temperature and stirred overnight. The residue was diluted with DCM and washed with 1.0-M HCl. The organic layer was washed with brine, dried(Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash chromatography (20percent ethyl acetate in hexane) to afford the title compound as a pale yellow oil (7.72, 100percent): 1H NMR (400 MHz, CD3OD) delta 1.19-1.88 (5 H, m), 2.15-2.28 (1H, m), 2.91-3.12 (1H, m), 3.70-3.73 (3H, 2s), 4.00-4.07 (1H, m), 4.82-4.87 (1H, m), 5.03-5.21 (2H, m), 7.24-7.39 (5H, m); 13C NMR (100 MHz, CD3OD) delta 22.0, 22.1 (CH2), 26.0, 26.1 (CH2), 28.1 (CH2), 43.4, 43.5 (CH2), 53.1 (CH3), 56.2, 56.5 (CH), 68.8, 68.9 (CH2), 129.2 (CH), 129.5 (CH), 129.9 (CH), 138.4 (C), 167.0 (CO), 173.7 (CO).
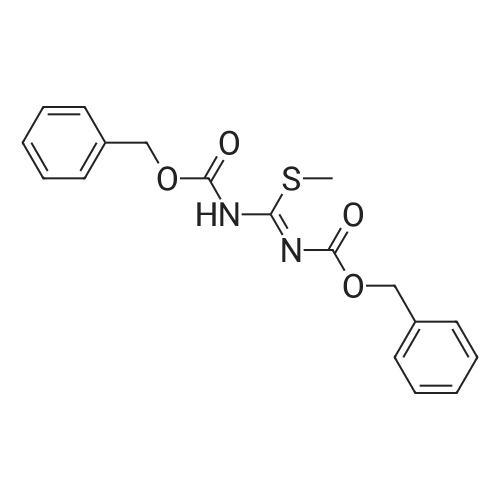
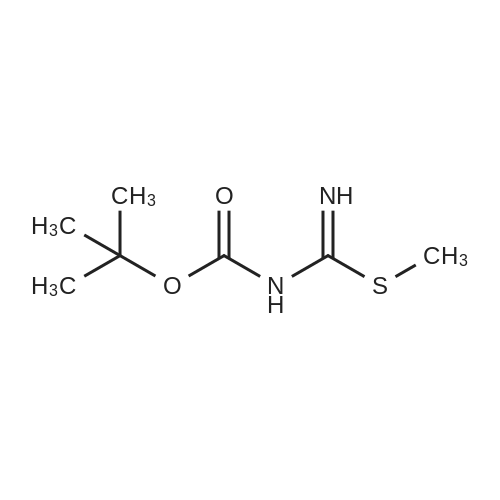
N-(benzyloxycarbonyl)-N'-(tert-butyloxycarbonyl)-S-methylisothiourea[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sodium hydroxide; In dichloromethane;
(b). N-(benzyloxycarbonyl)-N'-(tert-butyloxycarbonyl)-S-methylisothiourea <strong>[173998-77-1]N-(tert-butyloxycarbonyl)-S-methylisothiourea</strong> (2 g) was dissolved in dichloromethane (20 ml). To the solution, a 4N sodium hydroxide solution (2 ml) was added with stirring. The reaction mixture was placed in an ice bath; N-(benzyloxy-carbonyloxy)-succinimid (2.62 g) in dichloromethane (20 ml) was added dropwise together with a 2N sodium hydroxide solution to keep the pH around 11. After addition was complete, the reaction mixture was stirred overnight at room temperature. The dichloromethane layer was separated, and the aqueous layer was washed twice with dichloromethane. The combined organic layers were washed with water and dried over sodium sulfate, filtered and evaporated in vacuo. The residue was purified by chromatography on silica (eluent: heptane/ethyl acetate=3/2 v/v %) to yield N-(benzyloxy-carbonyl)-N'-(tert-butyloxycarbonyl)-S-methylisothiourea (3.15 g). TLC: Rf =0.78, silica gel, ethyl acetate/heptane=1/1 v/v %
With triethylamine; In 1,4-dioxane; water; ethyl acetate;
a. N-benzyloxycarbonyl-2-aminoadipic acid A solution of L-2-aminoadipic acid (920 mg, 5.71 mmol), triethylamine (2.85 mL, 20.5 mmol) and water (3 mL) in 12 mL of dioxane was cooled to 0 C. and N-benzyloxycarbonyloxysuccinimide (Cbz-OSu: 1.71 g, 6.85 mmol) was added. The reaction mixture was stirred at 0 C. for 2 hours and at ambient temperature for 0.5 hours. The reaction mixture was washed with ethyl acetate and then acidified with aqueous potassium hydrogen sulfate. The acidic solution was extracted with ethyl acetate and the ethyl acetate solution was washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to give 1.80 g of the title compound. MS (FAB+) m/e 296 (M+H)+.
With hydrogenchloride; sodium hydrogencarbonate; In water; acetone;
a N-[(Phenylmethoxy)carbonyl]-<strong>[672-15-1]L-homoserine</strong> N-(Benzyloxycarbonyloxy)succinimide (23.57 g., 94.58 mmol.) was added to a solution of <strong>[672-15-1]L-homoserine</strong> (10.24 g., 85.98 mmol.) and sodium bicarbonate (7.95 g., 94.58 mmol., 1.1 eq.) in a mixture of water (100 ml.) and acetone (100 ml.). The mixture was stirred at room temperature overnight. The acetone was removed under reduced pressure (rotovap) and the aqueous solution was washed with methylene chloride (2*75 ml.). The aqueous layer was then acidified to pH 2 by addition of 6N hydrochloric acid and extracted with ethyl acetate (2*250 ml.). The combined ethyl acetate layers were washed with water (2*100 ml.) and brine, dried over sodium sulfate, filtered, concentrated and dried in vacuo to afford 19.54 g. of title product as a white solid. TLC (ethyl acetate:n-butanol:acetic acid:water; 2:1:1:1) Rf =0.74
Example 13: Synthesis of 5-[5-Fluoro-2-oxo-1,2-dihydro-indol-(3Z)- ylidenemethyl]-2,4-dimethyl-1 H-pyrrole-3-carboxylic acid ((S)-2- dimethylcarbamoyl-2-hydroxy-ethyl)-amide; Synthesis of (S)-3-(benzyloxycarbonyl)-2-hydroxypropanoic acid:To the THF/water (50 mL/50 mL) solution of (S)-isoserine (2.429 g, 23.12 mmol) was added K2CO3 (3.834 g, 27.74 mmol) and N- (Benzyloxycarbonyloxy)-succinimide (5.76 g, 23.11 mmol). The reaction mixture was stirred at 25 0C overnight. The reaction mixture was concentrated and diluted with EtOAc and acidified with excess HCI. The aqueous layer was extracted with EtOAc, and the combined organic layers were washed with dilute HCI, water, brine and dried over sodium sulfate. The solvent was removed under reduced pressure to afford (S)-3-(benzyloxycarbonyl)-2- hydroxypropanoic acid (5.11 g, 92percent), which was used in the next step with no further purification. LC-ESIMS observed [M+H]+ 239.91 (calculated for Ci1H13NO5 239.08).
a) l,l-Dimethylethyl (3iS)-3-[([(phenylmethyl)oxy]carbonyl}amino)methyl]-l- piperidinecarboxylateTo a solution of 1,1-dimethylethyl (3.S)-3-(aminomethyl)-l- piperidinecarboxylate (2.0 g, 9.33 mmol) in DCM (12 ml) at 0 0C were added triethylamine (1.7 ml, 12.1 mmol) followed by N-(benzyloxycarbonyloxy)succinimide (2.56 g, 10.3 mmol). After a few minutes the cooling bath was removed and the reaction was stirred at room temperature for 2h. The reaction was diluted with ethyl acetate and washed with water (2x), IN HCl, <n="87"/>saturated aq. NaHCO3, brine, dried (Na2SO4) and concentrated. The product was purified on silica gel eluting with 10% ethyl acetate-DCM to give 3.48g of material containing a small amount of N-(benzyloxycarbonyloxy)succinimide which was used directly in next step. LC/MS (ES) m/e 349 (M+H)+.
With triethylamine; In dichloromethane; at 0 - 20℃;
TEA ( 6.91 g, 68.5 mmol) was added dropwise to an ice-cooled mixture of 3- bromopropylamine.hydrobromide (10.Og, 45.6 mmol) and N-(Benzyloxycarbonyloxy)- succinimide (11.22g, 47.9 mmol) in DCM (200 mL). The stirred mixture was allowed to warm to room temperature overnight, then washed with water (3x), brine, dried (MgSO4), filtered and concentrated, providing 11.43 g (92%) of N-(Benzyloxycarbonyl)- 3-bromopropylamine, as a pale yellow oil .
With triethylamine; In N,N-dimethyl-formamide; for 18h;
a) 1 ,1 -Dimethylethyl 3-[([(phenylmethyl)oxy]carbonyl}amino)methyl]-1 - azetidinecarboxylate To a solution of 1 ,1 -dimethylethyl 3-(aminomethyl)-1 -azetidinecarboxylate (5.0 g, 26.84 mmole) in DMF (40 mL) was added triethylamine (4.9 ml_, 34.9 mmole) and Cbz- succinamide (8.7 g, 34.9 mmole). After 18 hours, the reaction solution was concentrated under vacuum and redissolved in EtOAc (250 mL). The organic solution was washed with H2O (2 x 150 mL), brine (100 mL) and dried over Na2SC>4 and concentrated. The remaining residue was purified on silica (hexanes/EtOAc, 1 :1) to give the title compound (5.5 g, 64%) as a light yellow oil: LC-MS (ES) m/e 321 (M + H)+.
With aq. sodium hydroxide; In 1,2-dimethoxyethane; aqueous sodium hydroxide;
(a) Allyl 4-benzyloxycarbonylaminopiperidine-4-carboxylate. 4-Amino-1-tert-butoxycarbonylpiperidine-4-carboxylic acid (18.0 g) was dissolved in 0.03M aqueous sodium hydroxide (750 mL), and 1,2-dimethoxyethane (100 mL). The pH was adjusted to 9.5 with dilute hydrochloric acid, and the suspension was added to an ice-cold solution of N-(benzyloxycarbonyloxy)succinimide (28.8 g) in 1,2-dimethoxyethane (100 mL). The pH was kept at 9.5 by addition of aq. sodium hydroxide, and the mixture was stirred at room temperature. More N-(benzyloxycarbonyloxy)succinimide (3 g) was added after 7 h, then stirring continued overnight. After evaporation to remove the 1,2-dimethoxyethane, the aqueus residue was extracted with ether then acidified to pH 4 and extracted with ethyl acetate. This extract was washed with dilute hydrochloric acid, water and brine, dried and evaporated. Trituration of the oily residue gave 4-benzyloxycarbonylamino-1-tert-butoxycarbonylpiperidine-4-carboxylic acid (23 g).
In sodium hydroxide; 1,2-dimethoxyethane;
(a) Methyl 4-benzyloxycarbonylamino-1-tert-butoxycarbonylpiperidine-4-carboxylate. 4-Amino-1-tert-butoxycarbonylpiperidine-4-carboxylic acid (18.0 g) was dissolved in 0.03M aqueous sodium hydroxide (750 ml), and 1,2-dimethoxyethanol (DME, 100 ml). The pH was adjusted to 9.5 with dilute hydrochloric acid, and the suspension was added to an ice-cold solution of N-(benzyloxycarbonyloxy)succinimide (28.8 g) in DME (100 ml). The pH was kept at 9.5 by addition of aqueous sodium hydroxide, and the mixture was stirred at room temperature. More N-(benzyloxycarbonyloxy)succinimide (3 g) was added after 7 hours, then stirring was continued overnight. After evaporation, the aqueous residue was extracted with ether then acidified to pH4 and extracted with ethyl acetate. This extract was washed with dilute hydrochloric acid, water and brine, dried and evaporated. Trituration of the oily residue gave 4-benzyloxycarbonylamino-1-tert-butoxycarbonylpiperidine-4-carboxylic acid (23 g).
With sodium hydroxide; In tetrahydrofuran; methanol; water; at 0 - 20℃; for 17.5h;
N-(Benzyloxycarbonyloxy)succinimide (24.4 g, 1 equiv, 98 mmol) was added to a solution of <strong>[625-05-8]3-amino-3-methylbutanoic acid</strong> (16.0 g, 97.2 mmol) in methanol and water (200 mL: 100 mL) followed by the addition of THF (100 mL) to give a clear solution. <n="86"/>The solution was cooled in an ice bath for 15 min and a solution of 5 N sodium hydroxide (39 mL, 2 equiv) was added over 15 min. The solution was stirred at 0 0C for an additional hour and stirred at RT for 17 h. The organic solvent was removed and the aqueous was extracted with ethyl acetate (2x 200 mL). The aqueous layer was acidified with 6 N HCl (50 mL) and extracted with ethyl acetate (4x 200 mL). The organic layer was dried over anh. MgSO4, filtered, and concentrated to an orange yellow oil (16.2 g, 65.7percent). This oil was used without further purification. LRMS (ESI -ve) for Ci3H17NO4 (251.1); found: 250.
SOCI2 (1 62 mL) was added to 13 mL methanol at 0 C. To the solution was added D- aspartic acid (2.90 g) and the mixture was warmed to room temperature. After 3 hours, the mixture was cooled in an ice-water bath and 33 mL of aqueous 14% K3PO4 was added. MeOH was distilled away under vacuum. Toluene (16 mL) was added followed by Cbz-OSu (N-(benzyloxycarbonyloxy)succinimide) (5.97 g). The flask was cooled in an ice-water bath, and 8.65 g K3PO4 was charged. The mixture was stirred at room temperature for 14h.The organic layer was removed, and the aqueous layer was washed with toluene (20 mL). Isopropyl acetate (EPAc) (30 mL) was added to the aqueous layer. Concentrated HCl (8 mL) was then added to acidify the aqueous layer to pH 1. The aqueous layer was removed and the organic layer was washed twice with dilute HCl. <n="16"/>The IPAc solution was concentrated to about 13 mL and heptane (40 mL) was added to crystallize the product. The suspension was filtered, rinsing with heptane, giving 4.22 g (69% yield) of 1-2.
Step 1 To a solution of 2-aminoethylamine LVIII (150 mL; 2.25 mol) in chloroform (1.5 L), cooled to 0 C. was added a solution of carbobenzoxy N-hydroxysuccinimide (112.14 g; 0.45 mol) in water (0.5 L) with vigorously stirring for one hour. The solid was filtered and the solution was washed with brine (3*), once with water and dried over anhydrous MgSO4. The solvent was evaporated under reduced pressure and the residue was purified on a silica gel column (100:1?30:1 DCM:MeOH) to yield benzyl 2-aminoethylcarbamate LIX as a colorless viscous oil (350 g; 1.80 mol; 80% yield). 1H NMR (CDCl3) 2.79-2.83 (m, 2H), 3.21-3.28 (m, 2H), 5.10 (s, 2H), 5.19 (brs, 1H), 7.29-7.34 (m, 1H), 7.35-7.38 (m, 4H); ESIMS found for C10H14N2O2 m/z 195.2 (M+H).
In dichloromethane; for 2.5h;
A solution of N-(benzyloxycarbonyloxy)succinimide (ZOSu, 100 g, 401 mmol) in dichloromethane (500 mL) was added dropwise over 2 hours to a solution ofethylenediamine (1, 189 mL, 2.81 mol) in dichloromethane (750 mL). After 30 minutes the suspension was filtered and solids washed with dichloromethane. The filtrate was evaporated to dryness and the residue diluted with toluene (1.00 L) and water (0.50 L). The resulting mixture was filtered and the filtrate was separated to afford two phases. The aqueous phase contained the product; therefore it was extracted withdichloromethane (2 x 250 mL). All organic phases were combined, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was diluted with toluene (750 mL) and extracted with 2 M aqueous hydrochloric acid (500 mL) and 1 M aqueous hydrochloric acid (100 mL). Acidic aqueous phases were combined and basified with a solution of sodium hydroxide (60.0 g, 1.50 mol) in water (90 mL). The resulting mixture was extracted with dichloromethane (4 x 200 mL), dried over anhydrous sodium sulfate, filtered, concentrated in vacuo and diluted with hexanes (200 mL). 4 M Solution of hydrogen chloride in ether (100 mL, 400 mmol) was added to the solution, the resulting suspension was concentrated in vacuo and diluted with hexanes (1.00 L). Theprecipitated solid was filtered, washed with hexanes and dried in vacuo to give (2-amino- ethyl)-carbamic acid benzyl ester hydrochloride as white powder.Yield : 62.62 g (68%).RF (Si02, dichloromethane/methanol 4: 1) : 0.25 (free base).1H NMR spectrum (300 MHz, AcOD-d4, 80 C, dH) : 7.42-7.26 (m, 5 H); 5.16 (s, 2 H); 3.60 (t, J = 5.7 Hz, 2 H); 3.32 (t, J = 5.7 Hz, 2 H).
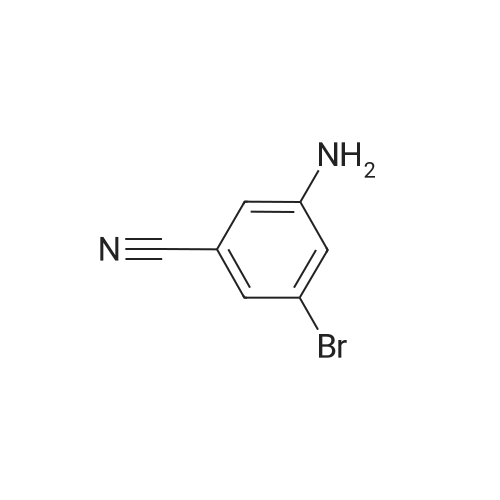
To a stirring of 965 mg (4.90 mmol) of <strong>[49674-16-0]3-amino-5-bromobenzonitrile</strong> in 15 mL of CH2Cl2 was added 1.27 g (5.09 mmol) of N-(benzyloxycarbonyloxy)succinimide (followed by 5 mL of CH2Cl2), and 1.5 mL (10.8 mmol) of Et3N. After 27 h, the solution was concentrated and 10 mL of 10% citric acid and EtOAc were added. The layers were <n="73"/>separated, and the organic layer was washed with 10 rnL of 10% citric acid, 20 rnL of H2O, and 10 rnL of brine, dried over Na2SO4, filtered, and concentrated. Purification by flash silica gel chromatography (25% EtOAc/hexanes) provided 773 mg of Cbz protected product(benzyl 3-bromo-5-cyanophenylcarbamate) with some impurity.
(R)-3-piperidinecarboxylic acid ethyl ester (+)-tartrate[ No CAS ]
[ 160706-62-7 ]
Yield
Reaction Conditions
Operation in experiment
85%
R (-) [ETHYINIPECONATE] tartaric acid salt (117 g, 382 [MMOL)] was dissolved in 2N [NAOH] (1200 ml, 2.40 mol) and cooled to 5 [C.] [Z-OSU] (100 g, 401 [MMOL)] dissolved in 100 [ML] THF was added to the chilled reaction. The mixture was allowed to stir at 5 [C] for 1 h. and at RT overnight. Approx. 100 ml of solvent was removed by rotary evaporation in vacuo, and the remaining solution was acidified to pH 2-3 with conc. HCI (app. 75 [ML).] The resulting crystal were isolated by filtration and dried in vacuo overnight. Yield 86 g (85 %) 1H-NMR (dmso-d6,400 MHz) [D :] 12.5 (s broad, 1H), (7.42-7. 30 (m, 5H), 5.1 (s, 2H), 4.0 (s, [1H),] 3. [80] (s, 1H), 3.12 (m, [1H),] 2.92 (t, 1H), 2.36 (m, 1H), 1.92 (m, 1H), 1.7-1. 48 (m, 2H), 1.48-1. 30 (m, [1 H).] HPLC-MS (Method B): [MILZ =] 264 (M+1), Rt = 3.30 min.
B) 3-[Benzyloxycarbonyl-((R)-2-tert-butoxycarbonylamino-1-methyl-ethyl)-amino]-propionic acid methyl ester; 72 g (413 mmol) <strong>[333743-54-7]((R)-2-amino-propyl)-carbamic acid tert-butyl ester</strong> were dissolved in MeOH (350 ml) in a jacketed reactor. A solution of 40 ml (438 mmol) methyl acrylate in MeOH (50 ml) was added over 10 min. After 15 h reaction at RT, 108 g (413 mmol) of N-benzyloxycarbonyloxy-succinimide were added in one portion. After 18 h, the reaction mixture was concentrated under reduced pressure affording a yellow oil (ca 270 g). The oil was re-dissolved in 300 mL MeOH and concentrated under reduced pressure (45 C./160-200 mbar) until constant weight to give 219 g of crude titled product as a yellow oil used directly in the next step.
EXAMPLE 1; 7V-Biphenyl-2-yl- 1 ,3-dioxo-2- [^r°ws-2-phenylcyclopr opyll hexahydr oimidazo [ 1 ,5-al pyrazine- 7qH)-carboxamide (A6); Step 1 : l-[(benzyloxy)carbonyll-4-(tert-butoxycarbonyl)piperazine-2-carboxylic acid (Al); A solution 0.164 M of <strong>[3022-15-9]2-piperazinecarboxylic acid dihydrochloride</strong> in a 1 :1 dioxane-water mixture was made basic (pH 11) with 50% aqueous NaOH. A 0.68 M solution of Boc-ON (1.1 eq) in dioxane was added dropwise at RT to the above mixture and the reaction mixture was stirred at RT overnight. The mixture was extracted with Et2O (x3) and acidified with cone. HCl to pH 2. The aqueous layer was extracted with EtOAc (x3). The aqueous solution was basified to pH 10 with 50% NaOH. A 0.59 M solution Lambda/-(benzyloxycarbonyloxy)succinimide (1.1 eq.) in dioxane was added to the mixture at 00C. The reaction mixture was stirred 3 h at RT. Dioxane was removed under reduced pressure. The basic solution was extracted with Et2O (x2) then acidified to pH 1 with cone. HCl, and extracted with EtOAc (x3). The combined organic layers were dried and evaporated. The residue was used as such in next step.
S-(-)-piperazine-2-carboxylic acid dihydrochloride[ No CAS ]
[ 150407-69-5 ]
Yield
Reaction Conditions
Operation in experiment
PREPARATIVE EXAMPLE 1; (1-tert-Butyl 3-methyl (3S)-l,3-piperazinedicarboxylate (AA3); Step 1 : (261-l-[(Benzyloxy)carbonyll-4-(tert-butoxycarbonyl)-2-piperazinecarboxylic acid(AAl); It was prepared from (25)-2-piperazinecarboxylic acid dihydrochloride with slight modifications as described in Molecular Discovery 1998, 4, 221-232. A solution of (25)-2-piperazinecarboxylic acid dihydrochloride in a dioxane-water mixture (1 :1, 0.164 M) was made basic (pH 11) with 50% aqueous NaOH. A 0.68 M solution of BOC-ON (1.1 eq.) in dioxane was added dropwise at RT to the above mixture and the reaction mixture was stirred at RT overnight. The mixture was extracted with Et2O (x3) and acidified with cone. HCl to pH 2. The aqueous layer was extracted with EtOAc (x3). The aqueous solution was basified to pH 10 with 50% NaOH. A solution Lambda/-(benzyloxycarbonyloxy)succinimide in dioxane (0.59 M) was added to the mixture at 00C. The reaction mixture was stirred 3 h at RT. Dioxane was removed under reduced pressure. The basic solution was extracted with Et2O (x2) then acidified to pH 1 with cone. HCl, and extracted with EtOAc (x3). The combined organic layers were dried and evaporated. The residue was used as such in next step.
Preparation of Intermediate 1 (Int. l); (Z)-tert-Butyl 1 -(4-(N'-hydroxycarbamimidoyl)-benzyl)azetidine-3 -carboxylate; Int.1-A. l-(Benzyloxycarbonyl)azetidine-3-carboxylic acid [00103] To a solution of azetidine-3-carboxylic acid (88 g, 0.871 mol) and sodium bicarbonate (161 g, 1.92 mol) in water (1.75 L) at room temperature was added a solution of benzyl 2,5-dioxopyrrolidin-l-ylcarbonate (239 g, 0.959 mol) in tetrahydrofuran (3.5 L). The reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the aqueous layer was washed with ethyl acetate (2 x 500 mL). The aqueous layer was acidified with a 1.0 N aqueous solution of hydrochloric acid and was then extracted with ethyl acetate (3 x 750 mL). The organic layer was washed with water, followed by brine, and dried over anhydrous sodium sulfate. Concentration under reduced pressure afforded 1- (benzyloxycarbonyl) azetidine-3-carboxylic acid as colorless oil (202 g, 99percent yield). The compound had an HPLC retention time = 2.27 min. - Column: YMC COMBISCREEN.(R). ODS-A 4.6 x 50 mm (4 min.); Solvent A = 10percent MeOH, 90percent H2O, 0.1percent TFA; Solvent B = 90percent MeOH, 10percent H2O, 0.1percent TFA. LC/MS M+1 = 236.15. 1H NMR (400 MHz, CDCl3) delta ppm 3.39 - 3.49 (m, IH), 4.22 (d, J=7.28 Hz, 4H), 5.11 (s, 2H), and 7.29 - 7.39 (m, 5H).
99%
Preparation of Intermediate 1 (Int. l); tert-Butyl 1 -(4-(N'-hydroxycarbamimidoyl)-benzyl)azetidine-3 -carboxylate; Int.1-A. l-(Benzyloxycarbonyl)azetidine-3-carboxylic acid; [00109] To a solution of azetidine-3-carboxylic acid (88 g, 0.871 mol) and sodium bicarbonate (161 g, 1.92 mol) in water (1.75 L) at room temperature was added a solution of benzyl 2,5-dioxopyrrolidin-l-ylcarbonate (239 g, 0.959 mol) in tetrahydrofuran (3.5 L). The reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the aqueous layer was washed with ethyl acetate (2 x 500 mL). The aqueous layer was acidified with a 1.0 N aqueous hydrochloric acid solution and was then extracted with ethyl acetate (3 x 750 mL). The organic layer was washed with water, followed by brine, and dried over anhydrous sodium sulfate. Concentration under reduced pressure afforded 1- (benzyloxycarbonyl) azetidine-3-carboxylic acid as colorless oil (202 g, 99percent yield). The compound had an HPLC retention time = 2.27 min. - Column: YMC COMBISCREEN.(R). ODS-A 4.6 x 50 mm (4 min.); Solvent A = 10percent MeOH, 90percent H2O, 0.1percent TFA; Solvent B = 90percent MeOH, 10percent H2O, 0.1percent TFA. LC/MS M+1 = 236.15. 1H NMR (400 MHz, CDCl3) delta ppm 3.39 - 3.49 (m, IH), 4.22 (d, J=7.28 Hz, 4H), 5.11 (s, 2H), and 7.29 - 7.39 (m, 5H).
99%
Preparation 25A: l-(Benzyloxycarbonyl)azetidine-3 -carboxylic acidCbZlM^-CO2Hv (25A) [00262] To a solution of azetidine-3-carboxylic acid (88 g, 0.871 mol) and sodium bicarbonate (161 g, 1.92 mol) in water (1.75 L) at room temperature was added a solution of benzyl 2,5-dioxopyrrolidin-l-ylcarbonate (239 g, 0.959 mol) in tetrahydrofuran (3.5 L). The reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the aqueous layer was washed with ethyl acetate (2 x 500 mL). The aqueous layer was acidified with a 1.0 N aqueous hydrochloric acid solution and was then extracted with ethyl acetate (3 x 750 mL). The organic layer was washed with water, followed by brine, and dried over anhydrous sodium sulfate.Concentration under reduced pressure afforded l-(benzyloxycarbonyl) azetidine-3- carboxylic acid as colorless oil (202 g, 99percent yield). The compound had an HPLC retention time = 2.27 min. - Column: YMC COMBISCREEN.(R). ODS-A 4.6 x 50 mm (4 min.); Solvent A = 10percent MeOH, 90percent H2O, 0.1percent TFA; Solvent B = 90percent MeOH, 10percent H2O, 0.1percent TFA. LC/MS M+1 = 236.15. 1H NMR (400 MHz, CDCl3) delta ppm 3.39 - 3.49 (m, IH), 4.22 (d, J=7.28 Hz, 4H), 5.11 (s, 2H), and 7.29 - 7.39 (m, 5H).
99%
With sodium hydrogencarbonate; In tetrahydrofuran; water; at 20℃;
INTERMEDIATE 6tert-Butyl azetidine-3 -carboxylate acetic acid saltStep A: l-(Benzyloxycarbonyl)azetidine-3-carboxylic acidCbzN-C°2H (I-6A)[00202] To a solution of azetidine-3 -carboxylic acid (88 g, 0.871 mol) and sodium bicarbonate (161 g, 1.92 mol) in water (1.75 L) at room temperature was added a solution of benzyl 2,5-dioxopyrrolidin-l-ylcarbonate (239 g, 0.959 mol) in THF (3.5 L). The reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the aqueous layer was washed with ethyl acetate (2 x 500 mL). The aqueous layer was acidified with a 1.0 N aqueous hydrochloric acid solution and was then extracted with ethyl acetate (3 x 750 mL). The organic layer was washed with water, followed by brine, and dried over anhydrous sodium sulfate. Concentration under reduced pressure afforded l-(benzyloxycarbonyl) azetidine-3 -carboxylic acid as colorless oil (202 g, 99percent yield). The compound had an HPLC retention time = 2.27 min (condition B); LC/MS M+1 = 236.15. XH NMR (400 MHz, CDC13) delta ppm 3.39 - 3.49 (m, 1H), 4.22 (d, J=7.28 Hz, 4H), 5.1 1 (s, 2H), and 7.29-7.39 (m, 5H).
99%
With sodium hydrogencarbonate; In tetrahydrofuran; water; at 20℃;
1-A: l-(Benzyloxycarbonyl)azetidine-3 -carboxylic acid[0119] To a solution of azetidine-3 -carboxylic acid (88 g, 0.871 mol) and sodium bicarbonate (161 g, 1.92 mol) in water (1.75 L) at room temperature was added a solution of benzyl 2,5-dioxopyrrolidin-l-ylcarbonate (239 g, 0.959 mol) in tetrahydrofuran (3.5 L). The reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the aqueous layer was washed with ethyl acetate (2 x 500 mL). The aqueous layer was acidified with a 1.0 N aqueous solution of hydrochloric acid and extracted with ethyl acetate (3 x 750 mL). The organic layer was washed with water, washed with brine, and dried over anhydrous sodium sulfate. Concentration under reduced pressure afforded l-(benzyloxycarbonyl) azetidine-3 -carboxylic acid as colorless oil (202 g, 99percent yield). The compound had an HPLC retention time = 2.27 min. - Column: Column: YMC Combiscreen ODS-A 4.6 x 50 mm (4 min.); Solvent A = 10percent MeOH, 90percent H20, 0.1percent TFA; Solvent B = 90percent MeOH, 10percent H20, 0.1percent TFA. LC/MS M+1 = 236.15. XH-NMR (400 MHz, CDC13) delta ppm 3.39-3.49 (m, 1H), 4.22 (d, J=7.28 Hz, 4H), 5.11 (s, 2H), and 7.29-7.39 (m, 5H).
99%
Preparation of Intermediate 1 (Int. l); tert-Butyl 1 -(4-(N'-hydroxycarbamimidoyl)-benzyl)azetidine-3 -carboxylate; Int. l-A. l-(Benzyloxycarbonyl)azetidine-3-carboxylic acid; [00101] To a solution of azetidine-3-carboxylic acid (88 g, 0.871 mol) and sodium bicarbonate (161 g, 1.92 mol) in water (1.75 L) at room temperature was added a solution of benzyl 2,5-dioxopyrrolidin-l-ylcarbonate (239 g, 0.959 mol) in tetrahydrofuran (3.5 L). The reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the aqueous layer was washed with ethyl acetate (2 x 500 mL). The aqueous layer was acidified with a 1.0 N aqueous solution of hydrochloric acid and extracted with ethyl acetate (3 x 750 mL). The organic layer was washed with water, washed with brine, and dried over anhydrous sodium sulfate. Concentration under reduced pressure afforded 1- (benzyloxycarbonyl) azetidine-3-carboxylic acid as colorless oil (202 g, 99percent yield). The compound had an HPLC retention time = 2.27 min. - Column: YMC COMBISCREEN.(R). ODS-A 4.6 x 50 mm (4 min.); Solvent A = 10percent MeOH, 90percent H2O, 0.1percent TFA; Solvent B = 90percent MeOH, 10percent H2O, 0.1percent TFA. LC/MS M+1 =236.15. 1H NMR (400 MHz, CDCl3) delta ppm 3.39 - 3.49 (m, IH), 4.22 (d, J=7.28 Hz, 4H), 5.11 (s, 2H), and 7.29 - 7.39 (m, 5H).
With triethylamine; In water; at 20℃; for 0.5h;
To azetidine-3-carboxylic acid (49.5 mmol, 5 g) in dioxane (150 ml), water (150 ml) and triethylamine (54.4 mmol, 7.58 ml) was added N-(benzyloxycarbonyloxy)succinimide (51.9 mmol, 12.94 g) and stirred at room temperature for half an hour. The reaction mixture was quenched with water and 30 ml of 2N hydrochloric acid and extracted three times with dichloromethane. The organic layers were combined, filtered through a phase separation filter and concentrated in vacuo. The residue was dissolved in dichloromethane and extracted three times with aqueous saturated sodium hydrogencarbonate solution. The aqueous layers were combined, acidified with 2N hydrochloric acid and extracted three times with dichloromethane. The organic layers were combined, washed with brine, filtered through a phase separation filter and concentrated in vacuo to give 1-(benzyloxycarbonyl)azetidine-3-carboxylic acid (10.27 g).
54a. 4-((Benzyloxy)carbonyl)-1 -(tert-butoxycarbonyl)piperazine-2- carboxylic acidThe tile compound was prepared according to the procedure of [Kempf, D. J.; Norbeck, D. W.; Sham, H. L U.S. Patent 5,455,351 , Oct 3, 1995]. Piperazine-2-carboxylic acid (10.0 g, 77.0 mmol) was dissolved in a 1 :1 solution of 1 ,4-dioxane:water (100 mL) at room temperature with vigorous stirring. The clear solution was adjusted to pH 11 by the addition of an aqueous solution of sodium hydroxide (80 mL of a 1Lambda/ solution). The pH was monitored in situ with a pH meter throughout the reaction. The reaction flask was fitted with an addition funnel that contained a solution of Lambda/-alpha- (benzyloxycarbonyloxy) succinamide (13.6 g, 55 mmol) in 1 ,4-dioxane (50 mL). The Lambda/-alpha-(benzyloxycarbonyloxy) succinamide solution was added over 45 minutes at room temperature and the pH was kept above 10 by the periodic addition of 1 Lambda/ sodium hydroxide. The pH of the solution was adjusted to 9.5 and 2-(teAf-butoxycarbonyloxyimino)-2-phenylacetonitrile (13.4 g, 55 mmol) was added as a solution in 1 ,4-dioxane (50 mL) over 10 minutes. The pH was maintained at 9.5 and the solution was stirred at room temperature for 17 hours. The solution was then acidified to pH 2 and the aqueous solution was washed with diethyl ether (3 x 150 mL). The aqueous solution was cooled to O0C and acidified by adding of concentrated hydrochloric acid. The acidic solution was extracted with ethyl acetate (5 x 150 mL). The combined organic phases were dried over sodium sulfate, filtered and concentrated in vacuo. The residue was triturated with a 1 :1 solution of dichloromethane: hexanes (150 mL) and the solvent was removed in vacuo to provide the product as a viscous yellow oil (15.7 g, 43 mmol, 80%). Rf = 0.60 (66:34 dichloromethane: ethyl acetate + 0.1% (v/v acetic acid); 1H-NMR (400 MHz, DMSO) delta 13.0 (br s, 1 H), 7.37-7.36 (m, 5H), 5.05 (s, 2H), 4.54-4.33 (m, 2H), 3.90-3.66 (m, 2H), 3.07-2.81 (m, 4H), 1.38 (s, 9H); Mass spectrum (ESI +ve) m/z 365.1 (MH+).
Synthesis and characterization of per N-cbz derivatives of neomycin, kanamycin, and apramycin have been reported.refPreviewPlaceHolder49 and refPreviewPlaceHolder[50] Synthesis of the per-N-cbz derivatives of these aminoglycosides used N-(benzyloxycarbonyloxy)succinimide (NHS-cbz) as previously reported for introducing cbz groups onto free amine groups of N-desulfonated heparin.refPreviewPlaceHolder17 Briefly, aminoglycoside (0.2-0.5 mmol) was dissolved in aqueous NaHCO3 and stirred at room temperature. Aliquots of NHS-cbz (1.2 equiv per amine moiety) dissolved in 4.5 mL DMF were then added to the above solution fraction-wise (0.9 mL) at 60-90 min intervals. During the reaction, a white solid precipitate was formed. The reaction was monitored using analytical HPLC: 20 muL of the reaction mixture and was diluted in 80 muL ACN and 40 muL water; 100 muL of this solution was injected and eluted with a gradient of 10-95% ACN in water (0.1% TFA) over 40 min at 1 mL/min. After complete acylation of amine groups, ?5-7 h, the reaction was diluted with water (4 mL) to give additional white precipitate. The precipitate was collected by centrifugation (20 min, 4 C, 3500 rpm) and decanting followed by washing the solid with cold water (10 mL), centrifugation and decanting. This wash procedure was repeated seven times followed by lyophilization of a final suspension to give dry white solid. In reactions where the N-acylation was incomplete, as detected by analytical HPLC, under-reacted product was removed by dissolving the product in ACN/water (3:1) and passing the solution through a column of amberlite cation (H+) exchange resin (4 mL bed volume).CommentIn some reactions, over-acylated product (having at least one O-cbz) was detected by ESI-MS; the desired product was then purified using semi-preparative HPLC. To this end, the product mixture was dissolved in 3:1 ACN/water and replicate aliquots were injected and separated. The per-N-cbz product was collected after about 20 min, depending on the parent aminoglycoside, when eluted with 67% ACN in water with 0.1% TFA, 7 mL/min. Product purified with cation exchange or semi-prep chromatography was rotary evaporated to remove organic solvent and the resulting aqueous suspension lyophilized to give a white solid.
benzyl 6,7-dihydro-1H-imidazo[4,5-c]pyridine-5(4H)-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
63%
With sodium carbonate; In 1,4-dioxane; water; at 0 - 20℃;
N-(Benzyloxycarbonyloxy)succinimide was added portion wise to a 0 C solution of 4,5,6,7-tetrahydro-lH-imidazo[4,5-c]pyridine dihydrochloride (530 mg, 2.70 mmol) and sodium bicarbonate (625.4 mg, 7.44 mmol) in l,4-dioxane/water (1 : 1, 20 mL). The resulting solution stirred overnight at room temperature. The reaction mixture was then poured into 50 mL of water, and extracted with 2x50 mL of ethyl acetate. The combined organic phases were washed with 100 mL of brine, dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The residue was purified via column chromatography on silica gel (eluting with 10: 1 dichloromethane/methanol) to afford benzyl 6,7-dihydro-lH-imidazo[4,5-c]pyridine-5(4H)- carboxylate (440 mg, 63%) as colorless oil. MS: (ESI, m/z): 258[M+H]+.
With sodium hydrogencarbonate; In 1,4-dioxane; water; at 20℃; for 16h;
A mixture of 2.0 g of <strong>[62002-31-7]4,5,6,7-tetrahydro-1H-imidazo[4,5-c]pyridine dihydrochloride</strong>, 2.36 g of NaHCO3 and 2.45 g of N-(benzyloxycarbonyloxy)succinimide in 80 ml of dioxane/water mixture (50/50; v/v) is stirred for 16 hours at RT. The reaction mixture is extracted with EtOAc, the organic phase is washed with a saturated solution of NaHCO3, with 0.1M HCl solution, with saturated NaCl solution, it is dried and the solvent is evaporated under vacuum. The residue is chromatographed on silica gel, eluting with DCM/MeOH mixture. 1.9 g of the expected compound is obtained.
With sodium hydrogencarbonate; In 1,4-dioxane; water; at 20℃; for 16h;
Preparation 7.71 -(2-Methoxyethyl)-4,5,6,7-tetrahydro-1 /-/-imidazo[4,5-c]pyridine.Step 1 : Benzyl 1 ,4,6,7-tetrahydro-5/-/-imidazo[4,5-c]pyridine-5-carboxylate.A mixture of 2.0 g of 4,5,6,7-tetrahydro-1 H-imidazo[4,5-c]pyridine dihydrochloride, 2.36 g of NaHCO3 and 2.45 g of N- (benzyloxycarbonyloxy)succinimide in 80 ml of dioxane/water mixture (50/50; v/v) is stirred for 16 hours at RT. The reaction mixture is extracted with EtOAc, the organic phase is washed with a saturated solution of NaHCO3, with 0.1 M HCI solution, with saturated NaCI solution, it is dried and the solvent is evaporated under vacuum. The residue is chromatographed on silica gel, eluting with DCM/MeOH mixture. 1 .9 g of the expected compound is obtained.
To 2-(2-aminoethoxyl)ethanol (20 g) dissolved in dichloromethane (200 ml) was added stepwise triethylamine (26.5 ml) and benzyloxy(carbonyloxy)succinimide (47.47 g). The reaction mixture was stirred overnight at room temperature. The solvent was distilled off under reduced pressure and the residue was dissolved in deionized water (500 ml). The insoluble layer was separated, and the aqueous layer was mixed with 50 g of NaCl and the pH adjusted to 5.0 with 5% H3PO4. The product was extracted with dichloromethane (100 ml, 50 ml, and 50 ml) and the extract was dried with MgSO4, filtered, and evaporated to dryness under reduced pressure. The residue was dried under vacuum overnight, then combined with the previously separated insoluble layer and dissolved in dichloromethane (300 ml). The solution was washed with 5-% aqueous NaCl solution (3×50 ml) and dried with MgSO4. The solvent was distilled under reduced pressure, and the residue was dried overnight under vacuum, giving 40 g of the desired product.
With sodium hydrogencarbonate; In tetrahydrofuran; water; at 20℃; for 1h;
Piperidin-4-ylmethanol (5.2 g, 45.1 mmol) was dissolved in 60 mE of THF and 60 mE of saturated NaHCO3 aq and then benzyl (2,5-dioxopyrrolidin- 1 -yl) carbonate (11.25 g, 45.1 mmol) added portionwise. The mixture was stirred for 1 h at RT, partitioned between AcOEt and water, washed twice with 1M HClaq and saturated NaClaq. Organic layer was dried over Na2504, evaporated under reduced pressure to afford the title compound (10.77 g, 43.2 mmol, 96% yield). The product obtained was used in the following steps without thrther purifications. UPLC-MS: 0.80 mm, 250.2 [M+H]+, method 1.
methyl 3-(benzyloxycarbonylamino)oxetane-3-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
62%
With sodium hydrogencarbonate; In tetrahydrofuran; water; at 0 - 22℃; for 24h;
To a suspension of benzyl (2,5-dioxopyrrolidin-1-yl) carbonate (2.34 g) in tetrahydrofuran (2.4 ml) was added at 0C a suspension of <strong>[1363383-31-6]methyl 3-aminooxetane-3-carboxylate</strong> (1 .0 g, prepared according to Eickmeier, C. et al. , US 20 3/0225593) in water (3.6 ml) followed by a suspension of sodium bicarbonate (1 .41 g) in water (7.2 ml) and stirring was continued at 22C for 24 h. The mixture was extracted with ethyl acetate, the organic layer was dried, evaporated and the residue purified by flash chromatography (silica gel, n-heptane/ethyl acetate 3: 1 ) to give the title compound (1 .1 1 g, 62%) as a colorless oil. MS (ESI , m/z): 266.1 [(M+H)+].
1-(((2-(4-amino-2,5-dioxo-2,3,4,5-tetrahydrobenzo[b][1,4]dioxocin-8-yl)ethyl)carbamoyl)oxy)ethyl isobutyrate hydrofluoride[ No CAS ]
1-(((2-(3-amino-2,5-dioxo-2,3,4,5-tetrahydrobenzo[b][1,4]dioxocin-8-yl)ethyl)carbamoyl)oxy)ethyl isobutyrate hydrofluoride[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In this example, a dopamine · HCl solution was prepared by adding dopamine · HCl (19 g) to acetone (200 mL). Water (200 mL) and sodium hydrogencarbonate (R0090, 50 g) were added to the dopamine · HCl solution to obtain a first mixture. Next, Nalpha- (benzyloxycarbonyloxy) succinimide (25 g) was added to the first mixture to obtain a second mixture. The second mixture was stirred at RT overnight and ethyl acetate (R0061, 500 mL) was added for extraction. The organic layer was separated, washed with water (2 × 100 mL), 20% citric acid (2 × 200 mL), and water (3 × 100 mL), respectively, and dried over sodium sulfate to give a third solution. Sodium sulfate was then filtered and washed with ethyl acetate to give a fourth mixture. The third solution and the fourth mixture were combined and evaporated to dryness. The obtained residue and Boc-L-aspartic acid (24 g) were dissolved in acetone (300 mL) to obtain a fifth mixture. 1-Ethyl- (3-dimethylaminopropyl) carbodiimide hydrochloride (40 g) and 4-dimethylaminopyridine (22 g) were added to the fifth mixture to obtain a sixth mixture. The sixth mixture was stirred at RT overnight and evaporated to dryness. Ethyl acetate (500 ml) was added to the residue to give a seventh mixture and washed with water (2 × 100 ml), 5% sodium bicarbonate (3 × 100 ml), water (100 ml), 20% citric acid (2 × 200 ml) And washed with water (3 × 100 mL), respectively, and dried over sodium sulfate to obtain an eighth solution. The sodium sulfate was filtered and washed with ethyl acetate to give a ninth solution. The eighth solution and the ninth solution were combined and evaporated to dryness. The residue was dissolved in methanol (R0084, 300 mL) to obtain a tenth mixture. Palladium activated carbon (10 g, 10%) was first added to the tenth mixture under nitrogen and then bubbled with hydrogen gas to remove the benzyloxycarbonyl group at RT. The resulting mixture (eleventh mixture) was filtered to remove palladium activated carbon and evaporated to dryness.The residue was suspended in DCM (200 mL) to give a twelfth mixture. Sodium hydrogencarbonate (20 g) and tetrabutylammonium hydrogen sulfate (11 g) were added to the twelfth mixture to give a thirteenth mixture. 1-Chloroethyl chloroformate (16 g) was then added to the thirteenth mixture to give a fourteenth mixture. The 14th mixture was stirred at RT overnight. The organic layer of the 14th mixture was then collected, washed with water (3 x 200 mL) and dried over anhydrous sodium sulfate to give a 15 th solution. The sodium sulfate was removed by filtration and washed with DCM to give a 16th solution. The 15th solution and the 16th solution were combined and evaporated to dryness. The residue was collected and dissolved in isobutyric acid (100 mL) to give a 17 th mixture. First, a mixture of diisopropylethylamine (60 mL) and isobutyric acid (36 mL) was prepared and then added to the 17 th mixture to obtain an 18 th mixture. The 18 th mixture was stirred at 55 C. for 48 h and ethyl acetate (500 mL) was added with stirring. The resulting organic layer was collected, washed with 5% sodium bicarbonate (3 × 100 mL) and water (3 × 100 mL), respectively, and dried over anhydrous sodium sulfate to give a nineteenth solution. The sodium sulfate was filtered and washed with ethyl acetate to give a twentieth solution. The 19th solution and the 20th solution were combined and concentrated to 300 mL. Anisole (20 g) was added to the concentrated solution and then HF gas (20 g) was bubbled to cause precipitation. The precipitated solid was collected and washed with ethyl acetate to give 1 - (((2- (4-amino-2,5-dioxo-2,3,4,5-tetrahydrobenzo [b] [1,4] dioxo- 8 - yl) ethyl) carbamoyl) oxy) ethyl isobutyrate fluorohydrochloride and 1 - (((2- (3 - amino - 2,5 - dioxo - 2, 3, 4, 5 - tetrahydrobenzo [b] [ 1, 4] dioxocin-8-yl) ethyl) carbamoyl) oxy) ethylisobutyrate fluorohydrochloride.
isopropyl (S)-2-amino-3-(3,4-dihydroxyphenyl)propanoate hydrochloride[ No CAS ]
(2S)-isopropyl-3-(3-amino-2,5-dioxo-2,3,4,5-tetrahydrobenzo[b][1,4]-dioxocin-8-yl)-2-(((1-(isobutyryloxy)ethoxy)carbonyl)amino)propanoate hydrobromide[ No CAS ]
(2S)-isopropyl-3-(4-amino-2,5-dioxo-2,3,4,5-tetrahydrobenzo[b][1,4]-dioxocin-8-yl)-2-(((1-(isobutyryloxy)ethoxy)carbonyl)amino)propanoate hydrobromide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In this example, (S)-2-amino-3-(3,4-dihydroxyphenyl)propanoic acid (20g) was added into isopropanol (200 mL)to provide a 1st mixture. HCl gas (20g) was bubbled into the1st mixture to provide a 2nd mixture. The 2nd mixture wasstirred for 2 days at 60 C., and then added with isopropylacetate (200 mL) to provide precipitation. The precipitatedSolid was collected and washed with isopropayl acetate toprovide isopropyl (S)-2-amino-3-(3,4-dihydroxyphenyl)propanoate hydrochloride. An isopropyl (S)-2-amino-3-(3,4-dihydroxyphenyl)propanoate hydrochloride solution was prepared by adding isopropyl (S)-2-amino-3-(3,4-dihydroxyphenyl)propanoatehydrochloride (28 g) into acetone (200 mL). Water (200 mL)and sodium bicarbonate (R0090, 50 g) were added into thedopamine.HCl solution to provide a 3rd mixture. ThenNC.-(benzyloxycarbonyloxy)succinimide (25 g) was addedto the 3rd mixture to provide a 4th mixture. The 4th mixturewas stirred at RT overnight, and added ethyl acetate (R0061,500 mL) for extraction. The organic layer was separated, andwashed with water (2x100 mL), 20% citric acid (2x200mL), and water (3x100 mL), respectively, and dried oversodium sulfate to provide a 5th solution. Then the sodiumsulfate was filtered and washed with ethyl acetate to providea 6th solution. The 5th solution and the 6th mixture werecombined, and evaporated to dryness. The obtained residueand Boc-L-aspartic acid (24 g) were dissolved in acetone(300 mL) to provide a 7th mixture. 1-Ethyl-(3-dimethylaminopropyl)carbodiimide hydrochloride (40 g) and 4-dimethylaminopyridine (22 g) were added into the 7th mixture toprovide an 8th mixture. The 8th mixture was stirred overnight at RT and evaporated to dryness. Ethyl acetate (500ml) was added into the residue to provide a 9th mixture,which was washed with water (2x100 mL), 5% sodium
Solid Phase Synthetic protocol: A solution of N-(benzyloxycarbonyloxy)succinimide (ZOSu, 100 g, 401 mmol) in dichloromethane (500 ml) was added dropwise over 2 hours to a solution of 5 ethylenediamine (1, 189 ml, 2.81 mol) in dichloromethane (750 ml). After 30 minutes the suspension was filtered and solids washed with dichloromethane. The filtrate was evaporated to dryness and the residue diluted with toluene (1.00 L) and water (0.50 L). The resulting mixture was filtered and the filtrate was separated to afford two phases. The aqueous phase contained the product; therefore it was extracted with 10 dichloromethane (2 x 250 ml). All organic phases were combined, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was diluted with toluene (750 ml) and extracted with 2 M aqueous hydrochloric acid (500 ml) and 1 M aqueous hydrochloric acid (100 ml). Acidic aqueous phases were combined and basified with a solution of sodium hydroxide (60.0 g, 1.50 mol) in water (90 ml). The resulting mixture 15 was extracted with dichloromethane (4 x 200 ml), dried over anhydrous sodium sulfate, filtered, concentrated in vacuo and diluted with hexanes (200 ml). 4 M Solution of hydrogen chloride in ether (100 ml, 400 mmol) was added to the solution, the resulting suspension was concentrated in vacuo and diluted with hexanes (1.00 L). The precipitated solid was filtered, washed with hexanes and dried in vacuo to give (2-amino20 ethyl)-carbamic acid benzyl ester hydrochloride as white powder. 25 Yield: 62.62 g (68%). RF (Si02, dichloromethane/methanol 4: 1): 0.25 (free base). 1H NMR spectrum (300 MHz, Ac0D-d4, 80 C, dH): 7.42-7.26 (m, 5 H); 5.16 (s, 2 H); 3.60 (t, J=5.7 Hz, 2 H); 3.32 (t, J=5.7 Hz, 2 H).
Example 10.2 (C16) Preparation of 16-[[(1S)-4-[2-[2-[2-[2-[2-[2-[2-[2-bromoacetyl)amino]ethylamino]-2-oxo-ethoxy]ethoxy]ethylamino]-2-oxo-ethoxy]ethoxy]ethylamino]-1-carboxy-4-oxo-butyl]amino]-16-oxo-hexadecanoic acid Solid Phase Synthetic protocol: A solution of N-(benzyloxycarbonyloxy)succinimide (ZOSu, 100 g, 401 mmol) in dichloromethane (500 mL) was added dropwise over 2 hours to a solution of ethylenediamine (1, 189 mL, 2.81 mol) in dichloromethane (750 mL). After 30 minutes the suspension was filtered and solids washed with dichloromethane. The filtrate was evaporated to dryness and the residue diluted with toluene (1.00 L) and water (0.50 L). The resulting mixture was filtered and the filtrate was separated to afford two phases. The aqueous phase contained the product; therefore it was extracted with dichloromethane (2*250 mL). All organic phases were combined, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was diluted with toluene (750 mL) and extracted with 2 M aqueous hydrochloric acid (500 mL) and 1 M aqueous hydrochloric acid (100 mL). Acidic aqueous phases were combined and basified with a solution of sodium hydroxide (60.0 g, 1.50 mol) in water (90 mL). The resulting mixture was extracted with dichloromethane (4*200 mL), dried over anhydrous sodium sulfate, filtered, concentrated in vacuo and diluted with hexanes (200 mL). 4 M Solution of hydrogen chloride in ether (100 mL, 400 mmol) was added to the solution, the resulting suspension was concentrated in vacuo and diluted with hexanes (1.00 L). The precipitated solid was filtered, washed with hexanes and dried in vacuo to give (2-amino-ethyl)-carbamic acid benzyl ester hydrochloride as white powder. Yield: 62.62 g (68%). RF (SiO2, dichloromethane/methanol 4:1): 0.25 (free base). 1H NMR spectrum (300 MHz, AcOD-d4, 80 C., dH): 7.42-7.26 (m, 5 H); 5.16 (s, 2 H); 3.60 (t, J=5.7 Hz, 2 H); 3.32 (t, J=5.7 Hz, 2 H).
A suspension of platinum (IV) oxide (180 mg, 0.79 mmol) and <strong>[37535-58-3](2R)-2-(tert-butoxycarbonylamino)-3-(4-pyridyl)propanoic acid</strong> (830 mg, 3.13 mmol) in MeOH (20 ml) and 1M HCl (3.1 ml) was stirred under a hydrogen atmosphere at room temperature for 24 h. The mixture was filtered through celite and the filtrate was evaporated to afford the product. The product was used in the next reaction without further purification. Consequently, a mixture of the product, N-(benzyloxycarbonyloxy)succinimide (0.93 g, 3.76 mmol), and NaHCO3 (0.78 g, 9.39 mmol) in CH3CN/H2O (40 ml/30 ml) was stirred at ambient temperature for 15 h. The organic solvent was removed in vacuo and the aqueous layer was washed with ether. The aqueous layer was collected and adjusted pH to 3 with 2M HCl and the white precipitation was extracted with EtOAc. The combined extracts were washed with H2O and brine, dried over MgSO4 and concentrated under reduced pressure to give 13a in 94 % yield. Rf=0.45 (CH2Cl2/MeOH=9/1). 1H NMR (600 MHz, CDCl3): delta 7.36-7.31 (m, 5H), 5.12 (s, 2H), 4.36 (m, 1H), 4.14 (br, 2H), 2.74 (br, 2H), 1.77-1.59 (m, 4H), 1.45 (s, 9H), 1.42 (m, 2H), 1.16 (m, 2H). HRMS (ESI/TOF) m/z: [M+Na]+ Calcd for C21H30N2Na1O6 429.20016 ; Found 429.19937.
3-(((benzyloxy)carbonyl)amino)oxetane-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With N-ethyl-N,N-diisopropylamine; In dichloromethane; at 20℃; for 24h;
To solution of 3-aminooxetane-3 -carboxylic acid (4.99 g, 42.6 mmol) and N,N- diisopropylethylamine (18 mL) in dichloromethane (120 mL) was added N- (benzyloxycarbonyloxy)succinimide (9.21 mL, 34.1 mmol) and the reaction mixture allowed to stir at room temperature for 24 h at which point the reaction volume was concentrated by two thirds under reduced pressure. This solution was then diluted with diethyl ether and washed with 1M aqueous hydrogen chloride then water, dried over sodium sulfate, filtered, and concentrated under reduced pressure to afford 3 -(((benzyl oxy)carbonyl)amino)ox etane-3 -carboxylic acid which was carrier on without further purification.
With potassium carbonate; In 1,4-dioxane; water; at 20℃;
potassium carbonate (2.1 g, 15 mmol), and N-(benzyloxycarbonyloxy)succinimide (1.4 g, 5.6 mmol) were treated with dioxane (8.0 mL) and water (8.0 mL). The mixture was stirred at room temperature overnight, and then partitioned between ethyl acetate and 0.5 M aqueous hydrochloric acid. The aqueous phase was extracted with ethyl acetate. The combined organic extracts were washed with sat. aq. sodium chloride solution, dried over anhydrous magnesium sulfate, filtered, and concentrated to a residue under reduced atmosphere. The residue was purified by silica flash chromatography to afford benzyl 3-hydroxyazetidine-1-carboxylate. LCMS-ESI+ (m/z): [M+H]+ calcd 208.10; found 207.76.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping