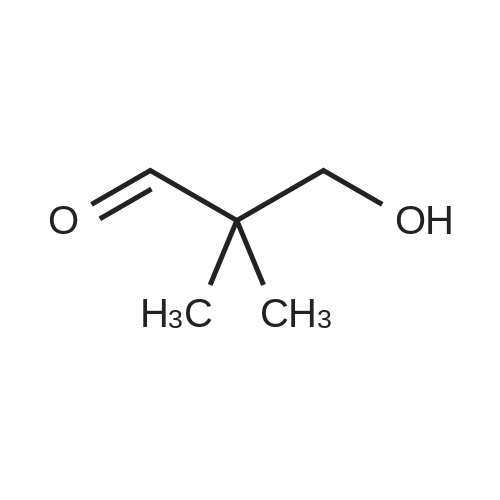
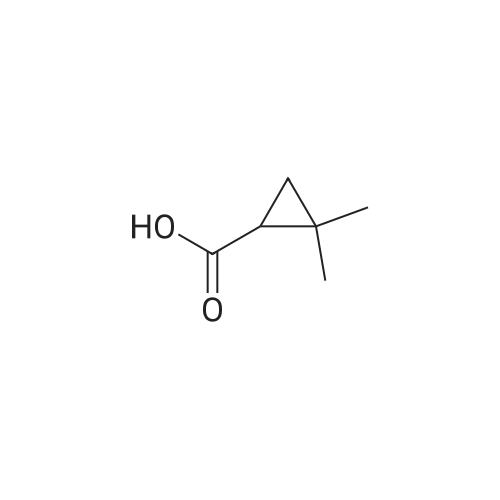
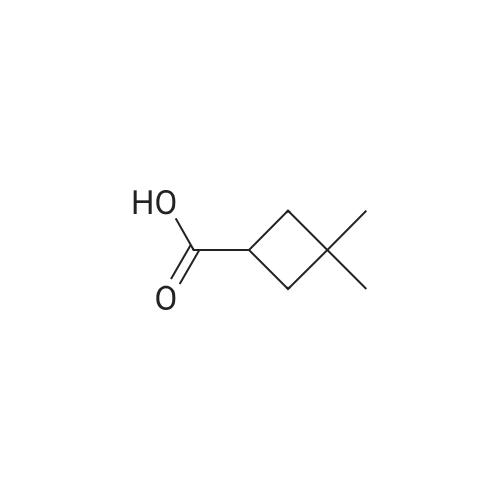
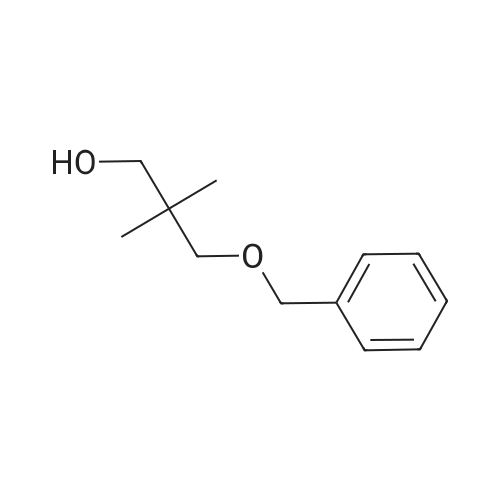
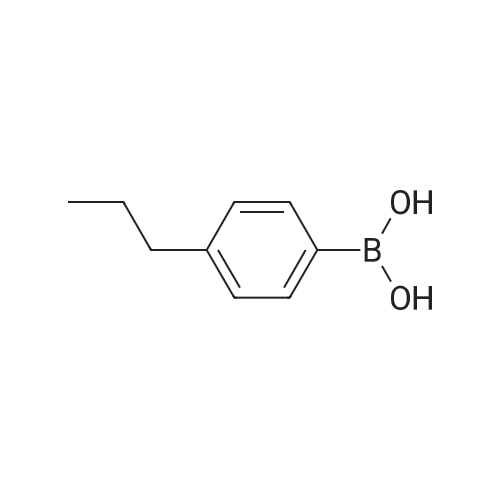
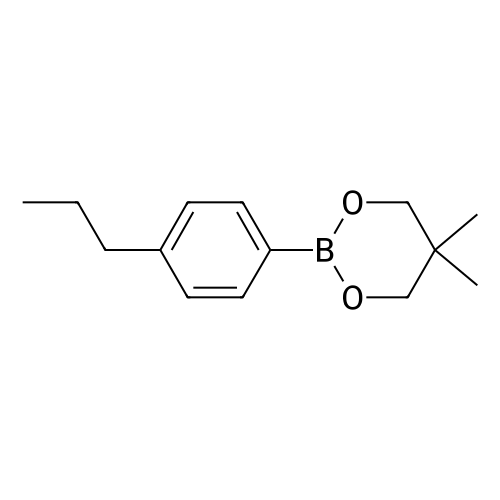
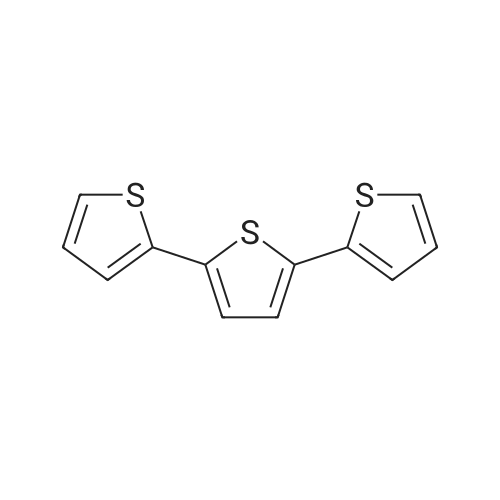
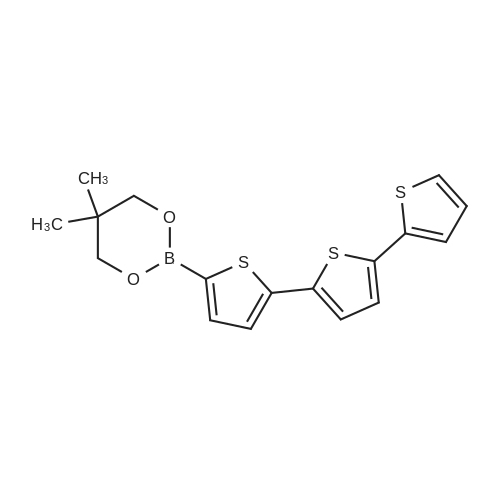
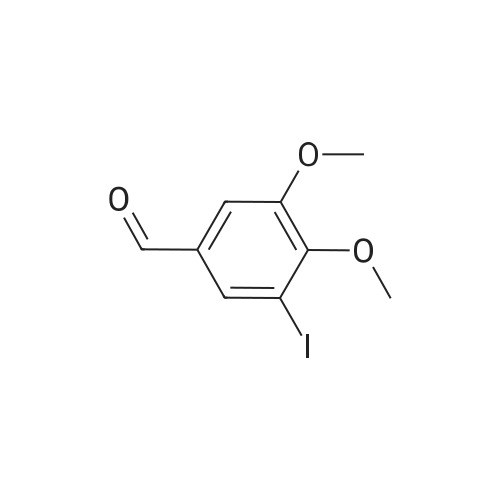
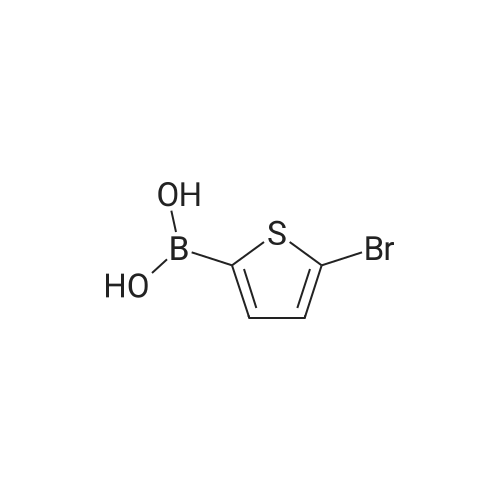
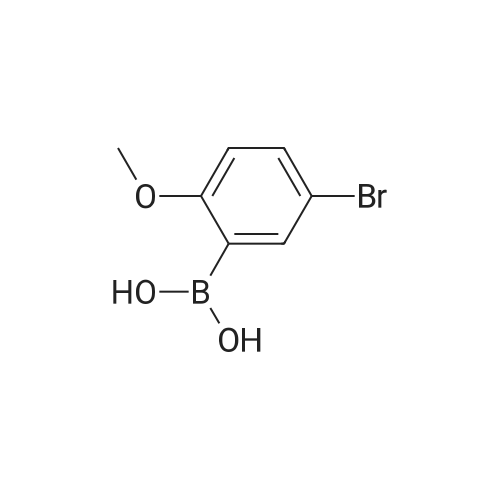
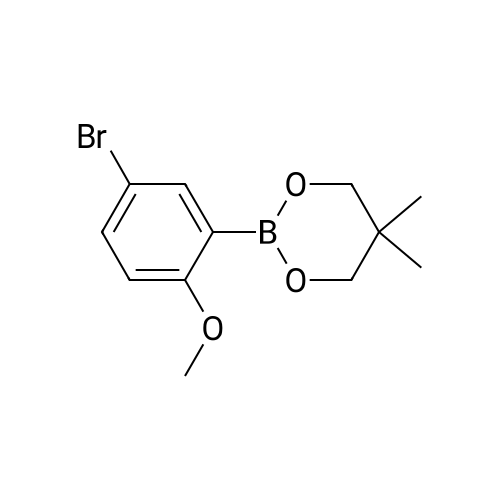
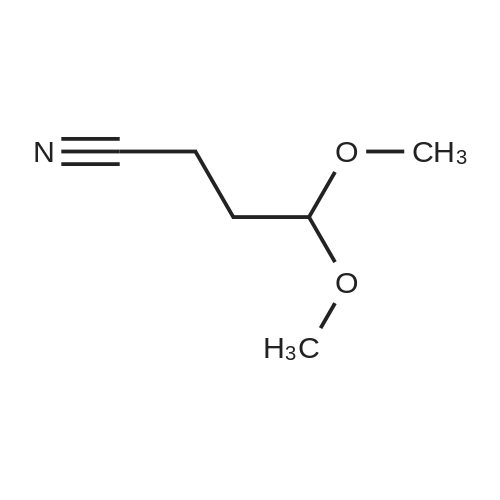
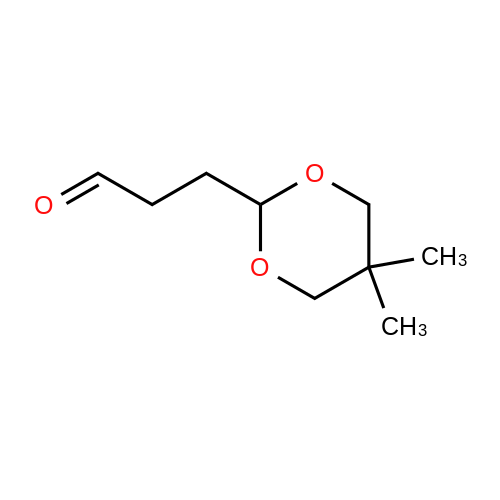
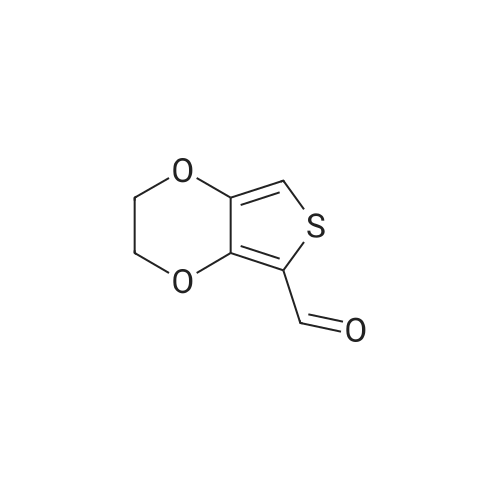
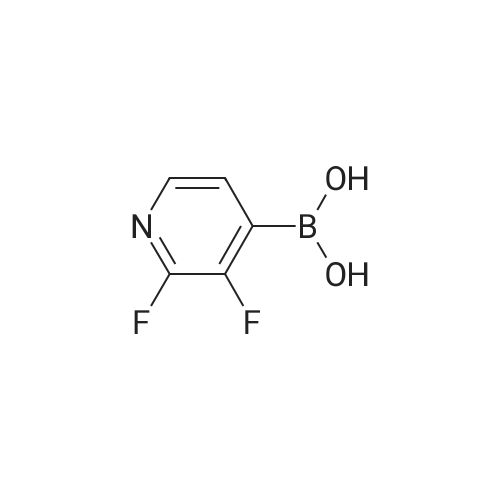
There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
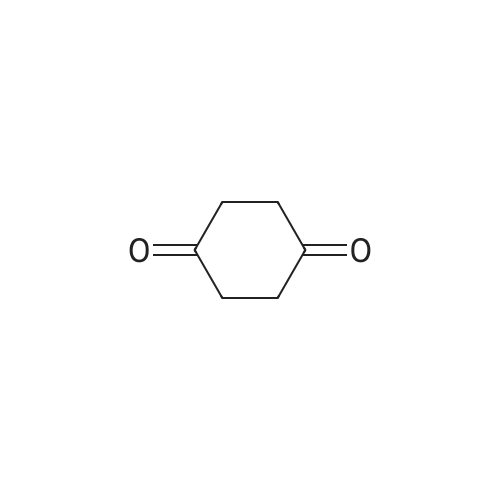
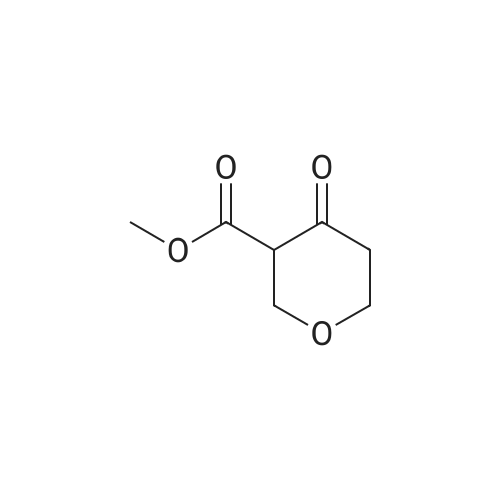
Structure of 126-30-7 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of Organic Chemistry, 1980, vol. 45, # 12, p. 2489 - 2498
[2] Journal of the American Chemical Society, 1977, vol. 99, # 2, p. 505 - 509
Stage #1: With sodium hydride In tetrahydrofuran; mineral oil at 20℃; for 0.0833333 h; Stage #2: With tetra-(n-butyl)ammonium iodide In tetrahydrofuran; mineral oil at 20℃;
Example 137C 3-(benzyloxy)-2,2-dimethylpropan-1-ol A solution of 2,2-dimethylpropane-1,3-diol (2 g, 19.20 mmol) in THF (30 mL) was treated with 60percent sodium hydride (0.256 g, 6.40 mmol) at room temperature, and the mixture stirred at room temperature for 5 minutes. Benzyl bromide (0.761 mL, 6.40 mmol) and tetrabutylammonium iodide (0.709 g, 1.920 mmol) were then added, and the reaction stirred overnight at room temperature. After this time, the mixture was diluted with ethyl acetate (150 mL) and water (50 mL), and the phases were separated. The aqueous layer was extracted with ethyl acetate (2*50 mL). The combined organics were washed with water (2*50 mL) and brine (50 mL) sequentially, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified on silica gel chromatography (10 to 50percent ethyl acetate-heptanes, eluent) to afford the title compound (0.992 g, 80percent). 1H NMR (400 MHz, CDCl3) δ 7.41-7.32 (m, 5H), 4.51 (s, 2H), 3.46 (d, J=5.7 Hz, 2H), 3.33 (s, 2H), 2.55 (t, J=5.9 Hz, 1H), 0.93 (s, 6H). MS (DCI+) m/z 195.0 (M+H).
77%
With tetra-(n-butyl)ammonium iodide In toluene at 0 - 20℃; for 16 h;
Into a 10-L 4-neck round-bottom flask, was placed 2,2-dimethylpropane-1,3-diol (200 g, 1920 mmol), toluene (1 L), 50percent KOH (aq. solution, 1L), n-Bu4NI (36 g, 97 mmol). Then (bromomethyl)benzene (328 g, 1920 mmol, 1.00 equiv.) was added at 0 °C. The resulting solution was stirred at room temperature for 16 h. The reaction was then quenched by the addition of 1 L of ice-water. The resulting solution was extracted with 2x4 L of ethyl acetate. The combined organic layer was washed with 2x4 L of brine, dried over anhydrous sodium sulfate and concentrated under vacuum. The crude product was purified by silica gel column chromatography to afford the title compound as light yellow oil (77percent). LCMS:mlz=195.0 [M+1]. ‘H-NMR (300 MHz, CDC13) ö: 7.37-7.25 (m, 5H), 4.47 (m, 3H), 3.18 (d, 1=6.4Hz, 4H), 0.82 (s, 6H).
64%
Stage #1: With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0℃; for 0.75 h; Inert atmosphere Stage #2: at 0 - 20℃; Inert atmosphere
General procedure: The diol (48 mmol, 1 eq) wasdissolved into DMF (120 mL) under argon. At 0°C NaH (1.1 eq, 60percent in mineral oil) was added inone portion and the mixture was stirred at 0°C during 45 min. BnBr (0.97 eq) was added dropwise,the mixture was allowed to warm at room temperature and stirred overnight. The reaction wasquenched with saturated NH4Cl solution (20 mL) and then diluted with water. The crude mixturewas extracted with Et2O and the organic layer was washed with brine, dried over anhydrous MgSO4,filtered and concentrated in vacuo. Benzylated compound was then purified by flashchromatography on silica gel.
62%
Stage #1: With sodium hydride In tetrahydrofuran at 0℃; for 1 h; Stage #2: at 0 - 20℃;
Step A: 3-Phenylmethoxy-neopentylol (31a) Adapting a procedure or a variation thereof according to Effenberger, et al., Tetrahedron: Asymmetry 1995, 6, 271-282, a 1000 mL round-bottomed flask equipped with a magnetic stirring bar and a rubber septum was charged under a nitrogen atmosphere with 2.40 g of a 60 wt-percent suspension of sodium hydride (NaH) in mineral oil (1.44 g, 60.0 mmol). The hydride was suspended in 50 mL of hexane and the supernatant was decanted, and the residue was dried under reduced pressure. Six-hundred (600) mL of anhydrous tetrahydrofuran (THF) were added under a nitrogen atmosphere and the suspension was cooled to ca. 0° C. (ice bath). 6.24 g (60 mmol) of commercially available neopentyl glycol (2,2-dimethyl-1,3-propandiol) was added and the reaction mixture was stirred for one hour at this temperature until the hydrogen evolution subsided. 5.9 mL of benzyl bromide (8.5 g, 50.0 mmol) was added to the stirred reaction mixture. The reaction mixture was stirred overnight with gradual warming to room temperature and the solvent was then partially removed under reduced pressure using a rotary evaporator. The reaction was quenched by addition of a one normal (1 N) aqueous solution of hydrogen chloride (HCl) and the product was extracted with ethyl acetate. The combined organic extracts were washed with water and brine, dried over anhydrous magnesium sulfate (MgSO4), filtered, and the solvents were removed under reduced pressure using a rotary evaporator. The residue was purified by silica gel chromatography using a mixture of ethyl acetate (EtOAc) and hexane (Hxn) as eluent (EtOAc/Hxn=1:4) to provide 6.0 g (62percent yield) of the title compound (31a) as a colorless liquid. Rf=0.34 (EtOAc/Hxn=1:4). 1H NMR (400 MHz, CDCl3): δ=0.953 (s, 6H), 2.62 (br. t, J=5.6 Hz, 1H), 3.34 (s, 2H), 3.47 (d, J=5.6 Hz, 2H), 4.54 (s, 2H), 7.27-7.38 (m, 5H) ppm. MS (ESI) m/z 195.10 (M+H)+, 217.10 (M+Na)+. The analytical data was consistent with the data given in the literature.
62%
With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0 - 20℃; for 12 h;
[00319] Into a 250-mL round-bottom flask, was placed 2,2-dimethylpropane-1,3-diol (10.4 g, 99.86 mmol) and N,N-dimethylformamide (100 mL). This was followed by the addition of 60percent sodium hydride (4 g, 100.00 mmol), in portions at 0°C. To this was added (bromomethyl)benzene (13.68 g, 79.98 mmol) at 0°C. The resulting solution was stuffed for 12 h at room temperature and then diluted with 200 mL of NH4C1 (sat. aq). The resulting solution was extracted with 2x200 mL of ethyl acetate and the organic layers were combined and dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by silica gel column with ethyl acetate/petroleum ether (1:10) to obtain 12 g (62percent) of 3-(benzyloxy)-2,2-dimethylpropan-1-ol as light yellow oil. ‘H NMR (300 MHz, DMSOd 6): 7.43-7.24 (m, 5H), 4.51-4.41 (m, 3H), 3.25-3.15 (m, 4H), 0.84 (s, 6H) ppm.
58%
Stage #1: With sodium hydride In tetrahydrofuran; mineral oil at 0℃; for 0.5 h; Inert atmosphere Stage #2: at 20℃;
[00509] 2,2-Dimethylpropane-l,3-diol 159 (4.24 g, 40 mmol) was dissolved in 120 mL of THF. To this mixture, was added NaH (1.92 g, 48 mmol, 60percent in mineral oil) at 0 °C under nitrogen atmosphere. After stirring at 0 °C for 30 min, benzyl bromide (6.84 g, 40 mmol) was added to this mixture, and the resulting mixture was stirred at room temperature overnight. The reaction mixture was then transferred into iced water and extracted with ethyl acetate (50 mL X 3). The combined organic layers were washed with brine, dried over anhydrous Na2S04, and concentrated under reduced pressure to obtain crude product, which was purified by silica gel chromatography (10percent ethyl acetate/petroleum ether) to give 4.5 g 3- (benzyloxy)-2,2-dimethylpropan-l-ol 160 (58percent yield). LCMS: m/z 195.1 [M+H]+, fR = 1.70 min.
51%
Stage #1: With potassium <i>tert</i>-butylate In 1,4-dioxane at 0 - 20℃; for 1 h; Inert atmosphere Stage #2: at 90℃; for 4 h; Inert atmosphere
A solution of 2,2-dimethyl-propane-l,3-diol (25.85 g, 248 mmol) in dioxane (400 mL) was cooled to 0°C. Potassium tert-butoxide (30 g, 267 mmol) was added portionwise to the cooled solution. The resulting mixture was stirred at room temperature for 1 h. (Bromomethyl)benzene (29.5 mL, 248 mmol) was added dropwise via addition funnel. The mixture was heated to 90°C and stirred for 4 h. The resulting mixture was concentrated in vacuo. The resulting residue was partitioned between water (200 mL) and ethyl acetate (200 mL) and extracted with ethyl acetate (3x200 mL). The organics were combined, washed with brine (100 mL), dried on sodium sulfate, and concentrated in vacuo. The crude material was purified by column chromatography (Si02, 5percent to 60percent ethyl acetate in heptane) to afford 3-benzyloxy-2,2-dimethyl-propan-1-ol as a yellow oil (24.84 g, 51percent). 1H NMR (CDCl3) : 7.28 - 7.42 (m, 5H), 4.52 (s, 2H), 3.47 (s, 2H), 3.34 (s, 2H), 0.94 (s, 6H). MS calcd. for C12H18O2 [(M+H)+] 195.3, obsd. 195.
12.50 g
Stage #1: With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0℃; for 0.5 h; Stage #2: at 20℃; for 16 h;
Sodium hydride (2.50 g, 105.62 mmol, 60percent in mineral oil) was suspended in dry DMF (150 ml) at 0° C. and a solution of 2,2-dimethylpropane-1,3-diol (10.0 g, 96.02 mmol) in DMF (50.0 ml) was added dropwise. The RM was stirred at 0° C. for 30 min. To the RM was added dropwise benzyl bromide (11.50 ml, 96.02 mmol) and the RM was allowed to warm to RT and stirred for 16 h. The RM was diluted with water (300 ml) and the organic product was extracted with EtOAc (3*200 ml). The organic layer was washed with brine (2*200 ml), dried over Na2SO4, filtered and the solvent evaporated in vacuo to give the crude product, which was purified by column chromatography (silica gel 60-120 mesh, 0-10percent EtOAc in PE) to get the title compound (12.50 g).
Reference:
[1] Tetrahedron Letters, 2009, vol. 50, # 13, p. 1466 - 1468
[2] Tetrahedron, 1986, vol. 42, # 21, p. 5941 - 5948
[3] Journal of the American Chemical Society, 2013, vol. 135, # 14, p. 5467 - 5474
[4] Patent: US2017/15675, 2017, A1, . Location in patent: Paragraph 1274
[5] Journal of Heterocyclic Chemistry, 2018, vol. 55, # 8, p. 1986 - 1990
[6] Chemistry - A European Journal, 2012, vol. 18, # 52, p. 16823 - 16827
[7] Patent: WO2016/44626, 2016, A1, . Location in patent: Paragraph 00435
[8] Angewandte Chemie - International Edition, 2000, vol. 39, # 1, p. 209 - 213
[9] Tetrahedron, 2016, vol. 72, # 2, p. 318 - 327
[10] Patent: US2009/82464, 2009, A1, . Location in patent: Page/Page column 45
[11] Patent: WO2014/153226, 2014, A1, . Location in patent: Paragraph 00319
[12] Patent: WO2015/42414, 2015, A1, . Location in patent: Paragraph 00509
[13] Patent: WO2013/189904, 2013, A1, . Location in patent: Page/Page column 47
[14] ACS Medicinal Chemistry Letters, 2015, vol. 6, # 9, p. 1019 - 1024
[15] Journal of Organic Chemistry, 1985, vol. 50, # 15, p. 2707 - 2711
[16] Tetrahedron: Asymmetry, 1995, vol. 6, # 1, p. 271 - 282
[17] Patent: US2017/101397, 2017, A1, . Location in patent: Paragraph 0468; 0469
[18] Patent: WO2018/108231, 2018, A1, . Location in patent: Page/Page column 50
9
[ 100-44-7 ]
[ 126-30-7 ]
[ 66582-32-9 ]
Reference:
[1] Tetrahedron Letters, 1997, vol. 38, # 8, p. 1359 - 1362
[2] European Journal of Organic Chemistry, 1999, # 11, p. 2817 - 2823
[3] Synlett, 2011, # 10, p. 1413 - 1418
[4] Tetrahedron Letters, 1977, p. 4257 - 4260
[5] Tetrahedron, 1988, vol. 44, # 13, p. 3899 - 3918
[6] Journal of pharmaceutical sciences, 1988, vol. 77, # 2, p. 149 - 152
10
[ 126-30-7 ]
[ 66582-32-9 ]
Reference:
[1] Tetrahedron Letters, 1983, vol. 24, # 46, p. 5139 - 5140
[2] Journal of Organic Chemistry, 1989, vol. 54, # 12, p. 2817 - 2825
11
[ 126-30-7 ]
[ 637-88-7 ]
[ 69225-59-8 ]
Yield
Reaction Conditions
Operation in experiment
74.5%
With sulfuric acid In water at 90℃;
Example 1; 3,3-Dimethyl-l,5-dioxaspiro[5.5]undecan-9-one.; A continuous extraction apparatus is assembled. A 500 mL extraction solvent pot is charged with 250 mLπ-hexane and 5.00 g sodium bicarbonate. An oil bath is heated to 90°C. A 500 mL reaction pot is charged with 82.5 g (0.792 mol, 2.33 equiv) neopentyl glycol, 338 mLH20, 0.79 mL (1.45 g, 14.8 mmol, 4.35 molpercent) of 98percent sulfuric acid, and 38.08 g (0.340 mol) of 1,4-cyclohexanedione. π-Hexane (85 mL) is then added to bring the pot volume to the extractor return sidearm. The extraction pot is immediately immersed in the oil bath and the reaction mixture stir rate is increased to the point where there is efficient mixing in the lower (aqueous) phase but not in the upper (π-hexane) phase in the extractor. The extraction is continued for 99 h.The suspension is cooled to 25°C and the precipitate is suction filtered, washed with 50 niLπ-hexane, and air dried 2 h at 25°C to afford 10.71 g of crude bisketal as a colorless solid. The bulk of the π-hexane is distilled from the combined mother liquors and the resulting suspension is cooled (95 g). Methanol (250 mL) is added and 163 mL of a mixture of the methanol-hexane azeotrope (28:72) and methanol are distilled to a head temperature of 60°C (bath 90°C). The suspension (168 g) is cooled to 25°C and water (100 mL) is added dropwise over 10 min. After stirring overnight, the precipitate is suction filtered, and air dried several h at 25°C to afford 7.22 g of additional crude bisketal as a colorless solid.The mother liquors are concentrated by distillation (dry ice-acetone cold finger condenser) at 30-35°C and 40-45 mm Hg ( 146 mL distillate collected) . The resulting suspension is cooled to 0-5°C and stirred for 90 min. The precipitate is suction filtered (mother liquors are used to complete the transfer) and air dried 24 h at 25°C to afford 50.17 g (74.5percent) of the monoketal 54 as a colorless solid.The combined crude bisketal crops ( 17.62 g) are resuspended in 200 mL water and stirred for I h. The insoluble material is suction filtered and air dried 6 h at 25°C to afford 13.06 g of bisketal as a colorless solid.
Reference:
[1] Journal of Fluorine Chemistry, 2009, vol. 130, # 4, p. 377 - 382
14
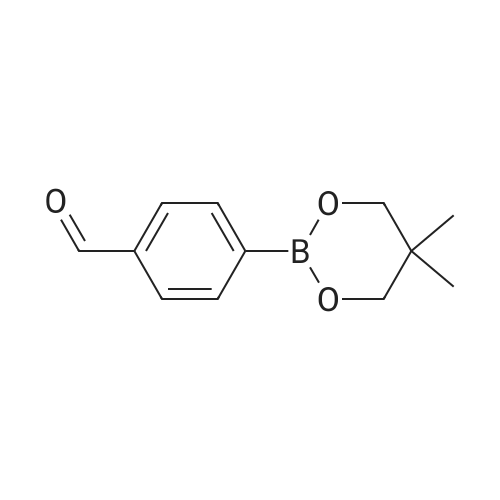
[ 87199-17-5 ]
[ 126-30-7 ]
[ 128376-65-8 ]
Yield
Reaction Conditions
Operation in experiment
95%
at 20℃; for 2 h;
To a solution of 4-formylphenylboronic acid (4.11 g) in anhydrous tetrahydrofuran (THF) (40 ml_) was added 2,2-dimethyl-1 ,3-propanediol (3.14 g) and the mixture was stirred at room temperature for 2 hours. The solvent was evaporated to dryness. The residue was dissolved in dichloromethane (120 ml_), washed with water (80 ml_ x 3), dried and evaporated under vacuum to obtain 4-(5,5-dimethyl-[1 ,3,2]dioxaborinan-2-yl)-benzaldehyde (5.66 g, 95percent yield).
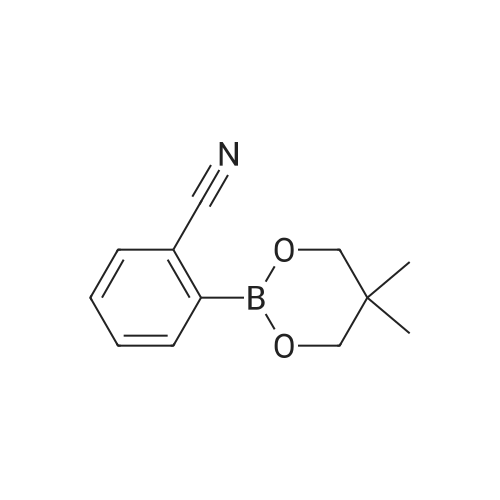
To a solution of TMP (3.28 mL, 19.4 mmol) in anhydrous THF (20 mL), n-BuLi (1.6 M in hexanes, 11.7 mL, 18.8 mmol) was added at -10 °C and the resulting mixture was stirred for 10 min. Then, B(Oi-Pr)3 (5.50 mL, 25 mmol) was added dropwise at -78 °C and stirred for additional 5 min before benzonitrile 1 (1.28 mL, 12.5 mmol) was added via a syringe in a single portion and the reaction mixture was stirred at -78 °C for 2 h. The solution was left to warm to rt while being stirred for 3 h and then quenched with saturated aqueous NH4Cl (60 mL). The resulting mixture was extracted with EtOAc (3 * 70 mL), the combined organic extracts were dried (Na2SO4) and concentrated in vacuo. The intermediate product 2 was dissolved in anhydrous Tol (50 mL) and 2,2-dimethyl-1,3-propandiol (1.57 g, 15 mmol) was added and then stirred overnight at rt. The organic phase was washed with H2O (3 * 30 mL) and the aqueous extracts were washed with CH2Cl2 (3 * 30 mL). The CH2Cl2-phase was washed with H2O (1 * 30 mL), combined with Tol extract, dried (Na2SO4) and concentrated in vacuo. Recrystallization of the crude product from heptane afforded pure 3 (58percent, 2 steps) as a white crystalline solid: M.p. 109-111 °C; ESI-MS (m/z): 216.64 [MH]; 1Η ΝΜR (400 MHz, CDCl3): δ 7.88 (d, 1Η, J = 7.5 Hz), 7.68 (d, 1Η, J = 7.5 Hz), 7.54 (td, 1Η, J = 7.5 Hz, 1.0 Hz), 7.48 (td, 1Η, J = 7.5 Hz, 1.0 Hz), 3.83 (s, 4H), 1.05 (s, 6H) ppm; 13C NMR (160 MHz, CDCl3): δ 135.07, 133.64, 131.44, 130.47, 119.63, 116.56, 72.49, 31.84, 21.83 ppm.
Reference:
[1] European Journal of Medicinal Chemistry, 2012, vol. 55, p. 358 - 374,17
[2] European Journal of Medicinal Chemistry, 2012, vol. 55, p. 358 - 374
With tetrakis(dimethylamido)diborane In toluene at 20 - 105℃; for 1.03333 h;
Tetrakis (dimethylamino) diboron (9.89 g, 52.65 mmol) was added to a solution of 2,2- dimethyl-1, 3-propanediol [neopentylglycol] (10.42 g, 100 mmol) in toluene (40 mL) at room temperature within two minutes. The mixture was heated to 105 C and an evolution of dimethylamine started to occur at 85 C. The mixture was heated for 60 minutes at 105 C then the toluene was removed to give a white solid: 11.39 g, 95.8percent. Recrystallisation from toluene gave bis (neopentylglycolato) boron (MF: CLOH20B204 ; FW: 225.89) 6.48 g, 54.5percent. mp 182.5-184. 5 C. 8 (CDCL3, 200 MHz) 0.94 (s, 12H); 3.58 (s, 6H) ppm. 13C 8 (CDCL3, 50 MHz) 22.3 (4x CH3) ; 31.9 (2x C), 71.7 (4x CH2) ppm. The mother liquor was kept for further recrystallization.
Reference:
[1] Journal of Organic Chemistry, 2006, vol. 71, # 6, p. 2518 - 2520
22
[ 121-43-7 ]
[ 126-30-7 ]
[ 862129-81-5 ]
Yield
Reaction Conditions
Operation in experiment
64%
With hydrogenchloride; magnesium; methyl iodide In tetrahydrofuran; water; ethyl acetate at 20℃; Reflux
Step two: adding metal magnesium (2.2 g, 92mmol) and 10 ml of tetrahydrofuran, add 2-3 drop iodine methane initiating the dropwise 13.2 g 3,6-dihydro -2H-pyran-4-polybromide dissolved in 60 ml tetrahydrofuran solution, reagent cheng Geshi preparing reflux reaction is omitted, then drop by adding boric acid three methyl ester (11.4 g, 0 . 11mol) in, after the reaction is complete, by adding 10percent hydrochloric acid solution to adjust PH= 3-4, organic layer by adding 110 ml of ethyl acetate and npg (10.0 g, 96mmol), stirring at room temperature until the reaction is complete. After laminating, an organic layer saturated salt water washing, solvent after evaporation to dryness, by adding normal heptane, cooling to -10 °C, filtering to obtain 10.0 g kind of white solid: 3,6-dihydro -2H-thiopyran-4-boronic acid new pentamethylene glycol ester, GC: 99.7percent, HNMR > 98percent, the yield is 64percent.
Reference:
[1] Patent: CN105503927, 2016, A, . Location in patent: Paragraph 0016
Example 137C 3-(benzyloxy)-2,2-dimethylpropan-1-ol A solution of 2,2-dimethylpropane-1,3-diol (2 g, 19.20 mmol) in THF (30 mL) was treated with 60% sodium hydride (0.256 g, 6.40 mmol) at room temperature, and the mixture stirred at room temperature for 5 minutes. Benzyl bromide (0.761 mL, 6.40 mmol) and tetrabutylammonium iodide (0.709 g, 1.920 mmol) were then added, and the reaction stirred overnight at room temperature. After this time, the mixture was diluted with ethyl acetate (150 mL) and water (50 mL), and the phases were separated. The aqueous layer was extracted with ethyl acetate (2*50 mL). The combined organics were washed with water (2*50 mL) and brine (50 mL) sequentially, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified on silica gel chromatography (10 to 50% ethyl acetate-heptanes, eluent) to afford the title compound (0.992 g, 80%). 1H NMR (400 MHz, CDCl3) delta 7.41-7.32 (m, 5H), 4.51 (s, 2H), 3.46 (d, J=5.7 Hz, 2H), 3.33 (s, 2H), 2.55 (t, J=5.9 Hz, 1H), 0.93 (s, 6H). MS (DCI+) m/z 195.0 (M+H).
77%
With tetra-(n-butyl)ammonium iodide; In toluene; at 0 - 20℃; for 16h;
Into a 10-L 4-neck round-bottom flask, was placed 2,2-dimethylpropane-1,3-diol (200 g, 1920 mmol), toluene (1 L), 50% KOH (aq. solution, 1L), n-Bu4NI (36 g, 97 mmol). Then (bromomethyl)benzene (328 g, 1920 mmol, 1.00 equiv.) was added at 0 C. The resulting solution was stirred at room temperature for 16 h. The reaction was then quenched by the addition of 1 L of ice-water. The resulting solution was extracted with 2x4 L of ethyl acetate. The combined organic layer was washed with 2x4 L of brine, dried over anhydrous sodium sulfate and concentrated under vacuum. The crude product was purified by silica gel column chromatography to afford the title compound as light yellow oil (77%). LCMS:mlz=195.0 [M+1]. ?H-NMR (300 MHz, CDC13) oe: 7.37-7.25 (m, 5H), 4.47 (m, 3H), 3.18 (d, 1=6.4Hz, 4H), 0.82 (s, 6H).
64%
General procedure: The diol (48 mmol, 1 eq) wasdissolved into DMF (120 mL) under argon. At 0C NaH (1.1 eq, 60% in mineral oil) was added inone portion and the mixture was stirred at 0C during 45 min. BnBr (0.97 eq) was added dropwise,the mixture was allowed to warm at room temperature and stirred overnight. The reaction wasquenched with saturated NH4Cl solution (20 mL) and then diluted with water. The crude mixturewas extracted with Et2O and the organic layer was washed with brine, dried over anhydrous MgSO4,filtered and concentrated in vacuo. Benzylated compound was then purified by flashchromatography on silica gel.
62%
Step A: 3-Phenylmethoxy-neopentylol (31a) Adapting a procedure or a variation thereof according to Effenberger, et al., Tetrahedron: Asymmetry 1995, 6, 271-282, a 1000 mL round-bottomed flask equipped with a magnetic stirring bar and a rubber septum was charged under a nitrogen atmosphere with 2.40 g of a 60 wt-% suspension of sodium hydride (NaH) in mineral oil (1.44 g, 60.0 mmol). The hydride was suspended in 50 mL of hexane and the supernatant was decanted, and the residue was dried under reduced pressure. Six-hundred (600) mL of anhydrous tetrahydrofuran (THF) were added under a nitrogen atmosphere and the suspension was cooled to ca. 0 C. (ice bath). 6.24 g (60 mmol) of commercially available neopentyl glycol (2,2-dimethyl-1,3-propandiol) was added and the reaction mixture was stirred for one hour at this temperature until the hydrogen evolution subsided. 5.9 mL of benzyl bromide (8.5 g, 50.0 mmol) was added to the stirred reaction mixture. The reaction mixture was stirred overnight with gradual warming to room temperature and the solvent was then partially removed under reduced pressure using a rotary evaporator. The reaction was quenched by addition of a one normal (1 N) aqueous solution of hydrogen chloride (HCl) and the product was extracted with ethyl acetate. The combined organic extracts were washed with water and brine, dried over anhydrous magnesium sulfate (MgSO4), filtered, and the solvents were removed under reduced pressure using a rotary evaporator. The residue was purified by silica gel chromatography using a mixture of ethyl acetate (EtOAc) and hexane (Hxn) as eluent (EtOAc/Hxn=1:4) to provide 6.0 g (62% yield) of the title compound (31a) as a colorless liquid. Rf=0.34 (EtOAc/Hxn=1:4). 1H NMR (400 MHz, CDCl3): delta=0.953 (s, 6H), 2.62 (br. t, J=5.6 Hz, 1H), 3.34 (s, 2H), 3.47 (d, J=5.6 Hz, 2H), 4.54 (s, 2H), 7.27-7.38 (m, 5H) ppm. MS (ESI) m/z 195.10 (M+H)+, 217.10 (M+Na)+. The analytical data was consistent with the data given in the literature.
62%
With sodium hydride; In N,N-dimethyl-formamide; mineral oil; at 0 - 20℃; for 12h;
[00319] Into a 250-mL round-bottom flask, was placed 2,2-dimethylpropane-1,3-diol (10.4 g, 99.86 mmol) and N,N-dimethylformamide (100 mL). This was followed by the addition of 60% sodium hydride (4 g, 100.00 mmol), in portions at 0C. To this was added (bromomethyl)benzene (13.68 g, 79.98 mmol) at 0C. The resulting solution was stuffed for 12 h at room temperature and then diluted with 200 mL of NH4C1 (sat. aq). The resulting solution was extracted with 2x200 mL of ethyl acetate and the organic layers were combined and dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by silica gel column with ethyl acetate/petroleum ether (1:10) to obtain 12 g (62%) of 3-(benzyloxy)-2,2-dimethylpropan-1-ol as light yellow oil. ?H NMR (300 MHz, DMSOd 6): 7.43-7.24 (m, 5H), 4.51-4.41 (m, 3H), 3.25-3.15 (m, 4H), 0.84 (s, 6H) ppm.
58%
[00509] 2,2-Dimethylpropane-l,3-diol 159 (4.24 g, 40 mmol) was dissolved in 120 mL of THF. To this mixture, was added NaH (1.92 g, 48 mmol, 60% in mineral oil) at 0 C under nitrogen atmosphere. After stirring at 0 C for 30 min, benzyl bromide (6.84 g, 40 mmol) was added to this mixture, and the resulting mixture was stirred at room temperature overnight. The reaction mixture was then transferred into iced water and extracted with ethyl acetate (50 mL X 3). The combined organic layers were washed with brine, dried over anhydrous Na2S04, and concentrated under reduced pressure to obtain crude product, which was purified by silica gel chromatography (10% ethyl acetate/petroleum ether) to give 4.5 g 3- (benzyloxy)-2,2-dimethylpropan-l-ol 160 (58% yield). LCMS: m/z 195.1 [M+H]+, fR = 1.70 min.
51%
A solution of 2,2-dimethyl-propane-l,3-diol (25.85 g, 248 mmol) in dioxane (400 mL) was cooled to 0C. Potassium tert-butoxide (30 g, 267 mmol) was added portionwise to the cooled solution. The resulting mixture was stirred at room temperature for 1 h. (Bromomethyl)benzene (29.5 mL, 248 mmol) was added dropwise via addition funnel. The mixture was heated to 90C and stirred for 4 h. The resulting mixture was concentrated in vacuo. The resulting residue was partitioned between water (200 mL) and ethyl acetate (200 mL) and extracted with ethyl acetate (3x200 mL). The organics were combined, washed with brine (100 mL), dried on sodium sulfate, and concentrated in vacuo. The crude material was purified by column chromatography (Si02, 5% to 60% ethyl acetate in heptane) to afford 3-benzyloxy-2,2-dimethyl-propan-1-ol as a yellow oil (24.84 g, 51%). 1H NMR (CDCl3) : 7.28 - 7.42 (m, 5H), 4.52 (s, 2H), 3.47 (s, 2H), 3.34 (s, 2H), 0.94 (s, 6H). MS calcd. for C12H18O2 [(M+H)+] 195.3, obsd. 195.
12.50 g
Sodium hydride (2.50 g, 105.62 mmol, 60% in mineral oil) was suspended in dry DMF (150 ml) at 0 C. and a solution of 2,2-dimethylpropane-1,3-diol (10.0 g, 96.02 mmol) in DMF (50.0 ml) was added dropwise. The RM was stirred at 0 C. for 30 min. To the RM was added dropwise benzyl bromide (11.50 ml, 96.02 mmol) and the RM was allowed to warm to RT and stirred for 16 h. The RM was diluted with water (300 ml) and the organic product was extracted with EtOAc (3*200 ml). The organic layer was washed with brine (2*200 ml), dried over Na2SO4, filtered and the solvent evaporated in vacuo to give the crude product, which was purified by column chromatography (silica gel 60-120 mesh, 0-10% EtOAc in PE) to get the title compound (12.50 g).
With potassium hydroxide; In toluene; at 0 - 20℃; for 12h;
To a stirred solution of 2,2-dimethyl propane-1,3-diol (100 g, 961.153 mmol) in toluene (1000 mL), potassium hydroxide (53.84 g, 961.5 mmol) and benzyl bromide (34 mL , 288.4 mmol) were added at 0 C. The resulting reaction mixture was stirred for 12h at RT. On completion, the reaction mixture was diluted with H20 (1000 mL) and extracted twice with EtOAc (2 x 1000 mL). The combined organic layers were washed with H20 (500 mL) and brine (500 mL), dried over anhydrous sodium sulfate and evaporated under vacuum. The resulting residue was purified by silica gel column chromatography (0-7% EtOAc in Pet ether as elute) to afford the title compound as colorless liquid.1H NMR (400MHz, CDCI3) delta = 7.40 - 7.23 (m, 5H), 4.51 (s, 2H), 3.46 (s, 2H), 3.32 (s, 2H), 2.60 (br s, 1H), 0.93 (s, 6H). LCMS: 86% (M + H) 234.
With pyridinium p-toluenesulfonate; In toluene;Dean-Stark; Reflux; Inert atmosphere;
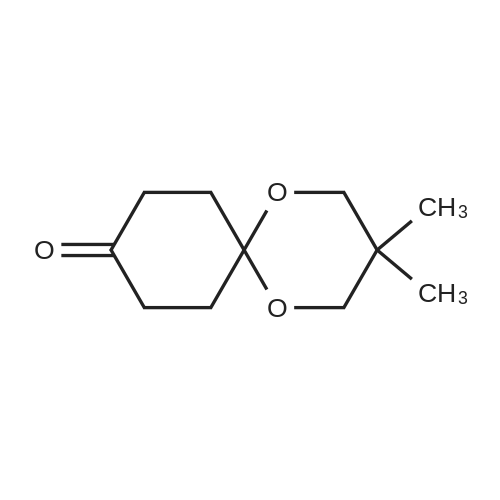
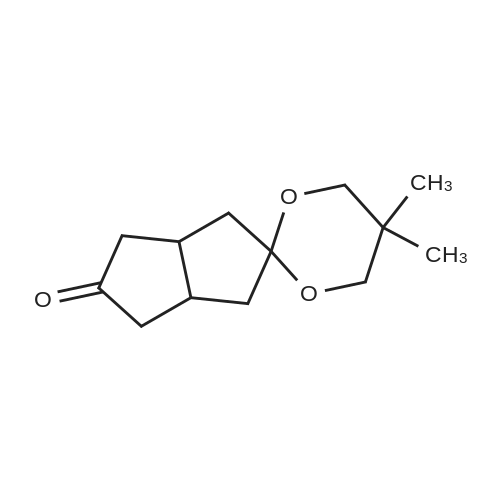
To a solution of <strong>[51716-63-3](3as,6as)-tetrahydropentalene-2,5(1H,3H)-dione</strong> (15 g, 108.57 mmol) in toluene (150 mL) was added 2, 2-dimethylpropane-1,3-diol (11.31 g, 108.57 mmol), followed by the addition of PPTS (250 mg) at rt. The reaction mixture was refluxed overnight with a Dean-Stark trap under N 2 atmosphere. After cooled to rt, the reaction mixture was concentrated under vacuum to remove the volatile. The residue was purified by silica gel flash column chromatography (PE: EtOAc = 20: 1) to give the title compound (16 g, yield: 66%) as a white solid.
8,8-dimethyl-6,10-dioxaspiro[4,5]decane-2-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In water; toluene;
2. 2-Carboxy-8,8-dimethyl-6,10-dioxaspiro[4,5]decane 72.8 g (0.70 mole) of 2,2-dimethylpropane-1,3-diol and 12.2 g (0.05 mole) of pyridinium-4-toluenesulphonate are added one after another to 40.0 g (0.28 mole) of methyl 3-oxocyclopentane carboxylate OCCME in 560 ml of absolute toluene. The mixture is boiled for 1.5 hours in a water separator; after 30 minutes the solid is dissolved. Approximately 8 ml of H2 O in total are separated out. After cooling, the solution is washed three times using 100 ml of H2 O each time, the toluene phase is dried over MgSO4 and evaporated in vacuo. The oily residue is fractionally distilled in vacuo at 0.6 mbar over a metallised pin tube column 20 cm long. Result: 51.0 g (79.8% of theory) of colourless oil, bp0.6 mbar: 97-103 C.
4-(1-hydroxymethyl-1-methylethyl)-3,6-dimethylpyridazine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With ammonium persulfate; sulfuric acid; silver nitrate; In water;
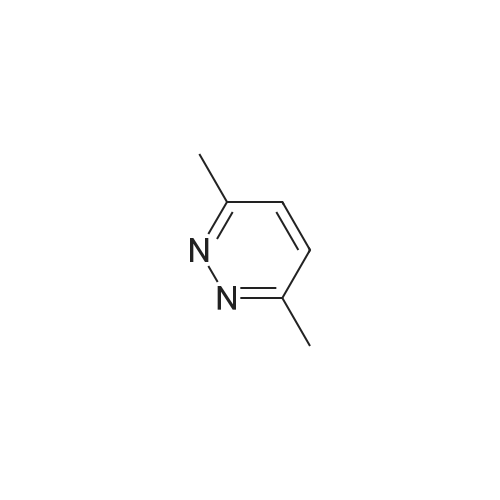
EXAMPLE 17 4-(1-chloromethyl-1-methylethyl)-<strong>[1632-74-2]3,6-dimethylpyridazine</strong> A 23.9 g. portion of 2,2-dimethyl-1,3-propanediol, 88 ml. of water, 11.3 g. of <strong>[1632-74-2]3,6-dimethylpyridazine</strong>, 3.6 g. of silver nitrate and 12.3 g. of concentrated sulfuric acid were combined at ambient temperature, and to the mixture was added 41.8 g. of ammonium persulfate dissolved in 68 ml. of water. The addition was dropwise over a period of only 15 minutes. The reaction temperature rose to 75, and the mixture was stirred at that temperature for 30 minutes. The reaction mixture was then worked up substantially as described in the examples above to obtain 1.2 g. of 4-(1-hydroxymethyl-1-methylethyl)-<strong>[1632-74-2]3,6-dimethylpyridazine</strong>.
3,6-dibromo-4-(1-hydroxymethyl-1-methylethyl)pyridazine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With ammonium persulfate; sulfuric acid; silver nitrate; In water;
EXAMPLE 15 3,6-dibromo-4-(1-bromomethyl-1-methylethyl)pyridazine Three g. of <strong>[17973-86-3]3,6-dibromopyridazine</strong>, 1 g. of silver nitrate, 3.2 g. of 2,2-dimethyl-1,3-propanediol, 7 ml. of water and 1.9 g. of concentrated sulfuric acid were stirred at 30 while 5.8 g. of ammonium persulfate dissolved in 15 ml. of water was added dropwise. The temperature rose to 60. The mixture was then cooled and extracted with 100 ml. of dichloromethane. The organic layer was washed with 10 ml. of water and dried over magnesium sulfate. The solvent was removed under vacuum, and the resulting solid was chromatographed on silica gel, eluding with 3:1 hexane:ethyl acetate in a high performance liquid chromatography device. The product-containing fractions were combined and evaporated under vacuum, and the product was recrystallized from benzene/hexane to obtain 1.4 g. of 3,6-dibromo-4-(1-hydroxymethyl-1-methylethyl)pyridazine m.p. 140-141.
bis-[4-(2',2'-dimethyl-cyclopropyl carboxy-amido phenol)]-methane[ No CAS ]
[ 22308-12-9 ]
Yield
Reaction Conditions
Operation in experiment
With potassium cyanide; sodium hydroxide; In pyridine; methanol; dichloromethane; water; ethylene glycol; N,N-dimethyl-formamide;
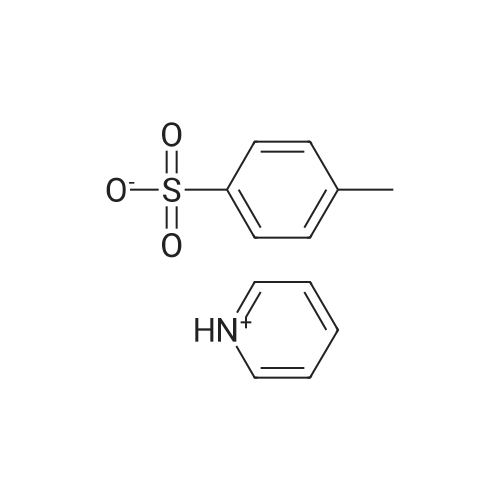
First, 2,2-dimethyl-1,3-propane-ditosylate was synthesised by adding p-toluenesulfonyl chloride (1000 g, 5.25 mol) at 0 C. to a solution of neopentyl glycol (218 g, 2.1 mol) in 500 ml pyridine with stirring. The mixture was stirred for 1.5 hr and then poured into 1500 ml water in a slow stream while stirring vigorously. It was stirred for an additional 1.5 hr and then filtered. The crude solid was recrystallized from acetone (2.0 L), filtered, washed with water (2*0.5 L), hexane (1*0.5 L) and dried. Snow white solid (814 g) m.p. 120-121 C. 2,2-Dimethyl-cyclopropyl nitrile was synthesised by stirring the 2,2-dimethyl-1,3-propane-ditosylate prepared above (412 g, 1.0 mol) with KCN (195.4 g, 3.0 mol) in 2.0 L of ethylene glycol with heating (E. R. Nelson et al., JACS, 1957, p. 3467). At around 80 C., a clear solution was formed. The desired product began to distill out at about 175 C. The distillation was continued until the temperature reached 200 C. The distillate (300 ml) formed two layers. The upper layer was separated and the lower layer was extracted with hexane (3*200 ml). The combined extracts were dried over Na2CO3, concentrated and re-distilled at normal pressure. The yield was 41.7 g (43.8%), b.p. 151-152 C. 2,2-Dimethyl-cyclopropyl carboxylic acid was prepared by mixing 2,2-dimethyl-cyclopropyl nitrile (41.7 g, 0.43 mol) with sodium hydroxide (44 g, 1.05 mol) in water (100 ml) and methanol (50 ml). The mixture was heated to reflux for 48 hr until a clear solution formed. Methanol was distilled off and the aqueous portion was extracted with ether (50 ml) and the aqueous layer was diluted with water (500 ml) and carefully acidified with conc. HCl. The acidified mixture was extracted with ether (5*300 ml), CH2Cl2 (5*300 ml). The extract was evaporated to yield a liquid which was distilled to give 44.9 g (91.6%) of oil, b.p. 55-57 C. at 0.3 mm. 2,2-Dimethyl-cyclopropyl carboxylic acid chloride was prepared by mixing 2,2-dimethyl-cyclopropyl carboxylic acid (20.0 g, 0.18 mol) in CH2Cl2 (100 ml) with 45.7 g (0.36 mol, 31.4 ml) of oxalyl chloride. The mixture was stirred for 1.0 hr and then a small amount of DMF was added to ensure the completion of the reaction. The mixture was then distilled to give 17.8 g (75%) of the desired product, b.p. 84-87 C. Compound III.1, Bis-[4-(2',2'-dimethyl-cyclopropyl carboxy-amido phenol)]-methane, was prepared by combining a solution of 4,4'-methylenedianiline (0.67 g, 3.4 mol) and diisopropyl-ethyl-amine (1.94 g, 2.61 ml, 0.019 mol) in THF (10 ml). The mixture was treated slowly with a solution of 2,2-dimethyl-cyclopropyl carboxylic acid chloride (1.0 g, 7.5 mmol) in THF (10 ml). The reaction mixture was stirred for 1.0 hr and then decomposed with water (250 ml). The precipitated solid was filtered and washed with 10% HCl (10 ml), 10% sodium hydroxide (10 ml), water and ether. The yield was 1.2 g, m.p. 207-210 C.
Reference example 1 [Synthesis of alkylphenyl borate 5(R = C5H11)]; THF (50 ml) is added to metal magnesium (1.22 g; 0.051mol), followed by agitation. After iodine (50 mg) is added thereto, a THF solution (10 ml) containing 4-bromopentylbenzene (4) (10.8 g; 0.048 mol) is slowly added dropwise thereto. Soon thereafter, a reaction is initiated. The THF solution (4) is added dropwise thereto at an appropriate speed to cause moderate reflux of the reaction solution. After the completion of dropwise addition, reflux of the solution is carried out for 1 hour. Then, it is cooled to -78°C and a THF solution (10 ml) containing trimethyl borate (5.9 g; 0.057 mol) is added dropwise thereto. Then, the temperature is increased to room temperature, followed by agitation for 3 hours. Subsequently, 2,2-dimethylpropane-1,3-diol (5.4 g; 0.052 mol) is added thereto, followed by agitation for 1 hour. Water is added to terminate the reaction and the water phase is removed using a separatory funnel. After drying with sodium sulfate, the solvent is removed by distillation and the residue is recrystalized with hexane (yield: 8.1g (0.031 mol; yield percentage: 65percent)).
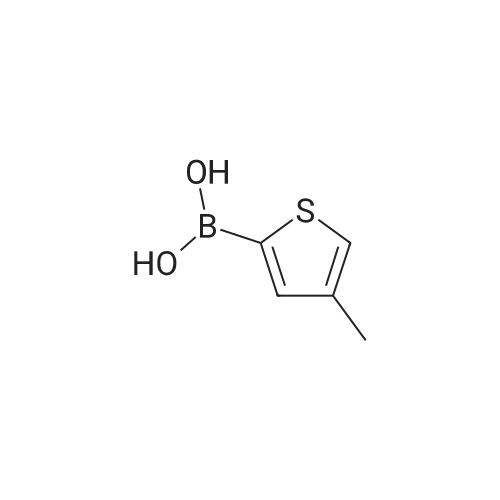
With toluene-4-sulfonic acid; In toluene; at 110℃; for 24h;
As shown in Scheme 2 below,To 100 mL of toluene in which 5.28 g (27.7 mmol) of p-toluenesulfonic acid (p-TsOH or p-TSA) was dissolved,20.0 g (138.7 mmol) of <strong>[51792-34-8]3,4-dimethoxy thiophene</strong>, compound (1) and 57.8 g (554.8 mmol) of 2,2-dimethyl-1,3-propanediol, compound (2) The reaction was carried out with stirring at 110 DEG C for 24 hours. after completion of the reaction, To the reaction was added a mixed solvent of water and ethyl acetate (EtOAc), the product was extracted into an organic layer, washed with brine,After removing water using anhydrous Na2SO4, filtered and concentrated. The product was purified by column chromatography on SiO2 (flash column chromatography,The eluate was separated into hexane: ethyl acetate (EtOAc) = 10: 1 mixture,3,4- (2,2-dimethylpropylene dioxy) thiophene(3,4- (2,2-Dimethylpropylenedioxy) thiophene,17.0 to 20.4 g (yield: 65 to 80percent) of Compound (3)) was obtained.1H-NMR data of the obtained compound are as follows: 1HNMR(CDCl3, Varian 400 MHz): delta 1.03 (6H, s), 3.72 (4H, s), 6.47 (2H, s).
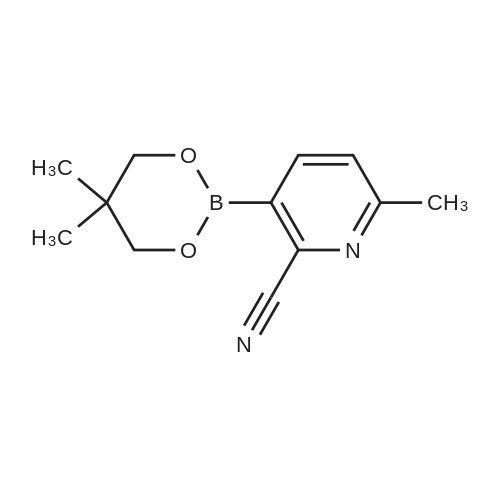
2,2,6,6-tetramethylpiperidine (3.49 ml, 20.52 mmol) was dissolved in dry THF (25ml) under argon and stirred at -30 0C; BuLi (13.33 ml, 21.33 mmol) 1.6 M in hexane was added over 5 min (the temperature never exceeded -25 0C). The yellow solution was stirred at -30 0C for 20 min, then chilled at -78 0C and tris(l-methylethyl) borate (4.38 ml, 18.96 mmol) was added over 5 min (the temperature never exceeded -73 0C). After 10 min at -78 0C, <strong>[1620-75-3]6-methyl-2-pyridinecarbonitrile</strong> (2.0 g, 16.93 mmol) dissolved in dry THF (14 ml) was added dropwise (over 20 min) maintaining internal temperature below -73 0C and the mixture became dark-brown. The mixture was stirred at -73 0C for 2 hours. The mixture was quenched with AcOH (2.374 ml, 41.5 mmol) dropwise at -73 0C (the temperature never exceeded -60 0C and the mixture became brilliant orange). The cooling bath was removed and the mixture left to reach the room temperature: during this period the mixture became thick and new THF (8 ml) had to be added in order to have a better stirring. The mixture was stirred 10 min at room temperature then 2,2-dimethyl-l,3-propanediol (2.409 g, 23.13 mmol) was added in one portion and the mixture stirred at room temperature overnight. The solvent was evaporated and the orange residue taken-up with DCM (100 ml) and 10 % water solution OfKH2PO4 (100 ml). The phases were separated and the water phase was back-extracted with DCM (50 ml). The combined organic phases were washed with 10 % water solution OfKH2PO4 (50 ml). The DCM was evaporated. The residue was dissolved in Et2O (100 ml) and extracted with NaOH 0.05 M (5 x 50 ml, boronic ester in water phase). The aqueous phases were joined together and the pH was adjusted between pH = 4 and pH = 5 with 10 % water solution OfKH2PO4 (50 ml). The so obtained yellow solution was extracted with EtOAc (3 x 200 mis). All the organics joined together were dried (Na2SO4) and evaporated the title compound D62 (2.29 g) of as yellow oil, that solidified on standing. C12Hi5BN2O2 requires 230. 1H NMR (400 MHz, CDCl3) delta ppm 7.97 - 8.15 (m, 1 H), 7.31 - 7.36 (m, 1 H), 3.85 (m, 4 H), 2.52 - 2.73 (s, 3 H), 0.97 - 1.10 (m, 6 H)
2,2,6,6-tetramethylpiperidine (3.49 ml, 20.52 mmol) was dissolved in dry THF (25ml) under argon and stirred at -30 0C; BuLi (13.33 ml, 21.33 mmol) 1.6 M in hexane was added over 5 min (the temperature never exceeded -25 0C). The yellow solution was stirred at -30 0C for 20 min, then chilled at -78 0C and tris(l-methylethyl) borate (4.38 ml, 18.96 mmol) was added over 5 min (the temperature never exceeded -73 0C).After 10 min at -78 0C, <strong>[1620-75-3]6-methyl-2-pyridinecarbonitrile</strong> (2.0 g, 16.93 mmol) dissolved in dry THF (14 ml) was added dropwise (over 20 min) maintaining internal temperature below -73 0C and the mixture became dark-brown. The mixture was stirred at -73 0C for 2 hours. The mixture was quenched with AcOH (2.374 ml, 41.5 mmol) dropwise at -73 0C (the temperature never exceeded -60 0C and the mixture became brilliant orange). The cooling bath was removed and the mixture left to reach the room temperature: during this period the mixture became thick and new THF (8 ml) had to be added in order to have a better stirring. The mixture was stirred 10 min at room temperature then 2,2-dimethyl-l,3-propanediol (2.409 g, 23.13 mmol) was added in one portion and the mixture stirred at room temperature overnight. The solvent was evaporated and the orange residue taken-up with DCM (100 ml) and 10 % water solution OfKH2PO4 (100 ml). The phases were separated and the water phase was back-extracted with DCM (50 ml). The combined organic phases were washed with 10 % water solution OfKH2PO4 (50 ml). The DCM was evaporated. The residue was dissolved in Et2O (100 ml) and extracted with NaOH 0.05 M (5 x 50 ml, boronic ester in water phase). The aqueous phases were joined together and the pH was adjusted between pH = 4 and pH = 5 with 10 % water solution OfKH2PO4 (50 ml). The so obtained yellow solution was extracted with AcOEt (3 x 200 mis). All the organics joined together were dried (Na2SO4) and evaporated the title compound D17 (2.29 g) of as yellow oil, that solidified on standing. C12Hi5BN2O2 requires 230. 1H NMR (400 MHz, CDCl3) delta ppm 7.97 - 8.15 (m, 1 H), 7.31 - 7.36 (m, 1 H), 3.85 (m, 4 H), 2.52 - 2.73 (s, 3 H), 0.97 - 1.10 (m, 6 H)
2,2,6,6-tetramethylpiperidine (3.49 ml, 20.52 mmol) was dissolved in dry THF (25 ml) under argon and stirred at -30 C.; BuLi (13.33 ml, 21.33 mmol) 1.6 M in hexane was added over 5 min (the temperature never exceeded -25 C.). The yellow solution was stirred at -30 C. for 20 min, then chilled at -78 C. and tris(1-methylethyl) borate (4.38 ml, 18.96 mmol) was added over 5 min (the temperature never exceeded -73 C.).After 10 min at -78 C., <strong>[1620-75-3]6-methyl-2-pyridinecarbonitrile</strong> (2.0 g, 16.93 mmol) dissolved in dry THF (14 ml) was added dropwise (over 20 min) maintaining internal temperature below -73 C. and the mixture became dark-brown. The mixture was stirred at -73 C. for 2 hours. The mixture was quenched with AcOH (2.374 ml, 41.5 mmol) dropwise at -73 C. (the temperature never exceeded -60 C. and the mixture became brilliant orange). The cooling bath was removed and the mixture left to reach the room temperature: during this period the mixture became thick and new THF (8 ml) had to be added in order to have a better stirring. The mixture was stirred 10 min at room temperature then 2,2-dimethyl-1,3-propanediol (2.409 g, 23.13 mmol) was added in one portion and the mixture stirred at room temperature overnight. The solvent was evaporated and the orange residue taken-up with DCM (100 ml) and 10% water solution of KH2PO4 (100 ml). The phases were separated and the water phase was back-extracted with DCM (50 ml). The combined organic phases were washed with 10% water solution of KH2PO4 (50 ml). The DCM was evaporated. The residue was dissolved in Et2O (100 ml) and extracted with NaOH 0.05 M (5×50 ml, boronic ester in water phase). The aqueous phases were joined together and the pH was adjusted between pH=4 and pH=5 with 10% water solution of KH2PO4 (50 ml). The so obtained yellow solution was extracted with EtOAc (3×200 mls). All the organics joined together were dried (Na2SO4) and evaporated the title compound D67 (2.29 g) of as yellow oil, that solidified on standing. C12H15BN2O2 requires 230. 1H NMR (400 MHz, CDCl3) delta ppm 7.97-8.15 (m, 1H), 7.31-7.36 (m, 1H), 3.85 (m, 4H), 2.52-2.73 (s, 3H), 0.97-1.10 (m, 6H).
2,2,6,6-tetramethylpiperidine (3.49 ml, 20.52 mmol) was dissolved in dry THF (25ml) under argon and stirred at -30 C; BuLi (13.33 ml, 21.33 mmol) 1.6 M in hexane was added over 5 min (the temperature never exceeded -25 C). The yellow solution was stirred at -30 C for 20 min, then chilled at -78 C and tris(l-methylethyl) borate (4.38 ml, 18.96 mmol) was added over 5 min (the temperature never exceeded -73 C).After 10 min at -78 C, <strong>[1620-75-3]6-methyl-2-pyridinecarbonitrile</strong> (2.0 g, 16.93 mmol) dissolved in dry THF (14 ml) was added dropwise (over 20 min) maintaining internal temperature below -73 C and the mixture became dark-brown. The mixture was stirred at -73 C for 2 hours. The mixture was quenched with AcOH (2.374 ml, 41.5 mmol) dropwise at -73 C (the temperature never exceeded -60 C and the mixture became brilliant orange). The cooling bath was removed and the mixture left to reach the room temperature: during this period the mixture became thick and new THF (8 ml) had to be added in order to have a better stirring. The mixture was stirred 10 min at room temperature then 2,2-dimethyl- 1,3 -propanediol (2.409 g, 23.13 mmol) was added in one portion and the mixture stirred at room temperature overnight. The solvent was evaporated and the orange residue taken-up with DCM (100 ml) and 10 % water solution of KH2PO4 (100 ml). The phases were separated and the water phase was back-extracted with DCM (50 ml). The combined organic phases were washed with 10 % water solution of KH2P04 (50 ml). The DCM was evaporated. The residue was dissolved in Et20 (100 ml) and extracted with NaOH 0.05 M (5 x 50 ml, boronic ester in water phase). The aqueous phases were joined together and the pH was adjusted between pH = 4 and pH = 5 with 10 % water solution of KH2P04 (50 ml). The so obtained yellow solution was extracted with EtOAc (3 x 200 mis). All the organics joined together were dried (Na2S04) and evaporated the title compound D31 (2.29 g) of as yellow oil, that solidified on standing. C12H15BN2O2 requires 230. 1H MR (400 MHz, CDC13) delta ppm 7.97 - 8.15 (m, 1 H), 7.31 - 7.36 (m, 1 H), 3.85 (m, 4 H), 2.52 - 2.73 (s, 3 H), 0.97 - 1.10 (m, 6 H).
2,2,6,6-tetramethylpiperidine (3.49 ml, 20.52 mmol) was dissolved in dry THF (25ml) under argon and stirred at -30 C; BuLi (13.33 ml, 21.33 mmol) 1.6 M in hexane was added over 5 min (the temperature never exceeded -25 C). The yellow solution was stirred at -30 C for 20 min, then chilled at -78 C and tris(l -methylethyl) borate (4.38 ml, 18.96 mmol) was added over 5 min (the temperature never exceeded -73 C).After 10 min at -78 C, 6-methyl-2 -pyridinecarbonitrile (2.0 g, 16.93 mmol) dissolved in dry THF (14 ml) was added dropwise (over 20 min) maintaining internal temperature below -73 C and the mixture became dark-brown. The mixture was stirred at -73 C for 2 hours. The mixture was quenched with AcOH (2.374 ml, 41.5 mmol) dropwise at -73 C (the temperature never exceeded -60 C and the mixture became brilliant orange). The cooling bath was removed and the mixture left to reach the room temperature: during this period the mixture became thick and new THF (8 ml) had to be added in order to have a better stirring. The mixture was stirred 10 min at room temperature then 2,2-dimethyl- 1,3 -propanediol (2.409 g, 23.13 mmol) was added in one portion and the mixture stirred at room temperature overnight. The solvent was evaporated and the orange residue taken-up with DCM (100 ml) and 10 % water solution of KH2PO4 (100 ml). The phases were separated and the water phase was back-extracted with DCM (50 ml). The combined organic phases were washed with 10 % water solution of KH2PO4 (50 ml). The DCM was evaporated. The residue was dissolved in Et20 (100 ml) and extracted with NaOH 0.05 M (5 x 50 ml, boronic ester in water phase). The aqueous phases were joined together and the pH was adjusted between pH = 4 and pH = 5 with 10 % water solution of KH2P04 (50 ml). The so obtained yellow solution was extracted with EtOAc (3 x 200 ml). All the organics joined together were dried (Na2S04) and evaporated the title compound D67 (2.29 g) of as yellow oil, that solidified on standing. C12H15BN2O2 requires 230. 1H MR (400 MHz, CDC13) delta ppm 7.97 - 8.15 (m, 1 H), 7.31 - 7.36 (m, 1 H), 3.85 (m, 4 H), 2.52 - 2.73 (s, 3 H), 0.97 - 1.10 (m, 6 H).
2,2,6,6-tetramethylpiperidine (8.72ml_, 51 .7mmol) was dissolved in dry THF (62ml_) under argon and stirred at -30 C; n-Butyl lithium (21 .33ml_, 53.32mmol) 2.5 M in hexane was added over 5 m. The yellow solution was stirred at -30 C for 20 min, then chilled at -78 C and tripropan-2-yl borate (10.94ml_, 47.4mmol) was added over 5 min. After 10 min at -78 C, <strong>[1620-75-3]6-methyl-2-pyridinecarbonitrile</strong> (5000mg, 42.32mmol) dissolved in dry THF (35ml_) was added dropwise (over 20 min) maintaining internal temperature below -73 C and the mixture became dark-brown. The mixture was stirred at -73 C for 6 hrs. The mixture was quenched with acetic acid (5.94ml_, 103.75mmol) dropwise at -73 C (the temperature never exceeded -60 C). The cooling bath was removed and the mixture left to reach the room temperature: during this period the mixture became thick and new THF (15 ml_) had to be added in order to have a better stirring. The mixture was stirred 10 min at RT then 2, 2-dimethylpropane-1 ,3-diol (6022.5mg, 57.83mmol) was added in one portion and the mixture stirred at RT overnight. The solvent was evaporated and the orange residue taken-up with DCM and 10 % water solution of Kappa2RhoOmicron4. The phases were separated and the water phase was back-extracted with DCM. The combined organic phases were washed with 10 % water solution of KH2PO4 (50 ml). The DCM was evaporated. The residue was dissolved in Et20 and extracted with NaOH 0.05 M (5 x 250 ml_, boronic ester in water phase). The aqueous phases were joined together and the pH was adjusted between pH = 4 and pH = 5 with 10 % water solution of KH2PO4 (50 ml_). The so obtained yellow solution was extracted with EtOAc and DCM. All the organics joined together were dried (Na2S04) and evaporated to afford 3-(5,5-dimethyl-1 ,3,2- dioxaborinan-2-yl)-<strong>[1620-75-3]6-methylpyridine-2-carbonitrile</strong> (Int a: 3.8 mg) which was used in the next step without purification.
2,2,6,6-tetramethylpiperidine (8.72mL, 51 .7mmol) was dissolved in dry THF (62mL) under argon and stirred at -30 C; n-Butyllithium (21.33mL, 53.32mmol) 2.5 M in hexane was added over 5 m. The yellow solution was stirred at -30 C for 20 min, then chilled at -78 C and tripropan-2-yl borate (10.94mL, 47.4mmol) was added over 5 min. After 10 min at -78 C, <strong>[1620-75-3]6-methyl-2-pyridinecarbonitrile</strong> (5000mg, 42.32mmol) dissolved in dry THF (35mL) was added dropwise (over 20 min) maintaining internal temperature below -73 C and the mixture became dark-brown. The mixture was stirred at -73 C for 6 hrs. The mixture was quenched with acetic acid (5.94mL, 103.75mmol) dropwise at -73 C (the temperature never exceeded -60 C). The cooling bath was removed and the mixture left to reach the room temperature: during this period the mixture became thick and new THF (15 mL) had to be added in order to have a better stirring. The mixture was stirred 10 min at RT then 2,2-dimethylpropane-1 ,3-diol (6022.5mg, 57.83mmol) was added in one portion and the mixture stirred at RT overnight. The solvent was evaporated and the orange residue taken-up with DCM and 10 % water solution of Kappa2RhoOmicron4. The phases were separated and the water phase was back-extracted with DCM. The combined organic phases were washed with 10 % water solution of KH2PO4 (50 ml). The DCM was evaporated. The residue was dissolved in Et20 and extracted with NaOH 0.05 M (5 x 250 ml_, boronic ester in water phase). The aqueous phases were joined together and the pH was adjusted between pH = 4 and pH = 5 with 10 % water solution of KH2PO4 (50 ml_). The so obtained yellow solution was extracted with EtOAc and DCM. All the organics joined together were dried (Na2S04) and evaporated to afford 3-(5,5-dimethyl-1 ,3,2- dioxaborinan-2-yl)-<strong>[1620-75-3]6-methylpyridine-2-carbonitrile</strong> (Int a: 3816 mg) which was used in the next step without purification.
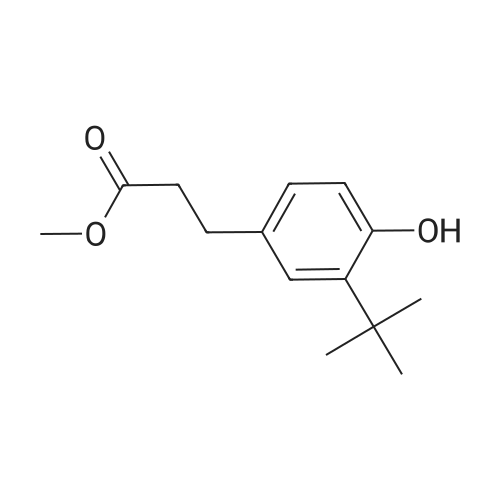
3-[3-tert-butyl-4-(5,5-dimethyl-1,3,2-dioxaphosphorinan-2-yloxy)-phenyl]-propionic acid methyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
82%
Example 1; Preparation of 3-[3-tert-butyl-4-(5,5-dimethyl -1 ,3,2-dioxaphosphorinan-2-yloxy)- phenyl]-propionic acid methyl ester;To a solution of 30 g (0.3 mole) of 2,2-dimethyl-1 ,3-propandiol in 700 ml of toluene is added dropwise at 60°C 26 ml. (0.3 mole) of phosphorus trichloride. After 55 min., the P31 NMR shows that the PCI3 (signal at 220ppm) is fully reacted and a new product is formed (signal at 148 ppm). The temperature is raised to 80°C. Now a solution of 71 g (0.3 mole) 3-(3-tert- butyl-4-hydroxy-phenyl)-propionic acid methyl ester and 200 ml of triethylamine in 300 ml of toluene are added dropwise (30 minutes). The reaction mixture turns into a suspension which is stirred for 2 hours 30 minutes. After 2 hours and 15 minutes the P31 NMR shows EPO <DP n="33"/>that the educt (signal at 148 ppm) is fully reacted. After filtration and removal of the toluene (rotary evaporator) the product is isolated. Yield: 90 g (0.244 mole = 82 percent of theory). The title compound is a crude yellow oil, molecular weight 368.41 (Ci9H29O5P), P31 NMR (400 MHz, CDCI3, reference is PCI3 (219 ppm) window is -230 to + 230 ppm) signals at: 1 15ppm (major peak). Total reaction time: 3 h 25 min. Filtration through a neutral alumina column provides a mobile colorless liquid. Calculated percentP: 8.41 ; found percentP: 8.61.
(3R,6S/3S,6R)-5,5-dimethylhydro-1'H-spiro[1,3-dioxane-2,2'-pentalen]-5'(3'H)-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
3.7 g
With toluene-4-sulfonic acid; In toluene; for 18h;Reflux;
[0398] 5 g of hydropentalene-2,5-dione (36.19 mmol, 1 eq.), 377 g of 2,2-dimethyl-1,3-propanediol (36.19 mmol, 1 eq.) and 0.069 g of para-toluenesulfonic acid (0.36 mmol, 0.01 eq.) in 50 mL of toluene are refluxed for 18 h. The medium is diluted in ethyl acetate and washed successively with aqueous 1N sodium hydroxide solution, water and brine. The organic phase is dried over sodium sulfate, filtered and evaporated. The residue is chromatographed on silica gel, eluting with a gradient of ethyl acetate in heptane ranging from 30% to 33%. 3.7 g of (3R,6S/3S,6R)-5,5-dimethylhydro-1?H-spiro[1,3-dioxane-2,2?-pentalen]-5?(3?H)-one are obtained.
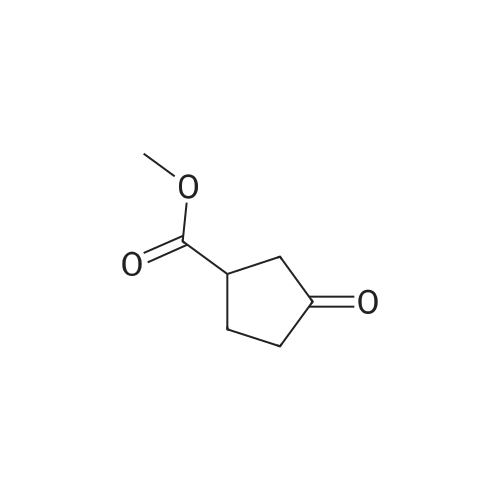
Example 204B 3,3-Dimethyl- l,5,9-trioxa-spiro[5.5]undecane-7-carboxylic acid methyl ester A mixture of Example 204A (5 g, 31 mmol), 2,2-dimethyl-propane-l,3-diol (4.27 g, 41 mmol) and toluene-4-sulfonic acid (200 mg) in toluene (60 mL) was refluxed overnight using Dean- Stark trap. After cooling to room temperature, the reaction mixture was quenched with a saturated NaHC(¾ (100 mL) solution. The aqeous layer was extracted with ethyl acetate (200 mL). The combined organic layers were washed with brine, dried over Na2S04, filtered, and concentrated in vacuo. The residue was purified by column chromatography (petroleum ether/ethyl acetate: 10/2) to provide the title compound (5 g, 65%) as a red oil.
In 1,4-dioxane; at 210.0℃; for 1.0h;Microwave irradiation;
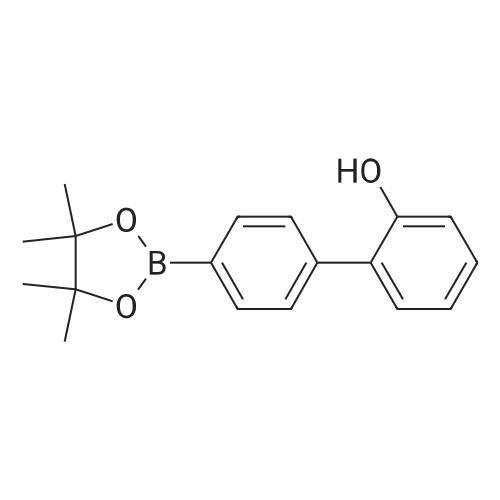
A mixture of 2,2-dimethylpropane-1,3-diol (4.0 g, 38 mmol) and 4'-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)biphenyl-2-ol (1.0 g, 3.4 mmol) in 1,4-dioxane (2 mL) was subjected to microwave irradiation at 210 C. for 1 hour. The cooled reaction mixture was partitioned between water (50 mL) and 1:1 EtOAc/heptane (25 mL:25 mL). The organic layer was washed with water (30 mL) and brine (30 mL), dried over Na2SO4 and concentrated in vacuo to give the title compound (0.95 g, 99% yield). GC/MS, M=282 at 5.31 min. 1H NMR (500 MHz, CDCl3) delta 7.94 (d, J=7.81 Hz, 2H), 7.48 (d, J=7.81 Hz, 2H), 7.31-7.26 (m, 2H), 7.03-6.98 (m, 2H), 3.82 (s, 4H), 1.08-1.04 (m, 6H).
To a solution of <strong>[552331-06-3]6-bromo-3-chloroisoquinoline</strong> (Frontier Scientific, 1.594 g, 6.57 mmol) and 2,2-dimethylpropane-1 ,3-diol (684 mg, 6.57 mmol) in cyclopentyl methyl ether (20 mL) was added cesium carbonate (2.354 g, 7.23 mmol) at 23C. The reaction mixture was heated to 120C for 18 h. The reaction mixture was cooled to RT, diluted with ethyl acetate (50 mL) and washed with water (30 mL), brine (30 mL) and the resulting organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The residue was purified by silica gel chromatography (80 g Isco Rf Gold Column, 0-100% ethyl acetate//so- hexanes gradient) to afford the title compound (461 mg, 23%) as a white solid
23%
With caesium carbonate; at 23 - 120℃; for 18h;
To a solution of <strong>[552331-06-3]6-bromo-3-chloroisoquinoline</strong> (Frontier Scientific, 1.594 g, 6.57 mmol) and 2,2-dimethylpropane-1,3-diol (684 mg, 6.57 mmol) in cyclopentyl methyl ether (20 mL) was added cesium carbonate (2.354 g, 7.23 mmol) at 23 C. The reaction mixture was heated to 120 C. for 18 h. The reaction mixture was cool to RT, diluted with ethyl acetate (50 mL) and washed with water (30 mL), brine (30 mL) and the resulting organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The residue was purified by silica gel chromatography (80 g Isco Rf Gold Column, 0-100% ethyl acetate/iso-hexanes gradient) to afford the title compound (461 mg, 23%) as a white solid.
3-((6-aminopyrazin-2-yl)oxy)-2,2-dimethylpropan-1-ol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
12.84%
To a stirred suspension of NaH (2.316 g, 57.9 mmol) in 1,4-Dioxane (20 mL) under nitrogen at 0C was added a solution of 2,2-dimethylpropane-l,3-diol (4.02 g, 38.6 mmol) in 1,4-Dioxane (20 mL) dropwise during 10 min at 0C. After 10 min added a solution of 6-chloropyrazin-2-amine (5 g, 38.6 mmol) in 1,4-Dioxane (20 mL) was added dropwise during 10 min at 0C.The reaction mixture was heated at 120 C for 48 hr. TLC indicates small amount starting material along with product. Reaction mixture was poured into ice cold water (60 mL), aqueous layer was extracted with EtOAc (2 x 100 mL). The organic layer was washed with brine (50 mL). The organic layer was dried over Na2S04, filtered and concentrated under reduced pressure to obtain crude product. Crude product was purified by column chromatography using 100-200 silica gel as a eluent (0-50% EtOAc in petether) to obtain 3-((6-aminopyrazin-2-yl)oxy)-2,2-dimethylpropan-l-ol (1 g, 4.95 mmol, 12.84 % yield), LCMS (m/z): 198.00 [M+H]+.
tert-butyl 4-(5,5-dimethyl-1,3,2-dioxaborinan-2-yl)piperidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
67%
2nd step: the metal lithium (1.0g, 0 . 15mol) and double (diisopropylamine) boron bromide (21.8g, 75mmol) by adding 2-methyl tetrahydrofuran (90 ml) in, temperature control -20 C to -10 C, dripping is dissolved in 2-methyl tetrahydrofuran (35 ml) of in N-Boc-1, 2, 5, 6-tetrahydro-pyridine-4-bromo (18.9g, 72mmol), stirring after 5 hours, the end of the detection reaction, adding npg (8.6g, 83mmol), heating to reflux reaction. Cooling, adding 10% hydrochloric acid aqueous solution to adjust PH= 5-6, organic layer by adding 10% palladium carbon (1.2g), after the replacement of nitrogen, filled with 1 atmospheric pressure hydrogen, after the reaction, drying of the organic layer, by adding 45 ml normal heptane cooling -10 C pulping, filtering to obtain 14.3g kind of white solid N-Boc-piperidin-4-boronic acid new pentamethylene glycol ester, GC: 98.5%, HNMR: > 98%, yield: 67%.
With hydrogenchloride; magnesium; methyl iodide; In tetrahydrofuran; water; ethyl acetate; at 20℃;pH 3-4;Reflux;
Step two: adding metal magnesium (2.2 g, 92mmol) and 10 ml of tetrahydrofuran, add 2-3 drop iodine methane initiating the dropwise 13.2 g 3,6-dihydro -2H-pyran-4-polybromide dissolved in 60 ml tetrahydrofuran solution, reagent cheng Geshi preparing reflux reaction is omitted, then drop by adding boric acid three methyl ester (11.4 g, 0 . 11mol) in, after the reaction is complete, by adding 10% hydrochloric acid solution to adjust PH= 3-4, organic layer by adding 110 ml of ethyl acetate and npg (10.0 g, 96mmol), stirring at room temperature until the reaction is complete. After laminating, an organic layer saturated salt water washing, solvent after evaporation to dryness, by adding normal heptane, cooling to -10 C, filtering to obtain 10.0 g kind of white solid: 3,6-dihydro -2H-thiopyran-4-boronic acid new pentamethylene glycol ester, GC: 99.7%, HNMR > 98%, the yield is 64%.
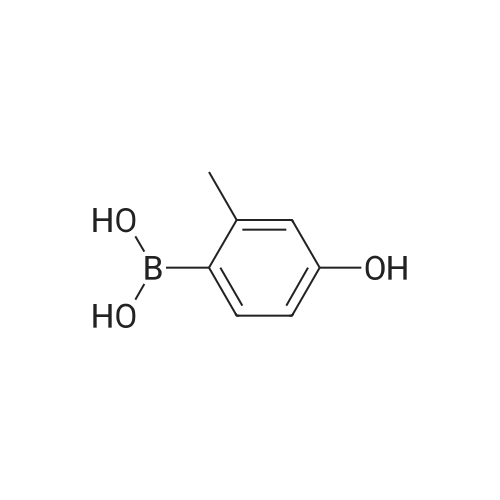
4-(5,5-dimethyl-1,3,2-dioxaborinan-2-yl)-3-methylphenol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With Amberlyst 15H+; In 2-methyltetrahydrofuran; at 20.0℃;Inert atmosphere;
4.675 g (4-hydroxy-2-methyl-phenyl)boronic acid (30.76 mmol), 3.204 neopentyl glycol(32.9 mmol), Amberlyst 15H and 150 mL 2-Me-THF were stirred at r.t. under N2atmosphere until no further conversion was observed. The mixture was then filteredthrough Celite and the filtrate was concentrated under reduced pressure to obtain4-(5 ,5-dimethyl- 1,3 ,2-dioxaborinan-2-yl)-3 -methyl-phenol. NMR (400 MHz, CDC13) oe:7.64 (m, 1H), 6.60 (m, 2H), 5.23 (br s, 1H), 3.75 (s, 4H), 2.47 (s, 3H), 1.01 (s, 6H)
Compound 2 (1.72 g, 10.00 mmol) and tetrahydrofuran (100 mL) were added using a four-necked flask, and argon bubbling was performed at room temperature for 30 minutes. After cooling to -78 C., lithium diisopropylamide (hexane suspension 10 w%, 49.1 mL, 30.0 mmol) was added and stirred for 20 minutes, trimethoxyborane (3.43 g, 33.0 mmol) was added and stirred for 1 hour did. Then, 2,2-dimethyl-1,3-propanediol (4.17 g, 40.0 mmol) was added and sulfuric acid was added dropwise until the pH reached 1 to 2, while stirring at room temperature, and the mixture was stirred for 3 hours. After filtration with celite, concentration, methanol / water was placed in the system, the precipitated solid was collected by filtration and recrystallized from hexane to obtain 1.11 g of white solid compound 4. (Yield 28.1%)
4-cyano-2-methylphenylboronic acid neopentyl glycol ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
82%
In toluene; at 120℃; for 1.0h;Molecular sieve;
A mixture of <strong>[313546-18-8]4-cyano-2-methylphenylboronic acid</strong> (906 mg, 5.6 mmol), 2,2-dimetyl-1,3-propanediol (641 mg, 6.15 mmol) and 3 molecular sieves (1 g) in anhydrous toluene (10 mL) was heated at 120 C. for 1 h and then was allowed to cool to room temperature. The mixture was filtered and the filtrate was concentrated in vacuo. The residue was purified by flash chromatography (SiO2, eluted with gradient from 20% to 60% EtOAc in hexanes). Yield: 1.054 g (82%) as yellowish solid.
General procedure: A 0.5-dram vial equipped with a magnetic stirring bar wascharged with stock solutions of 2-formylphenylboronicacid (0.10 M, 200 muL, 0.02 mmol), pinacol (0.10 M, 200 muL,0.02mmol) and 5c (0.25M, 80 muL, 0.02mmol). The solventswere removed in vacuo. CHCl3 (50 muL) was added undera positive pressure of argon. The reaction was stirred at 50°C for 30 min. The volatiles were then removed under highvacuum. [Pd(allyl)Cl]2 (0.13 M in CHCl3, 40 muL, 5.0 mumol)was added under a positive pressure of argon and thereaction stirred at 23°C for 15 min. Allyl acetate rac-7(0.01 M in CHCl3, 10 muL, 0.10 mmol), bis(trimethylsilyl)acetamide (49 muL, 0.20 mmol), diethyl malonate (30 muL,0.20 mmol) and potassium acetate (0.50 mg, 5.0 mumol)were added sequentially. The reaction was stirred at 23°Cfor 24 hours. The crude reaction mixture was then filteredthrough a short plug of Celite and rinsed three times withCH2Cl2. An aliquot of the reaction mixture (5.0 muL) wasused for conversion and ee analyses using HPLC. HPLCconditions: Chiralpak IA column, 2.0percent isopropanol/hexanes,0.5 mL/min, 254 nm, tr: 22.9 and 29.2min.
General procedure: A 0.5-dram vial equipped with a magnetic stirring bar wascharged with stock solutions of 2-formylphenylboronicacid (0.10 M, 200 muL, 0.02 mmol), pinacol (0.10 M, 200 muL,0.02mmol) and 5c (0.25M, 80 muL, 0.02mmol). The solventswere removed in vacuo. CHCl3 (50 muL) was added undera positive pressure of argon. The reaction was stirred at 50°C for 30 min. The volatiles were then removed under highvacuum. [Pd(allyl)Cl]2 (0.13 M in CHCl3, 40 muL, 5.0 mumol)was added under a positive pressure of argon and thereaction stirred at 23°C for 15 min. Allyl acetate rac-7(0.01 M in CHCl3, 10 muL, 0.10 mmol), bis(trimethylsilyl)acetamide (49 muL, 0.20 mmol), diethyl malonate (30 muL,0.20 mmol) and potassium acetate (0.50 mg, 5.0 mumol)were added sequentially. The reaction was stirred at 23°Cfor 24 hours. The crude reaction mixture was then filteredthrough a short plug of Celite and rinsed three times withCH2Cl2. An aliquot of the reaction mixture (5.0 muL) wasused for conversion and ee analyses using HPLC. HPLCconditions: Chiralpak IA column, 2.0percent isopropanol/hexanes,0.5 mL/min, 254 nm, tr: 22.9 and 29.2min.
General procedure: A 0.5-dram vial equipped with a magnetic stirring bar wascharged with stock solutions of 2-formylphenylboronicacid (0.10 M, 200 muL, 0.02 mmol), pinacol (0.10 M, 200 muL,0.02mmol) and 5c (0.25M, 80 muL, 0.02mmol). The solventswere removed in vacuo. CHCl3 (50 muL) was added undera positive pressure of argon. The reaction was stirred at 50C for 30 min. The volatiles were then removed under highvacuum. [Pd(allyl)Cl]2 (0.13 M in CHCl3, 40 muL, 5.0 mumol)was added under a positive pressure of argon and thereaction stirred at 23C for 15 min. Allyl acetate rac-7(0.01 M in CHCl3, 10 muL, 0.10 mmol), bis(trimethylsilyl)acetamide (49 muL, 0.20 mmol), diethyl malonate (30 muL,0.20 mmol) and potassium acetate (0.50 mg, 5.0 mumol)were added sequentially. The reaction was stirred at 23Cfor 24 hours. The crude reaction mixture was then filteredthrough a short plug of Celite and rinsed three times withCH2Cl2. An aliquot of the reaction mixture (5.0 muL) wasused for conversion and ee analyses using HPLC. HPLCconditions: Chiralpak IA column, 2.0% isopropanol/hexanes,0.5 mL/min, 254 nm, tr: 22.9 and 29.2min.
(3-(5,5-dimethyl-1,3-dioxan-2-yl)phenyl)methanol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
With toluene-4-sulfonic acid; In toluene;Reflux; Dean-Stark;
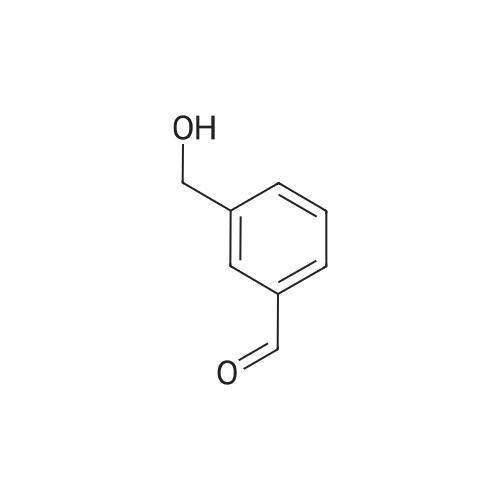
A solution of <strong>[52010-98-7]3-(hydroxymethyl)benzaldehyde</strong> (2.3 g, 16.89 mmol), 2,2- dimethylpropane-l,3-diol (5.25 g, 50.68 mmol) and TsOH (290.91 mg, 1.69 mmol) in toluene (60 mL) was refluxed for 2 h in a 100 mL flask with Dean-Stark trap. After cooling to rt, the mixture was concentrated to dryness. The residue was purified by column chromatography on silica gel (eluent: petroleum ether/ ethyl acetate from 5:1 to 1:1) to afford (3-(5,5-dimethyl-l,3-dioxan-2- yl)phenyl)methanol as an oil (3 g, 80%).
2-(3-bromo-5-chlorophenyl)-5,5-dimethyl-1,3,2-dioxaborinane[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
87%
In diethyl ether; at 20℃; for 4.0h;Inert atmosphere; Molecular sieve;
General procedure: To a Et2O solution of anorganoboronic acid (1.00 equiv) and 2,2-dimethylpropane-1,3-diol (neopentyl glycol)(1.02 equiv), 4A molecular sieves was added and the reaction mixture was stirred atroom temperature. After the reaction finished, the reaction mixture was filtered andconcentrated in vacuo. The residue was subjected to flash column chromatography(eluent: petroleum ether/ethyl acetate) or recrystallization to obtain the desired product
In diethyl ether; at 20℃;Inert atmosphere; Molecular sieve;
General procedure: To a Et2O solution of anorganoboronic acid (1.00 equiv) and 2,2-dimethylpropane-1,3-diol (neopentyl glycol)(1.02 equiv), 4A molecular sieves was added and the reaction mixture was stirred atroom temperature. After the reaction finished, the reaction mixture was filtered andconcentrated in vacuo. The residue was subjected to flash column chromatography(eluent: petroleum ether/ethyl acetate) or recrystallization to obtain the desired product
In diethyl ether; at 20℃;Inert atmosphere; Molecular sieve;
General procedure: To a Et2O solution of anorganoboronic acid (1.00 equiv) and 2,2-dimethylpropane-1,3-diol (neopentyl glycol)(1.02 equiv), 4A molecular sieves was added and the reaction mixture was stirred atroom temperature. After the reaction finished, the reaction mixture was filtered andconcentrated in vacuo. The residue was subjected to flash column chromatography(eluent: petroleum ether/ethyl acetate) or recrystallization to obtain the desired product






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +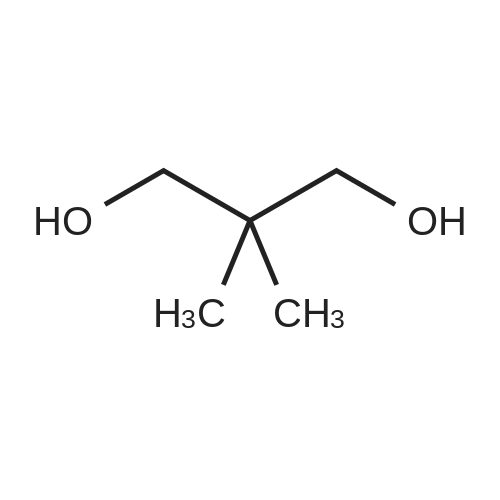

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping