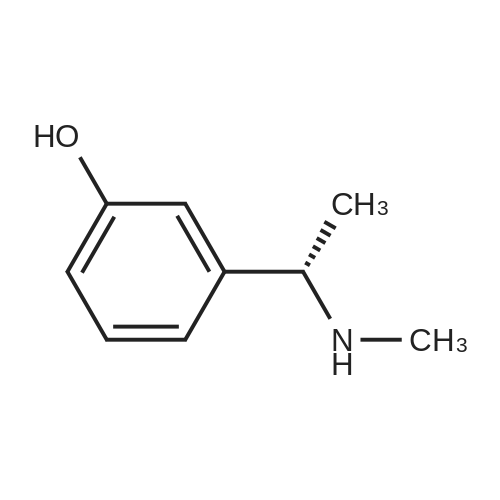
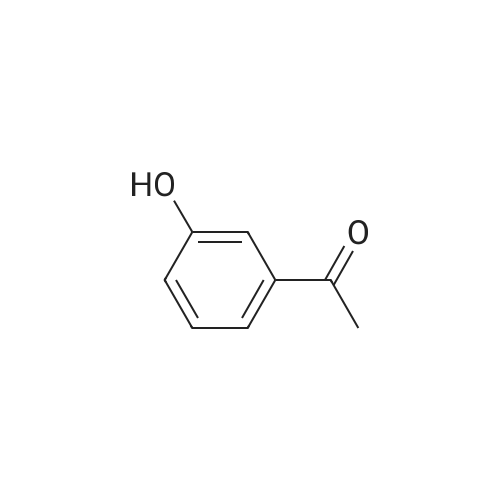
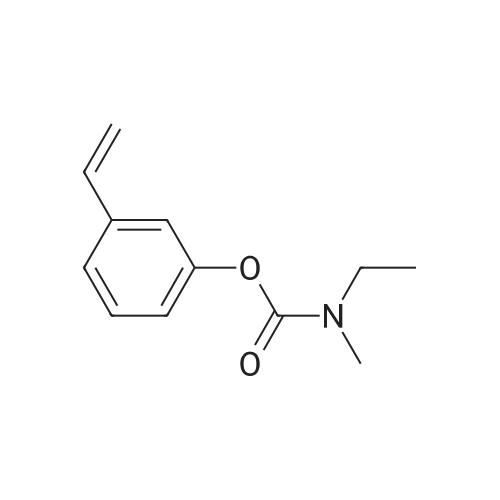
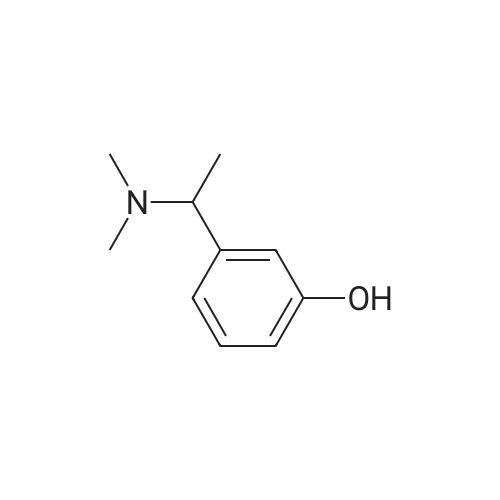
| 85% |
In acetone; at 60℃; for 1h; |
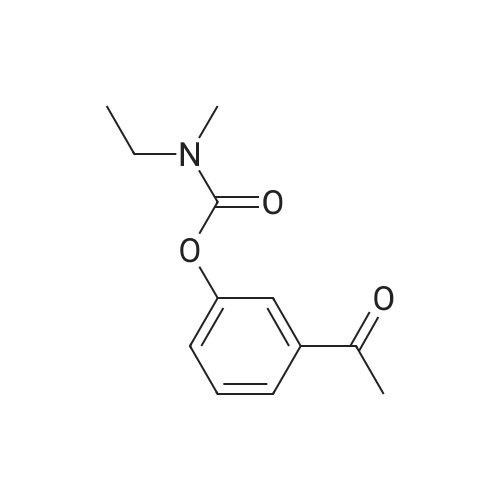
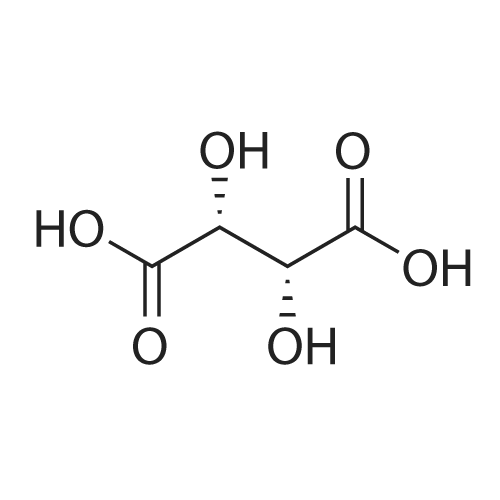
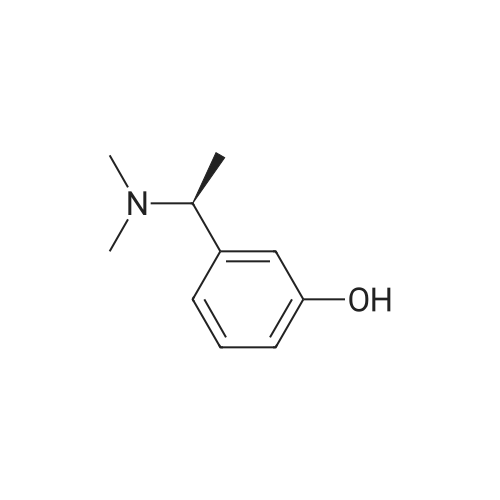
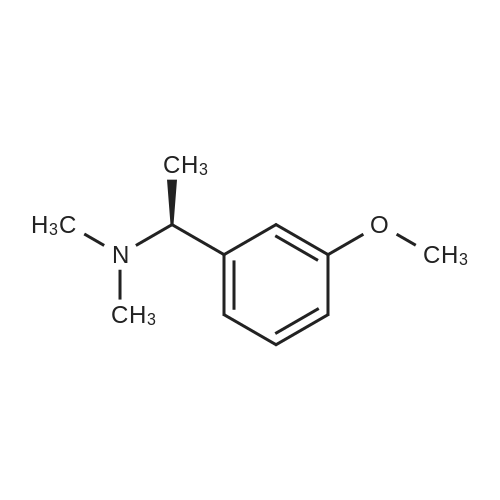
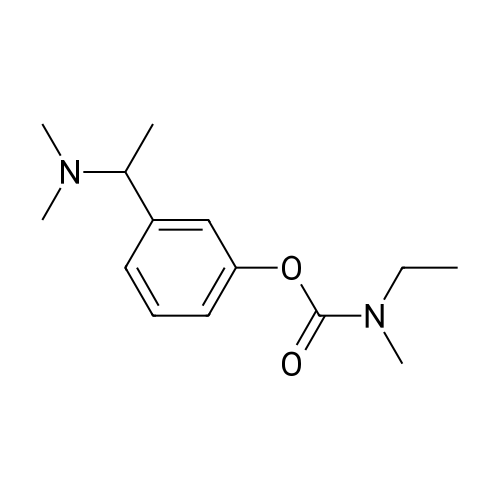
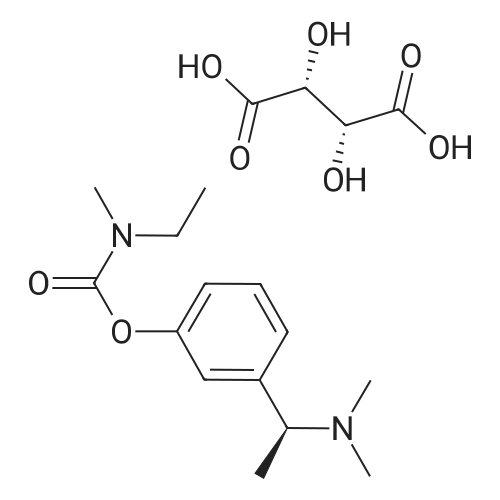
(-)-S-3-[l-(dimethylamino)ethyl]phenyl-N-ethyl-N-methylcarbamate (I) (100gms;1.0 mole) and L-tartaric acid (60gms; l.Omole) were added to acetone (1000ml) and the mixture heated at 600C for 1.0 hour to get a clear mixture, which was then cooled for EPO <DP n="14"/>complete precipitation of the tartrate salt of compound (I). The tartrate salt of compound was filtered and dried. Yield: 135 gms. %Yield: 85%. HPLC Purity: 99%. |
| 83% |
In acetone; at 20℃; for 0.666667h;Reflux; |
Embodiment 22 The Preparation of <strong>[123441-03-2]Rivastigmine</strong> (Formula I) Mix 5.0 g (0.02 mol) (S)-3-(1-(dimethylamino)ethyl)phenyl ethyl(methyl)carbamate (formula X) with 20 ml acetone and 3.0 g L-tartaric acid (0.02 mol) at room temperature, heat the mixture until it refluxes, and let it react for 40 min. After it is cooled to 40 C., add seed crystals and stir it at room temperature for 2 hours. Use ice bath for insulation for 5 hours and set it in a refrigerator overnight. Filter it, and set it in a vacuum oven at 40 C. for 10 hours to receive 6.64 g white crystals with a yield of 83%. HPLC purity?99.8%, ee value of ?99.8%. Optical rotation [alpha]20D=+ 6.0 (C=5, ethanol); mp 123.8-124.5 C. 1HNMR (CDCl3) delta: 1.16, 1.24 (2*t, 3H), 1.67 (d, 3H), 2.65 (s, 6H), 2.96, 3.05 (2*s, 3H), 3.37, 3.45 (2*q, 2H), 4.34 (q, 1H), 4.47 (s, 2H), 7.14 (t, 1H), 7.20 (s, 1H), 7.28 (d, 1H), 7.39 (t, 1H); MS (ESI) m/z: 251.2 ([M+1]+). |
| 83% |
In acetone; for 0.666667h;Reflux; |
Mix 5.0 g (0.02 mol) (S)-3-(1-(dimethylamino)ethyl)phenyl ethyl(methyl)carbamate (formulaX) with 20 ml acetone and 3.0 g L-tartaric acid (0.02 mol) at room temperature, heat the mixture until it refluxes, and let it react for 40 min. After it is cooled to 40 C, add seed crystals and stir it at room temperature for 2 hours. Use ice bath for insulation for 5 hours and set it in a refrigerator overnight. Filter it, and set it in a vacuum oven at 40 C for 10 hours to receive 6.64 g white crystals with a yield of 83%. HPLC purity > 99.8%, ee value of ? 99.8%. Optical rotation [alpha]20D = +6.0 (C = 5, ethanol); mp 123.8-124.5 C. 1HNMR (CDCl3) delta: 1.16,1.24 (2xt,3H), 1.67 (d,3H), 2.65 (s,6H), 2.96,3.05 (2xs,3H), 3.37,3.45 (2xq,2H), 4.34 (q,1H), 4.47 (s,2H), 7.14 (t,1H), 7.20 (s,1H), 7.28 (d,1H), 7.39 (t,1H); MS (ESI) m/z: 251.2 ([M+1]+). |
| 81.5% |
In isopropyl alcohol;Heating; |
Add 350 ml of isopropanol, 76 g of intermediate 2, 51.4 g of L(+) tartaric acid to a 1L reaction flask, stir and heat untilflow. Then cooled to room temperature, crystallized 2h, suction filtration, filter cake rinsed with a small amount of isopropanol, to obtain crude <strong>[123441-03-2]rivastigmin</strong>e <strong>[123441-03-2]rivastigmin</strong>e. willThe solid wet product obtained in the previous step was added to a 1L reaction flask, 350 ml of acetone was added, and the mixture was stirred at room temperature for 1 hour. Then filter, filter cake with a small amountRinse with acetone and dry to give 99.1 g of a white solid. Yield 81.5%, R isomer 0.11%. |
| 78% |
In methanol; acetone; at 40℃;Reflux; |
Example 10: Preparation of <strong>[123441-03-2]rivastigmin</strong>e (the compound represented by formula (VIII)); 117.5ml acetone and 2.83g (18.9mmol) L-tartaric acid were added into 4.72g (18.9mmol) the compound represented by formula (VII). The mixture was heated to 40C, followed by adding 11.8ml methanol, and refluxed for 40 min. After the mixture was cooled to 40C, seed crystal was added was and the mixture was stirred at room temperature for 2 hours. Then the mixture was settled in an ice bath for 5 hours followed by standing in a refrigerator overnight. After filtering and drying in a vacuum oven at 40C for 9 hours, white crystal (5.89g) was obtained with a yield of 78%. HPLC purity =99.8%, ee%=99.8% Optical rotation [alpha]20D = +6.0, C=5, ethanol; mp 122.3-124.1C. 1H NMR (CDCl3) delta ppm: 1.24, 1.16 (2×t, 3H), 1.67 (d, 3H), 2.65 (s, 6H), 2.96, 3.05(2xs, 3H), 3.37, 3.45(2×q, 2H), 4.34 (q, 1H), 4.47 (s, 2H), 7.14 (t, 1H), 7.20 (s, 1H), 7.28 (d, 1H), 7.39 (t, 1H); MS (ESI) m/z: 251.2. |
| 77% |
In methanol; at 8 - 55℃;Product distribution / selectivity; |
Biva. rtigmine bydrogen tartrate (9); Methanol (0. 4ml) was taken in a glass vial and L- (+)-tartaric acid (0.204g, 1. 36mmol, leq) was added to it. It was warmed to about 40-45C to get a clear solution. This was added to a flask containing 0. 34g <strong>[123441-03-2]rivastigmin</strong>e (1) (1. 36mmol, leq). The clear solution was heated to 55C for about 5 minutes and kept at 30C for 2 hours and at 8-10C overnight. The solvent was evaporated to dryness and the oil so obtained was triturated with hexanes (1 x 5ml, 2 x 10ml) to obtain a solid that was filtered off under vacuum on a sintered funnel under an atmosphere of nitrogen. Yield: 419mg, 77%. [a] D (c=5 methanol) +5. 02.'H NMR: 8 1.05-1. 18 (2 x t, 3H, N-CH2CH3), 8 1.53 (d, 3H, PhCCH3), 8 2. 54 (s, 6H, N- (CH3) , 8 2.87 and 2.99 (2 x s, 3H, N-CH3), 8 3. 24-3.43 (2 x q, 2H, N-CH2CH3), 8 4.22-4. 29 (q, 2H, CHCH3), 8 7.15 (d, 1H, Ar-H), 8 7.22 (s, 1H, Ar-H), 8 7.31 (d, 1H, Ar-H), 8 7.46 (t, 1H, Ar-H). |
| 76% |
In ethyl acetate; at 20 - 60℃; for 2h; |
L+Tartaric acid ( 8gm 0.052 mol) in Methanol 20 ml and Solution of III (13.5 gm 0.06 1 mol) inEthyl acetate 60 ml is added to above solution at 50-60C. The mass is stined for 2 hrs at 20-25C and filtered at 0-5C. The wet cake is refluxed in Acetone 100 ml. Final product is isolated after cooling to 0-5C followed by acetone wash. Obtained yield 76%.Chiral HPLC: R isomer 0.19% S Isomer 99.8% HPLC purity: 99.98% (figures 15 and 16). |
|
In acetone; for 1h;Heating / reflux;Product distribution / selectivity; |
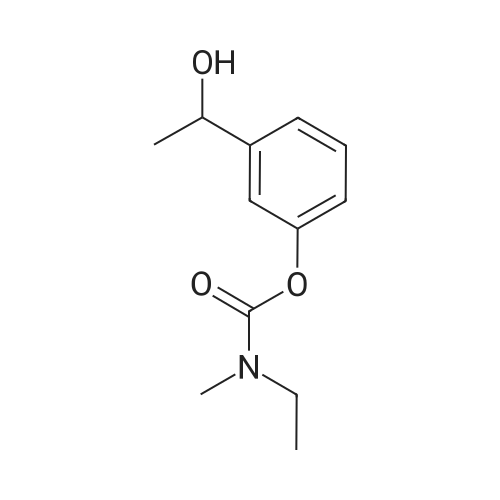
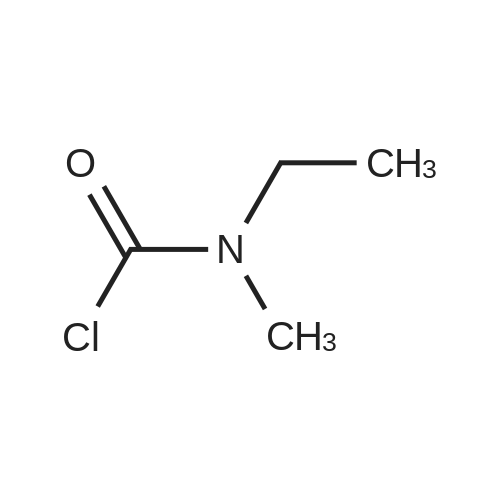
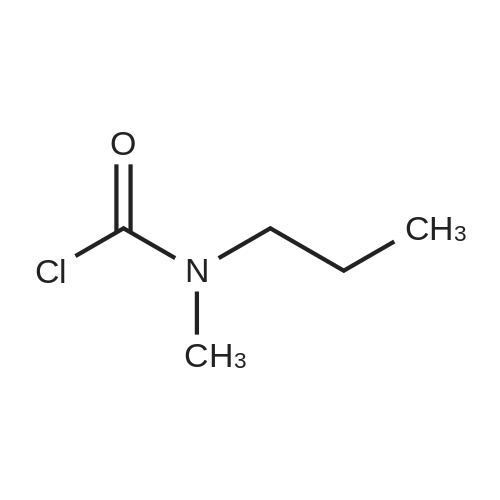
(S)-3-(l-dimethylaminoethyl)phenol hydrochloride (hydrochloride of the compound of Formula 8, 30 g, 0.148 mol) was suspended in acetonitrile (150 ml), and then N-ethyl-N-methylcarbamoyl chloride (36.1 g, 0.297 mol) was added thereto. The resulting solution was cooled to 0C. To the solution was added sodium hydride (29.7 g, 60%, 0.743 mol). The temperature was gradually allowed to rise to room temperature. The reaction mixture was stirred at room temperature for 24 hours. The completion of the reaction was confirmed by HPLC. The reaction mixture was extracted with ether, and organic layer was concentrated. Water and hydrochloric acid were added thereto. After the resulting mixture was stirred at room temperature for 2 hours, it was washed twice with ether. The obtained aqueous layer was concentrated and crystallized from ethylacetate, affording (S)-<strong>[123441-03-2]rivastigmin</strong>e hydrochloride (22.28g, 52%). The (S)-<strong>[123441-03-2]rivastigmin</strong>e hydrochloride (18.5 g, 64.5 mmol) was dissolved in water (60 ml), and then NaOH (2.58 g, 64.5 mmol, 1 eq.) was added thereto. The reaction mixture was stirred for 30 minutes. The mixture was extracted twice with ether, dried over anhydrous MgSO 4 , and concentrated under reduced pressure. The concentrate was distilled under vacuum at 116-128C to obtain <strong>[123441-03-2]rivastigmin</strong>e (10.7 g, 66.3%) as a pure distillate. To the distillate were sequentially added acetone (30 ml) and L-tartaric acid (6 g, 40 mmol, 1 eq.). The mixture was refluxed for one hour, cooled to 00C, and filtered to obtain a solid. The solid was washed, and dried to afford (S)-<strong>[123441-03-2]rivastigmin</strong>e tartrate salt*2 (16 g, 93.5% from the <strong>[123441-03-2]rivastigmin</strong>e hydrochloride, 99.8% ee).[59] In addition, the same results were obtained when(S)-3-(l-dimethylaminoethyl)phenol (Formula 8, Example 2) in the free-base form was used instead of (S)-3-(l-dimethylaminoethyl)phenol hydrochloride (hydrochloride salt of the compound of Formula 8).[60] n : (S)-<strong>[123441-03-2]rivastigmin</strong>e[61] ' H NuMR (CDCl3): 7.29 (IH, m), 7.01 (3H, m), 3.44 (2H, q), 3.24 (IH, q), 3.02(3H, d), 2.20 (6H, s), 1.35 (3H, d), 1.21 (3H, m)[62] *2 : (S)-<strong>[123441-03-2]rivastigmin</strong>e tartrate salt: m.p 124C |
|
In acetone; at 30 - 60℃; for 2.5h;Heating / reflux; |
EXAMPLE 6: PREPARATION OF RIVASTIGMINE TARTRATE (FORMULA I); 3 kg of <strong>[123441-03-2]rivastigmin</strong>e freebase of Formula II in 105 lit of acetone, 1.8 kg of L-(+)-Tartaric acid was charged and heated to about 60 C followed by stirring for about 30 minutes for complete dissolution. The resulting reaction solutions was passed through celite and wash the bed with 13.5 lit acetone to made particle free. The obtained clear solution was distilled off up to 50% of the initial volume and cooled to 30C. 12 g of <strong>[123441-03-2]rivastigmin</strong>e hydrogen tartrate was added and stirred for about 60 minutes. The reaction mixture was heated to reflux and stirred for about 60 minutes and cooled to about 30C and stirred for about 60 minutes for solid separation. The separated solid was filtered and washed the solid with 3 lit of acetone. Solid obtained was dried at about 60 C for about 9 hours to afford 4.10 kg of the title compound. Purity by HPLC: 97.37%. |
|
In acetone; at 30 - 60℃; for 2.5h;Heating / reflux; |
Example 6; Preparation Of <strong>[123441-03-2]Rivastigmine</strong> Tartrate (Formula I); 3 kg of <strong>[123441-03-2]rivastigmin</strong>e freebase of Formula II in 105 lit of acetone, 1.8 kg of L-(+)-Tartaric acid was charged and heated to about 60 C. followed by stirring for about 30 minutes for complete dissolution. The resulting reaction solutions was passed through celite and wash the bed with 13.5 lit acetone to made particle free. The obtained clear solution was distilled off up to 50% of the initial volume and cooled to 30 C. 12 g of <strong>[123441-03-2]rivastigmin</strong>e hydrogen tartrate was added and stirred for about 60 minutes. The reaction mixture was heated to reflux and stirred for about 60 minutes and cooled to about 30 C. and stirred for about 60 minutes for solid separation. The separated solid was filtered and washed the solid with 3 lit of acetone. Solid obtained was dried at about 60 C. for about 9 hours to afford 4.10 kg of the title compound.Purity by HPLC: 97.37%. |
|
In isopropyl alcohol; at 20 - 80℃;Product distribution / selectivity; |
EXAMPLE 6; Preparation of crystalline form of (S)-N-Ethyl-N-methyl-3-[1-(dimethylamino)ethyl]-phenyl carbamate (2R,3R)-hydrogen tartrate; (2R,3R)-Tartaric acid (9.0 g, 60.0 mmol) was added to a solution of <strong>[123441-03-2](S)-N-ethyl-N-methyl-3-[1-(dimethylamino)ethyl]phenyl carbamate</strong> (15.0 g, 60.0 mmol) in isopropyl alcohol (75 mL). The resulting mixture was heated to 75-80 C. and then cooled to 20-25 C. The resulting suspension was filtered and washed with isopropanol and the solid was dried under vacuum at 40-50 C. to give crystalline (S)-N-Ethyl-N-methyl-3-[1-(dimethylamino)ethyl]-phenyl carbamate (2R,3R)-hydrogen tartrate (<strong>[123441-03-2]Rivastigmine</strong> hydrogen tartrate) 1H NMR (CDCl3) delta 8.50-7.50 (s, 4H); 7.41 (t, J=7.8 Hz 1H); 7.30-7.16 (m, 3H); 4.44 (s, 2H); 4.34 (q, J=6.5 Hz, 1H); 3.50-3.34 (m, 2H); 3.02 (ad, 3H); 2.66 (s, 6H); 1.70 (d, J=6.8 Hz); 1.26-1.15 (m, 3H). PXRD (FIG. 1).; EXAMPLE 7 Preparation of (S)-N-Ethyl-N-methyl-3-[1-(dimethylamino)ethyl]-phenyl carbamate (2R,3R)-hydrogen tartrate from (S)-3-[1-(Dimethylamino)ethyl]phenol and 1-[(N-ethyl-methylamino)carbonyl]-3-methyl-1H-imidazolium methyl sulfate Triethylamine (3.6 g, 35.4 mmol) and 1-{([N-ethyl-(N-methyl)amino]carbonyl}-3-methyl-1H-imidazolium methyl sulfate (9.1 g, 32.6 mmol) were added slowly to the cooled solution of (S)-3-[1-(dimethylamino)ethyl]phenol (4.5 g, 27.23 mmol) in acetonitirile (25 mL). The reaction mixture was stirred at 75-80 C. until reaction completion as determined by 1H NMR. The reaction mixture was evaporated and the obtained residue was diluted with water (30 mL) and toluene (30 mL) and then basified with 50% aq. NaOH solution at internal temperature below 10 C. The phases were separated and the aqueous phase was extracted with toluene. The combined organic phases were washed with water. The organic solution was evaporated under vacuum to give <strong>[123441-03-2](S)-N-ethyl-N-methyl-3-[1-(dimethylamino)ethyl]phenyl carbamate</strong>. The residue dissolved in isopropanol (45 mL) and to the solution was added (2R,3R)-tartaric acid (4.08 g, 27.23 mmol) and heated to 70-80 C. The solution was cooled and the resulting suspension was filtered and dried to give (S)-N-Ethyl-N-methyl-3-[1-(dimethylamino)ethyl]-phenyl carbamate (2R,3R)-hydrogen tartrate (8.54 g, 78% yield). The 1H NMR spectrum of the product was identical to that of example 6. EXAMPLE 8 Preparation of amorphous form of (S)-N-Ethyl-N-methyl-3-[1-(dimethylamino)ethyl]phenyl carbamate hydrogen-(2R,3R)-tartrate The solution of (S)-N-Ethyl-N-methyl-3-[1-(dimethylamino)ethyl]-phenyl carbamate hydrogen-(2R,3R)-tartrate (<strong>[123441-03-2]Rivastigmine</strong> hydrogen tartrate) (2 g) in water (4 mL) was stirred at 20-25 C. for 0.5-1 hour. The mixture was evaporated at 45-60 C. under vacuum to give a white solid. The obtained material was further dried at 40-45 C. for 10-12 hours under vacuum to obtain amorphous <strong>[123441-03-2]Rivastigmine</strong> hydrogen tartrate. |
|
In acetone; at 50℃; for 0.5h; |
Example 2: Preparation <strong>[123441-03-2]Rivastigmine</strong> Hydrogentartarate salt22 gm of the <strong>[123441-03-2]Rivastigmine</strong> (+) DPTTA salt obtained as 2nd crop in Example 1 Step (c) was charged in a flask containing 150 ml of water and pH was adjusted to about 11 using about 20 ml of aqueous ammonia. The reaction mixture was extracted with dichloromethane(DCM) (250 ml) and the organic layer was washed with water (100 ml). The final organic layer was distilled completely to obtain 9.5 gm of <strong>[123441-03-2]Rivastigmine</strong> base as residue.25 ml of acetone was added to the above obtained residue and stirred for dissolution. 5.7 gm of L(+) tartaric acid was added to the solution, heated to about 50 C and stirred for about 30 minutes. The reaction mixture was cooled to 10-150C and stirred for 1 hour. The solid was filtered and washed with acetone (10 ml). The solid was dried 50-600C to obtain 12.5 gm of the title compound. |
|
In ethanol;Heating / reflux; |
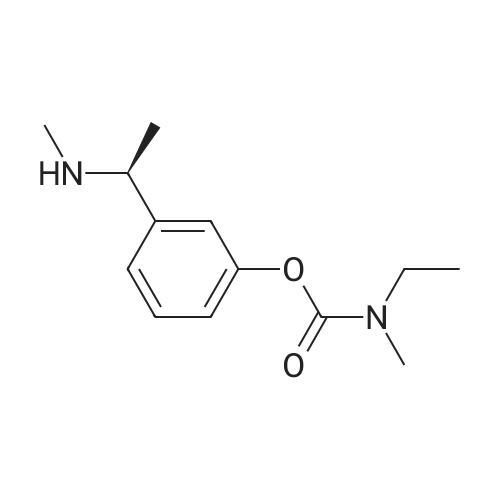
(iii) Synthesis of <strong>[123441-03-2]rivastigmin</strong>e hydrogentartrate An amount of 1.30 g (5.2 mmol) of 3-(1-(dimethylamino)ethyl)-phenyl ethyl(methyl)carbamate as obtained in step (i) and 2.10 g (5.2 mmol) of (+)-di-p-toluoyl-D-tartaric acid monohydrate were dissolved in 13 ml of methanol/water (2:1) solution under heating. The reaction mixture was refluxed for 1 hour and cooled to room temperature. The precipitate was filtered and four times recrystallized in a minimal volume of methanol/water solution to obtain optically pure di-p-toluoyl tartrate salt of (S)-3-(1-(dimethylamino)ethyl)phenyl ethyl-(methyl)carbamate. The salt was dissolved in 1 M NaOH and extracted into ether to obtain an optically pure oily residue of (S)-3-(1-(dimethylamino)ethyl)phenyl ethyl(methyl)carbamate which was dissolved in ethanol, mixed with hydrogentartrate, refluxed and cooled to room temperature. |
|
In acetone; at 4 - 60℃; for 19h; |
2 g S-(-)-<strong>[123441-03-2]Rivastigmine</strong> and 1.2 g L-(+)-tartaric acid were added to 20 ml acetone and the mixture was heated at 600C for 1 hour to get a clear mixture. The solution was allowed to slowly cool to ambient temperature and was stirred for 18 hours at 4C until complete precipitation of the tartrate salt. The solid was isolated by filtration, washed with acetone, and dried for 22 hours under vacuum at 400C. Isolated yield: 2.72g, white solidXRPD pattern: comparable to Fig.l. |
|
In acetone; at 0 - 50℃; for 1.5h; |
Example 7; Preparation of <strong>[123441-03-2]Rivastigmine</strong> hydrogentartrate salt170 ml of acetone was added to the above obtained residue and stirred for dissolution. 20.4 gm of L(+) tartaric acid was added to the solution, heated to about 50 C and stirred for about 30 minutes. The reaction mixture was cooled to 25-35 C and stirred for 30 minutes. The reaction mixture was further cooled to 0-5 C and stirred for 30 minutes. The solid was filtered and washed with acetone (20 ml). The solid was dried at 50-60C to obtain 46 gm of the title compound.Purity by HPLC: 99.84 % |
| 2.5 g |
In acetone; at 60℃; for 1h; |
Example-9: Preparation of <strong>[123441-03-2]Rivastigmine</strong> Tartrate To a 250 ml. RB flask, <strong>[123441-03-2]Rivastigmine</strong> base (2.5 g), acetone (87.5 ml) and L-(+)-tartaric acid (1.5 g) were added. Heated the reaction mass at 60 C. for 1 hour and gradually cooled to room temperature. Filtered the reaction mass through hyflow bed and washed with acetone (12 ml). Evaporated half of the acetone volume and stirred at room temperature for 1 hour. Filtered the solid, washed with acetone and dried under vacuum to obtained 2.5 g of <strong>[123441-03-2]Rivastigmine</strong> Tartrate (Yield: 62.6%, HPLC: 99.48%, Chiral HPLC: 99.56%). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping