|
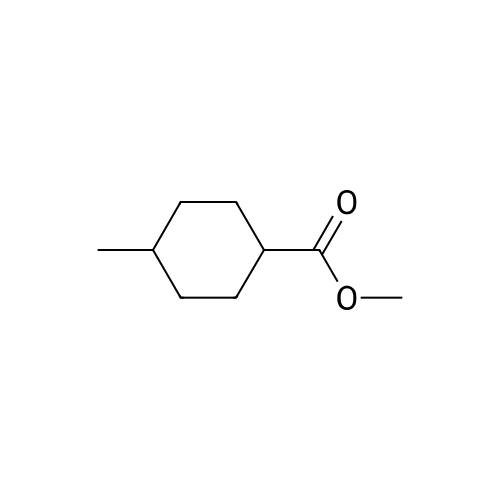
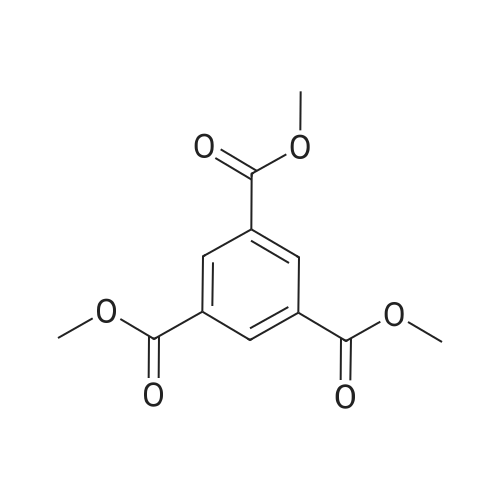
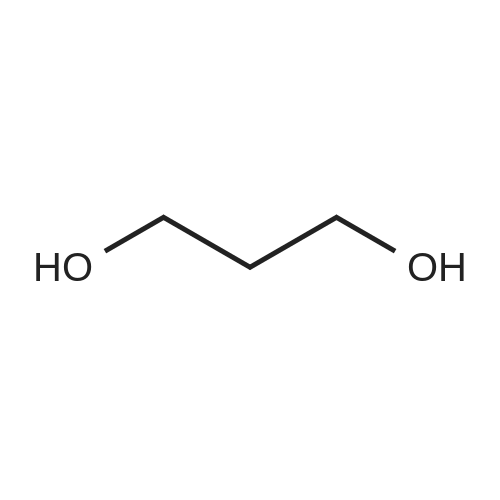
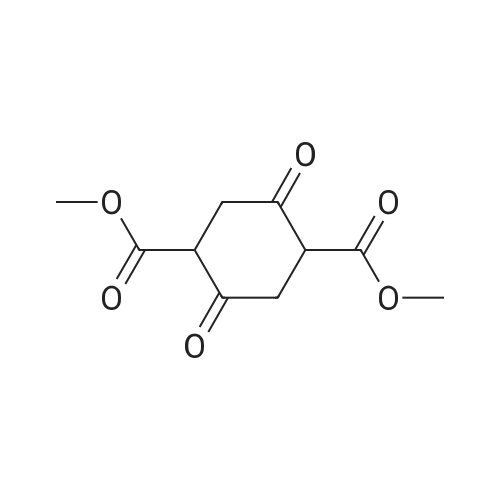
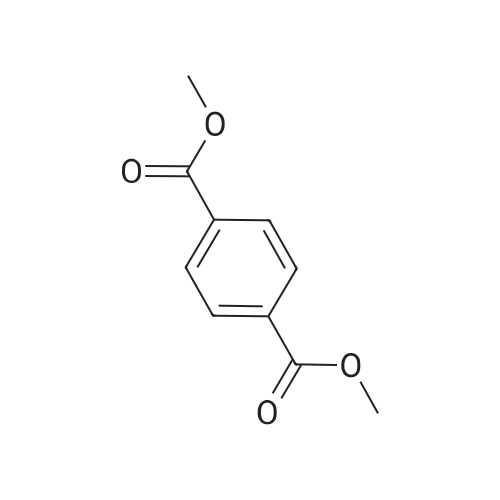
In ethylene glycol at 75 - 80℃; for 1h; |
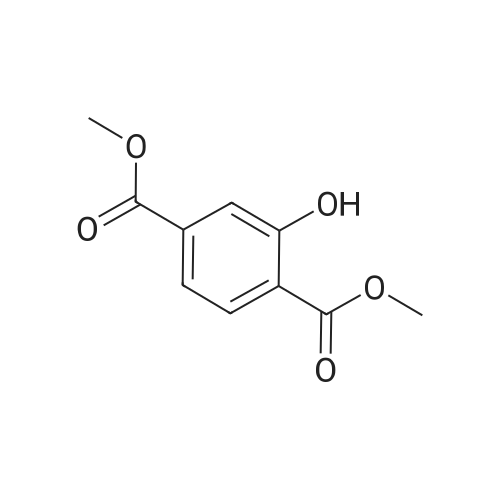
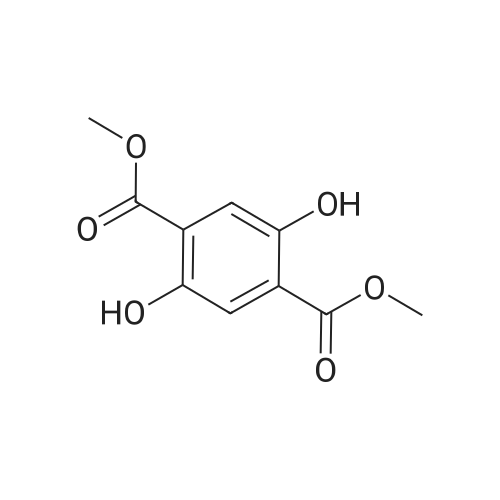
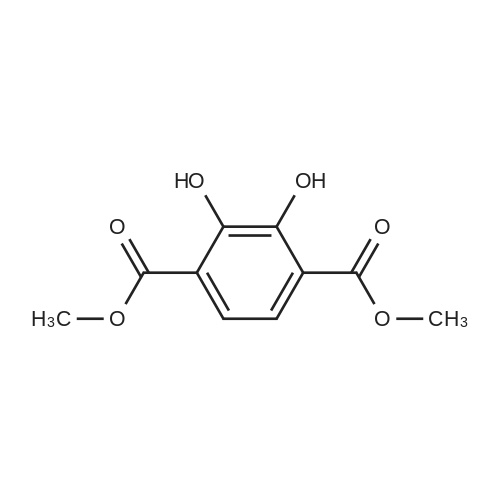
1-3
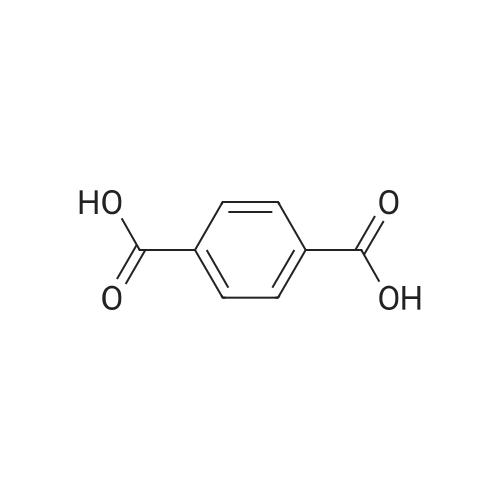
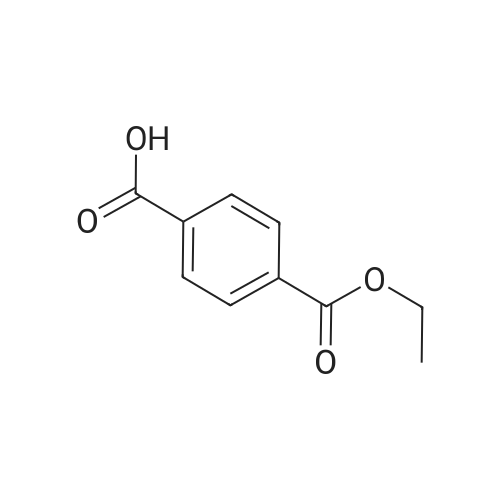
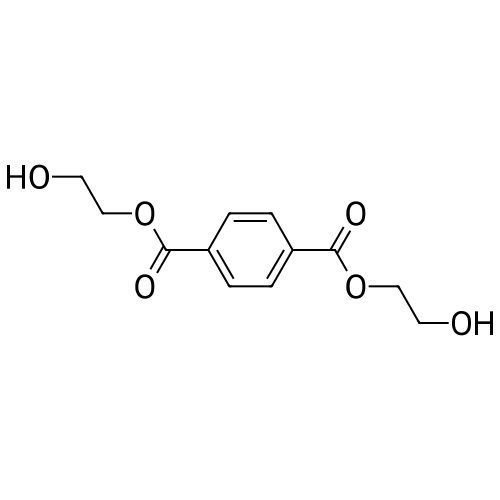
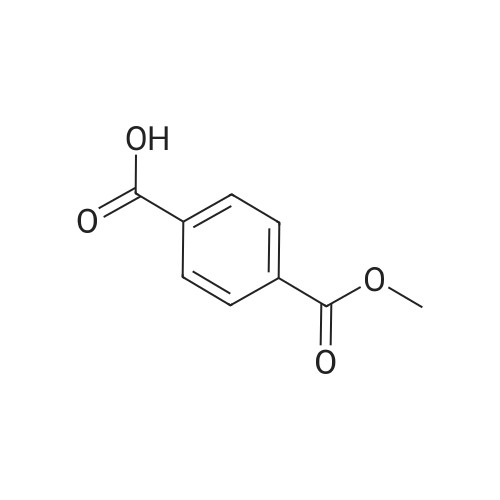
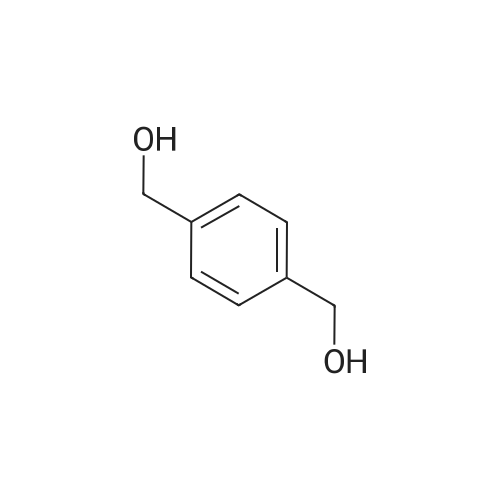
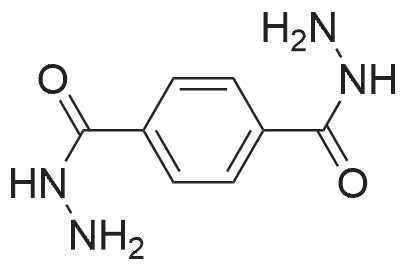
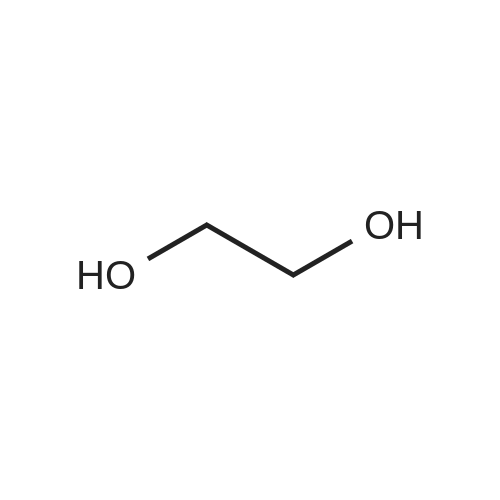
Example 1100 g of cut pieces of polyethylene terephthalate fabric dyed in black (nitrogen content in fabric prior to dye extraction: 900 ppm) which was a dyed polyester fiber to be subjected to the method of the invention, and 600 g of paraxylene were charged into a 2 L separable flask. A dye extraction step was carried out by performing heating and stirring under atmospheric pressure at 130° C. for 10 minutes. After the completion of extraction, as a solid liquid separation step, suction filtration with an aspirator was carried out, thereby to separate dye-containing paraxylene from the fabric from which the dye had been extracted (dye-extracted polyester fiber).Thereafter, the dye-extracted polyester fiber and 600 g of another paraxylene were charged into a separable flask, and extraction of the dye was carried out under the same conditions. After the completion of extraction, solid liquid separation was carried out again, thereby to separate dye-containing paraxylene and the fabric from which the dye had been extracted and removed.Subsequently, about 100 g of the fabric from which the dye had been extracted and removed and 600 g of another ethylene glycol were charged into a separable flask. A dye extraction step was carried out by performing heating and stirring under atmospheric pressure at 170° C. for 10 minutes. Most of the paraxylene contained in the fabric is evaporated through an exhaust tube by heating at this step. The evaporated paraxylene is recovered by a condenser. After the completion of extraction, a solid liquid separation step was carried out again, thereby to separate a dye-containing ethylene glycol from the fabric from which the dye had been extracted and removed.Subsequently, as a depolymerization reaction step, 100 g of the dye-extracted fabric was charged into a mixture of 400 g of ethylene glycol preheated up to 185° C., and 3 g of potassium carbonate as a depolymerization catalyst. That was allowed to react under atmospheric pressure at 185° C. for 4 hours, thereby to obtain a depolymerized solution containing bis-β-hydroxy ethylene terephthalate (BHET).Incidentally, after the depolymerization reaction at the depolymerization reaction step, solid matters were filtrated and removed by a wire gauze strainer with an opening of 350 μm3. In this solid matter removing step, it was possible to mainly remove different types of plastics other than polyester.The resulting depolymerized solution after filtration was fed to a distillation column, and a depolymerized solution concentration step of distilling away 300 g of ethylene glycol under the conditions of a column bottom temperature of 140 to 150° C. and a pressure of 13.3 kPa, and thereby concentrating the depolymerized solution was carried out. Then, to 200 g of the filtrated and concentrated depolymerized solution, 1.7 g of potassium carbonate as an ester interchange catalyst, and 200 g of methanol were added. Thus, an ester interchange reaction step was carried out under atmospheric pressure at 75 to 80° C. for 1 hour, thereby to obtain an ester interchange reaction product mixture.After the completion of the ester interchange reaction, the ester interchange reaction product mixture was cooled to 40° C. Then, a solid liquid separation step of achieving solid liquid separation between a cake containing crude dimethyl terephthalate as a main component and a filtrate containing methanol and crude ethylene glycol as main components was carried out by centrifugation.Then, crude dimethyl terephthalate was subjected to distillation under the conditions of a pressure of 6.7 kPa and a column bottom temperature of 180 to 200° C., thereby to obtain purified dimethyl terephthalate (step of separating useful components of DMT components). Further, similarly, crude ethylene glycol was subjected to distillation under the conditions of a pressure of 13.3 kPa and a column bottom temperature of 140 to 150° C., thereby to obtain purified ethylene glycol (step of separating useful components of EG components) Finally, as useful components, purified dimethyl terephthalate and purified ethylene glycol were obtained in a yield of 85 wt %, respectively.Purified dimethyl terephthalate recovered from the ester interchange reaction product mixture bore comparison with commercially available products in terms of the inspection items such as outward appearance, acid value, melt colorimetry, and sulfuric acid ash content. Whereas, the purified ethylene glycol recovered from the ester interchange reaction product mixture bore comparison with commercially available products in terms of the inspection items such as diethylene glycol content and moisture content. Further, any nitrogen content of the recovered purified dimethyl terephthalate and the recovered purified ethylene glycol was equal to, or lower than the lower detection limit. Thus, high purity useful components were obtained.Whereas, 1200 g of the dye-containing paraxylene obtained in the dye extraction step was distilled under the conditions of a column bottom temperature of 120 to 130° C. and a pressure of 40.0 kPa, so that 1100 g thereof was obtained as a distilled component. For such paraxylene obtained in the extracting solvent recovery step, the coloration due to mixing of a dye was not observed in outward appearance, and the nitrogen content was also equal to, or lower than the lower detection limit. As a result of these operations, it was possible to recover paraxylene in such a purity as to allow reuse thereof as an extracting solvent.Whereas, 600 g of the dye-containing ethylene glycol obtained in the dye extraction step was distilled under the conditions of a column bottom temperature of 140 to 150° C. and a pressure of 13.3 kPa, so that 536 g thereof was obtained as a distilled component. For such ethylene glycol obtained in the extracting solvent recovery step, the coloration due to mixing of a dye was not observed in outward appearance, and the nitrogen content was also equal to, or lower than the lower detection limit. As a result of these operations, it was possible to recover ethylene glycol in such a purity as to allow reuse thereof as an extracting solvent or a raw material for polyethylene terephthalate.Example 2100 g of cut pieces of polyethylene terephthalate fabric dyed in black (nitrogen content in fabric prior to dye extraction: 900 ppm) which was a dyed polyester fiber to be subjected to the method of the invention, and 600 g of ethylene glycol were charged into a 2 L separable flask. A dye extraction step was carried out by performing heating and stirring under atmospheric pressure at 170° C. for 10 minutes. After the completion of extraction, as a solid liquid separation step, suction filtration with an aspirator was carried out, thereby to separate a dye-containing ethylene glycol from the fabric from which the dye had been extracted (dye-extracted polyester fiber).Thereafter, the dye-extracted polyester fiber and 600 g of another paraxylene were charged into a separable flask, and for extraction of the dye, a step of extracting the dye was carried out by performing heating and stirring under atmospheric pressure at 130° C. for 10 minutes. After the completion of extraction, solid liquid separation was carried out again, thereby to separate a dye-containing paraxylene and the fabric from which the dye had been extracted and removed.Subsequently, about 100 g of the fabric from which the dye had been extracted and removed and 600 g of another paraxylene were charged into a separable flask. Then, extraction of the dye was carried out under the same conditions. After the completion of extraction, solid liquid separation was carried out again, thereby to separate a dye-containing paraxylene from the fabric from which the dye had been extracted and removed.Subsequently, as a depolymerization reaction step, 100 g of the dye-extracted fabric was charged into a mixture of 400 g of ethylene glycol preheated up to 185° C., and 3 g of potassium carbonate as a depolymerization catalyst. That was allowed to react under atmospheric pressure at 185° C. for 4 hours, thereby to obtain a depolymerized solution containing bis-β-hydroxy ethylene terephthalate (BHET). Thereafter, in the same manner as in Example 1, through the solid matter removing step, the depolymerized solution concentration step, the ester interchange reaction step, the solid liquid separation step, and step of separating useful components of DMT components, finally, dimethyl terephthalate was obtained in a yield of 87 wt %, and ethylene glycol was obtained in a yield of 84 wt % as useful components.The dimethyl terephthalate recovered from the ester interchange reaction product mixture bore comparison with commercially available products in terms of the inspection items such as outward appearance, acid value, melt colorimetry, and sulfuric acid ash content. Whereas, the ethylene glycol recovered from the ester interchange reaction product mixture bore comparison with commercially available products in terms of the inspection items such as diethylene glycol content and moisture content. Further, any nitrogen content of the recovered dimethyl terephthalate and the recovered ethylene glycol was equal to, or lower than the lower detection limit. Thus, high purity useful components were obtained.Whereas, 1200 g of the dye-containing paraxylene obtained in the dye extraction step was distilled under the conditions of a column bottom temperature of 120 to 130° C. and a pressure of 40.0 kPa, so that 1089 g thereof was obtained as a distilled component. For such paraxylene obtained in the extracting solvent recovery step, the coloration due to mixing of a dye was not observed in outward appearance, and the nitrogen content was also equal to, or lower than the lower detection limit. As a result of these operations, it was possible to recover paraxylene in such a purity as to allow reuse thereof as an extracting solvent.Whereas, 600 g of the dye-containing ethylene glycol obtained in the dye extraction step was distilled under the conditions of a column bottom temperature of 140 to 150° C. and a pressure of 13.3 kPa, so that 540 g thereof was obtained as a distilled component. For such ethylene glycol obtained in the extracting solvent recovery step, the coloration due to mixing of a dye was not observed in outward appearance, and the nitrogen content was also equal to, or lower than the lower detection limit. As a result of these operations, it was possible to recover ethylene glycol in such a purity as to allow reuse thereof as an extracting solvent or a raw material for polyethylene terephthalate.Example 3100 g of cut pieces of polyethylene terephthalate fabric dyed in black (nitrogen content in fabric prior to dye extraction: 900 ppm) which was a dyed polyester fiber to be subjected to the method of the invention, 600 g of paraxylene, and 300 g of ethylene glycol were charged at the same time into a 2 L separable flask. A dye extraction step was carried out by performing heating and stirring under atmospheric pressure at 135° C. for 10 minutes. After the completion of extraction, as a solid liquid separation step, suction filtration with an aspirator was carried out, thereby to separate a dye-containing ethylene glycol, paraxylene mixed solvent from the fabric from which the dye had been extracted (dye-extracted polyester fiber).Thereafter, the dye-extracted polyester fiber, 600 g of another paraxylene, and 300 g of another ethylene glycol were charged into a separable flask, and a step of extracting the dye was carried out by performing heating and stirring under atmospheric pressure at 135° C. for 10 minutes. After the completion of extraction, solid liquid separation was carried out, thereby to separate a dye-containing paraxylene, ethylene glycol mixed solution and the fabric from which the dye had been extracted and removed.Subsequently, as a depolymerization reaction step, 100 g of the dye-extracted fabric was charged into a mixture of 400 g of ethylene glycol preheated up to 185° C., and 3 g of potassium carbonate as a depolymerization catalyst. That was allowed to react under atmospheric pressure at 185° C. for 4 hours, thereby to obtain a depolymerized solution containing bis-β-hydroxy ethylene terephthalate (BHET). Thereafter, in the same manner as in Example 1, through the solid matter removing step, the depolymerized solution concentration step, the ester interchange reaction step, the solid liquid separation step, and the DMT distillation step, finally, dimethyl terephthalate was obtained in a yield of 83 wt %, and ethylene glycol was obtained in a yield of 82 wt % as useful components.The dimethyl terephthalate recovered from the ester interchange reaction product mixture bore comparison with commercially available products in terms of the inspection items such as outward appearance, acid value, melt colorimetry, and sulfuric acid ash content. Whereas, the ethylene glycol recovered from the ester interchange reaction product mixture bore comparison with commercially available products in terms of the inspection items such as diethylene glycol content and moisture content. Further, any nitrogen content of the recovered dimethyl-terephthalate and the recovered ethylene glycol was equal to, or lower than the lower detection limit. Thus, high purity useful components were obtained.Whereas, a mixture of about 1200 g of the paraxylene and about 600 g of ethylene glycol containing a dye obtained in the dye extraction step was allowed to stand still under ordinary temperatures, and was allowed to undergo phase separation into two layers. The paraxylene phase was distilled under the conditions of a column bottom temperature of 120 to 130° C. and a pressure of 40.0 kPa, so that 1087 g thereof was obtained as a distilled component. For such paraxylene obtained in the extracting solvent recovery step, the coloration due to mixing of a dye was not observed in outward appearance, and the nitrogen content was also equal to, or lower than the lower detection limit. As a result of these operations, it was possible to recover paraxylene in such a purity as to allow reuse thereof as an extracting solvent. Whereas, the ethylene glycol phase was distilled under the conditions of a column bottom temperature of 140 to 150° C. and a pressure of 13.3 kPa, so that 527 g thereof was obtained as a distilled component. For such ethylene glycol obtained in the extracting solvent recovery step, the coloration due to mixing of a dye was not observed in outward appearance, and the nitrogen content was also equal to, or lower than the lower detection limit. As a result of these operations, it was possible to recover ethylene glycol in such a purity as to allow reuse thereof as an extracting solvent or a raw material for polyethylene terephthalate.Comparative Example 1100 g of a polyethylene terephthalate fabric dyed in the same black as that used in Examples 1 to 3 was cut. Then, dimethyl terephthalate and ethylene glycol were recovered under the same conditions as in Example 1, except that the process did not go through the dye extraction step, and the solid liquid separation step. The dimethyl terephthalate recovered from the ester interchange reaction product mixture bore comparison with commercially available products in terms of the inspection items such as outward appearance, acid value, melt colorimetry, and sulfuric acid ash content. However, it contained 11 ppm by weight of nitrogen. Whereas, the ethylene glycol recovered from the ester interchange reaction product mixture bore comparison with commercially available products in terms of the inspection items such as diethylene glycol content and moisture content. However, it contained 45 ppm by weight of nitrogen. Thus, the degradation of the quality was observed.Comparative Example 2100 g of cut pieces of a polyethylene terephthalate fabric dyed in the same black as that used in Examples 1 to 3 and 1000 g of ethylene glycol were charged in a 5 L autoclave. Then, dimethyl terephthalate and ethylene glycol were recovered under the same conditions as in Examples 1 to 3, except that extraction of the dye was carried out by performing heating and stirring under a pressure of 430 kPa (absolute pressure) at a temperature of 240° C. for 30 minutes. The dimethyl terephthalate recovered from the ester interchange reaction product mixture bore comparison with commercially available products in terms of the inspection items such as outward appearance, acid value, melt colorimetry, and sulfuric acid ash content. The nitrogen content thereof was also equal to, or less than the lower detection limit. Thus, it was possible to obtain high purity useful components. Whereas, the ethylene glycol recovered from the ester interchange reaction product mixture bore comparison with commercially available products in terms of the inspection items such as diethylene glycol content and moisture content. However, it contained 15 ppm by weight of nitrogen. Thus, the degradation of the quality was observed. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping