There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
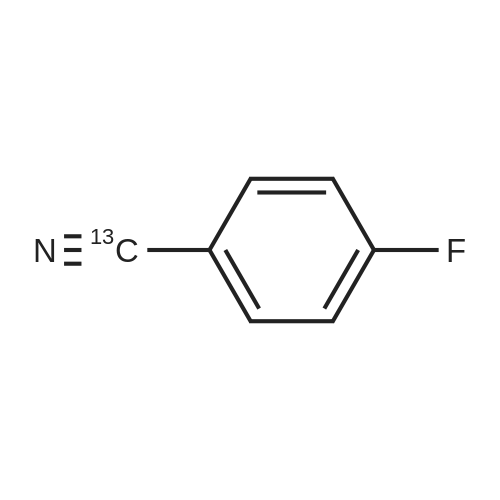
Structure of 1194-02-1 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Molecular Crystals and Liquid Crystals (1969-1991), 1985, vol. 124, p. 269 - 276
3
[ 288-32-4 ]
[ 1194-02-1 ]
[ 25372-03-6 ]
Yield
Reaction Conditions
Operation in experiment
93%
With sodium hydride In N,N-dimethyl-formamide at 20 - 100℃;
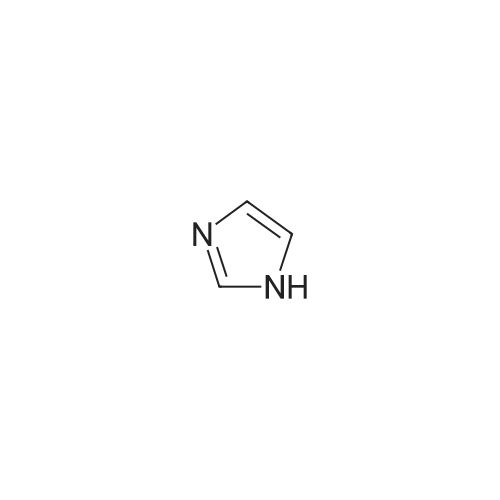
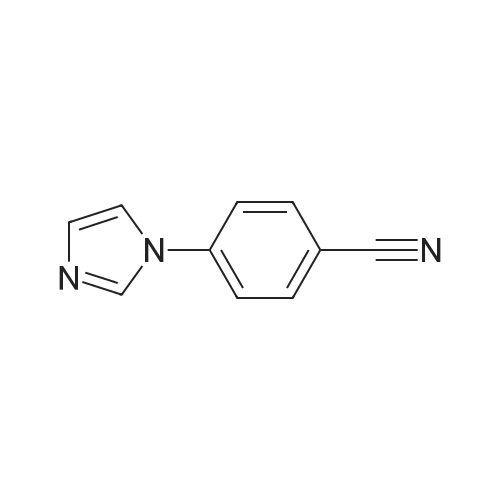
1500 ml of dry dimethylformamide (DMF) are placed in a 1000 ml four-necked flask while passing nitrogen over it and 72.67 g (0.6 mol) of 4-fluorocyanobenzene and 61.2 g (0.9 mol) of imidazole and finally 21.6 g (0.9 mol) of sodium hydride are added. The reaction mixture is heated to 100° C., stirred at this temperature for 4 hours and subsequently overnight at room temperature. The reaction mixture is then poured into water and the resulting mixture is extracted a number of times with dichloromethane. The organic phase is dried, evaporated on a rotary evaporator and finally dried further at 60° C. under reduced pressure. The yield is 94 g (93percent of theory).1H-NMR (400 MHz, CDCl3): δ=7.27 (s, 1H); 7.35 (s, 1H); 7.54 (d, J=8.8 Hz, 2H); 7.81 (d, J=8.8 Hz, 2H); 7.95 (s, 1H).
Reference:
[1] Synthetic Communications, 2008, vol. 38, # 4, p. 626 - 636
[2] Patent: WO2006/56418, 2006, A2, . Location in patent: Page/Page column 57
[3] Patent: US2009/18330, 2009, A1, . Location in patent: Page/Page column 14
[4] Advanced Synthesis and Catalysis, 2007, vol. 349, # 11-12, p. 1938 - 1942
[5] Monatshefte fur Chemie, 2004, vol. 135, # 4, p. 419 - 423
[6] Journal of Organic Chemistry, 2011, vol. 76, # 4, p. 1151 - 1154
[7] Bioorganic and Medicinal Chemistry, 2003, vol. 11, # 18, p. 3879 - 3887
[8] Tetrahedron Letters, 2009, vol. 50, # 12, p. 1286 - 1289
[9] Journal of Materials Chemistry A, 2017, vol. 5, # 2, p. 535 - 543
4
[ 1194-02-1 ]
[ 18156-74-6 ]
[ 25372-03-6 ]
Yield
Reaction Conditions
Operation in experiment
77%
Stage #1: With cesium fluoride In N,N-dimethyl-formamide for 0.166667 h; Inert atmosphere Stage #2: at 60℃; for 20 h;
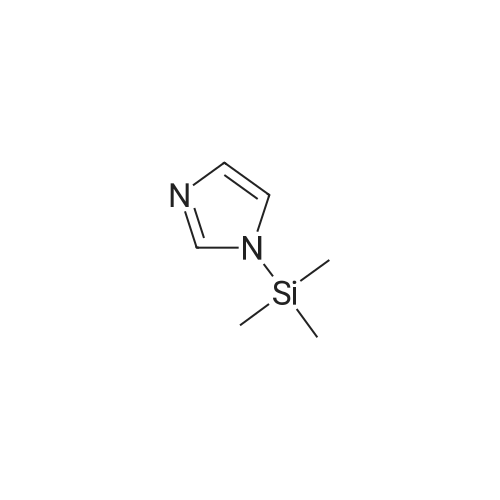
Under a nitrogen atmosphere 362 mg (2.38 mmol) of CsF, previously activated with NaOH, were suspended in 5 ml of DMF and stirred for 30 min. Then, 1.00 g (8.26 mmol) of 4-fluorobenzonitrile was added. After 10 min 1.20 ml (8.18 mmol) N-trimethylsilylimidazole were added and the mixture was stirred at 60° C. for 20 hr. For workup, most of the solvent was removed under vacuum (oil pump), and then 5 ml of water and 5 ml of CH2Cl2 were added to the reaction mixture. The organic phase was separated, the aqueous phase was extracted with CH2Cl2, the organic phases were combined, washed several times with water and dried over MgSO4. The solvent was removed under vacuum, and the resulting solid was rinsed twice with pentane.Yield: 1.07 g (6.31 mmol, 77percent), appearance: colorless solid. 1H NMR (CDCl3, 25° C., 400.13 MHz): δ=7.25 (s, 1H, ImH), 7.33 (s, 1H, ImH), 7.51-7.53 (m, 2H, PhH), 7.79-7.81 (m, 2H, PhH), 7.94 (s, 1H, ImH). 13C NMR (CDCl3, 25° C., 100.61 MHz): δ=111.3 (C-1), 117.7 (C-9), 117.9 (-CN), 121.5 (C-3, C-5), 131.6 (C-8), 134.2 (C-2, C-6), 135.4 (C-7) 140.6 (C-4).
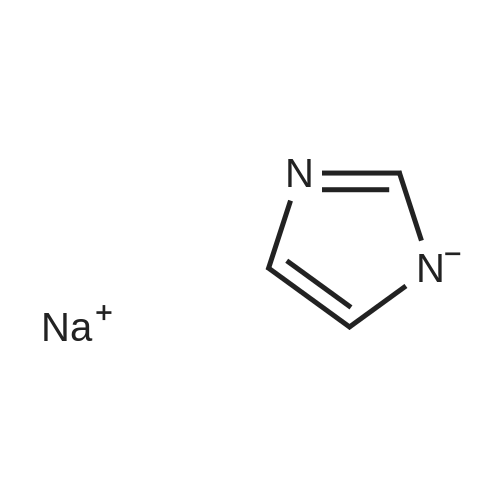
A mixture 4-fluorobenzonitrile (3 g, 25 MMOL) and imidazolyl sodium (2. 48 g, 27.5 MMOL) in DMF (50 mL) was stirred at 80 C under Ar for 12 h. Progress of reaction was monitored by TLC. The reaction mixture was concentrated in vacuo and the residue was diluted with 50 mL water and stirred. The aqueous mixture was extracted with EtOAc (2 x 50 mL). Combined EtOAc extracts was dried over anhydrous MGS04, concentrated, and the 4- (1-imidazolyl)-benzonitrile was isolated by column chromatography (3.6 g, 78percent). LCMS: MH+ = 170 ;'H NMR (CDCI3) 8 8.0 (s, 1 H), 7.5 (d, 2H), 7.4 (m, 3H), 7.3 (d, 1H)
Reference:
[1] Journal of Organic Chemistry, 2011, vol. 76, # 4, p. 1151 - 1154
7
[ 1194-02-1 ]
[ 25372-03-6 ]
Reference:
[1] Patent: US2004/209878, 2004, A1,
8
[ 288-32-4 ]
[ 1194-02-1 ]
[ 584-08-7 ]
[ 25372-03-6 ]
Reference:
[1] Patent: US5736548, 1998, A,
9
[ 288-13-1 ]
[ 1194-02-1 ]
[ 25699-83-6 ]
Yield
Reaction Conditions
Operation in experiment
87%
With potassium carbonate In N,N-dimethyl-formamide at 120℃; for 7 h;
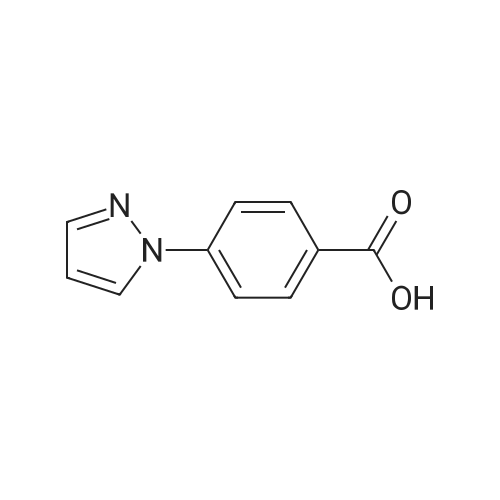
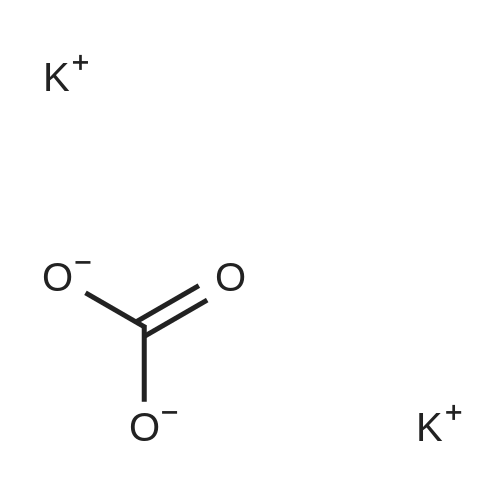
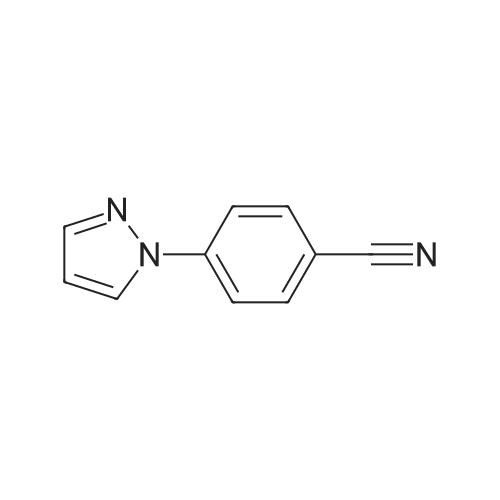
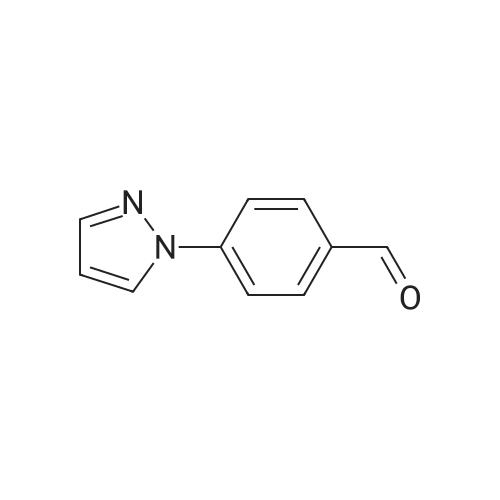
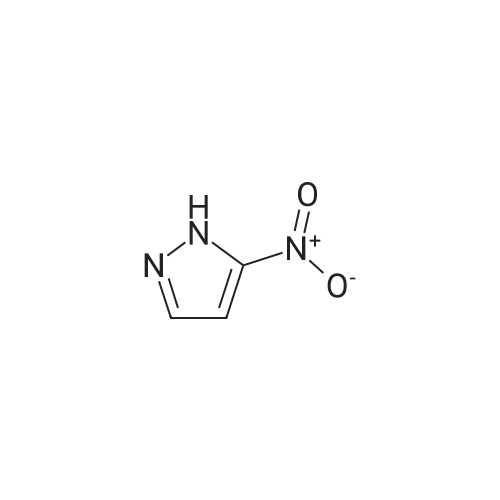
4-( 1 H-pyrazol- 1 -yl)- benzonitrile4-Fluorobenzonitrile (204.2 g), pyrazole (138.6 g, 1.22 eq) and potassium carbonate (281.5 g, 1.22 eq) in DMF (1110 ml) were heated at 120 0C for 7 hours. The suspension was cooled to 25 0C and water (2920 ml) added. The reaction was extracted with MTBE (3 x 1460 ml) and the combined extracts were washed with water (3 x 1460 ml) and saturated aqueous sodium chloride (1460 ml). The organic phase was concentrated at atmospheric pressure until the pot temperature rose to 65 0C. Heptane (1700 ml) was added over 30 minutes at 60-65 0C, and then a further 300 ml of distillate was collected. The solution was stirred at 60-65 0C for 15 minutes and then cooled to <5 0C. The slurry was filtered and washed with heptane (2 x 200 ml), and dried under vacuum to constant weight to give the title compound as a solid (245.3 g, 87percent). <n="47"/>1H NMR (400 MHz, CDCl3): 6.51 (q, IH), 7.71 (d, 2H), 7.75 (d, IH), 7.81 (d, 2H), 7.98 (d, IH).
83%
With potassium carbonate In N,N-dimethyl-formamide at 115 - 120℃; for 7 - 8 h; Inert atmosphere
5f: 4-(1H-pyrazol-1-yl)benzonitrile N,N-dimethylformamide (123.25 L) was charged to the vessel and analysed for moisture content (target<0.5percent). Potassium carbonate (34.01 kg) was then charged to the vessel followed by pyrazole (16.76 kg) and 4-fluorobenzonitrile (24.65 kg). The reaction mixture was heated to 115 to 120° C. and stirred at this temperature for between seven and eight hours under a nitrogen atmosphere. The reaction was monitored by GC (target<10percent 4-fluorobenzonitrile). The reaction was then cooled to 20-25° C. and quenched with water (369.7 L). Methyl tert-butyl ether (246.5 L) was then charged and the layers allowed to separate. The aqueous layer was washed with methyl tert-butyl ether (2 x147.9 L) and the organic layers combined. The organic layer was then washed with water (2 x 172.55 L) and aqueous brine (123.25 L, 24 wt percent). The organic phase was then concentrated to approximately 100 L at 60° C. or below at atmospheric pressure. n-Heptane (209 L) was then charged and the mixture concentrated to approximately 100 L at 60° C. or below at atmospheric pressure. The reaction was cooled to 0° C. and stirred for three hours at this temperature. The slurry was then filtered washing the filter cake with n-heptane (24.65 L). The resulting solid was dried under vacuum at 40° C. to yield the title product (28.6 kg, 99.32percent purity, 83percent yield). 1H NMR (300 MHz, DMSO-d6): 6.61 (dd, J=2.4, 1.9 Hz, 1H), 7.83 (d, J=1.4 Hz 1H), 7.94 (d, J=8.7 Hz, 2H), 8.04 (d, J=9.0 Hz, 2H), 8.65 (d, J=2.4 Hz, 1H).
80%
With potassium carbonate In N,N-dimethyl-formamide at 110℃; for 16 h;
Step 1. 4-(lH-Pyrazol-l-yl)benzonitrile (0847) [00269] A mixture of lH-pyrazole (15 g, 220.34 mmol), 4-fluorobenzonitrile (27 g, 222.93 mmol), potassium carbonate (60.7 g, 439.19 mmol) in DMF (200 mL) was stirred for 16 h at 110 °C. After cooling to ambient temperature, the reaction mixture was poured into water (500 mL) and the resulting solids were collected by filtration and dried under vacuum, resulting in 30 g (80percent) of 4-(lH-pyrazol-l-yl)benzonitrile. MS (ESI) m/z 170 [M+H]+.
62%
Stage #1: With sodium hydroxide In N,N-dimethyl-formamide at 80℃; for 0.5 h; Inert atmosphere Stage #2: at 110℃; for 3 h;
Into a 50-mL 3-necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen, was placed 1H-pyrazole (422 mg, 6.21 mmol, 1.50 equiv), sodium hydroxide (248 mg, 6.20 mmol, 1.50 equiv), and N,N-dimethylformamide (20 mL). The mixture was stirred and heated to 80° C. for 30 min, then to this was added 4-fluorobenzonitrile (500 mg, 4.13 mmol, 1.00 equiv). The resulting solution was stirred for 3 h at 110° C. in an oil bath. The resulting solution was diluted with 50 mL of H2O. The solids were collected by filtration and dried in an oven. The product was obtained as 0.43 g (62percent) of a white solid.
47%
With caesium carbonate In N,N-dimethyl-formamide for 2 h; Reflux
A 100-mE round-bottom flask was charged with4-fluorobenzonitrile (2 g, 16.51 mmol), cesium carbonate (16 g, 49.11 mmol), N,N-dimethylformamide (20 mE) and 1 H-pyrazole (2.24 g, 32.90 mmol). The resulting solution was refluxed for 2 h. The solids were removed by filtration and the filtrate was concentrated under vacuum. The residue was purified by column chromatography eluting with ethyl acetate/petroleum ether (1:10 to 1:4 v/v) to afford 4-(1H- pyrazol-i-yl)benzonitrile as a yellow oil (1.3 g, 47percent). LCMS: (ESI) mlz 170 [M+H].
47%
With caesium carbonate In N,N-dimethyl-formamide for 2 h; Reflux
A 100-mL round-bottom flask was charged with 4-fluorobenzonitrile (2 g, 16.51 mmol), cesium carbonate (16 g, 49.11 mmol), N,N-dimethylformamide (20 mL) and 1H-pyrazole (2.24 g, 32.90 mmol). The resulting solution was refluxed for 2 h. The solids were removed by filtration and the filtrate was concentrated under vacuum. The residue was purified by column chromatography eluting with ethyl acetate/petroleum ether (1:10 to 1:4 v/v) to afford 4-(1H-pyrazol-1-yl)benzonitrile as a yellow oil (1.3 g, 47percent). LCMS: (ESI) m/z 170 [M+H].
With potassium carbonate In butanone for 96 h; Reflux
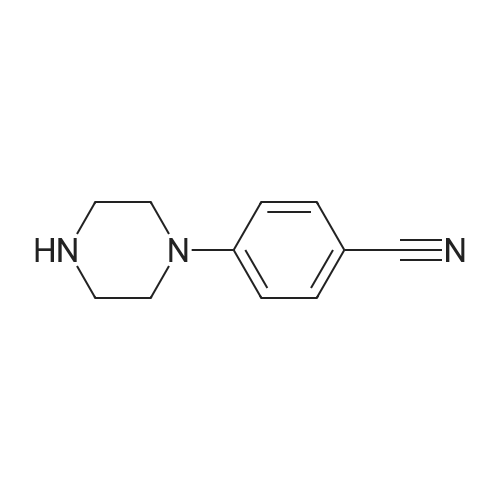
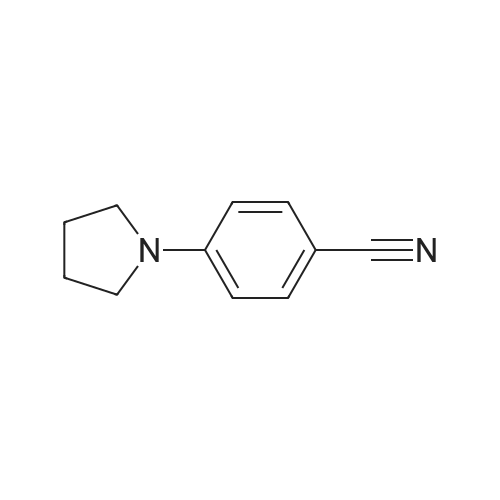
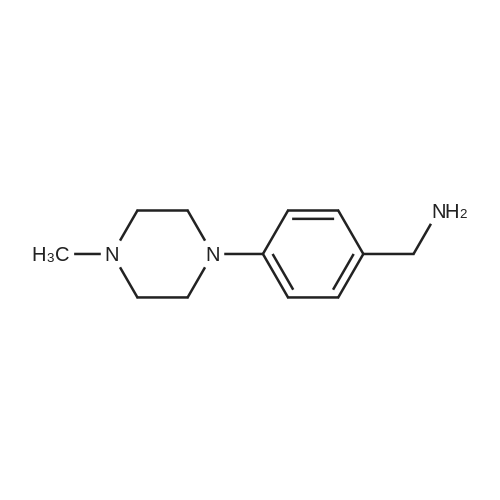
A mixture of 4-fluorobenzonitrile (3, 0.36 g, 3 mmol), piperazine (0.64 g, 7.4 mmol) and K2CO3 (0.85 g, 6.1 mmol) in ethylmethylketone (50 mL) was refluxed for 4 days. The completion of reaction was detected by TLC. The solvent was evaporated under reduced pressure and then chloroform and water was added. The organic layer was extracted. After evaporation of chloroform, the residue was crystallized from chloroform to give compound 4 as a cream solid. Yield: 95percent; m.p 82-85 °C; IR (KBr, cm-1): 3325, 2215, 1606. 1H NMR (500 MHz, CDCl3): 7.50 (d, 2H, J = 9.2 Hz, phenyl), 6.86 (d, 2H, J = 9.2 Hz, phenyl), 3.29 (t, 4H, J = 5.2 Hz, piperazine), 3.02 (t, 4H, J = 5.2 Hz, piperazine). MS (ESI): 188 [M + H+]. Anal. Calcd for C11H13N3: C, 70.56; H, 7.0; N, 22.44. Found: C, 70.35; H, 6.82; N, 22.73.
93.6%
With potassium carbonate In dimethyl sulfoxide
a) Potassium carbonate (20.7 g, 0.15 mol) was added to a solution of 4-fluorobenzonitrile (12.1 g, 0.1 mol) and piperazine (25.8 g, 0.3 mol) in dimethylsulfoxide (75 ml) and heated at 95° C. for 20 hours. The reaction was cooled and poured into water (1100 ml) and extracted into dichloromethane (4*400 ml). The organic solution was dried and the solvent removed in vacuo to yield 4-cyanophenylpiperazine (17.5 g, 93.6percent yield): 1H-NMR (CDCl3): 7.50 (d, 2H), 6.80 (d, 2H), 3.25 (m, 4H), 3.00 (m, 4H); MS (+ve ESI): 188 (M+H)+.
Reference:
[1] Journal of Medicinal Chemistry, 1994, vol. 37, # 23, p. 3882 - 3885
[2] European Journal of Medicinal Chemistry, 2011, vol. 46, # 6, p. 2602 - 2608
[3] Patent: US7235559, 2007, B1,
[4] Phosphorus, Sulfur and Silicon and the Related Elements, 2008, vol. 183, # 5, p. 1252 - 1263
12
[ 110-85-0 ]
[ 1194-02-1 ]
[ 101722-12-7 ]
[ 68104-63-2 ]
Reference:
[1] Organic Process Research and Development, 2018,
[2] Organic Process Research and Development, 2018,
[3] Organic Process Research and Development, 2018, vol. 22, # 9, p. 1086 - 1091
13
[ 1194-02-1 ]
[ 68104-63-2 ]
Reference:
[1] Journal of Medicinal Chemistry, 2011, vol. 54, # 8, p. 3086 - 3090
14
[ 109-01-3 ]
[ 1194-02-1 ]
[ 34334-28-6 ]
Yield
Reaction Conditions
Operation in experiment
93%
With potassium carbonate In dimethyl sulfoxide
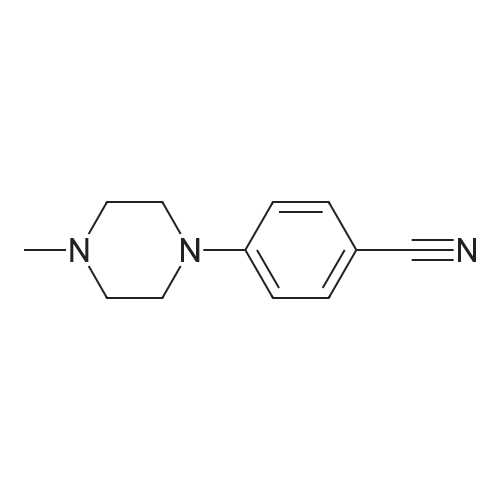
To a mixture of 4-FLUORO- benzonitrile 42B (1.0 g, 8. 25 MMOL) and K2CO3 (2.27 mg, 16.51 MMOL) in dimethyl sulfoxide (7 mL) was added 1-methyl piperazine (1.36 mL, 12. 38 MMOL) and the reaction continued as described above to afford amine 1.54 g of 45b in 93percent YIELDS. 1H-NMR (500 MHz, CDC . S) : No. 2.36 (3Hs, s), 2.55 (4Hs, t, J = 4. 88 Hz), 3.35 (4Hs, t, J = 4. 88 Hz), 6. 87 (2Hs, d, J = 8. 78 Hz), 7.49 (2Hs, d, J = 8. 78 Hz); ESI-MASS : 202. 1 (M+1).
84%
With potassium carbonate In N,N-dimethyl-formamide at 100℃; for 18 h;
To a solution of 4-fluorobenzonitrile (0.041 mol, 5 g) in DMF (10 ml) were added K2C03 (0.082 mol, 11.4 g) and 1 -methylpiperazine (0.062 mol, 6.15 g). The mixture was stirred over 1 8h at 100°C. The mixture was partitioned between water and ethyl acetate. The organic phase was washed with water and brine. The resulting solution was dried oversodium sulfate and evaporated to dryness. The title compound was obtained as a white solid and used without further purification (7 g, 84percent).(ESI+) MS: m/z 202.3 (MHj.
77%
at 90℃; for 15 h;
4-(4-Methylpiperazin-1-yl)benzamide 4-Fluorobenzonitrile (12 g, 99 mmol, 1 eq.) and 100 mL of DMF are placed in a 250 mL round-bottomed flask. N-Methylpiperazine (16 mL, 1.6 eq., 123 mmol) is added to this solution. The orange solution is heated at a temperature in the region of 90° C. for 15 hours. The solvent is then evaporated to dryness and the residue is diluted with 500 mL of diethyl ether. The solution is washed with sodium hydrogen carbonate solution (2*100 mL) and then with saturated sodium chloride solution (100 mL). After evaporation, the 4-(4-methylpiperazin-1-yl)benzonitrile (orange solid, 10 g, 77percent) is used without further purification in the following hydrolysis step. It is added at a temperature in the region of 0° C. to a solution of 98percent sulfuric acid (25 mL) and 5 mL of water. The purple solution is then heated at a temperature in the region of 100° C. for 8 hours. The solution is cooled and then hydrolyzed by pouring onto ice. The pH is adjusted to 9-10 with sodium hydroxide pellets. The precipitate obtained is filtered off and washed thoroughly with water and then with tetrahydrofuran, which partially dissolves the product. The water is separated out by settling. After evaporating the organic phase to dryness, a solid is obtained, which is purified by chromatography on silica gel, eluding with a methanol/dichloromethane mixture (15/85 by volume). 4-(4-Methylpiperazin-1-yl)benzamide (8.7 g, 80percent) is thus obtained in the form of a white solid, which is used directly in the following step.
Reference:
[1] Journal of Medicinal Chemistry, 2005, vol. 48, # 26, p. 8261 - 8269
[2] Patent: WO2005/7625, 2005, A2, . Location in patent: Page 63
[3] Phosphorus, Sulfur and Silicon and the Related Elements, 2008, vol. 183, # 5, p. 1252 - 1263
[4] Patent: WO2016/96709, 2016, A1, . Location in patent: Page/Page column 50
[5] Patent: US2008/146542, 2008, A1, . Location in patent: Page/Page column 36
[6] Bioorganic and Medicinal Chemistry Letters, 2005, vol. 15, # 5, p. 1529 - 1534
[7] Patent: EP1577288, 2005, A1, . Location in patent: Page/Page column 96-97
[8] Patent: US2002/193389, 2002, A1,
[9] Patent: US6127541, 2000, A,
[10] Patent: US2005/222408, 2005, A1,
[11] Journal of Medicinal Chemistry, 2014, vol. 57, # 12, p. 5293 - 5305
[12] ACS Medicinal Chemistry Letters, 2018, vol. 9, # 7, p. 741 - 745
15
[ 186650-77-1 ]
[ 1194-02-1 ]
[ 53617-35-9 ]
Reference:
[1] Patent: US6288061, 2001, B1,
16
[ 110-91-8 ]
[ 1194-02-1 ]
[ 7470-38-4 ]
Yield
Reaction Conditions
Operation in experiment
99%
With hydrogenchloride; sodium hydroxide In water
Example 10 Preparation of 4-(4-morpholinyl) Benzoic Acid Staring From 4-fluorobenzonitrile by the One-Pot Method Using Basic Hydrolysis: A mixture of 4-fluorobenzonitrile (5.04 g, 41.6 mmol) and morpholine (9.12 g, 104.6 mmol) is heated at 120° C. to achieve a complete conversion of 4-fluorobenzonitrile after 5 hours. Water (100 ml) and NaOH (4.1 g, 10 mmol) are added to the reaction mixture. The whole mixture is kept refluxing for another 5 h, cooled down to room temperature and made acidic by the addition of HCl (5percent) with efficient stirring. The precipitate is filtered off, washed with water and dried under vacuum (60° C.) to give 8.34 g of the title-compound. Yield: 99percent; m.p. 275-277° C.; MS 207 (100, M+); H1 NMR (DMSO): 12.33 (b, 1H), 7.78 (d, 2H), 6.95 (d, 2H), 3.72 (t, 2H), 3.23 (t, 2H); δ C3 NMR (CDCl3): δ 167.25, 153.90, 130.81, 119.91, 113.22, 65.87, 46.97.
Reference:
[1] Patent: US2003/171581, 2003, A1,
17
[ 110-91-8 ]
[ 934012-95-0 ]
[ 1194-02-1 ]
[ 7470-38-4 ]
Reference:
[1] Patent: US2003/171581, 2003, A1,
18
[ 1194-02-1 ]
[ 7470-38-4 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2005, vol. 15, # 5, p. 1529 - 1534
19
[ 74-96-4 ]
[ 1194-02-1 ]
[ 456-03-1 ]
Reference:
[1] European Journal of Pharmaceutical Sciences, 2011, vol. 43, # 5, p. 386 - 392
20
[ 15097-49-1 ]
[ 1194-02-1 ]
[ 10282-30-1 ]
Yield
Reaction Conditions
Operation in experiment
71%
Stage #1: With cesium fluoride In N,N-dimethyl-formamide for 0.166667 h; Inert atmosphere Stage #2: at 20℃; for 144 h;
Under a nitrogen atmosphere, 320 mg (2.11 mmol) of CsF were suspended in 5 ml of DMF and stirred for 30 min. Then, 1.00 g (8.26 mmol) of 4-fluorobenzonitrile were added. After 10 min 1.44 ml (8.25 mmol) of N-trimethylsilylpyrrolidine were added, and the mixture was stirred for 6 d at room temperature. For workup, 5 ml of water and 5 ml of CH2Cl2 were added to the reaction mixture. The organic phase was separated, the aqueous phase was extracted with CH2Cl2, the organic phases were combined, washed several times with water and dried over MgSO4, and the solvent was removed under vacuum.Yield: 1.01 g (5.87 mmol, 71percent), appearance: pale yellow solid. 1H NMR (CDCl3, 25° C., 400.13 MHz): δ=2.00-2.07 (m, 4H, H-8, H-9), 3.30-3.34 (m, 4H, H-7, H-10), 6.47-6.51 (m, 2H, H-3, H-5), 7.42-7.46 (m, 2H, H-2, H-6). 13C NMR (CDCl3, 25° C., 100.61 MHz): δ=25.5 (C-8, C-9), 47.6 (C-7, C-10), 96.7 (C-1), 111.6 (C-3, C-5), 121.1 (-CN), 133.6 (C-2, C-6), 150.1 (C-4).
Reference:
[1] Synthesis, 2003, # 11, p. 1643 - 1648
28
[ 1194-02-1 ]
[ 1009-35-4 ]
Reference:
[1] Journal of Organic Chemistry, 2011, vol. 76, # 19, p. 8088 - 8094
[2] Journal of the American Chemical Society, 1961, vol. 83, p. 4564 - 4571
[3] Journal of the Chemical Society, Perkin Transactions 2: Physical Organic Chemistry (1972-1999), 1972, p. 906 - 911
[4] Patent: US5360809, 1994, A,
[5] Patent: EP563001, 1993, A1,
29
[ 1194-02-1 ]
[ 4640-67-9 ]
Reference:
[1] Journal of Medicinal Chemistry, 1979, vol. 22, # 11, p. 1385 - 1389
30
[ 1194-02-1 ]
[ 1999-00-4 ]
Yield
Reaction Conditions
Operation in experiment
93%
Stage #1: at 0 - 25℃; for 26 h; Stage #2: With hydrogenchloride In tetrahydrofuran; water at 20 - 25℃; for 1 h;
Under nitrogen atmosphere, 30 ML of THF was added to 6.09 g (10 mmol, 1.0 equivalent) of (BrZnCH2COOEt*THF)2.. Under argon atmosphere, a solution of 1.21 g (10 mmol) of 4-fluorobenzonitrile in 5 ML of THF was added dropwise while stirring at 0.similar.5°C. The mixture was stirred at 20.similar.25°C for 26 hours. 15 ML of 10percent hydrochloric acid was added dropwise at 20°C or lower, and the mixture was stirred at 20.similar.25°C for 1 hour, followed by dilution with ethyl acetate.. Then, the layers were separated.. The organic layer was washed successively with 15 ML of 1N hydrochloric acid, 20 ML of an aqueous saturated sodium chloride solution, 20 ML (*2) of an aqueous saturated sodium bicarbonate solution, and 20 ML of an aqueous saturated sodium chloride solution.. After washing, the organic layer was dried with anhydrous magnesium sulfate.. Concentration under reduced pressure afforded 1.96 g of the desired product (yield 93percent).1H NMR (CDCl3), (ppm): δ [1.26 (t, J=7.1 Hz), 1.34 (t,J=7.1 Hz)] (3H), [3.96 (s), 5.61 (s), 12.6 (s)] (2H), 4.18-4.25 (2H, m), 7.07-8.02 (4H, m).
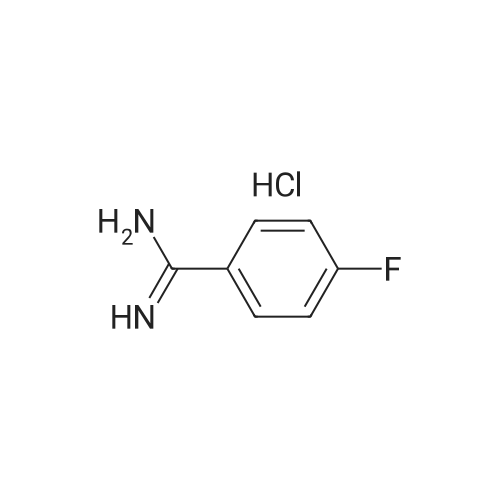
EXAMPLE 40 4-Fluorobenzamidine Hydrochloride 4-Fluorobenzonitrile (10 g, 83 mmol) is dissolved in a mixture of anhydrous ethanol (5 mL) and diethyl ether (70 mL). The reaction mixture is cooled to ice-bath temperature and saturated with gaseous hydrogen chloride for 90 minutes. The mixture is allowed to warm to ambient temperature and stirred overnight. The colorless precipitates are filtered off, washed with diethyl ether and dissolved in anhydrous ethanol (20 mL). Diethyl ether (100 mL) saturated with gaseous ammonia is added and the solution is stirred for 3 hours. The resulting suspension is filtered and the solvent of the filtrate is removed in vacuo. The residue is washed with diisopropyl ether. After drying colourless crystals (5.15 g, 35.5percent) of melting point 210° C. are obtained.
Reference:
[1] Magnetic Resonance in Chemistry, 1987, vol. 25, p. 923 - 927
[2] Patent: US5849758, 1998, A,
[3] Journal of Medicinal Chemistry, 1990, vol. 33, # 4, p. 1230 - 1241
[4] Patent: US2003/139415, 2003, A1,
[5] Tetrahedron, 2014, vol. 70, # 8, p. 1635 - 1645
38
[ 92292-00-7 ]
[ 1194-02-1 ]
[ 38646-68-3 ]
Reference:
[1] Patent: US6288061, 2001, B1,
39
[ 1194-02-1 ]
[ 100-83-4 ]
[ 90178-72-6 ]
Reference:
[1] Liebigs Annalen der Chemie, 1984, vol. 1984, # 3, p. 409 - 425
[2] Patent: WO2006/123725, 2006, A1, . Location in patent: Page/Page column 46
40
[ 1194-02-1 ]
[ 99662-34-7 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2006, vol. 16, # 11, p. 2955 - 2959
41
[ 1194-02-1 ]
[ 146137-79-3 ]
Yield
Reaction Conditions
Operation in experiment
79%
With n-butyllithium; acetic acid; diisopropylamine In tetrahydrofuran; <i>N</i>-methyl-acetamide; hexane; water
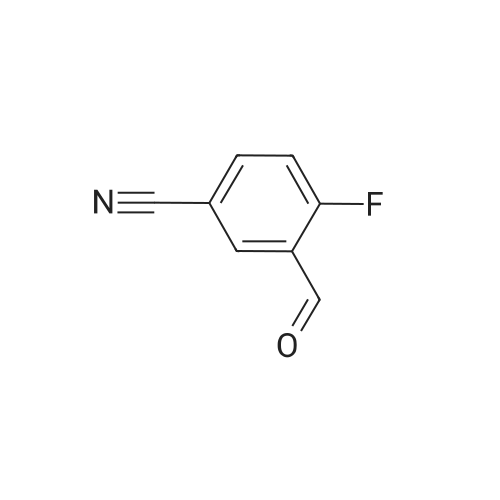
EXAMPLE 112 2-Fluoro-5-cyano-benzaldehyde To a stirred, cooled solution (0° C.) of diisopropylamine (15.4 mL, 0.11 mol) in anhydrous tetrahydrofuran (200 mL) n-butyllithium (40 mL of 2.5M in hexane, 0.11 mol) was added from a dropping funnel over a period of 30 min. under argon. The mixture was stirred at that temperature for 30 min. and then cooled to -78° C. A solution of 4-fluoro-benzonitrile (12.1g, 0.1 mol) in dry THF (50 mL) was then added dropwise over 15 min. via syringe and stirred for 1 hour at -78° C. Dimethylformamide (8 mL) was added dropwise from a syringe and the stirring was continued for another 20 min. The reaction was quenched by the rapid addition of acetic acid (20 mL) followed by water (500 mL) and the product was extracted with diethyl ether (2*500 mL). The combined organic layers were washed with 1N HCl, water, saturated sodium chloride and then dried over anhydrous magnesium sulfate, and evaporated to give 2-fluoro-5-cyano-benzaldehyde (11.8 g, 79percent yield) as a light yellow solid. 1H NMR (CDCl3): δ10.32 (s, 1H), 8.18 (dd, 1H, J=6.5, 2.5 Hz), 7.88 (m, 1H), 7.33 (t, 1H, J=9.5 Hz).
74.6%
With acetic acid; diisopropylamine In <i>N</i>-methyl-acetamide; ethyl acetate
A. 4-Fluoro-3-Formylbenzenecarbonitrile Lithium diisopropyi amide (LDA) (22 mL, 49.56 mmol, 2.0 N commercial solution in heptanes) was added to tetrahydrofliran (50 mL), cooled to 78° C. and under nitrogen. 4-Fluorobenzonitrile was weighed out (5.0 g, 41.3 imnol), placed under nitrogen and dissolved in 25 mL of dry tetrahydrofliran. This solution was added dropwise to the solution of LDA. The resulting solution was stirred at -78° C. for one hour before quenching with 4 mL of dimethylformamide. The temperature was maintained for 10 min before adding 8 mL of acetic acid and 20 mL of distilled water. The crude product was extracted with ethyl acetate. Purification by column chromatography (SiO2, 20percent ethyl acetate in hexanes) afforded 4.6 g of pure product as a white solid (74.6percent yield). -A second batch of the title compound (3.5 g, 56.8 percent/yield) was prepared 20 using 5 g of benzonitrile (41.3 mmol): 1H NMR (CDCl3) δ 10.3 (s, 1H), 8.21 (dd, 1H), 7.91 (d of q, 1H), 7.35 (t, 1H); ES-MS M+was not detected.
Stage #1: With sodium hydride In N,N-dimethyl-formamide at 20℃; for 1 h; Stage #2: at 60℃; for 0.5 h;
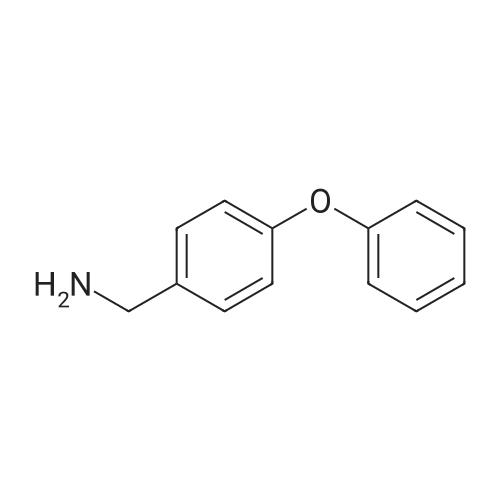
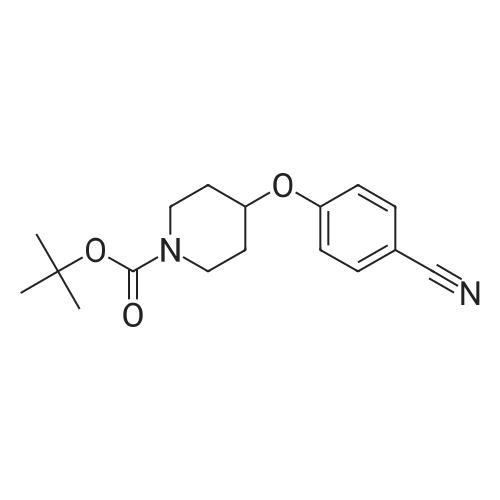
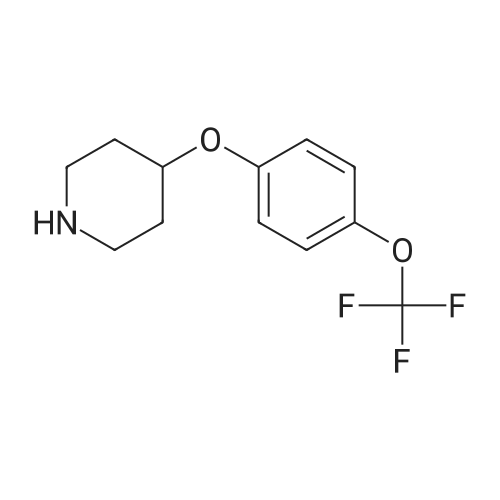
Step A: tert-Butyl-4-(4-cvanophenyloxy)- 1 -piperidinecarboxylate <n="102"/>To tert-butyl 4-hydroxy- 1 -piperidinecarboxylate (24.1 g, 100. 0 mmol) in dimethylformamide (250 mL) is added sodium hydride (60percent emulsion in mineral oil) (8.00 g, 200.0 mmol) and the mixture is stirred at rt for 1 h. 4-Fluorobenzonitrile (12.1 g, 100.0 mmol) is added, the mixture is heated to 60 0C for 30 minutes, diluted with ethyl acetate and the reaction is quenched by the addition of water. The organic phase is evaporated in vacuo and purified on silica gel to give the desired product (27.5 g, 91percent).
Stage #1: With sodium hydride In DMF (N,N-dimethyl-formamide) at 50℃; for 0.75 h; Stage #2: at 50℃; for 2 h;
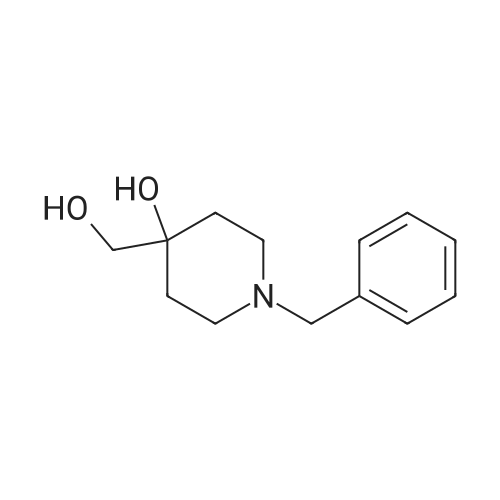
Example 9 4- (1-Benzyl-piperidin-4-yloxy)-benzamide Step 1 4- (4-Cyano-phenoxy)-piperidine-1-carboxylic acid tert-butyl ester Add a solution of N-Boc-4-hydroxypiperidine (3.0 g, 14.9 mmol) in DMF (5 mL) to a suspension of sodium hydride (894 mg, 22.4 mmol) in DMF (17 mL). Stir the reaction mixture while heating at 50°C for 45 min. Then add a solution of 4-fluoro- benzonitrile (2.16 g, 17.9 mmol) in DMF (5 mL). Stir and heat at 50°C for 2h. Let cool to room temperature and quench with water (0.5 mL). Evaporate DMF. Redissolved the resulting residue in EtOAc/hexanes (2/1,20 mL) and wash with water (3x15 mL). Dry the organic layer over magnesium sulfate, filter and concentrate. Purify by chromatography (EtOAc/hexanes 20percent and EtOAc/hexanes 10percent) to yield the title compound (2.32 g, 52percent).
Reference:
[1] Journal of Medicinal Chemistry, 2005, vol. 48, # 26, p. 8261 - 8269
[2] Journal of Medicinal Chemistry, 2014, vol. 57, # 12, p. 5293 - 5305
[3] ACS Medicinal Chemistry Letters, 2018, vol. 9, # 7, p. 741 - 745
49
[ 1194-02-1 ]
[ 114365-04-7 ]
Reference:
[1] Journal of Medicinal Chemistry, 2007, vol. 50, # 15, p. 3651 - 3660
50
[ 121-43-7 ]
[ 1194-02-1 ]
[ 468718-30-1 ]
[ 468717-46-6 ]
Yield
Reaction Conditions
Operation in experiment
70%
With hydrogenchloride; n-butyllithium; diisopropylamine In tetrahydrofuran
EXAMPLE 163 3-(2-Dimethylaminomethyl-cyclopropyl)-1H-indazole-5-carbonitrile n-BuLi (1.9 M, 26.3 mL, 50 mmol) was added dropwise to a solution of diisopropylamine (7.71 mL, 55 mmol) in 100 mL anhydrous THF under N2 at 0° C. After 10 minutes, the reaction was cooled to -78° C. A solution of 4-fluorobenzonitrile (6.06 g, 50 mmol) in 20 mL anhydrous THF was added dropwise, at such a rate to maintain an internal temperature of -78° C. After stirring for 1 h at this temperature, trimethyl borate (8.41 mL, 75 mmol) was added dropwise at such a rate to maintain an internal temperature of -78° C. The reaction was stirred and gradually warmed to room temperature over 16 h. The reaction was cooled to 10° C. and 25 mL 6N HCl was added. After stirring at room temperature for 4 h, the reaction was partitioned between water and ethyl acetate. The organic layer was washed three times with 100 mL 2N NaOH. The aqueous layers were pooled and adjusted to pH 6 with 6N HCl. The white solid which forms was extracted by three washes of 100 mL Ethyl acetate. The organic layers were pooled, dried over sodium sulfate, concentrated and dried under high vacuum to give 2-fluoro-5-cyanophenyl boronic acid (5.74 g, 70percent). 1H NMR (500 MHz, acetone-d6) 8.08 (1 H, dd, J=2.14, 5.5 Hz), 7.90 (1 H, m), 7.31 (1 H, t, J=8.85 Hz). Anal. calcd. for C7H5BFNO2: C, 50.97; H, 3.05; N, 8.49. Found: C, 51.19; H, 3.19; N, 8.26.
Reference:
[1] Patent: US2003/73849, 2003, A1,
51
[ 1194-02-1 ]
[ 252017-04-2 ]
Reference:
[1] Organic Process Research and Development, 2012, vol. 16, # 3, p. 447 - 460
With potassium carbonate In N,N-dimethyl-formamide; toluene at 115℃; for 8 h; Inert atmosphere
In a round bottom flask, under nitrogen atmosphere, 2-bromo-5-hydroxybenzaldehyde (100 g, 497 mmol) and 4-fluorobenzonitrile (301 .2 g, 2487 mmol) were dissolved in DMF (250 ml) and toluene (500 ml). To the resulting dark solution, potassium carbonate (206.2 g, 1492 mmol) was added and the resulting mixture was stirred for 8 hours at 1 15 °C. After the reaction was completed by TLC, the heterogeneous mixture was filtered. The solvents and 4-fluoro-benzonitrile were distilled under reduced pressure from the obtained filtrate. Ethyl acetate (800 ml), water (500 ml) and brine (40 ml) were added and the formed phases were separated at 65-70 °C. The aqueous phase was extracted with ethyl acetate (150 ml) at 65-70 °C and the combined organic phase was washed with a mixture of water (225 ml) and brine (180 ml) at 65-70 °C. The obtained organic phase was evaporated under reduced pressure to give a brown solid. The solid was purified twice by recrystallization in a hot mixture of ethyl acetate and toluene to give 4-(4-bromo-3- formylphenoxy)benzonitrile (compound A) (107.5 g). Yield: 71 percent Purity by HPLC: >99percent 1H-NMR (200 MHz, CDCI3, δ ppm): 10.32 (1 H, s); 7.62-7.72 (3H, m); 7.57-7.59 (1 H, dd; J= 3, J= 1 ); 7.18- 7.23 (1 H, dd, J=8 Hz, J=3 Hz), 7.01 -7.08 (2H, m) 13C-NMR (50 MHz, CDCI3, δ ppm): 190.69; 160.08; 155.13; 135.59; 134.89; 134.39; 126.86; 121.81 ; 120.28; 1 18.68; 1 18.33; 107.32 DSC: Endothermic peak at 1 1 1.9 °C.
50 g
With potassium carbonate In N,N-dimethyl-formamide at 75 - 85℃; for 20 h;
A mixture of 2-bromo-5-hydroxybenzaldehyde (50g), 4-fluorobenzonitrile (75.3 lg) and potassium carbonate (103g) in dimethylformamide (500mL) was stirred at about 75°C to about 85°C for about 20h. The reaction mixture was filtered and the filtrate was quenched with water at about room temperature, stirred for about 2h, filtered and dried. The crude product was purified by ethyl acetate. Yield: 50g HPLC purity: >99percent
EXAMPLE 40 4-Fluorobenzamidine Hydrochloride 4-Fluorobenzonitrile (10 g, 83 mmol) is dissolved in a mixture of anhydrous ethanol (5 mL) and diethyl ether (70 mL). The reaction mixture is cooled to ice-bath temperature and saturated with gaseous hydrogen chloride for 90 minutes. The mixture is allowed to warm to ambient temperature and stirred overnight. The colorless precipitates are filtered off, washed with diethyl ether and dissolved in anhydrous ethanol (20 mL). Diethyl ether (100 mL) saturated with gaseous ammonia is added and the solution is stirred for 3 hours. The resulting suspension is filtered and the solvent of the filtrate is removed in vacuo. The residue is washed with diisopropyl ether. After drying colourless crystals (5.15 g, 35.5percent) of melting point 210° C. are obtained.
2.9 g (100%)
EXAMPLE 4A 4-Fluorobenzenecarboximidamide Hydrochloride In analogy to the procedure for Example 3A, 2,0 g (16,5 mmol) 4-fluorobenzonitrile and proportionate amounts of the other reagents are used. Yield: 2.9 g (100percent) 1H-NMR (DMSO-d6, 200 MHz): delta=7,5 (m, 2H), 8,0 (m, 2H) ppm.
A 1 L four-necked flask was fitted with a constant pressure dropping funnel and mechanical agitation.N2 is replaced three times, and N2 is protected by flow. Turn on mechanical agitation,Add 400 ml of THF and 182 ml of LDA (2 mol/L);40 g of p-fluorobenzonitrile (formula IV, 0.33 mol) was dissolved in 200 ml of THF.Transfer to a constant pressure dropping funnel and start to cool down.Temperature control at -75 ° C ~ -85 ° C dropwise addition of p-fluorobenzonitrile in THF solution; after the end of the addition,The temperature is controlled at -75 ° C ~ -85 ° C for 1 h,At the same time, 26.6 g of DMF was diluted with 30 ml of THF and transferred to a constant pressure dropping funnel. Temperature control at -75 ° C ~ -85 ° C dropwise addition of DMF in THF solution, after the end of the addition,The temperature control was stirred at -75 ° C ~ -85 ° C for 1 h until the reaction with p-fluorobenzonitrile was complete.After the reaction was completed, it was quickly added to 64 ml of acetic acid and quenched, and 400 g of water was added.The layers were separated and the aqueous extracted with EtOAc EtOAc. The organic phase was combined, washed with 400 ml of diluted HCl (1 mol/L), and the organic layer was washed with 400 ml of brine, and the organic39 g of the compound of formula III are obtained as a bright yellow solid
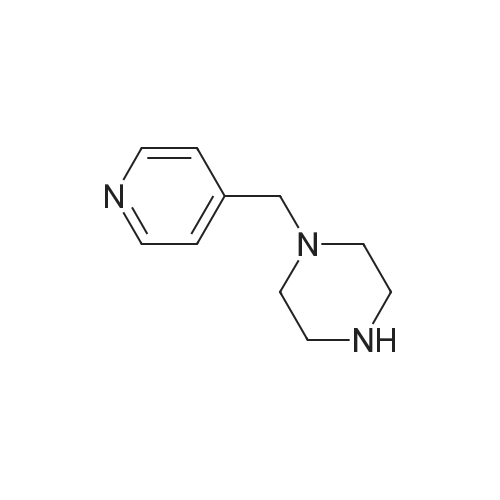
With sodium hydride; In N,N-dimethyl-formamide; at 20 - 100℃;
1500 ml of dry dimethylformamide (DMF) are placed in a 1000 ml four-necked flask while passing nitrogen over it and 72.67 g (0.6 mol) of 4-fluorocyanobenzene and 61.2 g (0.9 mol) of imidazole and finally 21.6 g (0.9 mol) of sodium hydride are added. The reaction mixture is heated to 100° C., stirred at this temperature for 4 hours and subsequently overnight at room temperature. The reaction mixture is then poured into water and the resulting mixture is extracted a number of times with dichloromethane. The organic phase is dried, evaporated on a rotary evaporator and finally dried further at 60° C. under reduced pressure. The yield is 94 g (93percent of theory).1H-NMR (400 MHz, CDCl3): delta=7.27 (s, 1H); 7.35 (s, 1H); 7.54 (d, J=8.8 Hz, 2H); 7.81 (d, J=8.8 Hz, 2H); 7.95 (s, 1H).
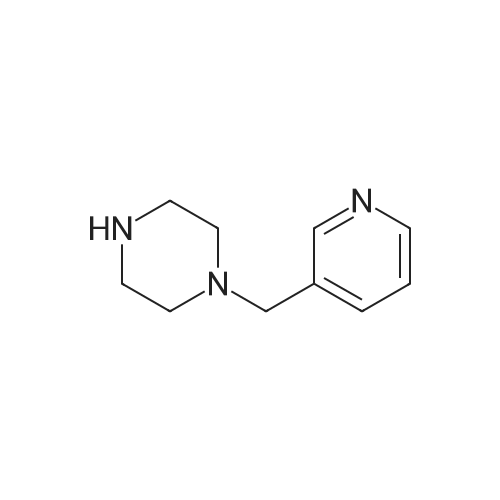
With potassium carbonate; In N,N-dimethyl-formamide; at 80℃;
A suspension of l-(pyridin-3-ylmethyl) piperazine (549 mg, 3.10 mmol), 4- fluorobenzonitrile (375 mg, 3.10 mmol), and K2C03 (643 mg, 4.65 mmol) in DMF (50 mL) was stirred at 80C overnight. The reaction mixture was cooled to room temperature and filtered, and the filtrate was washed with ethyl acetate (30 mL). The combined filtrate and washings were evaporated to dryness under reduced pressure. The crude residue was purified by MPLC on silica gel (3% MeOH in CH2Ck) to afford the titled compound (326 mg, 38%) as a yellow solid. IH NMR (CDC13/TMS) 8 2.59 (apparent t, 4 H, J= 5.1 Hz), 3.33 (apparent t, 4 H, J= 5.1 Hz), 3.57 (s, 2H), 6. 85 (m, 2H), 7.27 (m, 1H), 7. 48 (m, 2H), 7.69 (m, 1H), 8. 53 (m, 1H), 8. 57 (m, 1H)
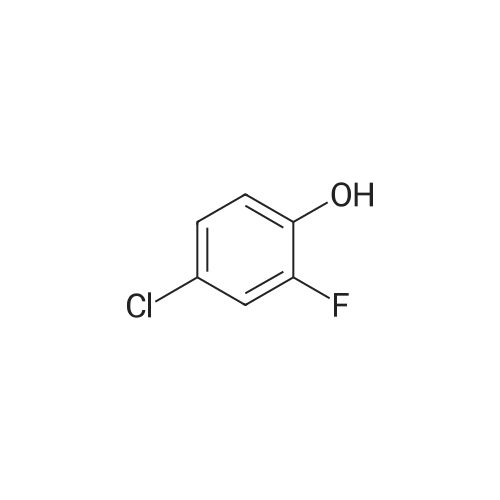
With potassium carbonate; In DMF (N,N-dimethyl-formamide);Heating / reflux;
A mixture of 4-fluorobenzonitrile (5.0 g, 41.3 mmol), <strong>[348-62-9]4-chloro-2-fluorophenol</strong> (4.7 mL, 44 mmol), and potassium carbonate (13.8 g, 99.8 mmol) in DMF (100 ml) was refluxed overnight. After cooling to room temperature, the mixture was diluted with ethyl acetate, washed twice with a 2N aqueous sodium hydroxide solution, washed with water, dried over sodium sulfate, filtered, and evaporated under reduced pressure to give very light yellow solid. The weight of crude product was 7.56 g (74%). 1H NMR (CDCl3): delta 7.61 (d, J = 8.1 Hz, 2H), 7.27 - 7.07 (m, 3H), 6.98 (d, J = 8.7 Hz, 2H).
With potassium carbonate; In N,N-dimethyl-formamide;
a) 4-(4-Chloro-2-fluorophenoxy)benzonitrile A mixture of 4-fluorobenzonitrile (5.0 g, 41.3 mmol), <strong>[348-62-9]4-chloro-2-fluorophenol</strong> (4.7 mL, 44 mmol), and potassium carbonate (13.8 g, 99.8 mmol) in DMF (100 ml) was refluxed overnight. After cooling to room temperature, the mixture was diluted with ethyl acetate, washed twice with a 2N aqueous sodium hydroxide solution, washed with water, dried over sodium sulfate, filtered, and evaporated under reduced pressure to give very light yellow solid. The weight of crude product was 7.56 g (74%). 1H NMR (CDCl3): delta 7.61 (d, J=8.1 Hz, 2H), 7.27-7.07 (m, 3H), 6.98 (d, J=8.7 Hz, 2H).
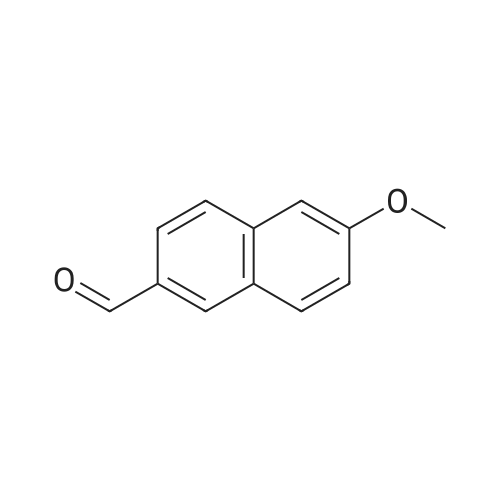
A. 4-Fluoro-3-[hydroxy-(6-methoxy-naphthalen-2-yl)-methyl]-benzonitrile To a cooled solution (-78 C.) of LDA (24.5 mL, 49 mmol) in THF (40 mL) was added 4-fluorobenzonitrile (5.45 g, 45 mmol) in THF (30 mL) and 6-methoxy-2-naphthaldehyde (9.13 g, 49 mmol) in THF (40 mL). The reaction was stirred at -78 C. for 1 hour and then allowed to warm to room temperature over a period of 2 hours. The reaction was quenched with ice and THF was removed in vacuo. The aqueous solution was then extracted with ethyl acetate. The organic phase was dried over magnesium sulfate, filtered and the solvent was removed in vacuo. The crude material was purified by column chromatography (SiO2, 4:1 hexanes: ethyl acetate) to provide the title compound (5.5 g, 40% yield). ES-MS (m/z) 308 [M+1]+.
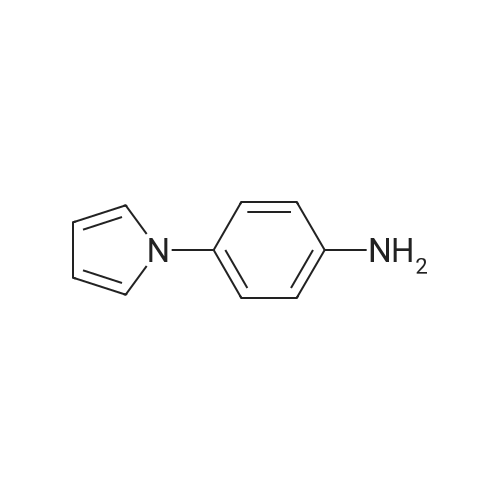
A mixture 4-fluorobenzonitrile (3 g, 25 MMOL) and imidazolyl sodium (2. 48 g, 27.5 MMOL) in DMF (50 mL) was stirred at 80 C under Ar for 12 h. Progress of reaction was monitored by TLC. The reaction mixture was concentrated in vacuo and the residue was diluted with 50 mL water and stirred. The aqueous mixture was extracted with EtOAc (2 x 50 mL). Combined EtOAc extracts was dried over anhydrous MGS04, concentrated, and the 4- (1-imidazolyl)-benzonitrile was isolated by column chromatography (3.6 g, 78percent). LCMS: MH+ = 170 ;'H NMR (CDCI3) 8 8.0 (s, 1 H), 7.5 (d, 2H), 7.4 (m, 3H), 7.3 (d, 1H)
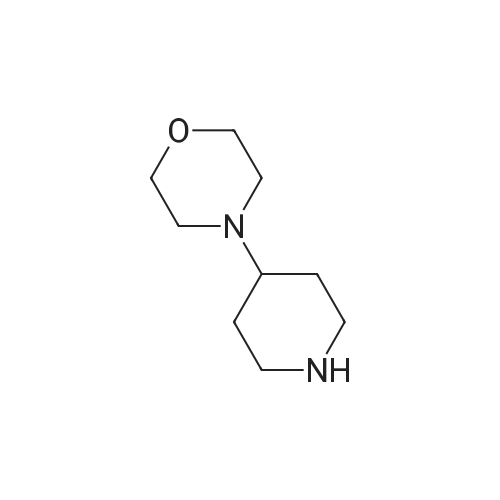
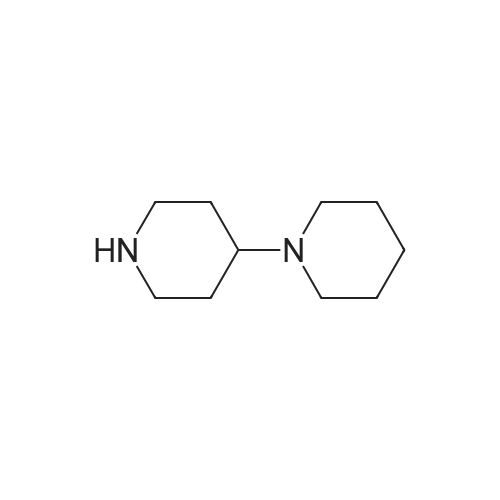
4-[(4-piperidin-1-yl)piperidin-1-yl]benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
81%
With potassium carbonate; In dimethyl sulfoxide; at 95℃; for 3h;
Potassium carbonate (4.14 g, 30 mmols) and (4-piperidin-1-yl)piperidine (6.05 g, 36 mmols) were added to dimethylsulfoxide solution (10 ml) of 4-fluorobenzonitrile (3.63 g, 30 mmols), and stirred at 95°C for 3 hours. The reaction mixture was cooled to room temperature, and poured into ice-water (300 ml) and stirred. The insoluble matter formed was taken out through filtration, and dried to obtain the entitled compound (635 g, 81 percent).
With potassium carbonate; In N,N-dimethyl-formamide; at 130℃; for 6h;
A mixture of <strong>[85310-69-6]pyrrolidin-3-ylmethanol</strong> (1.0 g, 9.9 mmol), 4-fluorobenzonitrile (1.2 g, 9.9 mmol), K2CO3 (2.0 g, 14 mmol), and DMF (10 mL) was heated at 130 0C for 6 h under N2 and then allowed to cool to it The reaction was concentrated, diluted with water (50 mL), and extracted with ethyl acetate (100 mL x 3). The organic extracts were washed with brine (100 mL), dried (MgSO4), filtered, and concentrated to give a brown oil. MS (ESI): 203.0 (M+Eta). Similar procedures as outlined in examples 26 & 1 were followed using the above alcohol. 1H NMR (500 MHz, DMSO) delta 7.85 (d, 2H), 7.26 (d, IH), 7.21 (d, IH), 6.90 (d, IH), 6.71 (d, 2H), 6.38 (d, IH), 4.05-3.96 (m, 2H), 3.90 (s, 2H), 3.58 (dd, IH), 3.51- 3.45 (m, IH), 3.40 (dd, IH), 3.29 (dd, IH), 2.88-2.79 (m, IH), 2.78 (t, 2H), 2.26-2.20 (m, EPO <DP n="47"/>IH), 2.00-1.94 (m, IH), 1.64-1.55 (m, 2H), 0.92 (s, 9H), 0.92 (t, 3H). MS (ESI): 473.1 (M+H).
With hydrogenchloride; n-butyllithium; diisopropylamine; In tetrahydrofuran;
EXAMPLE 163 3-(2-Dimethylaminomethyl-cyclopropyl)-1H-indazole-5-carbonitrile n-BuLi (1.9 M, 26.3 mL, 50 mmol) was added dropwise to a solution of diisopropylamine (7.71 mL, 55 mmol) in 100 mL anhydrous THF under N2 at 0 C. After 10 minutes, the reaction was cooled to -78 C. A solution of 4-fluorobenzonitrile (6.06 g, 50 mmol) in 20 mL anhydrous THF was added dropwise, at such a rate to maintain an internal temperature of -78 C. After stirring for 1 h at this temperature, trimethyl borate (8.41 mL, 75 mmol) was added dropwise at such a rate to maintain an internal temperature of -78 C. The reaction was stirred and gradually warmed to room temperature over 16 h. The reaction was cooled to 10 C. and 25 mL 6N HCl was added. After stirring at room temperature for 4 h, the reaction was partitioned between water and ethyl acetate. The organic layer was washed three times with 100 mL 2N NaOH. The aqueous layers were pooled and adjusted to pH 6 with 6N HCl. The white solid which forms was extracted by three washes of 100 mL Ethyl acetate. The organic layers were pooled, dried over sodium sulfate, concentrated and dried under high vacuum to give 2-fluoro-5-cyanophenyl boronic acid (5.74 g, 70%). 1H NMR (500 MHz, acetone-d6) 8.08 (1 H, dd, J=2.14, 5.5 Hz), 7.90 (1 H, m), 7.31 (1 H, t, J=8.85 Hz). Anal. calcd. for C7H5BFNO2: C, 50.97; H, 3.05; N, 8.49. Found: C, 51.19; H, 3.19; N, 8.26.
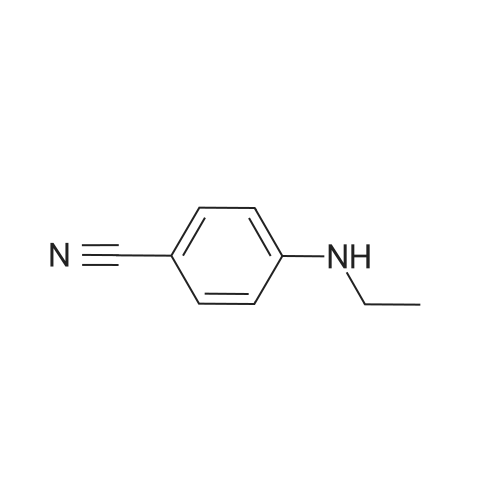
REFERENCE EXAMPLE 6 4-Ethylaminobenzonitrile By using the same method as described in Reference Example 1, the subject compound is synthesised from 4-fluorobenzonitrile and a 70%-ethylamine aqueous solution. 1H-NMR (CDCl3) delta (ppm): 1.27 (3H, t, J=7.3 Hz), 3.13-3.23 (2H, m), 4.22 (1H, br), 6.54 (2H, d, J=8.6 Hz), 7.40 (2H, d, J=8.6 Hz).
With n-butyllithium; acetic acid; diisopropylamine; In tetrahydrofuran; N-methyl-acetamide; hexane; water;
EXAMPLE 112 2-Fluoro-5-cyano-benzaldehyde To a stirred, cooled solution (0° C.) of diisopropylamine (15.4 mL, 0.11 mol) in anhydrous tetrahydrofuran (200 mL) n-butyllithium (40 mL of 2.5M in hexane, 0.11 mol) was added from a dropping funnel over a period of 30 min. under argon. The mixture was stirred at that temperature for 30 min. and then cooled to -78° C. A solution of 4-fluoro-benzonitrile (12.1g, 0.1 mol) in dry THF (50 mL) was then added dropwise over 15 min. via syringe and stirred for 1 hour at -78° C. Dimethylformamide (8 mL) was added dropwise from a syringe and the stirring was continued for another 20 min. The reaction was quenched by the rapid addition of acetic acid (20 mL) followed by water (500 mL) and the product was extracted with diethyl ether (2*500 mL). The combined organic layers were washed with 1N HCl, water, saturated sodium chloride and then dried over anhydrous magnesium sulfate, and evaporated to give 2-fluoro-5-cyano-benzaldehyde (11.8 g, 79percent yield) as a light yellow solid. 1H NMR (CDCl3): delta10.32 (s, 1H), 8.18 (dd, 1H, J=6.5, 2.5 Hz), 7.88 (m, 1H), 7.33 (t, 1H, J=9.5 Hz).
74.6%
With acetic acid; diisopropylamine;SiO2; In N-methyl-acetamide; ethyl acetate;
A. 4-Fluoro-3-Formylbenzenecarbonitrile Lithium diisopropyi amide (LDA) (22 mL, 49.56 mmol, 2.0 N commercial solution in heptanes) was added to tetrahydrofliran (50 mL), cooled to 78° C. and under nitrogen. 4-Fluorobenzonitrile was weighed out (5.0 g, 41.3 imnol), placed under nitrogen and dissolved in 25 mL of dry tetrahydrofliran. This solution was added dropwise to the solution of LDA. The resulting solution was stirred at -78° C. for one hour before quenching with 4 mL of dimethylformamide. The temperature was maintained for 10 min before adding 8 mL of acetic acid and 20 mL of distilled water. The crude product was extracted with ethyl acetate. Purification by column chromatography (SiO2, 20percent ethyl acetate in hexanes) afforded 4.6 g of pure product as a white solid (74.6percent yield). -A second batch of the title compound (3.5 g, 56.8 percent/yield) was prepared 20 using 5 g of benzonitrile (41.3 mmol): 1H NMR (CDCl3) delta 10.3 (s, 1H), 8.21 (dd, 1H), 7.91 (d of q, 1H), 7.35 (t, 1H); ES-MS M+was not detected.
With acetic acid; diisopropylamine; lithium diisopropyl amide;SiO2; In tetrahydrofuran; N-methyl-acetamide; ethyl acetate;
A. 4-Fluoro-3-formylbenzenecarbonitrile Lithium diisopropyl amide (LDA) (22 mL, 49.56 mmol, 2.0 N commercial solution in heptanes) was added to tetrahydrofuran (50 mL), cooled to 78° C. and under nitrogen. 4-Fluorobenzonitrile was weighed out (5.0 g, 41.3 mmol), placed under nitrogen and dissolved in 25 mL of dry tetrahydrofuran. This solution was added dropwise to the solution of LDA. The resulting solution was stirred at -78° C. for one hour before quenching with 4 mL of dimethylformamide. The temperature was maintained for 10 min before adding 8 mL of acetic acid and 20 mL of distilled water. The crude product was extracted with ethyl acetate. Purification by column chromatography (SiO2, 20percent ethyl acetate in hexanes) afforded 4.6 g of pure product as a white solid (74.6percent yield). A second batch of the title compound (3.5 g, 56.8percent yield) was prepared 20 using 5 g of benzonitrile (41.3 mmol): 1H NMR (CDCl3) delta 10.3 (s, 1H), 8.21 (dd, 1H), 7.91 (d of q, 1H), 7.35 (t, 1H); ES-MS M+ was not detected.
With acetic acid; diisopropylamine; lithium diisopropyl amide;SiO2; In tetrahydrofuran; N-methyl-acetamide; ethyl acetate;
A. 4-Fluoro-3-formylbenzenecarbonitrile Lithium diisopropyl amide (LDA) (22 mL, 49.56 mmol, 2.0 N commercial solution in heptanes) was added to tetrahydrofuran (50 mL), cooled to 78° C. and under nitrogen. 4-Fluorobenzonitrile was weighed out (5.0 g, 41.3 mmol), placed under nitrogen and dissolved in 25 mL of dry tetrahydrofuran. This solution was added dropwise to the solution of LDA. The resulting solution was stirred at -78° C. for one hour before quenching with 4 mL of dimethylformamide. The temperature was maintained for 10 min before adding 8 mL of acetic acid and 20 mL of distilled water. The crude product was extracted with ethyl acetate. Purification by column chromatography (SiO2, 20percent ethyl acetate in hexanes) afforded 4.6 g of pure product as a white solid (74.6percent yield). A second batch of the title compound (3.5 g, 56.8percent yield) was prepared 20 using 5 g of benzonitrile (41.3 mmol): 1H NMR (CDCl3) delta 10.3 (s, 1H), 8.21 (dd, 1H), 7.91 (d of q, 1H), 7.35 (t, 1H); ES-MS M+was not detected.
REFERENCE EXAMPLE 6 4-Ethylaminobenzonitrile According to a similar manner to that in Reference Example 1, the title compound was synthesised from 4-fluorobenzonitrile and 70% aqueous ethylamine solution. 1 H-NMR (CDCl3) delta (ppm): 1.27(3H, t, J=7.3 Hz), 3.13-3.23(2H, m), 4.22(1H, br), 6.54(2H, d, J=8.6 Hz), 7.40(2H, d, J=8.6 Hz).
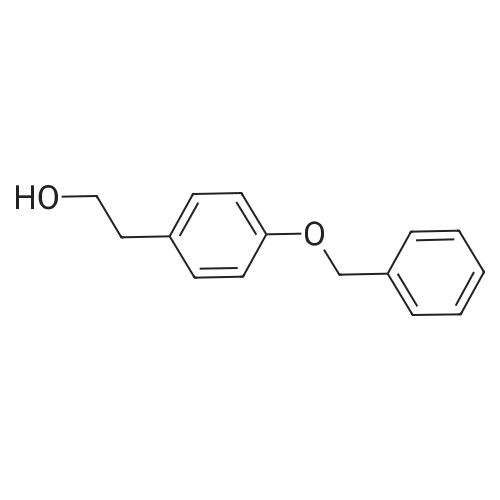
4-[(4-Benzyloxyphenyl)ethoxy]benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In tetrahydrofuran;
B. 4-[(4-Benzyloxyphenyl)ethoxy]benzonitrile To a suspension of sodium hydride (60percent, 2.2 g, 55 mmol) in dry tetrahydrofuran (250 ml) was added a solution of 1-benzyl-oxy-4-(2-hydroxyethyl)benzene (11.4 g, 50 mmol) in tetrahydrofuran (50 ml). The reaction was heated to 40° C. for 1 hour. 4-Fluorobenzonitrile (6.7 g, 55 mmol) was added and the mixture was heated to reflux for 5 hours, cooled and neutralized with concentrated HCl. The precipitate was filtered, the filtrate was concentrated to dryness and the resulting solid was recrystallized from ethanol (12 g).
(b) Alternatively, the product can be obtained as follows: To a mixture of imidazole (5.13 g, 75.35 mmol), K2 CO3 (11.45 g) and DMSO (50 ml) at room temperature was added to 4-fluorobenzonitrile (10.04 g) in one portion. The reaction mixture was stirred at room temperature for 1 hour, and then was warmed on a steam for 2 hours. The reaction mixture was cooled to room temperature, and poured into cold water. A precipitate formed which was collected by filtration and washed with water and recrystallized from CHCl3 /hexane to afford 4.07 g of 4-(1-imidazolyl)benzonitrile, m.p. 146°-148° C.
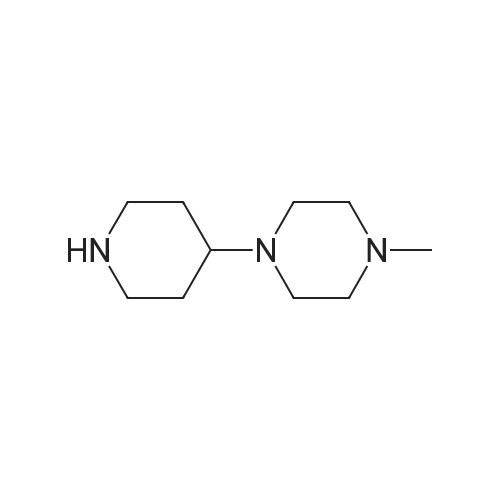
4-[4-(4-methylpiperazin-1-yl)piperidin-1yl]benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Starting Material Synthetic Example 161 In the same manner as in Starting Material Synthetic Example 68, the objective 4-[4-(4-methylpiperazin-1-yl)piperidin-1yl]benzonitrile is obtained from 4-fluorobenzonitrile and 4-(4-methylpiperazin-1-yl)piperidine.
Starting Material Synthetic Example 68 4-Fluorobenzonitrile (25.5 g), 4-methylhomopiperazine (26.2 ml) and potassium carbonate (28 g) were heated in DMSO for 15 hr. After heating, the reaction mixture was poured into water (1 L). The precipitated crystals were collected by filtration to give the objective 4-(4-methylhomopiperazin-1-yl)benzonitrile (34.6 g), melting point 82-84 C.
In N,N-dimethyl-formamide; at 90℃; for 36h;
4-(4-Methyl[1,4]diazepan-1-yl)benzamide 4-Fluorobenzonitrile (5 g, 41 mmol, 1 eq.) and 35 mL of dimethylformamide are placed in a 100 mL round-bottomed flask. N-Methylhomopiperazine (5.18 g, 45 mmol, 1.1 eq.) is added to this solution. The solution is heated at a temperature in the region of 90 C. for 36 hours and the solvent is then evaporated to dryness. The residue is diluted with 150 mL of ethyl acetate and 30 mL of water. The aqueous phase is extracted with ethyl acetate and the combined organic phases are dried over magnesium sulfate. After evaporation, the residual oil is purified by chromatography on a column of silica gel, eluding with a 0-10% methanol/dichloromethane gradient (Rf=0.2, 10/90 methanol/dichloromethane (v/v)). The 4-(4-methyl[1,4]diazepan-1-yl)benzonitrile (yellow solid) is placed directly in a mixture of 20 mL of 98% sulfuric acid and 2 mL of water. This orange solution is heated at a temperature in the region of 100 C. for 18 hours, and then cooled and hydrolyzed by pouring it onto ice. The pH is adjusted to 9-10 with sodium hydroxide pellets. The yellow precipitate obtained is filtered off and washed thoroughly with water and then dried under vacuum. 4-(4-Methyl[1,4]diazepan-1-yl)benzamide (3.7 g, 38%) is thus obtained in the form of a yellow solid, which is used directly in the following step.
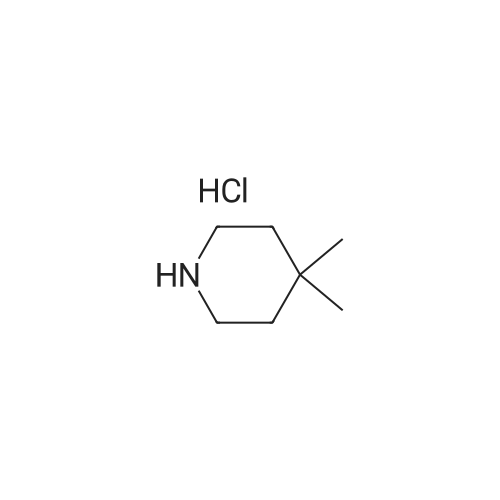
Starting Material Synthetic Example 76 In the same manner as in Starting Material Synthetic Example 68, the objective 4-(4,4-dimethylpiperidin-1-yl)benzonitrile (34.7 g), melting point 115-117 C., was obtained from 4-fluorobenzonitrile (23 g) and 4,4-dimethylpiperidine hydrochloride (28.5 g).
EXAMPLE 7; 8-fluoro-7-methoxydibenzo[b,d]furan-2-carbonitrile; A. 4-(4-fluoro-3-methoxyphenoxy)benzonitrile; This compound was prepared according to the method of Sawyer, et al. (Sawyer, J. Scott; Schmittling, Elisabeth A.; Palkowitz, Jayne A.; Smith, William J., III. Journal of Organic Chemistry (1998), 63(18), 6338-6343) from <strong>[117902-15-5]4-fluoro-3-methoxyphenol</strong> (Belanger, Patrice C.; Lau, C. K.; Williams, Haydn W. R.; Dufresne, C.; Scheigetz, John. Canadian Journal of Chemistry (1988), 66(6), 1479-82) (9.71 g, 68.3 mmol) and 4-fluorobenzonitrile (8.27 g, 68.3 mmol), which provided 4-(4-fluoro-3-methoxyphenoxy)benzonitrile (11.69 g, 70percent) as an orange oil.
Step A A mixture 4-fluorobenzonitrile (3 g, 25 mmol) and imidazolyl sodium (2.48 g, 27.5 mmol) in DMF (50 mL) was stirred at 80° C. under Ar for 12 h. Progress of reaction was monitored by TLC. The reaction mixture was concentrated in vacuo and the residue was diluted with 50 mL water and stirred. The aqueous mixture was extracted with EtOAc (2*50 mL). Combined EtOAc extracts was dried over anhydrous MgSO4, concentrated, and the 4-(1-imidazolyl)-benzonitrile was isolated by column chromatography (3.6 g, 78percent). LCMS: MH+=170; 1H NMR (CDCl3) delta 8.0 (s, 1H), 7.5 (d, 2H), 7.4 (m, 3H), 7.3 (d, 1H)
lambda/-(l-methylethyl)-2-propanamine, lithium salt (1 :1) (0.035 mol) was cooled to -78 0C. A solution of <strong>[23806-24-8]3-methyl-2-thiophenecarboxylic acid</strong> (2.5 g, 0.0175 mol) in THF (100 ml) was added drop wise and the mixture was stirred for 1 hour at 0 0C. Then the mixture was cooled to -78 0C and a solution of 4-fluorobenzonitrile (2.1 g, 0.0175 mol) in THF (100 ml) was added dropwise. The reaction mixture was stirred overnight at room temperature and then H2O was added dropwise. The mixture was extracted with DCM. The separated organic layer was washed with H2O, dried, filtered and the solvent was evaporated. The residue was stirred in DIPE. The precipitate was filtered off and dried. Yield: 1.9 g of intermediate 64.
With lithium diisopropyl amide; In tetrahydrofuran; n-heptane; ethylbenzene; at -78℃; for 2h;
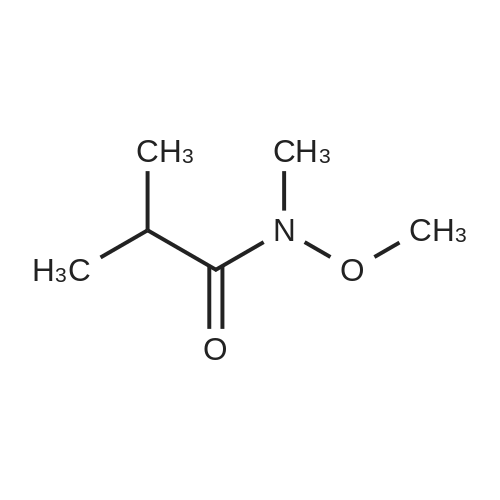
H. 4-Fluoro-3-(2-methylpropanoyl)benzenecarbonitrile. 4-Fluorobenzonitrile (2.31 g, 19.08 mmol) and N-methoxy-2-methyl-N-methylpropanamide (3 g, 22.9 mmol) were dissolved in THF (300 mL). The mixture was cooled to -78 C. and LDA (1.8 M in THF/heptane/ethylbenzene, 23.3 mL, 41.98 mmol) was added dropwise. The reaction was stirred at -78 C. for 2 hours and quenched by the addition of water. The reaction was extracted with ethyl acetate (3×100 mL), the organic layers combined and dried over magnesium sulfate. Solvent was removed under reduced pressure and the crude material was purified using column chromatography (SiO2, 8:2 n-Hexanes:ethyl acetate) to provide the title compound (1.39 g, 38%). 1H NMR (300 MHz, CD3OD) delta 8.14 (dd, J=6.59, 2.20, 1H), 7.97 (m, 1H), 7.44 (dd, J=10.44, 8.51, 1H), 3.40 (m, 1H), 1.17 (d, J=6.87, 6H).
60% sodium hydride (0.30 g) was added to a solution of the compound (1.66 g) obtained in Step 2 in anhydrous dimethylformamide (8 mL) under cooling with ice-water, and stirred at room temperature for thirty minutes under nitrogen atmosphere. 4-fluorobenzonitrile (0.91 g) was added to the mixture and stirred at room temperature for two days. The resulting mixture was poured into water (30 mL) and extracted with ethyl acetate. The organic layer was washed with brine and dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure. The obtained residue was purified by silica gel column chromatography (Chromatorex NH)(eluent; ethyl acetate : hexane = 1 : 1) to obtain the titled compound (2.2 g).
The compound prepared in the preceding step is solubilized in DMF, 8 g NaH is added and stirred at AT for 1 h, 20.6 g of 4-fluorobenzonitrile are added and stirring continued for 4 h at AT. After evaporation to dryness, the residue is redissolved in water, extracted with TBME, the organic phase is extracted with acid water (HCl), this aqueous phase is basified and the product is extracted with TBME. The final organic layer is dried over MgSO4 and evaporated in vacuo. 35.5 g of the desired product are obtained.
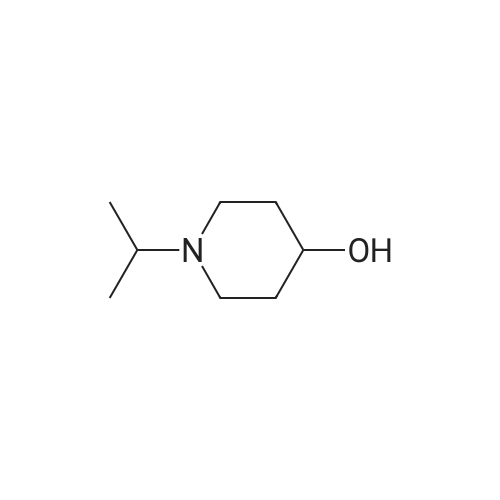
7.2 g of <strong>[5570-78-5]1-isopropyl-piperidin-4-ol</strong> obtained as described in Preparation 2, step A, are solubilized in DMF (20 ml), this solution is added to a suspension of NaH (3.4 eq) in DMF (50 ml) and stirred 1 h at AT, then 3-fluorobenzonitrile (1.2 eq) in 5 ml DMF is added and heated for 14 h at 60 C. The reaction medium is concentrated to dryness, the residue obtained redissolved in TBME, washed with water, the organic layer is dried over MgSO4, filtered and concentrated. 4.5 g of desired product are isolated in solid form after chromatography on silica eluting with a DCM/MeOH/NH4OH mixture (90:10:0.1 v/v/v).
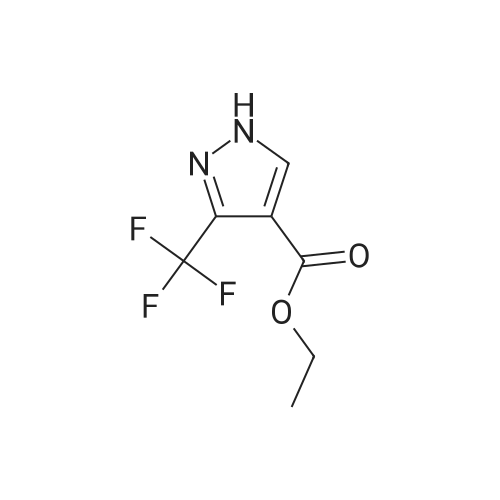
A solution of ethyl 3-trifluoromethylpyrazole-4-carboxylate (8.5 g, 41 mmol) in DMF (200 ml) was treated portionwise with sodium hydride (60% oil dispersion, 1.95 g, 48 mmol) and the reaction mixture stirred at room temperature under Ar for I h. 4-fluorobenzonitrile (5.3 g, 44 mmol) was added and the mixture stirred at 110C for 16h. After cooling to room temperature, the reaction was quenched with water, then concentrated in vacuo. The residue was purified by silica chromatography affording a white solid (11.1g, 88%). 1H NMR (400 MHz, CDCl3): delta (ppm): 8.56 (s, 1H), 7.90 (d, 2H), 7.84 (d, 2H), 4.37 (q, 2H), 1.39 (t, 3H).
With sodium carbonate; In N,N-dimethyl-formamide; for 24h;Reflux;
General procedure: A suitable phenol or alcohol (1Meq.), 4-fluorobenzonitrile (1Meq.) and Na2CO3 (2Meq.) were suspended in DMF (3.5mL/mmol) and heated. The reaction was followed by TLC, hexane:diethyl ether (9:1), and after completion (approximately 24h), the desired product extracted with CH2Cl2 (3×50mL). The crude product was purified by flash chromatography, using hexane:diethyl ether (100:1) as eluent.
80%
With caesium carbonate; In N,N-dimethyl-formamide; for 24h;Reflux;
General procedure: A DMF (40 mL) solution containing 4-fluorobenzonitrile (10) (1.0 equiv), m-substituted phenol (11?15) (1.1 equiv), and K2CO3or Cs2CO3(1.0 equiv) was stirred at reflux (24 h). The mixture was cooled, and then H2O (400 mL) was added and the reaction mixture extracted with CH2Cl2(3 x 100 mL). The organic layers were combined, washed with brine (3 x 100 mL), dried (Na2SO4), and evaporated under reduced pressure. The residue was purified by column chromatography using SiO2and 1:30 EtOAc and hexanes as the eluent giving the desired product as a colorless oil.
With caesium carbonate; In N,N-dimethyl-formamide; at 100℃; for 4h;
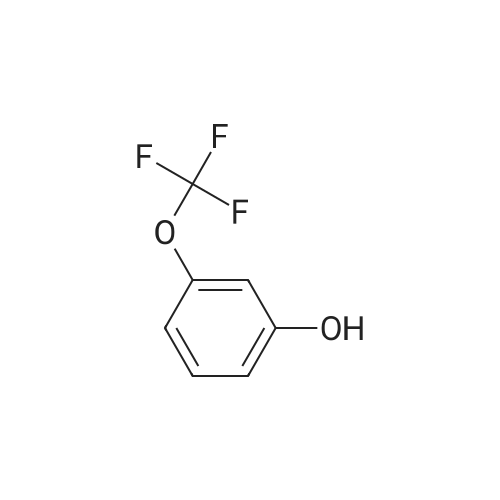
726 mg of 4-fluorobenzonitrile, 890 mg of <strong>[827-99-6]3-trifluoromethoxyphenol</strong>, and 1.95 g of cesium carbonate were added to 6 ml of DMF. The obtained mixture was stirred at 100°C for 4 hours. Thereafter, diluted hydrochloric acid was added to the reaction mixture, and it was then extracted with ethyl acetate. The organic layer was washed with a saturated saline solution, and it was then dried over magnesium sulfate. The resultant was concentrated by centrifugation under reduced pressure. Sodium hydroxide was added to the residue, and the mixture was then extracted with MTBE. The organic layer was washed with water. The resultant was dried over sodium sulfate, and it was then concentrated by centrifugation under reduced pressure, so as to obtain 1.32 g of 4-(3-trifluoromethoxyphenoxy)benzonitrile.
With sodium hydride; In N,N-dimethyl-formamide; mineral oil; at 60℃; for 6h;Inert atmosphere;
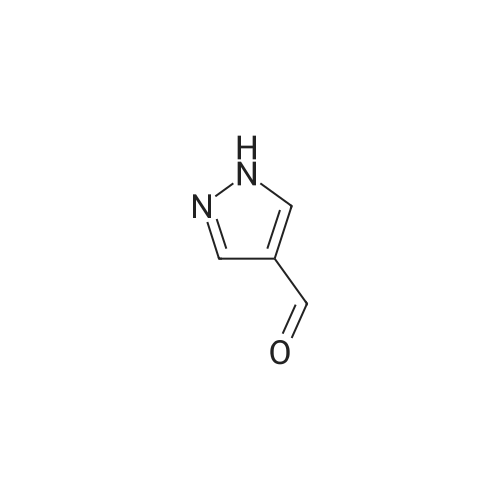
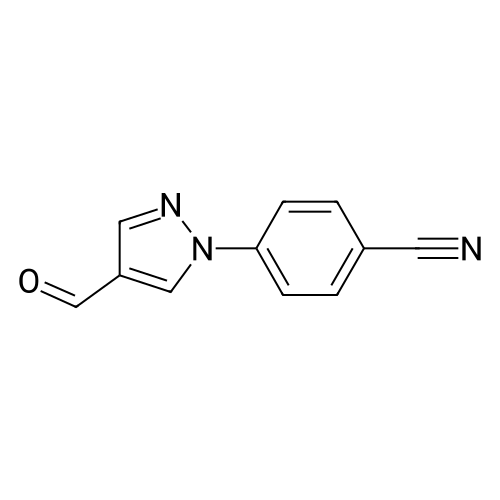
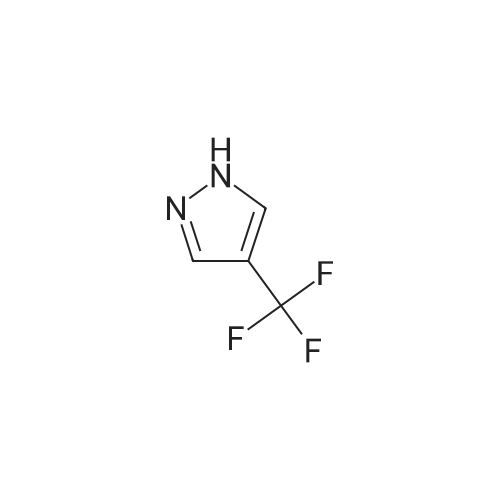
To a mixture of 4-fluorobenzonitrile (2.49 g, 20.6 mmol) and 4-formylpyrazole (1.98 g, 20.6 mmo) in 50 mL of DMF at 0 0C was added 95percent sodium hydride (0.54 g, 22.7 mmol) under nitrogen. The mixture was heated to 60 0C for 6 hours and cooled to room temperature, Water was added carefully and the mixture was then extracted with ethyl acetate. The combined extracts were washed with water and brine, and then dried over sodium sulfate, filtered, and concentrated in vacuo to provide crude 4~(4-formyMH-pyrazol-l-yl)benzonitrile.
A mixture of 4-fluoro-benzonitrile ( 121 mg, 1 mmol), ethylamine (90 mg , 2 mmol), and 2C03 (276 mg, 2 mmol) in DMSO ( 10 m was irradiated with microwave in a Biotage Smith synthesizer at 150 C for 20 min. After the reaction was completed as indicated by TLC, the mixture was filtered, and washed with water (2 x 50 mL) and ether (2 x 50 mL). The aqueous layer was separated and adjusted to about pH 1 with I N aq. HC1 solution. The aqueous layer was then extracted with ethyl acetate (3 x 60 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and evaporated to give the title compound(130 mg, 90 %).
With potassium carbonate; In dimethyl sulfoxide; at 100℃; for 18h;Inert atmosphere;
To a solution of <strong>[6294-93-5]4-chloro-3-(trifluoromethyl)phenol</strong> (31.5 g, 160 mmol) in DMSO (200 ml) was added K2CO3 (46.5 g, 337 mmol) at room temperature under nitrogen. After 5 minutes stirring, 4-fluorobenzonitrile (19.4 g, 160 mmol) was added to the mixture in one portion and the resulting mixture was stirred at 100° C. for 18 hours. The reaction mixture was allowed to cool to room temperature and poured into ice-cold 1N aqueous NaOH solution with vigorous stirring. The precipitated solid was collected by filtration and under vacuum to afford 50 g of the title compound.1H NMR (400 MHz; d6-DMSO): delta 7.21 (d, 2H), 7.36-7.48 (m, 1H), 7.60 (s, 1H), 7.75 (d, 1H), 7.80-7.89 (m, 2H).LCMS Rt=3.60 minutes MS m/z 298 [MH]+.
D57: 4- { [4-Chloro-3-(trifluoromethyl)phenyl] oxy}benzonitrile The mixture of <strong>[6294-93-5]4-chloro-3-(trifluoromethyl)phenol</strong> (2g, 10.18 mmol), K2C03 (2.109 g, 15.26 mmol) and 4-fluorobenzonitrile (1.232 g, 10.18 mmol) in DMF (20 ml), was heated at 120°C overnight. It was diluted with water and extracted with EtOAc. The organic phases were collected, washed with water and brine, dried over Na2S04 and concentrated to afford the title compound as a brown oil. LCMS: rt = 3.81 min, [M+H+] =298
Under a nitrogen atmosphere 362 mg (2.38 mmol) of CsF, previously activated with NaOH, were suspended in 5 ml of DMF and stirred for 30 min. Then, 1.00 g (8.26 mmol) of 4-fluorobenzonitrile was added. After 10 min 1.20 ml (8.18 mmol) N-trimethylsilylimidazole were added and the mixture was stirred at 60° C. for 20 hr. For workup, most of the solvent was removed under vacuum (oil pump), and then 5 ml of water and 5 ml of CH2Cl2 were added to the reaction mixture. The organic phase was separated, the aqueous phase was extracted with CH2Cl2, the organic phases were combined, washed several times with water and dried over MgSO4. The solvent was removed under vacuum, and the resulting solid was rinsed twice with pentane.Yield: 1.07 g (6.31 mmol, 77percent), appearance: colorless solid. 1H NMR (CDCl3, 25° C., 400.13 MHz): delta=7.25 (s, 1H, ImH), 7.33 (s, 1H, ImH), 7.51-7.53 (m, 2H, PhH), 7.79-7.81 (m, 2H, PhH), 7.94 (s, 1H, ImH). 13C NMR (CDCl3, 25° C., 100.61 MHz): delta=111.3 (C-1), 117.7 (C-9), 117.9 (-CN), 121.5 (C-3, C-5), 131.6 (C-8), 134.2 (C-2, C-6), 135.4 (C-7) 140.6 (C-4).
Intermediate (13): 4-(4-(trifluoromethyl)-1H-pyrazol-1-yl)benzonitrile To a 0 C. solution of <strong>[52222-73-8]4-(trifluoromethyl)-1H-pyrazole</strong> (1 g, 7 mmol) in N,N-dimethylformamide (10 mL) was added 60 wt % sodium hydride (132 mg, 3.31 mmol). The mixture was stirred at 0 C. for 30 minutes. 4-fluorobenzonitrile (979 mg, 8.08 mmol) was added and the reaction was heated to 80 C. overnight. Saturated ammonium chloride was added and the mixture extracted with ethyl acetate. The organic layer was dried over sodium sulfate, filtered and concentrated. Purification by column chromatography gave 4-(4-(trifluoromethyl)-1H-pyrazol-1-yl)benzonitrile (1.2 g, 72%). 1H NMR (400 MHz, CD3OD, delta): 8.84 (s, 1H), 7.96 (s, 1H), 7.95 (d, 2H), 7.78 (d, 2H).
With potassium carbonate; In dimethyl sulfoxide; at 100℃; for 6h;
4-Trifluoromethylphenyl imidazole (4.0 g, 19 mmol), 4-fluorobenzonitrile (1.2 g, 8.5 mmol) and potassium carbonate (1.5 g, 10.9 mmol) were combined in DMSO (15 mL) and heated at 100 0C for 6 h. The cooled solution was then poured onto water (H2O; 100 mL), and the resulting solid was filtered and air-dried to give the imidazole nitrile (4.65 g) as a white solid: mp 252 0C; 1H NMR (300 MHz, CDCl3) delta 8.05 (s, IH), 7.95 (d, J = 8 Hz, 2H), 7.85 (d, J = 8 Hz, 2H), 7.72 (s, IH), 7.72 (d, J = 8 Hz, 2H), 7.62 (d, J = 8 Hz, 2H); ESIMS m/z 314.1 (M+H). Anal. Calcd. for Ci6Hi0F3N3O2: C, 65.18; H, 3.22; N, 13.41. Found: C, 64.49; H, 3.23; N, 13.08. A portion of the nitrile (3.8 g) was reduced in the presence of DIBAL under conditions described previously to give the corresponding aldehyde (2.41 g): mp 141 0C; 1H NMR (300 MHz, CDCl3) delta 10.1 (s, IH), 8.10 (d, J = 8 Hz, 2H), 8.05 (s, IH), 7.95 (d, J= 8 Hz, 2H), 7.75 (s, IH), 7.7 (m, 4H); ESIMS m/z 317.1 (M+H).
With potassium carbonate; In dimethyl sulfoxide; at 100℃; for 6h;
Example 11 : Preparation of 4-[4-(4-trifluoromethylphenyl)-lH-imidazol-l-yl]-benzonitrile.4-Trifluoromethylphenyl imidazole (4.0 g, 19 mmol), 4-fluorobenzonitrile (1.2 g, 8.5 mmol) and potassium carbonate (1.5 g, 10.9 mmol) were combined in DMSO (15 mL) and heated at 100 0C for 6 h. The cooled solution was then poured onto water (100 mL), and the resulting solid was filtered and air-dried to give the imidazole nitrile (4.65 g) as a white solid: mp 252 0C; 1H NMR (300 MHz, CDCl3) delta 8.05 (s, IH), 7.95 (d, J= 8 Hz, 2H), 7.85 (d, 2 H), 7.72 (s, IH), 7.72 (d, J= 8 Hz, 2 H), 7.62 (d, J= 8 Hz, 2H); ESIMS m/z 314.1 (M+H). Anal. Calcd. for Ci6Hi0F3N3O2: C, 65.18; H, 3.22; N, 13.41. Found: C, 64.49; H, 3.23; N, 13.08.
With potassium carbonate; In dimethyl sulfoxide; at 110℃; for 48h;
To a solution of <strong>[37900-81-5]3-chloro-4-(trifluoromethyl)phenol</strong> (200 mg, 1 .02 mmol) in dimethylsulfoxide (7 mL) was added potassium carbonate (281 mg, 2.04 mmol) followed by 4-fluorobenzonitrile (123 mg, 1 .02 mmol). The reaction mixture was stirred at 1 10 °C for 48 hours. The reaction mixture was cooled to room temperature and diluted with ethyl acetate (25 ml_) and water (25 ml_). The organic phase was separated and washed with water (2 x 15 ml_), dried over anhydrous magnesium sulfate, filtered and evaporated in vacuo to obtain the title compound as a colourless gum (335 mg) which was used in the next step without further purification. 1H NMR (400 MHz, CDCI3): delta ppm 7.01 -7.07 (m, 2H), 7.15-7.19 (m, 1 H), 7.39 (d, 1 H), 7.53 (d, 1 H), 7.64-7.69 (m, 2H). LCMS Rt = 1 .80 minutes MS m/z mass ion not observed
With potassium carbonate; In N,N-dimethyl-formamide; at 100℃; for 5h;Inert atmosphere;
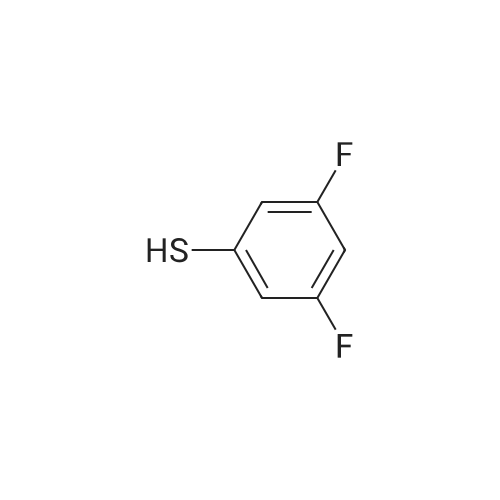
A mixture of 4-fluorobenzonitrile (1.5 g, 12.39 mmol, 1.00 equiv), <strong>[99389-26-1]3,5-difluorobenzene-1-thiol</strong> (2.17 g, 14.85 mmol, 1.20 equiv), and potassium carbonate (5.13 g, 37.12 mmol, 3.00 equiv) were combined in DMF (50 mL). The reaction mixture was stirred under nitrogen for 5 h at 100 C. The resulting solution was diluted with 150 mL of water and then extracted with 3x50 mL of ethyl acetate. The combined organic layers were washed with 3x100 mL of brine, dried over anhydrous sodium sulfate, and concentrated under vacuum. The residue was purified on a silica gel column eluted with ethyl acetate/petroleum ether (1 :5) to give 2 g (65%) of the title compound as a yellow solid. 1H NMR (300 MHz, CDCl3) delta 7.60-7.57(d, J = 8.7 Hz, 2H), 7.37-7.34 (d, J = 8.7 Hz, 2H), 7.04-6.89 (m, 2H), 6.84-6.77 (m, 1 H).
With lithium hydroxide monohydrate; silver trifluoromethanesulfonate; Selectfluor; In ethyl acetate; at 55℃;Sealed tube;
General procedure: Trifluoroborate (0.50 mmol, 1.0 equiv), LiOH·H2O (25.2 mg, 0.60 mmol, 1.2 equiv), Selectfluor (212 mg, 0.60 mmol, 1.2 equiv), AgOTf (386 mg, 1.5 mmol, 3.0 equiv) were weighed into a 20 mL microwave vial. EtOAc (5 mL) was added, and sealed with a microwave cap and the mixture was allowed to stir at 55 C for 5-15 h. The resulting solution was cooled to room temperature. For the compounds reported with isolated yields (2a, 2b, 2c, 2d, 2e, 2g, 2h, 2j, 2k, 2l, 2m, 2o, 2p, 2t, 2u, 2x, 2v, 2y, 2ab, 2ac, and 2ag) the reaction mixture was diluted with MTBE or hexane (5 mL) and H2O (4.0 mL). Then organic phase was separated, the aqueous phase was extracted with MTBE (2*5 mL). The combined organic phases were dried over anhydrous Na2SO4. The filtrate was concentrated in rotavapor and the residue was purified by column chromatography on Combiflash with hexanes/EtOAc to afford the desired compounds. The volatile and low yielding products were not isolated and their yields were determined only by 19F NMR of the reaction mixture. For the compounds reported with 19F NMR yields, 4-fluorobenzonitrile (0.50 mmol) was added as reference to the reaction mixture, stirred for 5 min, and then diluted with MTBE or hexane (5 mL) and H2O (3.0 mL). The layers were separated and an an aliquote of the organic phase was withdrawn for the 19F NMR measurement in CDCl3.
10%Spectr.
With iron(III) chloride; potassium fluoride; Selectfluor; In ethyl acetate; at 55℃;
General procedure: FeCl3 (60.8 mg, 0.375 mmol, 1.5 equiv), Selectfluor (177 mg, 0.5mmol, 2.0 equiv), and KF (43.6 mg, 0.75 mmol, 3.0 equiv) wereweighed into a 10-mL microwave vial. EtOAc (2.5 mL) was added, andthe vial was sealed with a septum. The mixture stirred at 25 C for 5min. Then, potassium aryltrifluoroborate (0.25 mmol, 1.0 equiv) wasadded to the mixture, and the vial was sealed with microwave capand the mixture stirred at 55 C for 10-15 h. The resulting solutionwas cooled to r.t.The volatile products were not isolated and their yields were determinedonly by 19F NMR of the reaction mixture. For the compoundsreported with 19F NMR yields, 4-fluorobenzonitrile (0.25 mmol) wasadded as reference to the mixture, stirred for 5 min, and then dilutedwith t-BuOMe or hexane (2.5 mL) and H2O (3.0 mL). The layers wereseparated and an organic aliquot was withdrawn for the 19F NMRmeasurement in either in CDCl3 or DMSO-d6. The 19F NMR spectroscopicdata were identical to those reported previously in the literature.The identity of the product was further confirmed by GC-MSanalysis. For the compounds reported as isolated yields, the mixture was dilutedwith t-BuOMe or hexane (2.5 mL) and H2O (4.0 mL). Then organicphase was separated, the aqueous phase was extracted with t-BuOMe(2 × 5 mL). The combined organic phases were dried (anhyd Na2SO4),the solvent was removed at 1.0 bar and the residue was purified bycolumn chromatography (Combiflash, hexanes) to afford the desiredcompounds. The identity of the product was confirmed by 1H NMRand GC-MS analyses.
With potassium carbonate; In dimethyl sulfoxide; at 80℃; for 8h;
General procedure: To a stirred solution of 3 (4, 5) (0.10 mmol) in DMSO (10 ml) were added ZH (0.12 mmol) and K2CO3 (27.6 mg 0.20 mmol). After stirring at 80 °C for 8 hours, the mixture was cooled to room temperature and poured into water, the precipitation was filtered and dried directly for next step. To a solution of above solid (0.10 mmol) in dry THF (10 ml) was added LiAlH4 (11.4 mg, 0.30 mmol) at 0 °C. After stirring at room temperature for 4 h, the mixture was quenched with water and extracted by CH2Cl2 (10 ml). The extraction was dried over anhydrous MgSO4 and filtered. The filtration was concentrated for next step. To a solution of above crude solid (0.12 mmol) in dry CH2Cl2 (10 ml) were added compound 6 (7-11) (0.10 mmol), TEA (22.2mg, 0.22 mmol) and BopCl (30.4 mg, 0.12 mmol). After stirring at room temperature for 12 h, the mixture was washed with brine and dried over anhydrous MgSO4, filtered and concentrated. The residue was purified by silica gel column (CH2Cl2 : MeOH = 100 : 1) to yield compounds 1, 2, A, B, C.
tert-butyl 4-[5-(4-fluorophenyl)-4H-1,2,4-triazol-3-yl]piperidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
49.6%
With potassium carbonate; In butan-1-ol; at 150℃; for 3h;
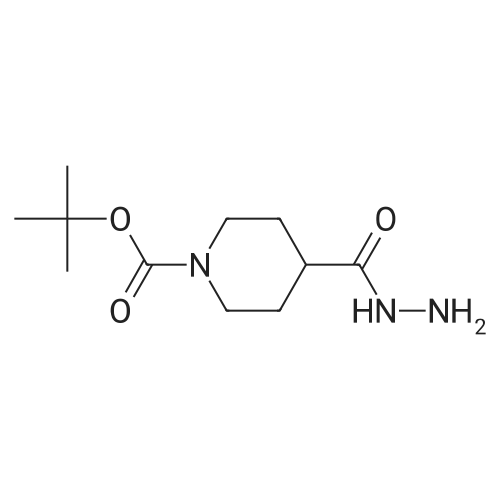
In a microwave vial, tert-butyl 4-(hydrazinecarbonyl) piperidine-1-carboxylate 9 (0.3 g, 3 mmol), 4-fluorobenzonitrile 10 (0.448 g, 4 mmol), 1-butanol (3.385 mL), and K2CO3 (0.085 g, 0.6 mmol) were combined and heated at 150 C. for 3 hours. The reaction mixture was allowed to cool, and diluted with water. The reaction mixture was extracted with ethyl acetate three times. The combined organic layers were washed with brine, dried over sodium sulfate and evaporated under reduced pressure. The crude residue was purified via flash chromatography on silica gel with 0-30% MeOH in dichloromethane (DCM) as an eluent. The product 11 (0.223 g, 49.6%) was isolated as a light yellow oil. MS ESI [M+H+]+=347.2.
With water; In aq. phosphate buffer; at 25℃; for 6h;pH 2.7 - 3.2;Electrochemical reaction;
General procedure: A conventional two compartment glass or PTFE H-cell separated by a Nafion-117 membrane was used for electrochemical HDF experiments. The cathode was fabricated from a piece of Rh/Ni foam, Rh/Agmesh, Rh/Cu foam, or Rh/carbon felt (projected area: 2 × 3 cm2), whereas the anode was fashioned from a graphite sheet (2 × 3 cm2). 30 mL phosphate buffer solutions (20 mM) served as both the catholyte and the anolyte. Unless otherwise noted, the catholyte was stirred with a stirring velocity of 350 rpm during electrolysis. The concentrations of reactants and their products were determined by an Agilent 7890 A gas chromatograph (GC) and a Waters HPLC system, using standard calibration curves. The fluoride ion (F-) concentration was determined by a REX PHSJ-4F pH/mV meter combined with F- selective electrode (REXPF-202-C). The specific electric energy consumption (SEEC, kW h m-3catholyte) of the HDF process was calculated from the following equation: SEEC I *U*t/1000*V where I is the applied current (A), U is the average cell voltage (3 V), t is HDF time (h), and V is the volume of the catholyte (3 × 10-5 m3). Unless otherwise noted, all of the above-mentioned experimentswere performed under air atmosphere, at 25C.
With potassium carbonate; In dimethyl sulfoxide; at 100℃;
A mixture of compound 3 (300 mg, 1.7 mmoi), 4-fluorobenzonitriie (181 mg,1.5 mrnol). and K2C03 (414 mg. 3 mmoi) in DM50 (10 ml) was stirred at 100C overnight.To the mixture was added EA (lOOmI) and aqueous saturated citric acid (30 ml), and the resulting reaction mixture was stirred for 30 nun. The organic layer was separated, washed by aqueous saiurated NaCI (50 ml*2). dried and concentrated, and purified by silica gel column to yield compound 4 (300 mg, 72%) as a yellow solid.
With potassium carbonate; In dimethyl sulfoxide; at 120℃; for 6h;
A mixture of the resulting product and anhydrous K2CO3 (1.57g, 11.34mmol) in DMSO (23mL) was added 4-florobenzonitrile (1.14g, 9.45mmol). The reaction was stirred at 120C for 6h. After the reaction was finished, the mixture was poured into 100mL water with stirring and the precipitated solid was filtrated for further purification through flash column chromatography to afford the title compound 8e (0.95g, 69.23%). 1H NMR (400MHz, CDCl3) delta (ppm) 7.57 (d, J=8.4Hz, 2H), 7.15-7.17 (m, 4H), 6.92 (d, J=8.8Hz, 2H), 4.57 (m, 1H), 3.62-3.67 (m, 2H), 3.34-3.39 (m, 2H), 2.26 (m, 2H), 2.01 (m, 2H). MS (ESI) m/z 363 [M+H]+.
tert-butyl 2-(4-(aminomethyl)phenyl)-2,6-dihydropyrrolo[3,4-c]pyrazole-5(4H)-carboxylate[ No CAS ]
tert-butyl 1-(4-(aminomethyl)phenyl)-4,6-dihydropyrrolo[3,4-c]pyrazole-5(1H)-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
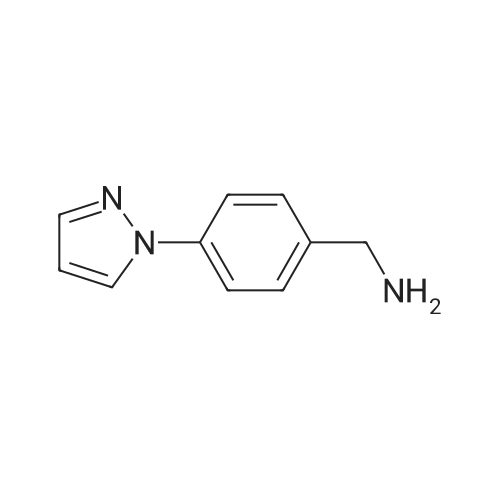
Step 1. 4-(lH-Pyrazol-l-yl)benzonitrile (0847) [00269] A mixture of lH-pyrazole (15 g, 220.34 mmol), 4-fluorobenzonitrile (27 g, 222.93 mmol), potassium carbonate (60.7 g, 439.19 mmol) in DMF (200 mL) was stirred for 16 h at 110 C. After cooling to ambient temperature, the reaction mixture was poured into water (500 mL) and the resulting solids were collected by filtration and dried under vacuum, resulting in 30 g (80%) of 4-(lH-pyrazol-l-yl)benzonitrile. MS (ESI) m/z 170 [M+H]+. Step 2. (4-(lH-Pyrazol-l-yl)phenyl)methanamine (0849) [00270] A mixture of 4-(lH-pyrazol-l-yl)benzonitrile (15 g, 88.66 mmol), Raney nickel (10 g), palladium on carbon (10 wt. %, 1 g) and lithium hydroxide (1 g, 41.75 mmol) in EtOAc (200 mL) was evacuated and back-filled with hydrogen several times and then charged with hydrogen. The resulting mixture was stirred for 16 h at ambient temperature, then was filtered and concentrated under vacuum. The residue was purified by silica gel chromatography (eluting with a gradient of 1-10% DCM/MeOH) resulting in 8 g (52%) of (4-(lH-pyrazol-l- yl)phenyl)methanamine. MS (ESI) m/z 174 [M+H]+.
tert-butyl 2-(4-fluorophenyl)-4-hydroxy-5-(4-nitrophenyl)-1H-pyrrole-3-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
General procedure: To a stirred suspension of commercial zinc dust (1.26 g, 19.5 mmol) was added methanesulfonic acid (3.7 mg, 0.37mmol) in anhydrous THF(20 mL). After 10 min of reflux, benzonitrile (1.0 g, 9.7 mmol) was added all at once. Whilemaintaining reflux temperature, ethyl bromoacetate (2.43 g, 14.5 mmol) was added over 1 h with use of a syringepump, and the reaction mixture was further heated at reflux for 1h. The reaction mixture was cooled to roomtemperature, and then arylglyoxals (1.62 g, 10.6 mmol) was added. After being stirred for 0.5h at r t, the reactionmixture was quenched with saturated aqueous NH4Cl at room temperature and extracted with ethyl acetate (3×30mL). The combined organic layer was dried with anhydrous MgSO4, filtered, and concentrated under reducedpressure. The residue was purified by column chromatography (EtOAc/Hexane, 1:7 v/v) to afford product 4.
(S)-4-(3-(methylamino)-1-(thiophen-2-yl)propoxy)benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
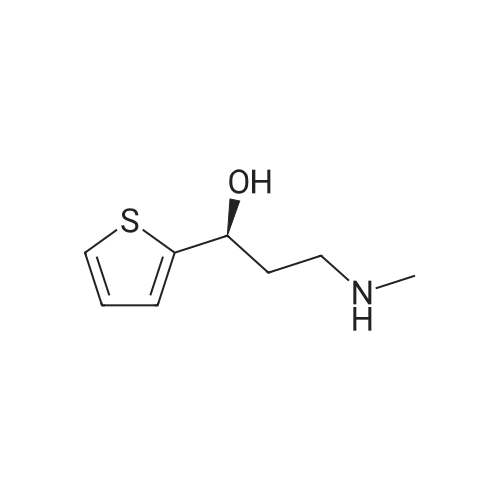
To a solution of (S)-3-(methylamino)-1-(thiophen-2-yl)propan-1 -ol (1 .6 g, 9.34 mmol) in DMA (20 mL), NaH (561 mg, 60% suspension in mineral oil, 14.01 mmol) was added and the solution was stirred at rt for 30 min. A solution of 4-fluorobenzonitrile (2.26 g, 18.69 mmol) in DMA (10 mL) was added and the mixture was heated at 90 C for 2 h. Water was added and extracted with EtOAc. The organic layer was dried with Na2S04 and the solvent was removed under vacuum to afford the title compound, that was used in the next step without further purification. HPLC-MS (Method B): Ret, 3.64 min; ESI+-MS m/z, 273.1 (M+H).
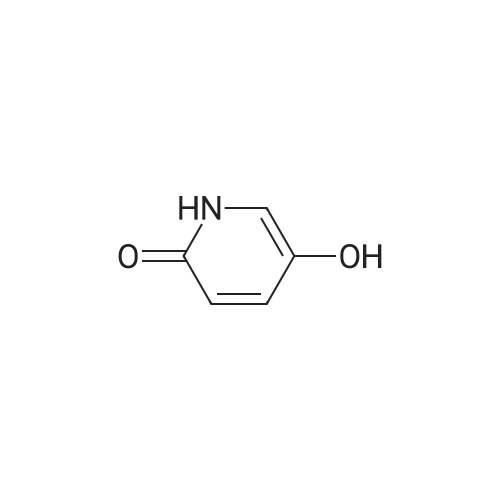
4-((6-oxo-1,6-dihydropyridin-3-yl)oxy)benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
49.7%
With potassium carbonate; In N,N-dimethyl-formamide; at 110℃;
A 50 mL flask was charged with <strong>[5154-01-8]pyridine-2,5-diol</strong> (III) (2.0 g, 18.00 mmol), 4-fluorobenzonitrile (2.18 g, 18.00 mmol), potassium carbonate (4.98 g, 36.0 mmol) and DMF (25.7 mL). The reaction was heated at 110 C overnight, and then allowed to cool to room temperature. The reaction was quenched with water (50 mL) and diluted with ethyl acetate.The layers were separated and the aqueous layer was extracted with ethyl acetate. Thecombined organic layers were washed with brine (3x). The organic layer was then dried over anhydrous sodium sulfate, filtered, and concentrated. DCM was added resulting in a brown precipitate which was collected via vacuum filtration. The wetcake was dried to give 4-((6- oxo-1,6-dihydropyridin-3-yl)oxy)benzonitrile (II) as a tan solid (2 g, 8.95 mmol, 49.7%yield). mp: 223 -226 C. ?H NMR (500 MHz, DMSO-d6) 11.66 (s, 1H), 7.86-7.76 (m,2H), 7.57 (d, J = 3.2 Hz, 1H), 7.43 (dd, J = 9.7, 3.2 Hz, 1H), 7.20 - 6.99 (m, 2H), 6.49 (d, J =9.7 Hz, 1H). ?3C NMR (126 MHz, DMSO-d6) 162.3, 161.7, 137.1, 136.9, 135.0, 129.8,120.2, 119.2, 117.2, 105.2. HRMS-ESI (mlz) [M+Hj calcd for C,2H8N202, 213.0659; found,213.0653.
With potassium tert-butylate; In dimethyl sulfoxide; at 150℃; for 0.416667h;Cooling with ice; Microwave irradiation;
To the DMSO (1 mL)solution of <strong>[6358-06-1]5-amino-2-chlorophenol</strong> (0.25 g, 1.75 mmol), t-BuOK (0.245 g, 2.19 mmol) was added at ice-bath. 4-fluorobenzonitrile(0.176, 1.45 mmol) were added to the solution and stirred at room temperature for 5 minutes and thenheated to 150°C for 20 minutes by microwave machine. Ice water was added, and the suspension was extracted withEthyl acetate (3x100 mL). The collected organic phases were washed with brine, dried over MgSO4 and filtered. Thefiltrate was concentrated and then purified with chromatography to obtain the intermediate of 4-(5-amino-2-chlorophenoxy)benzonitrile.4-(5-amino-2-chlorophenoxy)benzonitrile: 1H NMR (400 MHz, MeOD): delta 7.68 (d, J = 8.8 Hz, 2H), 7.18 (d, J = 8.8 Hz,1H), 6.99 (d, J = 8.8 Hz, 2H), 6.58 (dd, J = 8.4 Hz, 2.8 Hz, 1H), 6.49 (d, J = 2.8 Hz, 1H).
With potassium carbonate; In dimethyl sulfoxide; at 120℃; for 0.5h;Microwave irradiation;
A suspension of <strong>[896464-16-7]tert-butyl-2,7-diazaspiro[3.5]nonane-7-carboxylate</strong> (CAS [896464-16-7], 2.6 g, 11.49 mmol), 4-Fluorobenzonitrile ( CAS [1194-02-1], 2.78 g, 22.98 mmol) and potassium carbonate (7.94 g, 57.44 mmol) in DMSO (40 mL) was heated at 120 C using a single mode microwave (Biotage Initiator60) with a power output ranging from 0 to 400 W for 30 min [fixed hold time]. The mixture was diluted in EtOAc, washed with water (3x), brine (3x), dried over MgS04, filtered off and evaporated. The crude product was purified by preparative LC (irregular silica, 15-40 muiotaeta, 24 g, Grace,liquid loading, mobile phase gradient: Heptane/EtOAc from 90/10 to 80/20). The fractions containing product were combined and the solvent was removed in vacuo to give 2.9 g of intermediate A as a white solid (77%).
With potassium carbonate; In N,N-dimethyl-formamide; at 110℃; for 12h;
Add to the round bottom flask with mechanical stirringA 161g nitrile fluorophenyl,2-hydroxymethyl-5-hydroxybenzeneboronic acid half ester 200g,Potassium carbonate 221g,N,N-dimethylformamide 2000mL,Raise the temperature to 110 C for 12 h,HPLC monitors the progress of the reaction,After the raw materials are completely converted,Filter to remove solids,The filtrate was poured into ice water and quenched.Extracted with ethyl acetate,Dry with anhydrous sodium sulfate,Concentrated to a crude product of gramboroline,Then beat with n-hexane, filter,Dry the gram of Bora.
76.9%
With potassium carbonate; In dimethyl sulfoxide; at 60 - 70℃; for 4h;Inert atmosphere;
2.1 g (17.3 mmol) of p-fluorobenzonitrile was added to the reaction flask. 2g (13.3mmol) benzo[c][1,2]oxaborol-1,5(3H)-diol (VI), 5.5g K2CO3 (40mmol), 20mL DMSO, nitrogen, The reaction mixture was heated to 60-70 C for 4 h, and quenched with water. The mixture was stirred with EtOAc EtOAc. The liquid was added to 10 mL of dichloromethane, and 5 mL of n-heptane was stirred and crystallized at room temperature to obtain 2.5 g of an off-white solid, which was 5-(4-cyanophenoxy)-1,3-dihydro-1-hydroxy-2,1- Benzooxaborole (I), molar yield 76.9%.
With potassium carbonate; In dimethyl sulfoxide; at 80℃; for 4h;
General procedure: To a stirred solution of compound 1 (121.0 mg, 1.0mmol) and2a-e (1.0 mmol) in anhydrous DMSO (10 mL) was added anhydrous K2CO3 (165.6 mg, 1.2 mmol). After stirring 4 h at 80 C, the reactionwas poured into ice, the precipitationwas filtrated to obtain 3a-e ina yield of 45%-75%.
With potassium carbonate; In dimethyl sulfoxide; at 80℃; for 4h;
General procedure: To a stirred solution of compound 1 (121.0 mg, 1.0mmol) and2a-e (1.0 mmol) in anhydrous DMSO (10 mL) was added anhydrous K2CO3 (165.6 mg, 1.2 mmol). After stirring 4 h at 80 C, the reactionwas poured into ice, the precipitationwas filtrated to obtain 3a-e ina yield of 45%-75%.
With potassium carbonate; In N,N-dimethyl-formamide; at 80 - 90℃;
2-bromo-5-hydroxybenzyl alcohol II (20.3 g, 0.1 mol) was sequentially added to a three-necked flask.N,N-dimethylformamide (50 mL), potassium carbonate (27.6 g, 0.2 mol)After the addition of p-fluorobenzonitrile (14.5 g, 0.12 mol), the reaction system was warmed to 80-90 C, and the reaction was completed by TLC. The mixture was slowly added to water (250 mL) and quenched.Filtration, drying to give a white solid, intermediate III, HPLC purity 93.5%, yield 96.2%.
54%
With potassium carbonate; In dimethyl sulfoxide; at 125℃; for 4h;Inert atmosphere;
27.2 g (197 mmol) of K2CO3, 11.9 g (98.5 mmol) of 4-fluorobenzonitrile of formula (IV), 20.0 g (98.5 mmol) of 2-bromo-5-hydroxy-benzyl alcohol of formula (III), obtained as described in Example 1, and 80 mL of DMSO are placed under nitrogen in a previously anhydrified flask. The mixture is heated to 125 C. and maintained at said temperature for 4 hours. 240 mL of H2O are added and the precipitate is filtered off and washed with H2O until reaching a neutral pH value. The solid is dried and analyzed by HPLC (yield of 70%). The mixture is then dissolved in 70 mL hot toluene and allowed to crystallize while cooling the solution down to room temperature obtaining 16.3 g of 4-(4-bromo-3-hydroxymethyl-phenoxy)-benzonitrile of formula (II) with a yield of 54%. 1H-NMR (DMSO-d6, 300 MHz) delta (ppm): 7.85-7.80 (2H, m) 7.62 (1H, d, J=8.4 Hz), 7.22 (1H, d, J=3.0 Hz), 7.13-7.11 (2H, m) 6.98 (1H, dd, J=3.0, J=8.4 Hz), 4.47 (2H, s).
1-bromo-4-trimethylsilanoxy-2-trimethylsilanoxymethylbenzene[ No CAS ]
[ 906673-45-8 ]
Yield
Reaction Conditions
Operation in experiment
19.7 g
With potassium carbonate; In dimethyl sulfoxide; at 105℃; for 2h;Inert atmosphere;
27.1 g (197 mmol) of K2CO3, 12.13 g (100.3 mmol) of 4-fluorobenzonitrile of formula (IV), 34.2 g (98.3 mmol) of 1-bromo-4-trimethylsilanoxy-2-trimethylsilanoxymethyl benzene of formula (III), obtained as described in Example 2, and 60 mL of DMSO are placed under nitrogen in a previously anhydrified flask. The reaction mixture is heated to 105 C. and kept at the same temperature for 2 hours. Then, 120 mL of toluene and 240 mL of H2O are added, the phases are separated and the organic phase is washed with a solution of 10% K2CO3. The organic phase is allowed to cool down to room temperature and the product is allowed to crystallize providing 19.7 g 4-(4-bromo-3-hydroxymethyl-phenoxy)-benzonitrile of formula (II) with a yield of 66%. 1H-NMR (DMSO-d6, 300 MHz) delta (ppm): 7.85-7.80 (2H, m) 7.62 (1H, d, J=8.4 Hz), 7.22 (1H, d, J=3.0 Hz), 7.13-7.11 (2H, m) 6.98 (1H, dd, J=3.0, J=8.4 Hz), 4.47 (2H, s)






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +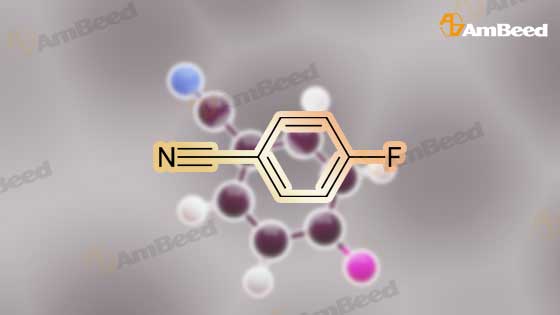

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping