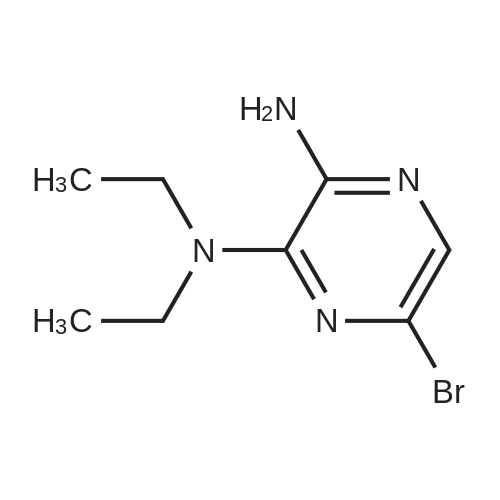
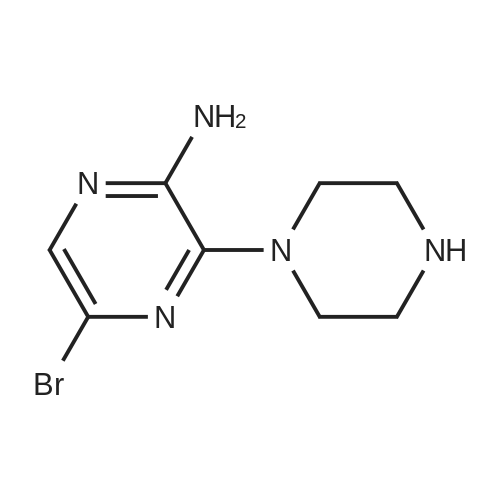
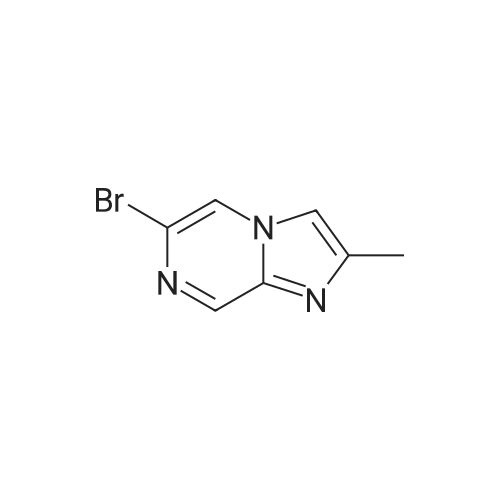
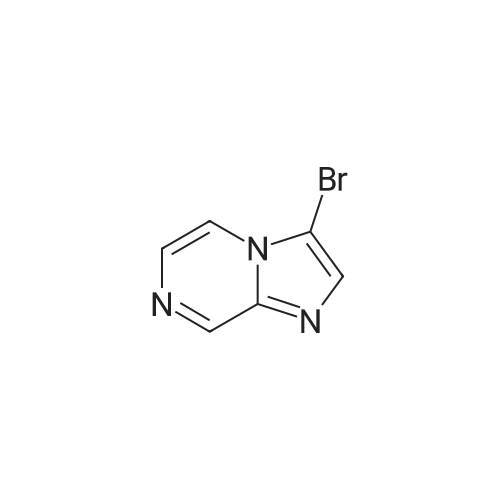
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
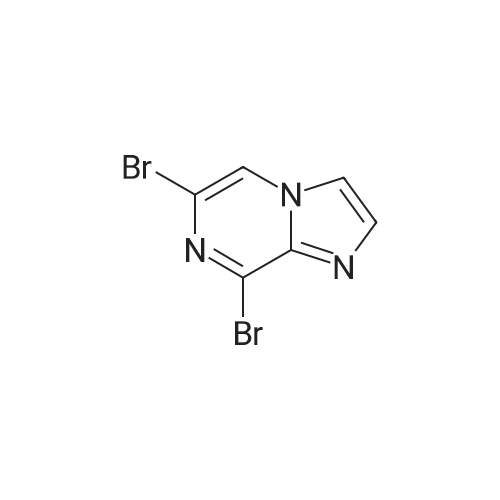
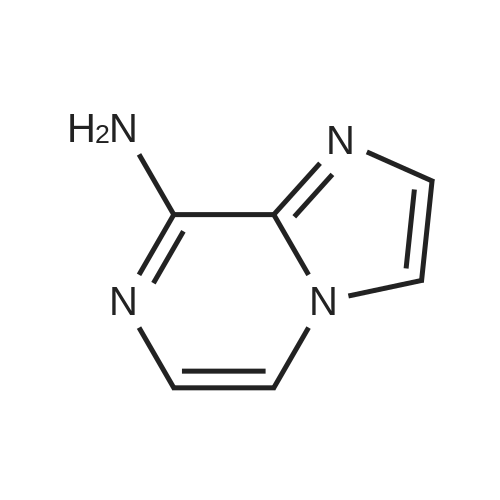
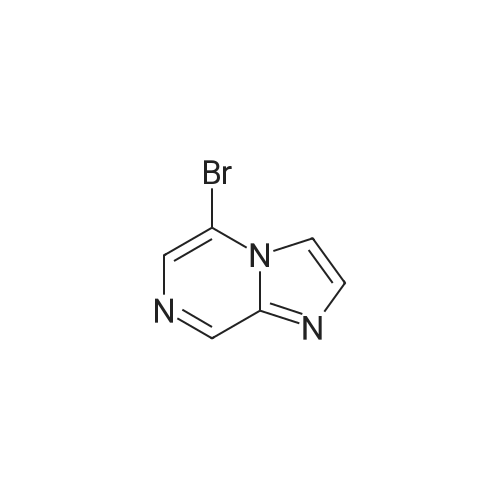
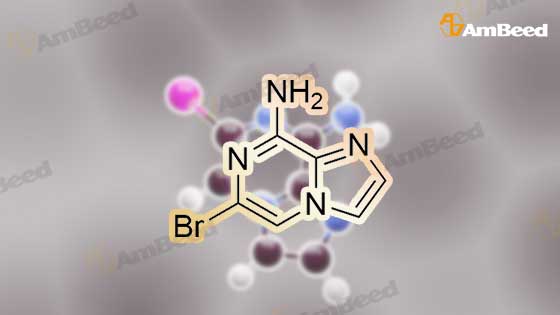
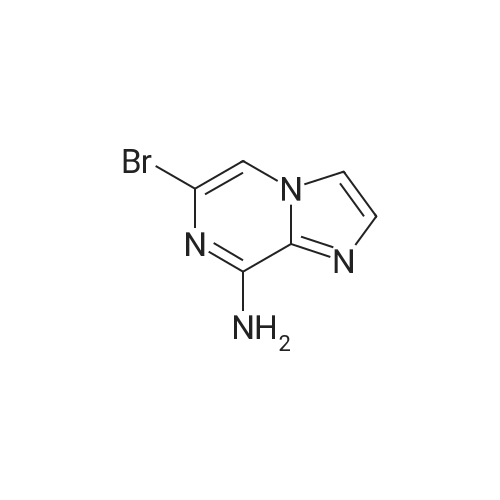
A mixture of the product from Preparative Example 29 (1.80 g, 6.45 mmol) and concentrated aqueous NH4OH (27.0 mL) was stirred in a closed pressure vessel at 90° C. for 24 hr. The solvent was evaporated and the residue was purified by column chromatography on silica gel with EtOAc. White solid (1.01 g, 73percent) was obtained. LCMS: MH+=213.
Reference:
[1] Journal of Medicinal Chemistry, 1992, vol. 35, # 18, p. 3353 - 3358
[2] Patent: US2004/63715, 2004, A1, . Location in patent: Page/Page column 27
[3] Patent: WO2004/22562, 2004, A1, . Location in patent: Page 28
A mixture of the product from Preparative Example 29 (1.80 g, 6.45 mmol) and concentrated aqueous NH4OH (27.0 mL) was stirred in a closed pressure vessel at 90 C. for 24 hr. The solvent was evaporated and the residue was purified by column chromatography on silica gel with EtOAc. White solid (1.01 g, 73%) was obtained. LCMS: MH+=213.
With ammonia; In water; at 80 - 90℃; for 24.0h;
[8-AMINO-6-BROMOIMIDAZO [1,] 2-a] pyrazine (4). Procedure 1: A mixture of 1.00 eq. of 6, 8-imidazo [1, 2-a] pyrazine 3 in 28 % [AMMONIA/WATER] solution or 40% aqueous methyl amine is heated to between 80 to [90C] for 24 hr. The resulting mixture is partitioned between CH2C12 and [H20.] The aqueous layer is extracted with [CH2C12] and the combined organic extracts are dried over [NA2S04.] The solvent is removed under reduced pressure and the resulting residue is crystallized from ethanol to yield 4.
2.21 g
With ammonium hydroxide; at 90℃; for 8.0h;
A solution of 6,8-dibromoimidazo[1 ,2-a]pyrazine (2 g, 7.22 mmol) in ammonium hydroxide so-lution (28-30%, l5mL) was heated at 9000 for 8h then the reaction mixture was concentratedunder reduced pressure to afford the title product as a light yellow solid (2.21 g) that was used without further purification. ESI-MS: 213.15/215.15 [M+H]+.
6-phenyl-imidazo[1,2-a]pyrazin-8-ylamine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; water; for 24.0h;Heating / reflux;
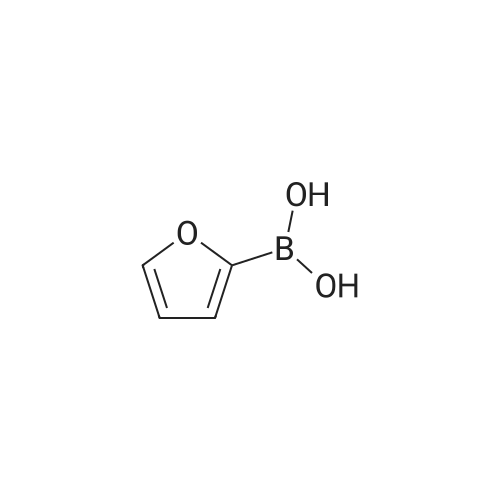
A mixture of the product from Preparative Example 30 (500 mg, 2.36 mmol), phenyl boronic acid (431 mg, 3.53 mmol), Pd(PPh3)4 (277 mg, 0.24 mmol), and Na2CO3 (2.50 g, 23.6 mmol) in 1,2-dimethoxyethane (30 mL) and H2O (8 mL) was stirred and refluxed under N2 for 24 hr. The mixture was poured into H2O (500 mL), extracted with CH2Cl2 (4×50 mL) and the extracts were dried over Na2SO4 and filtered. The solvent was evaporated and the residue was purified by column chromatography on silica gel with PhCH3/7N NH3 in MeOH (10:1). This afforded a slightly impure product as a pale orange solid, which was used for the next step.
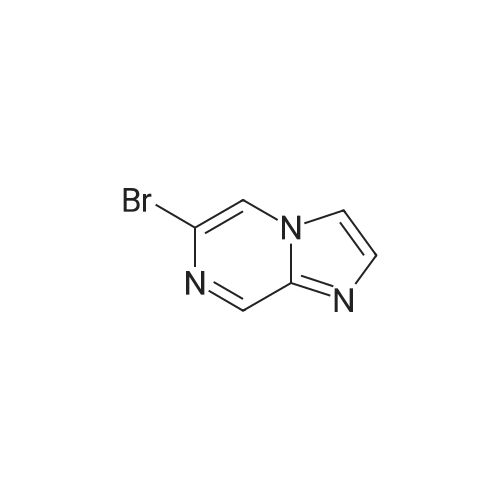
Step 1 : Preparation of 6-bromo-8-[bis[(l,l -dimethylethoxy)carbonyl]amino]imidazo[l,2- a]pyrazine6-bromo-8-[bis[(l , 1 -dimethylethoxy)carbonyl] amino] imidazo[l ,2-a]pyrazine was prepared from 6-bromoimidazo[l ,2-a]pyrazin-8-amine according to reference procedure for similar compound (Lind, Kenneth E. etc. WO2008005457).
(1R,3s,5S)-tert-butyl 3-(8-(tert-butoxycarbonylamino)-3-(6-phenylpyridin-3-yl)imidazo[1,2-a]pyrazin-6-yl)-8-azabicyclo[3.2.1]octane-8-carboxylate[ No CAS ]
6-(furan-2-yl)imidazo[1,2-a]pyrazin-8-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
100%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In 1,4-dioxane; water; at 90℃;
General procedure: In a pressure tube were mixed 5-bromo-6-chloropyrazin-2-amine (4.32 g, 20.73 mmol), phenylboronic acid (2.78 g, 22.80 mmol, 1 .1 equiv.) and Na2003 (4.39 g, 41 .45 mmol, 2 equiv.) in a4:1 mixture of 1 ,4-dioxane : water (50 mL). The reaction mixture was sparged with argon andPd(PPh3)4 (1 .20 g, 1 .04 mmol, 5 mol%) was then added. The pressure tube was capped andthe reaction mixture was heated at 10000 overnight. After that time, the reaction mixture wascooled down to r.t., filtered and concentrated under reduced pressure. The obtained crude ma-terial was purified by flash chromatography on silica eluting with hexane: EtOAc = 1:0 - 1:1 to lead to the title product as a white solid (2.11 g, 50%). ESI-MS: 206.05 [M+H]+. 1 H NMR (300 MHz, DMSO-d6) 6 7.93 (s, 1 H), 7.66 - 7.59 (m, 2H), 7.47 - 7.32 (m, 3H), 7.00 (br s, 2H).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping