* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With 5%-palladium/activated carbon; hydrogen In tetrahydrofuran at 50℃;
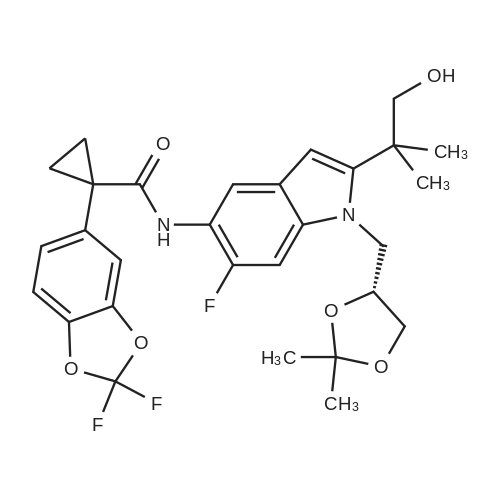
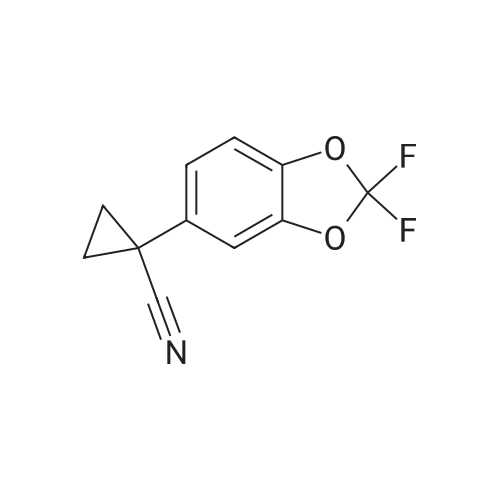
Synthesis of Compound 1 Method A [0235] A 20 L autoclave was flushed three times with nitrogen gas and then charged with palladium on carbon (Evonik E 101 NN/W, 5percent Pd, 60percent wet, 200 g, 0.075 mol, 0.04 equiv). The autoclave was then flushed with nitrogen three times. A solution of crude benzyl protected Compound 1 (1.3 kg, 1.9 mol) in THF (8 L, 6 vol) was added to the autoclave via suction. The vessel was capped and then flushed three times with nitrogen gas. With gentle stirring, the vessel was flushed three times with hydrogen gas, evacuating to atmosphere by diluting with nitrogen. The autoclave was pressurized to 3 Bar with hydrogen and the agitation rate was increased to 800 rpm. Rapid hydrogen uptake was observed (dissolution). Once uptake subsided, the vessel was heated to 50° C. [0236] For safety purposes, the thermostat was shut off at the end of every work-day. The vessel was pressurized to 4 Bar with hydrogen and then isolated from the hydrogen tank. [0237] After 2 full days of reaction, more Pd/C (60 g, 0.023 mol, 0.01 equiv) was added to the mixture. This was done by flushing three times with nitrogen gas and then adding the catalyst through the solids addition port. Resuming the reaction was done as before. After 4 full days, the reaction was deemed complete by HPLC by the disappearance of not only the starting material but also of the peak corresponding to a mono-benzylated intermediate. [0238] The reaction mixture was filtered through a Celite pad. The vessel and filter cake were washed with THF (2 L, 1.5 vol). The Celite pad was then wetted with water and the cake discarded appropriately. The combined filtrate and THF wash were concentrated using a rotary evaporator yielding the crude product as a black oil, 1 kg. [0239] The equivalents and volumes in the following purification are based on 1 kg of crude material. The crude black oil was dissolved in 1:1 ethyl acetate-heptane. The mixture was charged to a pad of silica gel (1.5 kg, 1.5 wt. equiv) in a fritted funnel that had been saturated with 1:1 ethyl acetate-heptane. The silica pad was flushed first with 1:1 ethyl acetate-heptane (6 L, 6 vol) and then with pure ethyl acetate (14 L, 14 vol). The eluent was collected in 4 fractions which were analyzed by HPLC. [0240] The equivalents and volumes in the following purification are based on 0.6 kg of crude material. Fraction 3 was concentrated by rotary evaporation to give a brown foam (600 g) and then redissolved in MTBE (1.8 L, 3 vol). The dark brown solution was stirred overnight at ambient temperature, during which time, crystallization occurred. Heptane (55 mL, 0.1 vol) was added and the mixture was stirred overnight. The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (900 mL, 1.5 vol). The filter cake was air-dried for 1 h and then vacuum dried at ambient temperature for 16 h, furnishing 253 g of VXc-661 as an off-white solid. [0241] The equivalents and volumes for the following purification are based on 1.4 kg of crude material. Fractions 2 and 3 from the above silica gel filtration as well as material from a previous reaction were combined and concentrated to give 1.4 kg of a black oil. The mixture was resubmitted to the silica gel filtration (1.5 kg of silica gel, eluted with 3.5 L, 2.3 vol of 1:1 ethyl acetate-heptane then 9 L, 6 vol of pure ethyl acetate) described above, which upon concentration gave a tan foamy solid (390 g). [0242] The equivalents and volumes for the following purification are based on 390 g of crude material. The tan solid was insoluble in MTBE, so was dissolved in methanol (1.2 L, 3 vol). Using a 4 L Morton reactor equipped with a long-path distillation head, the mixture was distilled down to 2 vol. MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 2 vol. A second portion of MTBE (1.6 L, 4 vol) was added and the mixture was distilled back down to 2 vol. A third portion of MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 3 vol. Analysis of the distillate by GC revealed it to consist of 6percent methanol. The thermostat was set to 48° C. (below the boiling temp of the MTBE-methanol azeotrope, which is 52° C.). The mixture was cooled to 20° C. over 2 h, during which time a relatively fast crystallization occurred. After stirring the mixture for 2 h, heptane (20 mL, 0.05 vol) was added and the mixture was stirred overnight (16 h). The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (800 mL, 2 vol). The filter cake was air-dried for 1 h and then vacuum dried at ambient temperature for 16 h, furnishing 130 g of Compound 1 as an off-white solid.
130 g
With 5%-palladium/activated carbon; hydrogen In tetrahydrofuran at 50℃; Inert atmosphere; Sealed tube
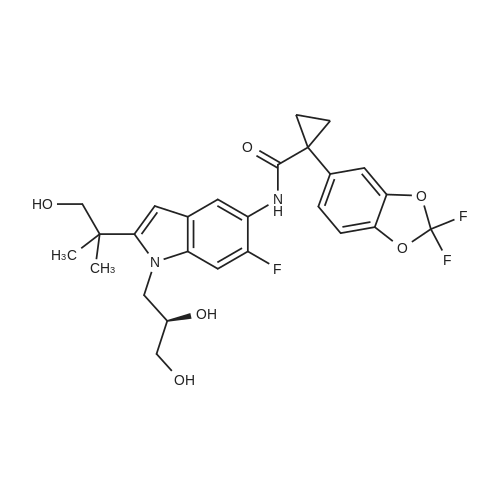
Synthesis of (R)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide [0323] Method A [0324] A 20 L autoclave was flushed three times with nitrogen gas and then charged with palladium on carbon (Evonik E 101 NN/W, 5percent Pd, 60percent wet, 200 g, 0.075 mol, 0.04 equiv). The autoclave was then flushed with nitrogen three times. A solution of crude benzyl protected (R)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide (1.3 kg, 1.9 mol) in THF (8 L, 6 vol) was added to the autoclave via suction. The vessel was capped and then flushed three times with nitrogen gas. With gentle stirring, the vessel was flushed three times with hydrogen gas, evacuating to atmosphere by diluting with nitrogen. The autoclave was pressurized to 3 Bar with hydrogen and the agitation rate was increased to 800 rpm. Rapid hydrogen uptake was observed (dissolution). Once uptake subsided, the vessel was heated to 50° C. [0325] For safety purposes, the thermostat was shut off at the end of every work-day. The vessel was pressurized to 4 Bar with hydrogen and then isolated from the hydrogen tank. [0326] After 2 full days of reaction, more Pd/C (60 g, 0.023 mol, 0.01 equiv) was added to the mixture. This was done by flushing three times with nitrogen gas and then adding the catalyst through the solids addition port. Resuming the reaction was done as before. After 4 full days, the reaction was deemed complete by HPLC by the disappearance of not only the starting material but also of the peak corresponding to a mono-benzylated intermediate. [0327] The reaction mixture was filtered through a Celite pad. The vessel and filter cake were washed with THF (2 L, 1.5 vol). The Celite pad was then wetted with water and the cake discarded appropriately. The combined filtrate and THF wash were concentrated using a rotary evaporator yielding the crude product as a black oil, 1 kg. [0328] The equivalents and volumes in the following purification are based on 1 kg of crude material. The crude black oil was dissolved in 1:1 ethyl acetate-heptane. The mixture was charged to a pad of silica gel (1.5 kg, 1.5 wt. equiv) in a fitted funnel that had been saturated with 1:1 ethyl acetate-heptane. The silica pad was flushed first with 1:1 ethyl acetate-heptane (6 L, 6 vol) and then with pure ethyl acetate (14 L, 14 vol). The eluent was collected in 4 fractions which were analyzed by HPLC. [0329] The equivalents and volumes in the following purification are based on 0.6 kg of crude material. Fraction 3 was concentrated by rotary evaporation to give a brown foam (600 g) and then redissolved in MTBE (1.8 L, 3 vol). The dark brown solution was stirred overnight at ambient temperature, during which time, crystallization occurred. Heptane (55 mL, 0.1 vol) was added and the mixture was stirred overnight. The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (900 mL, 1.5 vol). The filter cake was air-dried for 1 h and then vacuum dried at ambient temperature for 16 h, furnishing 253 g of (R)-1-(2,2-difluorobenzo [d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide as an off-white solid. [0330] The equivalents and volumes for the following purification are based on 1.4 kg of crude material. Fractions 2 and 3 from the above silica gel filtration as well as material from a previous reaction were combined and concentrated to give 1.4 kg of a black oil. The mixture was resubmitted to the silica gel filtration (1.5 kg of silica gel, eluted with 3.5 L, 2.3 vol of 1:1 ethyl acetate-heptane then 9 L, 6 vol of pure ethyl acetate) described above, which upon concentration gave a tan foamy solid (390 g). [0331] The equivalents and volumes for the following purification are based on 390 g of crude material. The tan solid was insoluble in MTBE, so was dissolved in methanol (1.2 L, 3 vol). Using a 4 L Morton reactor equipped with a long-path distillation head, the mixture was distilled down to 2 vol. MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 2 vol. A second portion of MTBE (1.6 L, 4 vol) was added and the mixture was distilled back down to 2 vol. A third portion of MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 3 vol. Analysis of the distillate by GC revealed it to consist of 6percent methanol. The thermostat was set to 48° C. (below the boiling temp of the MTBE-methanol azeotrope, which is 52° C.). The mixture was cooled to 20° C. over 2 h, during which time a relatively fast crystallization occurred. After stirring the mixture for 2 h, heptane (20 mL, 0.05 vol) was added and the mixture was stirred overnight (16 h). The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (800 mL, 2 vol). The filter cake was air-dried for 1 hand then vacuum dried at ambient temperature for 16 h, furnishing 130 g of (R)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide as an off-white solid.
1.02 kg
With 5%-palladium/activated carbon; hydrogen In tetrahydrofuran at 50℃; Inert atmosphere; Large scale
[00372] Method B [00373] Benzyl protected Compound 1 was dissolved in THF (3 vol) and then stripped to dryness to remove any residual solvent. Benzyl protected Compound 1 was redissolved in THF (4 vol) and added to the hydrogenator containing 5 wtpercent Pd/C (2.5 molpercent, 60percent wet, Degussa E5 El 01 N /W). The internal temperature of the reaction was adjusted to 50 °C, and flushed with N2 (x5) followed by hydrogen (x3). The hydrogenator pressure was adjusted to 3 Bar of hydrogen and the mixture was stirred rapidly (>1100 rpm). At the end of the reaction, the catalyst was filtered through a pad of Celite and washed with THF (1 vol). The filtrate was concentrated in vacuo to obtain a brown foamy residue. The resulting residue was dissolved in MTBE (5 vol) and 0.5N HCl solution (2 vol) and distilled water (1 vol) were added. The mixture was stirred for NLT 30 min and the resulting layers were separated. The organic phase was washed with 10 wtpercent K2CO3 solution (2 vol x2) followed by a brine wash. The organic layer was added to a flask containing silica gel (25 wtpercent), Deloxan-THP II (5wtpercent, 75percent wet), and Na2SO4 and stirred overnight. The resulting mixture was filtered through a pad of Celite and washed with 10percentTHF/MTBE (3 vol). The filtrate was concentrated in vacuo to afford crude Compound 1 as pale tan foam. [00374] Compound 1 recovery from the mother liquor: Option A. [00375] Silica gel pad filtration: The mother liquor was concentrated in vacuo to obtain a brown foam, dissolved in dichloromethane (2 vol), and filtered through a pad of silica (3x weight of the crude Compound 1). The silica pad was washed with ethyl acetate/heptane (1 :1, 13 vol) and the filtrate was discarded. The silica pad was washed with 10percent THF/ethyl acetate (10 vol) and the filtrate was concentrated in vacuo to afford Compound 1 as pale tan foam. The above crystallization procedure was followed to isolate the remaining Compound 1. {00376] Compound 1 recovery from the mother liquor: Option B, [00377] Silica gel column chromatography: After chromatography on silica gel (50percent ethyl acetate/hexaties to 100percent ethyl acetate), the desired compound was isolated as pale tan foam. The above crystallization procedure was followed to isolate the remaining Compound 1. [00378] Additional Recrystaliization of Compound 1 [0379] Solid Compound 1 (135 kg) was suspended in IPA (5.4 L, 4 vol) and then heated to 82 °C. Upon complete dissolution (visual), heptane (540 mL, 0.4 vol) was added slowly. The mixture was cooled to 58 °C The mixture was then cooled slowly to 51 °C, during which time crystallization occurs. The heat source was shut down and the recrystalfeation mixture was allowed to cool naturally overnight. The mixture was filtered using a benchtop Buclmer funnel and the filter cake was washed with IPA (2.7 L, 2 vol). The filler cake was dried in the tunnel under air flow for 8 h and then was oven-dried in vacuo at 45-50 °C overnight to give 1.02 kg of recrystallized Compound 1 ,
130 g
With 5%-palladium/activated carbon; hydrogen In tetrahydrofuran at 50℃; for 96 h; Inert atmosphere; Autoclave
A 20 L autoclave was flushed three times with nitrogen gas and then charged with palladium on carbon (Evonik E 101 NN/W, 5percent Pd, 60percent wet, 200 g, 0.075 mol, 0.04 equiv). The autoclave was then flushed with nitrogen three times. A solution of crude benzyl protected Compound 3 (1.3 kg, about 1.9 mol) in THF (8 L, 6 vol) was added to the autoclave via suction. The vessel was capped and then flushed three times with nitrogen gas. With gentle stirring, the vessel was flushed three times with hydrogen gas, evacuating to atmosphere by diluting with nitrogen. The autoclave was pressurized to 3 Bar with hydrogen and the agitation rate was increased to 800 rpm. Rapid hydrogen uptake was observed (dissolution). Once uptake subsided, the vessel was heated to 50° C. For safety purposes, the thermostat was shut off at the end of every work-day. The vessel was pressurized to 4 Bar with hydrogen and then isolated from the hydrogen tank. (2007) After 2 full days of reaction, more Pd/C (60 g, 0.023 mol, 0.01 equiv) was added to the mixture. This was done by flushing three times with nitrogen gas and then adding the catalyst through the solids addition port. Resuming the reaction was done as before. After 4 full days, the reaction was deemed complete by HPLC by the disappearance of not only the starting material, but also the peak corresponding to a mono-benzylated intermediate. The reaction mixture was filtered through a Celite® pad. The vessel and filter cake were washed with THF (2 L, 1.5 vol). The Celite® pad was then wetted with water and the cake discarded appropriately. The combined filtrate and THF wash were concentrated using a rotary evaporator yielding the crude product as a black oil, 1 kg. (2009) The equivalents and volumes in the following purification are based on 1 kg of crude material. The crude black oil was dissolved in 1:1 ethyl acetate-heptane. The mixture was charged to a pad of silica gel (1.5 kg, 1.5 wt. equiv) in a flitted funnel that had been saturated with 1:1 ethyl acetate-heptane. The silica pad was flushed first with 1:1 ethyl acetate-heptane (6 L, 6 vol) and then with pure ethyl acetate (14 L, 14 vol). The eluent was collected in 4 fractions that were analyzed by HPLC. (2010) The equivalents and volumes in the following purification are based on 0.6 kg of crude material. Fraction 3 was concentrated by rotary evaporation to give a brown foam (600 g) and then redissolved in MTBE (1.8 L, 3 vol). The dark brown solution was stirred overnight at ambient temperature, during which time, crystallization occurred. Heptane (55 mL, 0.1 vol) was added and the mixture was stirred overnight. The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (900 mL, 1.5 vol). The filter cake was air-dried for 1 h and then vacuum dried at ambient temperature for 16 h, furnishing 253 g of Compound 3 as an off-white solid. (2011) The equivalents and volumes for the following purification are based on 1.4 kg of crude material. Fractions 2 and 3 from the above silica gel filtration as well as material from a previous reaction were combined and concentrated to give 1.4 kg of a black oil. The mixture was resubmitted to the silica gel filtration (1.5 kg of silica gel, eluted with 3.5 L, 2.3 vol of 1:1 ethyl acetate-heptane then 9 L, 6 vol of pure ethyl acetate) described above, which upon concentration gave a tan foamy solid (390 g). The equivalents and volumes for the following purification are based on 390 g of crude material. The tan solid was insoluble in MTBE, so was dissolved in methanol (1.2 L, 3 vol). Using a 4 L Morton reactor equipped with a long-path distillation head, the mixture was distilled down to 2 vol. MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 2 vol. A second portion of MTBE (1.6 L, 4 vol) was added and the mixture was distilled back down to 2 vol. A third portion of MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 3 vol. Analysis of the distillate by GC revealed it to consist of about 6percent methanol. The thermostat was set to 48° C. (below the boiling temp of the MTBE-methanol azeotrope, which is 52° C.). The mixture was cooled to 20° C. over 2 h, during which time a relatively fast crystallization occurred. After stirring the mixture for 2 h, heptane (20 mL, 0.05 vol) was added and the mixture was stirred overnight (16 h). The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (800 mL, 2 vol). The filter cake was air-dried for 1 h and then vacuum dried at ambient temperature for 16 h, furnishing 130 g of Compound 3 as an off-white solid.
With hydrogen;5%-palladium/activated carbon; In tetrahydrofuran; at 50℃; under 2250.23 - 3000.3 Torr; for 96.0h;autoclave; Inert atmosphere;Product distribution / selectivity;
00238] Synthesis of Compound 1.Gamma002391 Method A[00240] A 20 L autoclave was flushed three times with nitrogen gas and then charged with palladium on carbon (Evonik E 101 N/W, 5% Pd, 60% wet, 200 g, 0.075 mol, 0.04 equiv). The autoclave was then flushed with nitrogen three times. A solution of crude benzyl protected Compound 1 (1.3 kg, -1.9 mol) in THF (8 L, 6 vol) was added to the autoclave via suction. The vessel was capped and then flushed three times with nitrogen gas. With gentle stirring, the vessel was flushed three times with hydrogen gas, evacuating to atmosphere by diluting with nitrogen. The autoclave was pressurized to 3 Bar with hydrogen and the agitation rate was increased to 800 rpm. Rapid hydrogen uptake was observed (dissolution). Once uptake subsided, the vessel was heated to 50 C.[00241] For safety purposes, the thermostat was shut off at the end of every work-day. The vessel was pressurized to 4 Bar with hydrogen and then isolated from the hydrogen tank.[00242] After 2 full days of reaction, more Pd / C (60 g, 0.023 mol, 0.01 equiv) was added to the mixture. This was done by flushing three times with nitrogen gas and then adding the catalyst through the solids addition port. Resuming the reaction was done as before. After 4 full days, the reaction was deemed complete by HPLC by the disappearance of not only the starting material but also of the peak corresponding to a mono-benzylated intermediate. [00243] The reaction mixture was filtered through a Celite pad. The vessel and filter cake were washed with THF (2 L, 1.5 vol). The Celite pad was then wetted with water and the cake discarded appropriately. The combined filtrate and THF wash were concentrated using a rotary evaporator yielding the crude product as a black oil, 1 kg.[00244] The equivalents and volumes in the following purification are based on 1 kg of crude material. The crude black oil was dissolved in 1 : 1 ethyl acetate-heptane. The mixture was charged to a pad of silica gel (1.5 kg, 1.5 wt. equiv) in a fritted funnel that had been saturated with 1 : 1 ethyl acetate -heptane. The silica pad was flushed first with 1 : 1 ethyl acetate- heptane (6 L, 6 vol) and then with pure ethyl acetate (14 L, 14 vol). The eluent was collected in 4 fractions which were analyzed by HPLC.[00245] The equivalents and volumes in the following purification are based on 0.6 kg of crude material. Fraction 3 was concentrated by rotary evaporation to give a brown foam (600 g) and then redissolved in MTBE (1.8 L, 3 vol). The dark brown solution was stirred overnight at ambient temperature, during which time, crystallization occurred. Heptane (55 mL, 0.1 vol) was added and the mixture was stirred overnight. The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (900 mL, 1.5 vol). The filter cake was air-dried for 1 h and then vacuum dried at ambient temperature for 16 h, furnishing 253 g of VXc-661 as an off- white solid.[00246] The equivalents and volumes for the following purification are based on 1.4 kg of crude material. Fractions 2 and 3 from the above silica gel filtration as well as material from a previous reaction were combined and concentrated to give 1.4 kg of a black oil. The mixture was resubmitted to the silica gel filtration (1.5 kg of silica gel, eluted with 3.5 L, 2.3 vol of 1 : 1 ethyl acetate-heptane then 9 L, 6 vol of pure ethyl acetate) described above, which upon concentration gave a tan foamy solid (390 g).[00247] The equivalents and volumes for the following purification are based on 390 g of crude material. The tan solid was insoluble in MTBE, so was dissolved in methanol (1.2 L, 3 vol). Using a 4 L Morton reactor equipped with a long-path distillation head, the mixture was distilled down to 2 vol. MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 2 vol. A second portion of MTBE (1.6 L, 4 vol) was added and the mixture was distilled back down to 2 vol. A third portion of MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 3 vol. Analysis of the distillate by GC revealed it to consist of ~6% methanol. The thermostat was set to 48 C (below the boiling temp of the MTBE- methanol azeotrope, which is 52 C). The mixture was cooled to 20 C over 2 h, during which time a relatively fast crystallization occurred. After stirring the mixture for 2 h, heptane (20 mL, 0.05 vol) was added and the mixture was stirred overnight (16 h). The mixture was filtered using a Buchner funnel and the filter cake was washed with 3: 1 MTBE-heptane (800 mL, 2 vol). The filter cake was air-dried for 1 h and then vacuum dried at ambient temperature for 16 h, furnishing 130 g of Compound 1 as an off-white solid.
130 g
With 5%-palladium/activated carbon; hydrogen; In tetrahydrofuran; at 50℃; under 2250.23 Torr;
Synthesis of Compound 1 Method A [0235] A 20 L autoclave was flushed three times with nitrogen gas and then charged with palladium on carbon (Evonik E 101 NN/W, 5% Pd, 60% wet, 200 g, 0.075 mol, 0.04 equiv). The autoclave was then flushed with nitrogen three times. A solution of crude benzyl protected Compound 1 (1.3 kg, 1.9 mol) in THF (8 L, 6 vol) was added to the autoclave via suction. The vessel was capped and then flushed three times with nitrogen gas. With gentle stirring, the vessel was flushed three times with hydrogen gas, evacuating to atmosphere by diluting with nitrogen. The autoclave was pressurized to 3 Bar with hydrogen and the agitation rate was increased to 800 rpm. Rapid hydrogen uptake was observed (dissolution). Once uptake subsided, the vessel was heated to 50 C. [0236] For safety purposes, the thermostat was shut off at the end of every work-day. The vessel was pressurized to 4 Bar with hydrogen and then isolated from the hydrogen tank. [0237] After 2 full days of reaction, more Pd/C (60 g, 0.023 mol, 0.01 equiv) was added to the mixture. This was done by flushing three times with nitrogen gas and then adding the catalyst through the solids addition port. Resuming the reaction was done as before. After 4 full days, the reaction was deemed complete by HPLC by the disappearance of not only the starting material but also of the peak corresponding to a mono-benzylated intermediate. [0238] The reaction mixture was filtered through a Celite pad. The vessel and filter cake were washed with THF (2 L, 1.5 vol). The Celite pad was then wetted with water and the cake discarded appropriately. The combined filtrate and THF wash were concentrated using a rotary evaporator yielding the crude product as a black oil, 1 kg. [0239] The equivalents and volumes in the following purification are based on 1 kg of crude material. The crude black oil was dissolved in 1:1 ethyl acetate-heptane. The mixture was charged to a pad of silica gel (1.5 kg, 1.5 wt. equiv) in a fritted funnel that had been saturated with 1:1 ethyl acetate-heptane. The silica pad was flushed first with 1:1 ethyl acetate-heptane (6 L, 6 vol) and then with pure ethyl acetate (14 L, 14 vol). The eluent was collected in 4 fractions which were analyzed by HPLC. [0240] The equivalents and volumes in the following purification are based on 0.6 kg of crude material. Fraction 3 was concentrated by rotary evaporation to give a brown foam (600 g) and then redissolved in MTBE (1.8 L, 3 vol). The dark brown solution was stirred overnight at ambient temperature, during which time, crystallization occurred. Heptane (55 mL, 0.1 vol) was added and the mixture was stirred overnight. The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (900 mL, 1.5 vol). The filter cake was air-dried for 1 h and then vacuum dried at ambient temperature for 16 h, furnishing 253 g of VXc-661 as an off-white solid. [0241] The equivalents and volumes for the following purification are based on 1.4 kg of crude material. Fractions 2 and 3 from the above silica gel filtration as well as material from a previous reaction were combined and concentrated to give 1.4 kg of a black oil. The mixture was resubmitted to the silica gel filtration (1.5 kg of silica gel, eluted with 3.5 L, 2.3 vol of 1:1 ethyl acetate-heptane then 9 L, 6 vol of pure ethyl acetate) described above, which upon concentration gave a tan foamy solid (390 g). [0242] The equivalents and volumes for the following purification are based on 390 g of crude material. The tan solid was insoluble in MTBE, so was dissolved in methanol (1.2 L, 3 vol). Using a 4 L Morton reactor equipped with a long-path distillation head, the mixture was distilled down to 2 vol. MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 2 vol. A second portion of MTBE (1.6 L, 4 vol) was added and the mixture was distilled back down to 2 vol. A third portion of MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 3 vol. Analysis of the distillate by GC revealed it to consist of 6% methanol. The thermostat was set to 48 C. (below the boiling temp of the MTBE-methanol azeotrope, which is 52 C.). The mixture was cooled to 20 C. over 2 h, during which time a relatively fast crystallization occurred. After stirring the mixture for 2 h, heptane (20 mL, 0.05 vol) was added and the mixture was stirred overnight (16 h). The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (800 mL, 2 vol). The filter cake was air-dried for 1 h and then vacuum dried at ambient temperature for 16 h, furnishing 130 g of Compound 1 as an off-white solid.
130 g
With 5%-palladium/activated carbon; hydrogen; In tetrahydrofuran; at 50℃; under 2250.23 Torr;Inert atmosphere; Sealed tube;
Synthesis of (R)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide [0323] Method A [0324] A 20 L autoclave was flushed three times with nitrogen gas and then charged with palladium on carbon (Evonik E 101 NN/W, 5% Pd, 60% wet, 200 g, 0.075 mol, 0.04 equiv). The autoclave was then flushed with nitrogen three times. A solution of crude benzyl protected (R)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide (1.3 kg, 1.9 mol) in THF (8 L, 6 vol) was added to the autoclave via suction. The vessel was capped and then flushed three times with nitrogen gas. With gentle stirring, the vessel was flushed three times with hydrogen gas, evacuating to atmosphere by diluting with nitrogen. The autoclave was pressurized to 3 Bar with hydrogen and the agitation rate was increased to 800 rpm. Rapid hydrogen uptake was observed (dissolution). Once uptake subsided, the vessel was heated to 50 C. [0325] For safety purposes, the thermostat was shut off at the end of every work-day. The vessel was pressurized to 4 Bar with hydrogen and then isolated from the hydrogen tank. [0326] After 2 full days of reaction, more Pd/C (60 g, 0.023 mol, 0.01 equiv) was added to the mixture. This was done by flushing three times with nitrogen gas and then adding the catalyst through the solids addition port. Resuming the reaction was done as before. After 4 full days, the reaction was deemed complete by HPLC by the disappearance of not only the starting material but also of the peak corresponding to a mono-benzylated intermediate. [0327] The reaction mixture was filtered through a Celite pad. The vessel and filter cake were washed with THF (2 L, 1.5 vol). The Celite pad was then wetted with water and the cake discarded appropriately. The combined filtrate and THF wash were concentrated using a rotary evaporator yielding the crude product as a black oil, 1 kg. [0328] The equivalents and volumes in the following purification are based on 1 kg of crude material. The crude black oil was dissolved in 1:1 ethyl acetate-heptane. The mixture was charged to a pad of silica gel (1.5 kg, 1.5 wt. equiv) in a fitted funnel that had been saturated with 1:1 ethyl acetate-heptane. The silica pad was flushed first with 1:1 ethyl acetate-heptane (6 L, 6 vol) and then with pure ethyl acetate (14 L, 14 vol). The eluent was collected in 4 fractions which were analyzed by HPLC. [0329] The equivalents and volumes in the following purification are based on 0.6 kg of crude material. Fraction 3 was concentrated by rotary evaporation to give a brown foam (600 g) and then redissolved in MTBE (1.8 L, 3 vol). The dark brown solution was stirred overnight at ambient temperature, during which time, crystallization occurred. Heptane (55 mL, 0.1 vol) was added and the mixture was stirred overnight. The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (900 mL, 1.5 vol). The filter cake was air-dried for 1 h and then vacuum dried at ambient temperature for 16 h, furnishing 253 g of (R)-1-(2,2-difluorobenzo [d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide as an off-white solid. [0330] The equivalents and volumes for the following purification are based on 1.4 kg of crude material. Fractions 2 and 3 from the above silica gel filtration as well as material from a previous reaction were combined and concentrated to give 1.4 kg of a black oil. The mixture was resubmitted to the silica gel filtration (1.5 kg of silica gel, eluted with 3.5 L, 2.3 vol of 1:1 ethyl acetate-heptane then 9 L, 6 vol of pure ethyl acetate) described above, which upon concentration gave a tan foamy solid (390 g). [0331] The equivalents and volumes for the following purification are based on 390 g of crude material. The tan solid was insoluble in MTBE, so was dissolved in methanol (1.2 L, 3 vol). Using a 4 L Morton reactor equipped with a long-path distillation head, the mixture was distilled down to 2 vol. MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 2 vol. A second portion of MTBE (1.6 L, 4 vol) was added and the mixture was distilled back down to 2 vol. A third portion of MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 3 vol. Analysis of the distillate by GC revealed it to consist of 6% methanol. The thermostat was set to 48 C. (below the boiling temp of the MTBE-methanol azeotrope, which is 52 C.). The mixture was cooled to 20 C. over 2 h, during which time a relatively fast crystallization occurred. After stirring the mixture for 2 h, heptane (20 mL, 0.05 vol) was added and the mixture was stirred overnight (16 h). The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (800 mL, 2 vol). The fi...
With hydrogenchloride; 5%-palladium/activated carbon; hydrogen; In methanol;Inert atmosphere;
5% palladium on charcoal (-50%, wet, 0.01 eq) was charged to an appropriatehydrogenation vessel. The (R)-N-(1-(3-(ben.zyloxy)-2-hydrox:ypropyl)-2-(1-(benzyloxy)-2-methylpropan-2-yl)-6-t1uoro-1H-indol-5-yl)-l-(2,2-dit1uorobenzo[ dJ[ 1 ,3]dioxol-5-yl)cyclopropanecarboxamide solution in methanol (2 vol) obtained above was added carefully,followed by a 3 M solution of HCl in methanoL The vessel was purged with nitrogen gas andthen with hydrogen gas. The mixture was stirred vigorously until the reaction was complete, asdetermined by HPLC analysis. Typical reaction time was 3-5 h. The reaction mixture wasfiltered through Celite and the cake was washed with methanol (2 vol). A solvent swap intoisopropanol (3 vol) was performed. Crude Compound 3 was crystallized from 75% IPA-heptane(4 vol, i.e., 1 vol heptane added to the 3 vol ofiPA) and the resulting crystals were matured in50% IPA-heptane (i.e., 2 vol of heptane added to the mixture). Typical yields of Compound 3from the two-step acylation I hydrogenolysis procedure range from 68% to 84%. Compound 3can be recrystallized from IPA-heptane following the same procedure just described.
1.02 kg
With 5%-palladium/activated carbon; hydrogen; In tetrahydrofuran; at 50℃; under 2250.23 Torr;Inert atmosphere; Large scale;
[00372] Method B [00373] Benzyl protected Compound 1 was dissolved in THF (3 vol) and then stripped to dryness to remove any residual solvent. Benzyl protected Compound 1 was redissolved in THF (4 vol) and added to the hydrogenator containing 5 wt% Pd/C (2.5 mol%, 60% wet, Degussa E5 El 01 N /W). The internal temperature of the reaction was adjusted to 50 C, and flushed with N2 (x5) followed by hydrogen (x3). The hydrogenator pressure was adjusted to 3 Bar of hydrogen and the mixture was stirred rapidly (>1100 rpm). At the end of the reaction, the catalyst was filtered through a pad of Celite and washed with THF (1 vol). The filtrate was concentrated in vacuo to obtain a brown foamy residue. The resulting residue was dissolved in MTBE (5 vol) and 0.5N HCl solution (2 vol) and distilled water (1 vol) were added. The mixture was stirred for NLT 30 min and the resulting layers were separated. The organic phase was washed with 10 wt% K2CO3 solution (2 vol x2) followed by a brine wash. The organic layer was added to a flask containing silica gel (25 wt%), Deloxan-THP II (5wt%, 75% wet), and Na2SO4 and stirred overnight. The resulting mixture was filtered through a pad of Celite and washed with 10%THF/MTBE (3 vol). The filtrate was concentrated in vacuo to afford crude Compound 1 as pale tan foam. [00374] Compound 1 recovery from the mother liquor: Option A. [00375] Silica gel pad filtration: The mother liquor was concentrated in vacuo to obtain a brown foam, dissolved in dichloromethane (2 vol), and filtered through a pad of silica (3x weight of the crude Compound 1). The silica pad was washed with ethyl acetate/heptane (1 :1, 13 vol) and the filtrate was discarded. The silica pad was washed with 10% THF/ethyl acetate (10 vol) and the filtrate was concentrated in vacuo to afford Compound 1 as pale tan foam. The above crystallization procedure was followed to isolate the remaining Compound 1. {00376] Compound 1 recovery from the mother liquor: Option B, [00377] Silica gel column chromatography: After chromatography on silica gel (50% ethyl acetate/hexaties to 100% ethyl acetate), the desired compound was isolated as pale tan foam. The above crystallization procedure was followed to isolate the remaining Compound 1. [00378] Additional Recrystaliization of Compound 1 [0379] Solid Compound 1 (135 kg) was suspended in IPA (5.4 L, 4 vol) and then heated to 82 C. Upon complete dissolution (visual), heptane (540 mL, 0.4 vol) was added slowly. The mixture was cooled to 58 C The mixture was then cooled slowly to 51 C, during which time crystallization occurs. The heat source was shut down and the recrystalfeation mixture was allowed to cool naturally overnight. The mixture was filtered using a benchtop Buclmer funnel and the filter cake was washed with IPA (2.7 L, 2 vol). The filler cake was dried in the tunnel under air flow for 8 h and then was oven-dried in vacuo at 45-50 C overnight to give 1.02 kg of recrystallized Compound 1 ,
130 g
With 5%-palladium/activated carbon; hydrogen; In tetrahydrofuran; at 50℃; under 2250.23 Torr; for 96.0h;Inert atmosphere; Autoclave;
A 20 L autoclave was flushed three times with nitrogen gas and then charged with palladium on carbon (Evonik E 101 NN/W, 5% Pd, 60% wet, 200 g, 0.075 mol, 0.04 equiv). The autoclave was then flushed with nitrogen three times. A solution of crude benzyl protected Compound 3 (1.3 kg, about 1.9 mol) in THF (8 L, 6 vol) was added to the autoclave via suction. The vessel was capped and then flushed three times with nitrogen gas. With gentle stirring, the vessel was flushed three times with hydrogen gas, evacuating to atmosphere by diluting with nitrogen. The autoclave was pressurized to 3 Bar with hydrogen and the agitation rate was increased to 800 rpm. Rapid hydrogen uptake was observed (dissolution). Once uptake subsided, the vessel was heated to 50 C. For safety purposes, the thermostat was shut off at the end of every work-day. The vessel was pressurized to 4 Bar with hydrogen and then isolated from the hydrogen tank. (2007) After 2 full days of reaction, more Pd/C (60 g, 0.023 mol, 0.01 equiv) was added to the mixture. This was done by flushing three times with nitrogen gas and then adding the catalyst through the solids addition port. Resuming the reaction was done as before. After 4 full days, the reaction was deemed complete by HPLC by the disappearance of not only the starting material, but also the peak corresponding to a mono-benzylated intermediate. The reaction mixture was filtered through a Celite pad. The vessel and filter cake were washed with THF (2 L, 1.5 vol). The Celite pad was then wetted with water and the cake discarded appropriately. The combined filtrate and THF wash were concentrated using a rotary evaporator yielding the crude product as a black oil, 1 kg. (2009) The equivalents and volumes in the following purification are based on 1 kg of crude material. The crude black oil was dissolved in 1:1 ethyl acetate-heptane. The mixture was charged to a pad of silica gel (1.5 kg, 1.5 wt. equiv) in a flitted funnel that had been saturated with 1:1 ethyl acetate-heptane. The silica pad was flushed first with 1:1 ethyl acetate-heptane (6 L, 6 vol) and then with pure ethyl acetate (14 L, 14 vol). The eluent was collected in 4 fractions that were analyzed by HPLC. (2010) The equivalents and volumes in the following purification are based on 0.6 kg of crude material. Fraction 3 was concentrated by rotary evaporation to give a brown foam (600 g) and then redissolved in MTBE (1.8 L, 3 vol). The dark brown solution was stirred overnight at ambient temperature, during which time, crystallization occurred. Heptane (55 mL, 0.1 vol) was added and the mixture was stirred overnight. The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (900 mL, 1.5 vol). The filter cake was air-dried for 1 h and then vacuum dried at ambient temperature for 16 h, furnishing 253 g of Compound 3 as an off-white solid. (2011) The equivalents and volumes for the following purification are based on 1.4 kg of crude material. Fractions 2 and 3 from the above silica gel filtration as well as material from a previous reaction were combined and concentrated to give 1.4 kg of a black oil. The mixture was resubmitted to the silica gel filtration (1.5 kg of silica gel, eluted with 3.5 L, 2.3 vol of 1:1 ethyl acetate-heptane then 9 L, 6 vol of pure ethyl acetate) described above, which upon concentration gave a tan foamy solid (390 g). The equivalents and volumes for the following purification are based on 390 g of crude material. The tan solid was insoluble in MTBE, so was dissolved in methanol (1.2 L, 3 vol). Using a 4 L Morton reactor equipped with a long-path distillation head, the mixture was distilled down to 2 vol. MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 2 vol. A second portion of MTBE (1.6 L, 4 vol) was added and the mixture was distilled back down to 2 vol. A third portion of MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 3 vol. Analysis of the distillate by GC revealed it to consist of about 6% methanol. The thermostat was set to 48 C. (below the boiling temp of the MTBE-methanol azeotrope, which is 52 C.). The mixture was cooled to 20 C. over 2 h, during which time a relatively fast crystallization occurred. After stirring the mixture for 2 h, heptane (20 mL, 0.05 vol) was added and the mixture was stirred overnight (16 h). The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (800 mL, 2 vol). The filter cake was air-dried for 1 h and then vacuum dried at ambient temperature for 16 h, furnishing 130 g of Compound 3 as an off-white solid.
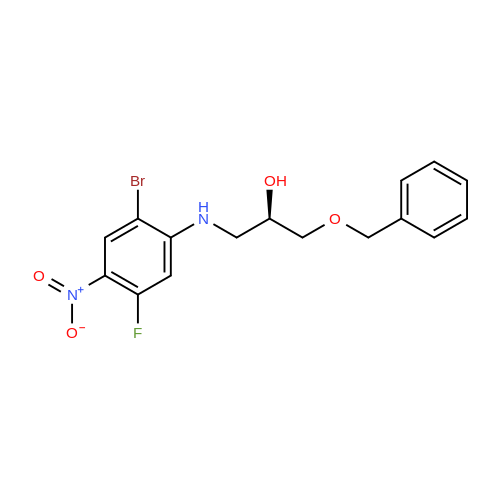
N-benzylglycolated-5-amino-2-(2-benzyloxy-1,1-dimethylethyl)-6-fluoroindole[ No CAS ]
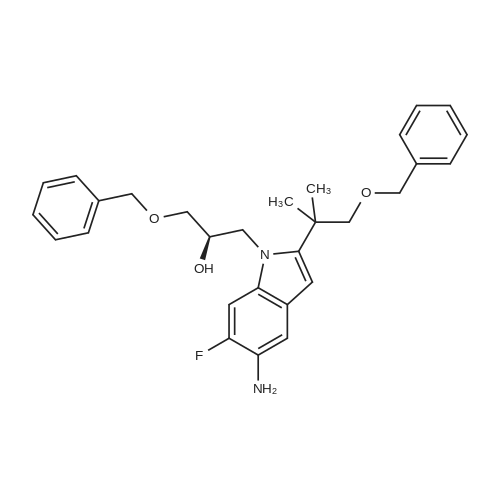
[ 1152311-62-0 ]
Yield
Reaction Conditions
Operation in experiment
With thionyl chloride; triethylamine; In dichloromethane; water; toluene;
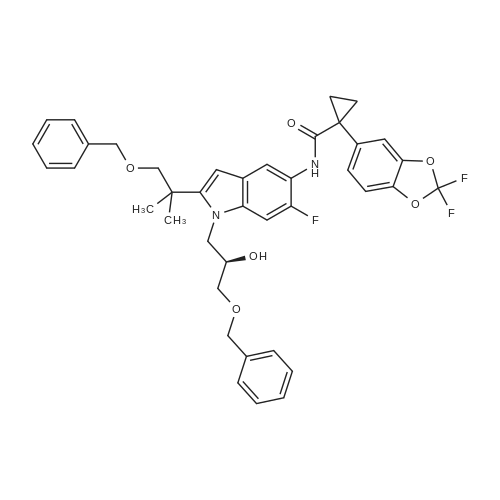
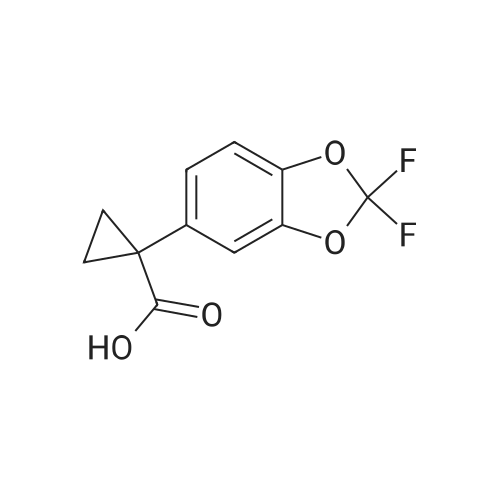
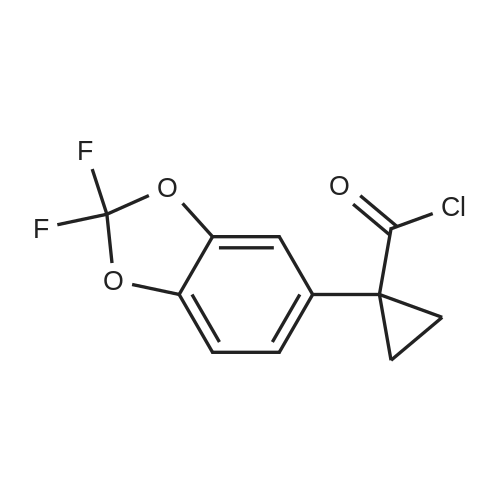
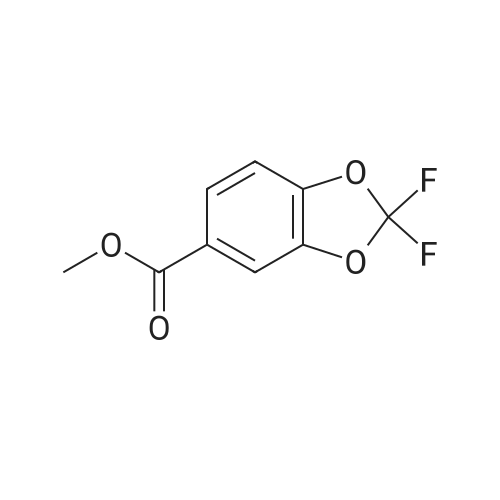
Synthesis of Benzyl Protected Compound 1. 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarboxylic acid (1.3 equiv) was slurried in toluene (2.5 vol, based on 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarboxylic acid) and the mixture was heated to 60 C. SOCl2 (1.7 equiv) was added via addition funnel. The resulting mixture was stirred for 2 hr. The toluene and the excess SOCl2 were distilled off using rotavop. Additional toluene (2.5 vol, based on 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarboxylic acid) was added and distilled again. The crude acid chloride was dissolved in dichloromethane (2 vol) and added via addition funnel to a mixture of N-benzylglycolated-5-amino-2-(2-benzyloxy-1,1-dimethylethyl)-6-fluoroindole (1.0 equiv), and triethylamine (2.0 equiv) in dichloromethane (7 vol) while maintaining 0-3 C. (internal temperature). The resulting mixture was stirred at 0 C. for 4 hrs and then warmed to room temperature overnight. Distilled water (5 vol) was added to the reaction mixture and stirred for NLT 30 min and the layers were separated. The organic phase was washed with 20 wt % K2CO3 (4 vol*2) followed by a brine wash (4 vol) and concentrated to afford crude benzyl protected Compound 1 as a thick brown oil, which was purified further using silica pad filtration. Silica gel pad filtration: Crude benzyl protected Compound 1 was dissolved in ethyl acetate (3 vol) in the presence of activated carbon Darco-G (10 wt %, based on theoretical yield of benzyl protected Compound 1) and stirred at room temperature overnight.
Synthesis of 1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-((4R)-8-fluoro-2-hydroxy-4-(hydroxymethyl)-1,1-dimethyl-1,2,4,5-tetrahydro-[1,4]oxazepino[4,5-a]indol-9-yl)cyclopropanecarboxamide [0339] <strong>[1152311-62-0](R)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide</strong> (11.5 mmol, 1 equiv) was suspended in DCM (51 mL, 8.5 vol). A solution of Dess-Martin periodinane (0.3 M in DCM, 12.8 mmol, 1.1 equiv) was added at ambient temperature. The mixture was stirred until the reaction was deemed complete by HPLC. A 5% aqueous solution of sodium sulfite was added and the mixture was stirred for up to 4 h. The phases were separated and then the organic phase was washed with 1 N HCl, brine and was then concentrated by rotary evaporation. The residue was purified by chromatography. The yield of purified material was between 7 and 15%.
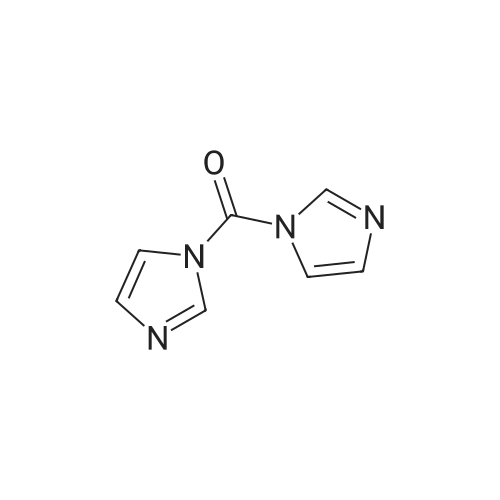
Step 1. Preparation of [0346] [0347] <strong>[1152311-62-0](R)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide</strong> (50 g, 1.0 eq) was suspended in dichloromethane (700 mL, 14 vol) and then cooled to -10 C. Solid carbonyl diimidazole (CDI, 34.2 g, 2.2 eq) was added. The reaction was monitored for completion by HPLC. Water (1 L, 20 vol) was added to the mixture and the phases were allowed to separate. The organic phase was solvent swapped into THF and the total volume was adjusted to 500 mL (10 vol). 2 M HCl (400 mL, 8 vol) was added to the THF solution. The mixture was stirred until all peaks coalesced into a single peak by HPLC (approximately 4 h). Toluene (700 mL, 14 vol) was added to the mixture, causing phase separation. The organic phase was washed with water (400 mL, 8 vol). The organic phase was concentrated at reduced pressure to give a light tan foam. The foam was suspended in isopropyl acetate (IPA, 700 mL, 14 vol) and heated to 80 C. n-Heptane (236 mL, 4.7 vol) was added at a rate to maintain the temperature at greater than 75 C. The mixture was cooled to 20 C. at a rate of 10-15 C. per hour. Crystallization occurred at approximately 65 C. The mixture was then filtered. The solid was washed with 1:1 IPA-heptane (120 mL, 2.4 vol) and vacuum-dried at 55 C. for 6 hours.
With 10% Pd/C; hydrogen In methanol at 30 - 35℃; for 4h;
12 Example 12: Preparation of the compound of formula I
Add to 200ml stainless steel autoclave80 grams of methanol,6.1 g (0.01 mol) obtained from Example 10The compound of formula XI1,0.2g 10wt% palladium-carbon catalyst,After nitrogen replacement three times,Add hydrogen,Keep the system pressure at 0.2-0.3MPa,React at 30-35°C for 4 hours.Replace with nitrogen three times,Filter to remove palladium carbon,Wash the filter cake twice with methanol,20 grams of methanol each time,Combine the filtrate,Add 0.5 g of activated carbon to the obtained filtrate,Stir at 65-70 to decolorize for 1 hour,Filter while it is hot,Distilling the filtrate to recover 90 grams of methanol,The remainder is cooled to 10-15,Crystallize, filter,The filter cake is dried,5.1 g of the compound of formula I are obtained,The yield was 98.2%, and the liquid phase purity was 99.9%.
With hydrogen In acetonitrile at 40 - 45℃; for 4h;
13 Example 13: Preparation of the compound of formula I
Add to 200ml stainless steel autoclave80 grams of acetonitrile,6.3 g (0.01 mol) obtained from Example 11Compound of formula XI2,0.5 g of 50wt% Raney nickel catalyst,After nitrogen replacement three times,Add hydrogen,Keep the system pressure at 0.3-0.4MPa,React at 40-45°C for 4 hours.Replace with nitrogen three times,Filter to remove Raney nickel catalyst,Wash the filter cake twice with acetonitrile,20 grams each time,Combine the filtrate,Add to the filtrate0.5 grams of activated carbon,Stir at 65-70 to decolorize for 1 hour,Filter while it is hot,Distilling the filtrate to recover 90 grams of acetonitrile,The remainder is cooled to 10-15,Crystallize, filter,The filter cake is dried,5.0 g of the compound of formula I are obtained,The yield was 96.1%,The purity of the liquid phase is 99.8%.
With palladium on activated charcoal; ammonium formate In methanol at 25 - 85℃;
11 Example-11: Preparation of Tezacaftor [Formula-1]
Ammonium formate (1.6 g) was added to the solution of R)-N-(2-(1-(benzyloxy)-2-methylpropan-2-yl)- 1 -(2,3-dihydroxypropyl)-6-fluoro- 1 H-indol-5-yl)- 1-(2,2-difluoro benzo[d] [ 1 ,3]dioxol-5-yl) cyclopropanecarboxamide of formula- 13 (0.8 g) in methanol (20 ml) at 25-30°C. Pd/C was added to the mixture, heated the resultant mixture to 80-85°C and stirred. Cooled the reaction mixture to 25-30°C and filtered. Distilled off the solvent from the filtrate. Water and ethyl acetate were added to the resultant residue. Distilled off the solvent from the resultant organic phase to get title compound.Yield: 0.2 g.
4.6 g
With hydrogenchloride; 5%-palladium/activated carbon; hydrogen In methanol at 50 - 55℃; for 3h; Autoclave;
26; 27; 28 EXAMPLE-26: Preparation of Tezacaftor of Formula I:
Compound of Formula Xlb (7.0 gms) was charged in to an Autoclave. MeOH (50 mL), methanolic HC1 (0.7 mL) and 5% Pd/C (-50% wet; l.Og) were added to the Autoclave.The autoclave was pressurized with hydrogen gas (4-5kg /cm ) and the temperature was gradually raised to 50-55°C, maintained for 3 hrs under hydrogen atmosphere. The reaction mass was cooled to ambient temperature and the catalyst was filtered. The filtrate was concentrated under reduced pressure and the obtained residue (foamy solid) was crystallized from a mixture of isopropyl alcohol and heptane to afford Tezacaftor of Formula I. Yield= 4.6gms. Purity by HPLC 95.7% LC-MS m/z: 521(M+1). lH NMR (300 MHz, DMSO-d6) d 8.31 (s, 1H, NH), 7.52 (d, J=1.5Hz, 1H, Ar-H), 7.42- 7.30 (m, 4H, Ar-H), 6.22 (s, 1H, Ar-H), 5.00 (d, J=5.1 Hz, 1H, -OH), 4.89 (t, J=5.5 Hz, 1H, OH), 4.74 (t, J=5.7Hz, 1H, OH), 4.40 (dd, Jl=3Hz, J2=2.4Hz, 1H, N-CH2), 4.14-4.06 (m, 1H, N-CH2), 3.90 (brs, 1H, CH), 3.62-3.57 (m, 2H, Isopropyl-CH2), 3.45-3.39 (m, 2H, CH2OH), 1.46 (q, J=3.6Hz, 2H,Cyclopropyl-CH2), 1.35 (s, 3H, CH3), 1.32 (s, 3H, CHs) 1.12 (q, J=3.6Hz, 2H, Cyclopropyl-CH2).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping