| 90% |
With trifluoroacetic acid; In acetonitrile; at 40 - 50℃;Product distribution / selectivity; |
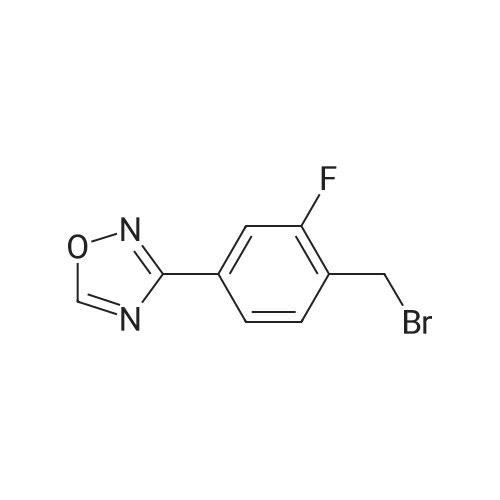
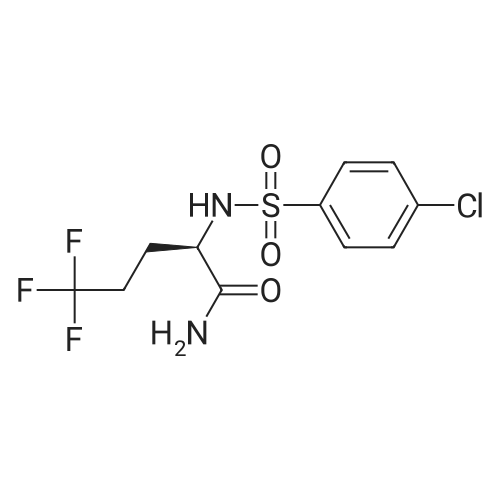
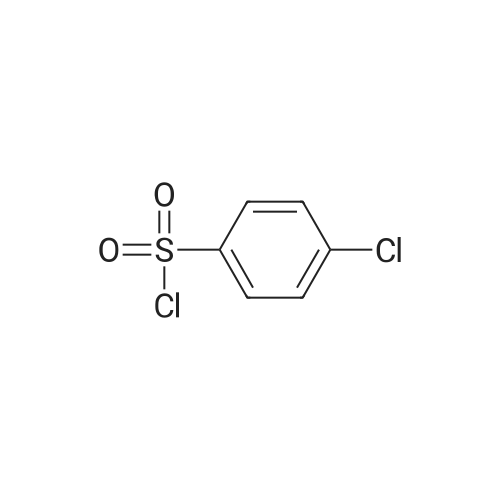
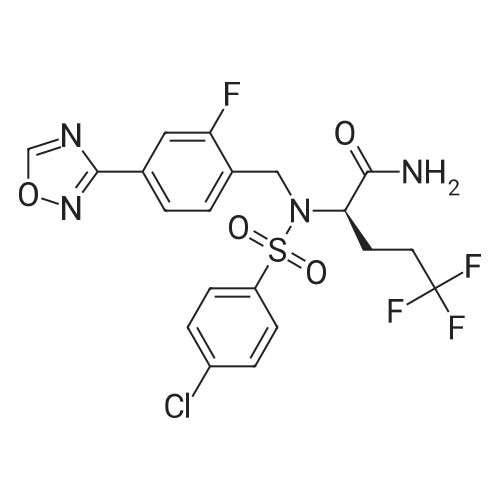
Step C. (2R)-2-[[(4-Chlorophenyl)sulfonyl][[2-fluoro-4-(1,2,4-oxadiazol-3-yl)phenyl]methyl]amino]-5,5,5-trifluoropentanamide (R)-2-(4-Chloro-N-(2-fluoro-4-(N'-hydroxycarbamimidoyl)benzyl)-phenylsulfonamido)-5,5,5-trifluoropentanamide (246 g) was charged to a reactor followed by dry acetonitrile (509 mL), triethyl orthoformate (120 mL), and trifluoroacetic acid (7 mL). The solution was heated to 40-50 C. until the reaction was complete by HPLC (<0.15 relative AP starting material). Methanol (1.48 L) was charged in one portion, followed by water (1.034 L), keeping the batch at 45-50 C. The batch was then cooled to 15-20 C. and filtered. The cake was washed with 2:6:5 MeCN:MeOH:water and tray dried under vacuum at 50-60 C., giving the title compound as a white solid (228 g, 90% yield). 1H NMR, (CDCl3, 300 MHz) delta: 1.40-1.58 (m, 1H) 1.75-1.90 (m, 1H) 1.92-2.07 (m, 1H) 2.10 -2.26 (m, 1H) 4.37 (dd, J=8.67, 6.22 Hz, 1H) 4.48 (d, J=15.64 Hz, 1H) 4.64 (d, J=15.82 Hz, 1H) 5.54 (s, 1H) 6.33 (s, 1H) 7.44-7.54 (m, 2H) 7.62 (t, J=7.72 Hz, 1H) 7.68-7.76 (m, 3H) 7.85 (dd, J=7.91, 1.51 Hz, 1H) 8.76 (s, 1H). 13C NMR, (DMSO-d6, 75 MHz) delta: 170.34, 167.75, 165.80, 159.64 (d, J=244.5 Hz, 1C), 138.19, 137.64, 131.25 (d, J=3.75 Hz, 1C), 129.31, 129.23, 129.05 (d, J=14.25 Hz, 1C), 126.74 (q, J=274.5 Hz, 1C), 126.91, 126.80, 123.12 (d, J=3.75 Hz, 1C), 113.7 (d, J=24.0 Hz, 1C), 57.92, 41.38 (d, J=4.5 Hz, 1C), 30.04 (d, J=30.0 Hz, 1C), 22.90. 19F NMR, (CDCl3, 282 MHz) delta: -116.3, -66.5. IR (KBr): 3454, 334, 3286, 2952, 1705, 1432, 1325, 1260, 1167, 1084, 828 cm-1. Anal. Calcd. for C20H17ClF4N4O4S Calc. C, 46.11; H, 3.29; N, 10.71; S, 6.15; F, 14.58; Cl, 6.80. Found C, 46.06; H, 3.24; N, 10.71; S, 6.25; F, 14.60; Cl, 6.88. |
| 69.3% |
With boron trifluoride diethyl etherate; In 1,1-dichloroethane; at 70℃; for 1.0h;Product distribution / selectivity; |
Step C. (2R)-2-[[(4-Chlorophenyl)sulfonyl][[2-fluoro-4-(1,2,4-oxadiazol-3-yl)phenyl]methyl]amino]-5,5,5-trifluoropentanamide; To a solution of (R)-2-(4-chloro-N-(4-cyano-2-fluorobenzyl)phenylsulfonamido)-5,5,5-trifluoropentanamide (6.5 g, 13.6 mmol) in EtOH (70 mL) was added NH2OH (50% in H2O, 2.6 mL 40.8 mmol). The resulting mixture was stirred at 80 C. under nitrogen for 1 h and then cooled to rt. The solvents were evaporated under reduced pressure. The residue was dissolved in EtOAc and washed with water and dried over Na2SO4. Evaporation of the solvent gave a white solid which was recrystallized from EtOAc and hexanes to afford the intermediate amide oxime as a white solid (6.93 g, quantitative yield). To a mixture of the intermediate (R)-2-(4-chloro-N-(2-fluoro-4-(N'-hydroxycarbamimidoyl)benzyl)phenylsulfonamido)-5,5,5-trifluoropentanamide (6.93 g, 13.6 mmol) and triethyl orthoformate (6.77 mL, 40.8 mmol) in dichloroethane (30 mL) was added BF3.OEt2 (0.17 mL, 1.36 mmol). The resulting mixture was stirred at 70 C. for 1 h and then cooled to room temperature. Chromatography (silica gel, biotage, 40+M column, 3% to 80% EtOAc in hexanes, 651 mL) provided the title compound as a white solid. (4.9 g, 69.3% yield). The above 4.9 g of product was combined with a second lot of 9.8 g of (2R)-2-[[(4-chlorophenyl)sulfonyl][[2-fluoro-4-(1,2,4-oxadiazol-3-yl)phenyl]methyl]amino]-5,5,5-trifluoropentanamide (prepared by the procedure described in Example 1). To the combined lots (14.7 g) was added isopropyl alcohol (75 mL). The mixture was refluxed until almost complete dissolution, and then filtered. The filtrate was stirred at rt for 16 h. and filtered. A white fine crystalline white solid was obtained after drying to a constant mass to afford (2R)-2-[[(4-chlorophenyl)sulfonyl][[2-fluoro-4-(1,2,4-oxadiazol-3-yl)phenyl]methyl]amino]-5,5,5-trifluoropentanamide (13.7 g). 1H NMR (CDCl3, 500 MHz) delta 8.77 (s, 1H), 7.90 (dd, J=8.0, 1.5 Hz, 1H), 7.77-7.71 (m, 3H), 7.64 (dd, J=7.5, 7.5 Hz, 1H), 7.51 (d, J=8.5 Hz, 2H), 6.34 (s, 1H), 5.28 (s, 1H), 4.66 (d, J=15.6 Hz, 1H), 4.51 (d, J=15.6 Hz, 1H), 4.39 (dd, J=8.9, 6.3 Hz, 1H), 2.25-1.82 (m, 3H), 1.54-1.47 (m, 1H); ESI MS m/z 521 [C20H17ClF4N4O4S+H]+. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping