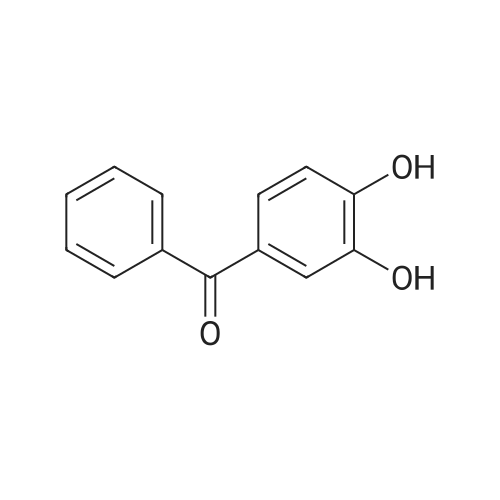
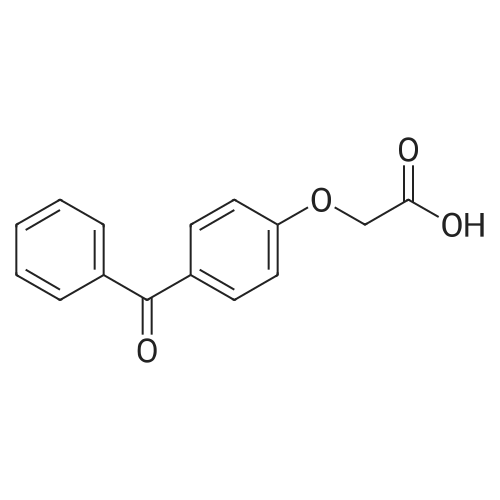
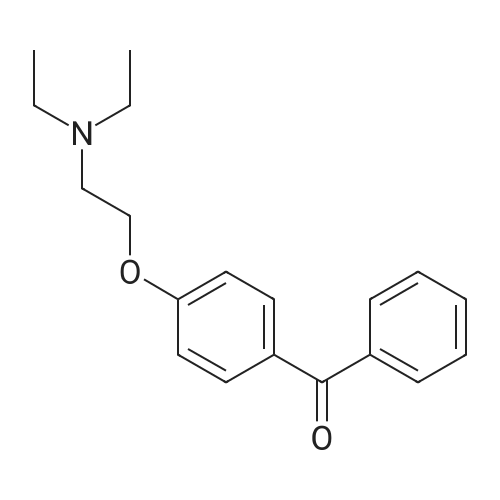
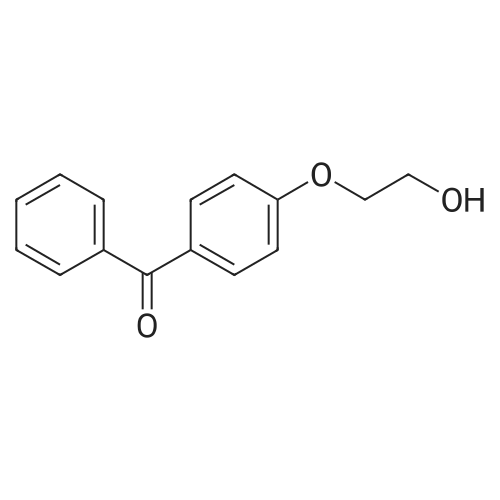
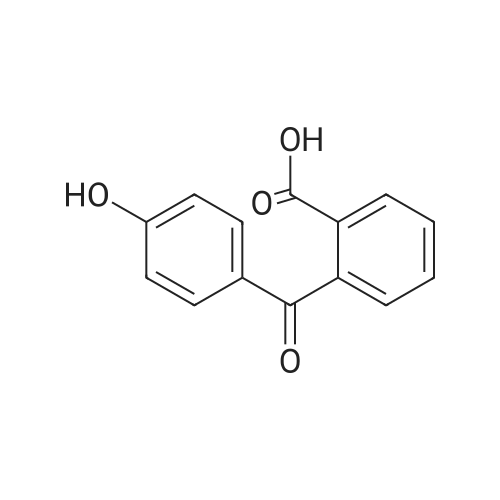
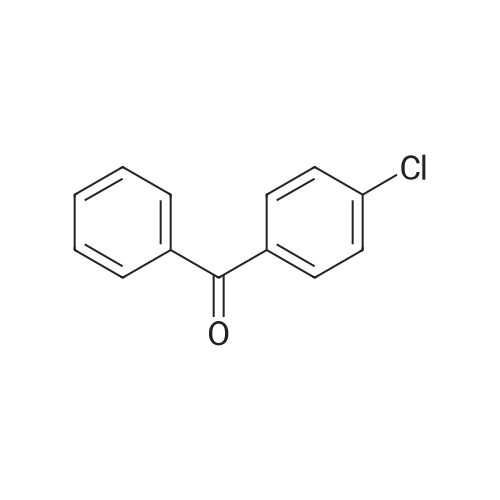
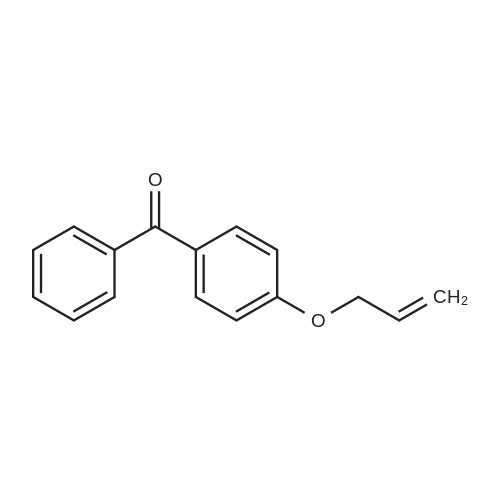
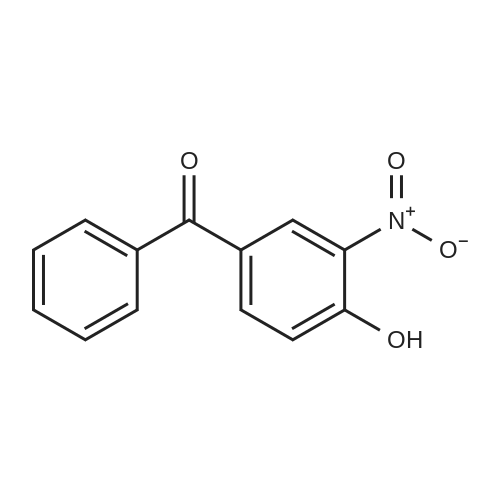
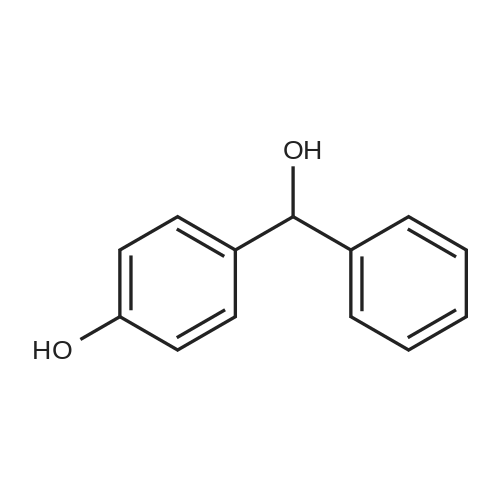
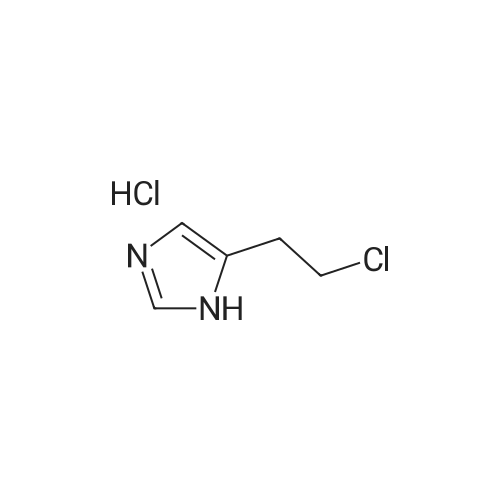
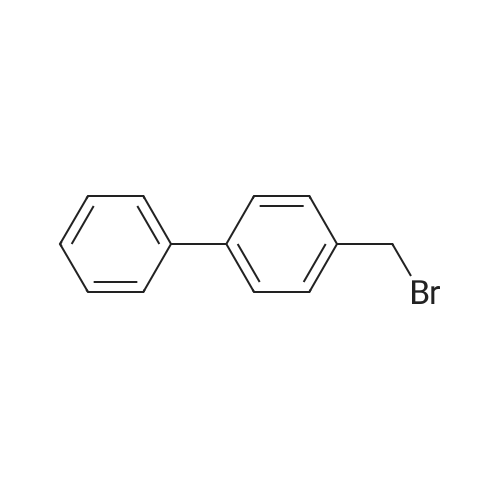
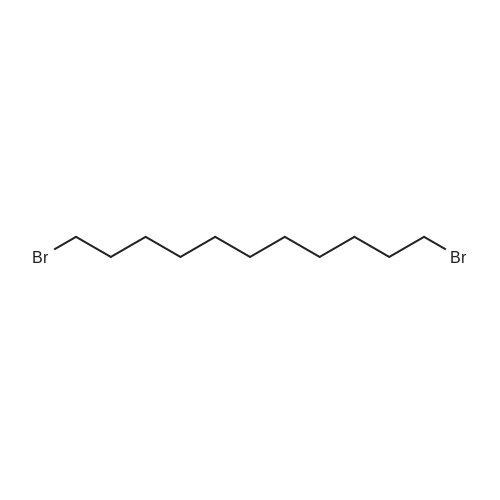
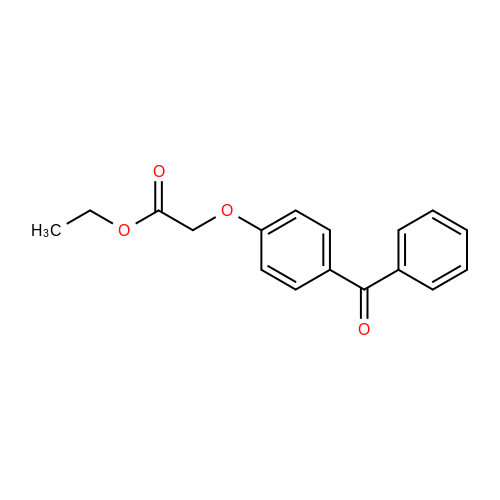
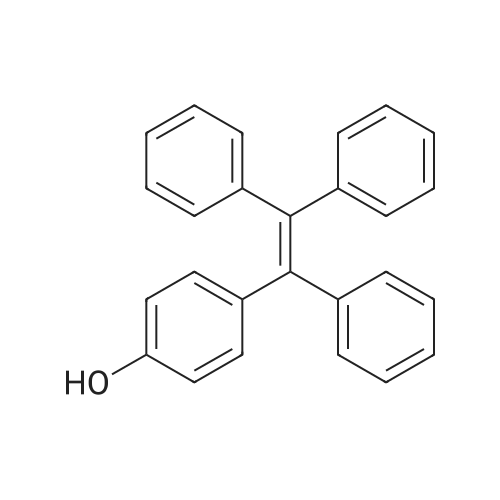
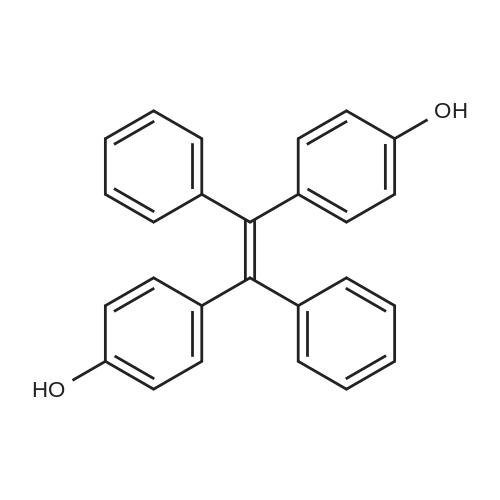
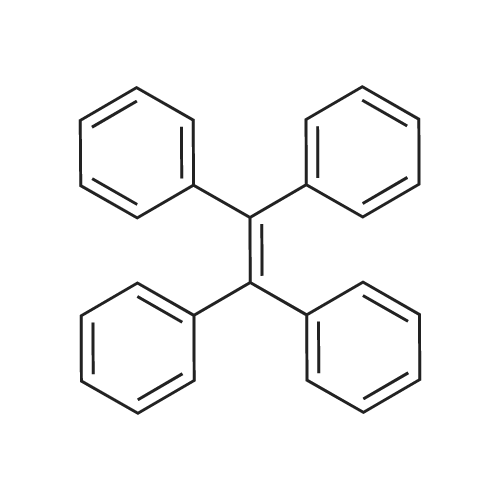
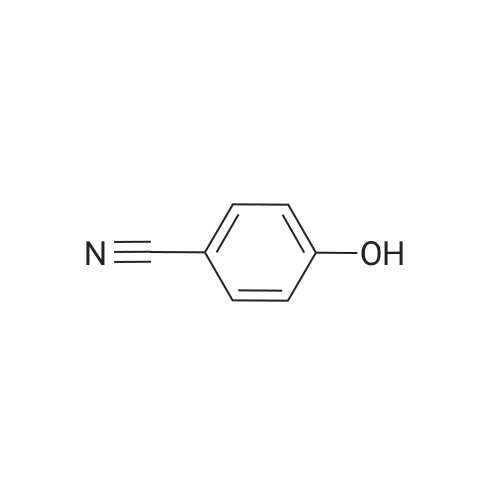
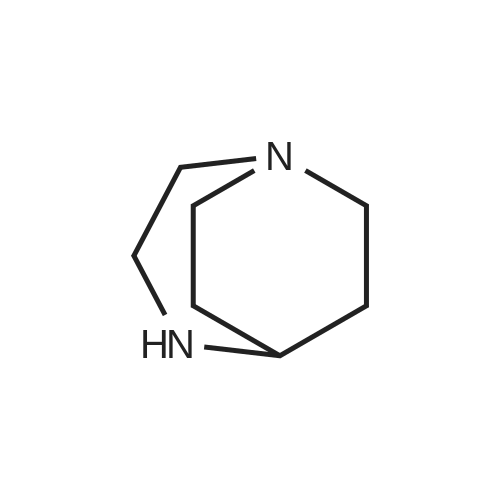
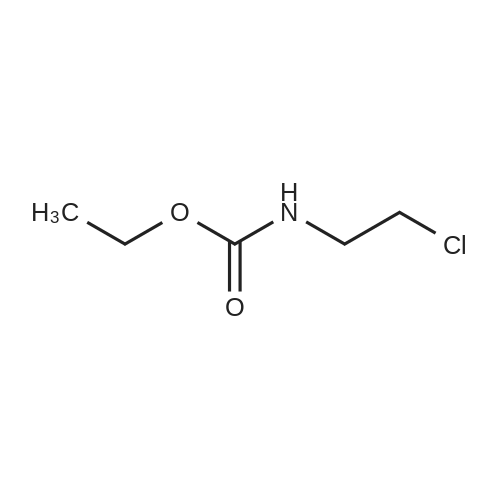
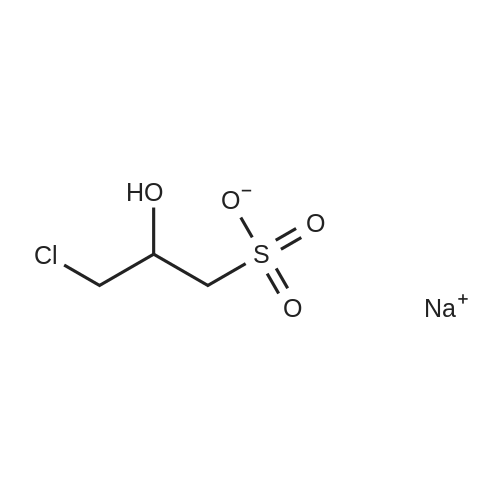
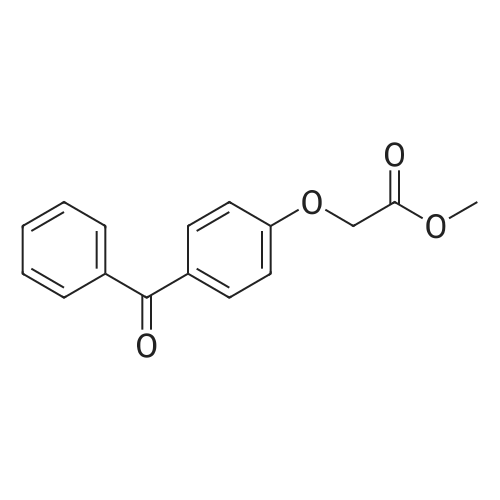
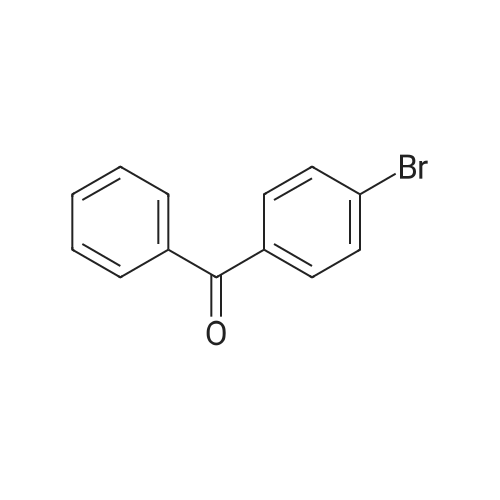
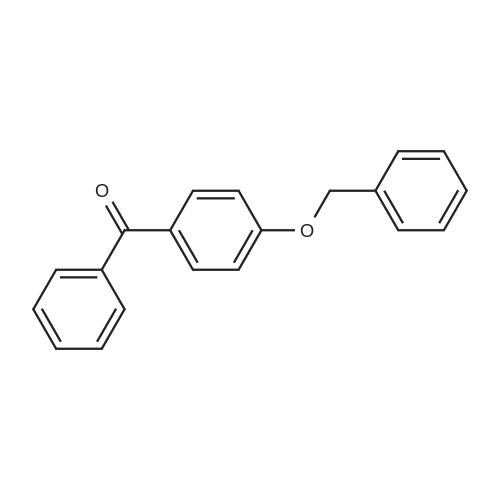
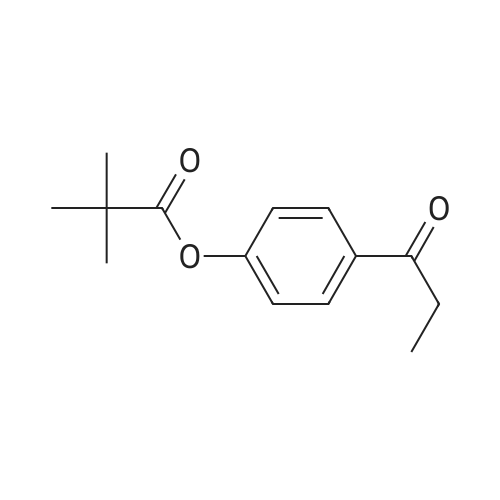
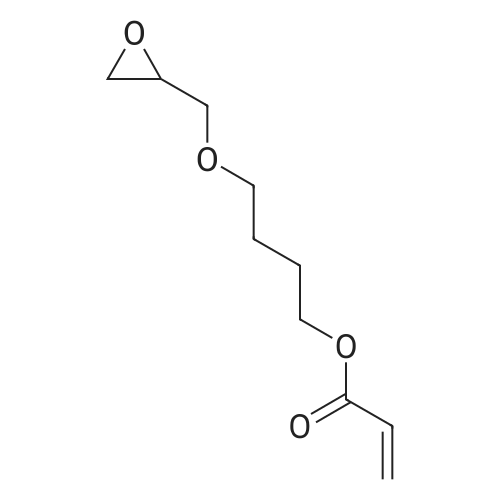
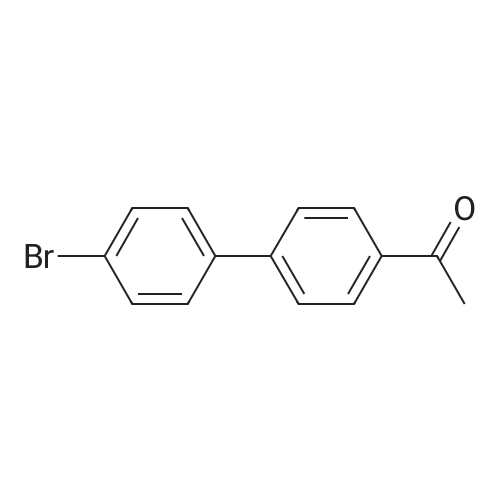
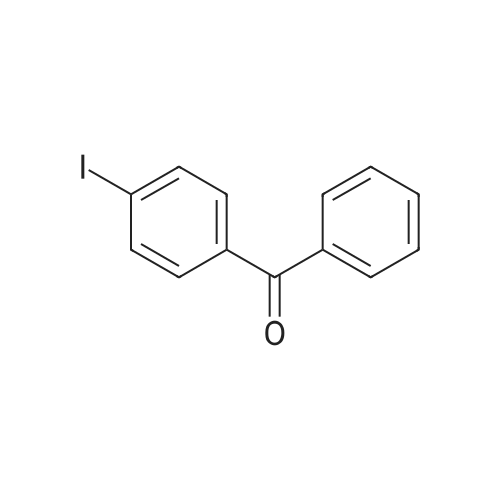
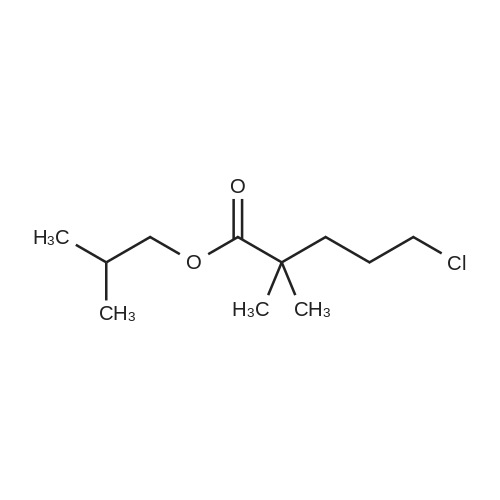
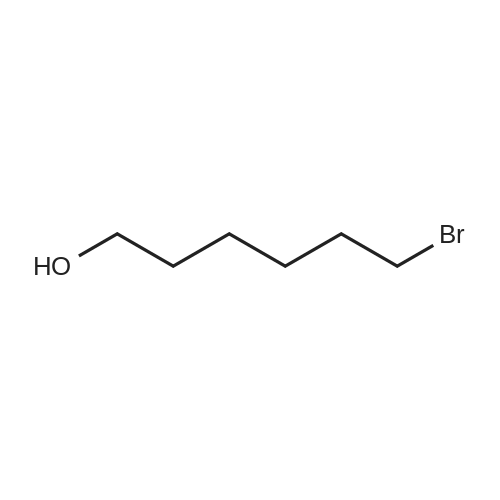
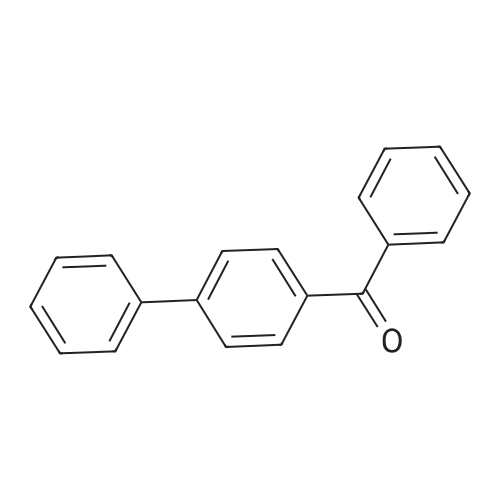
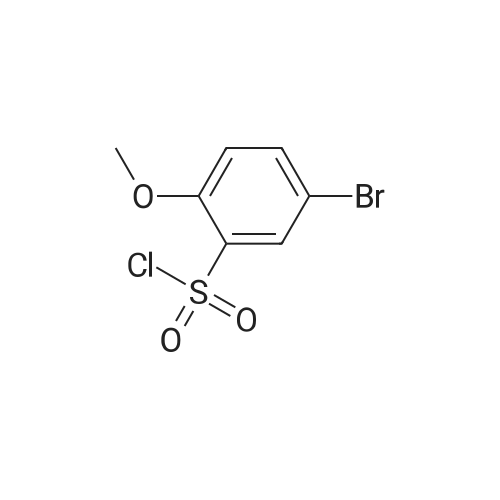
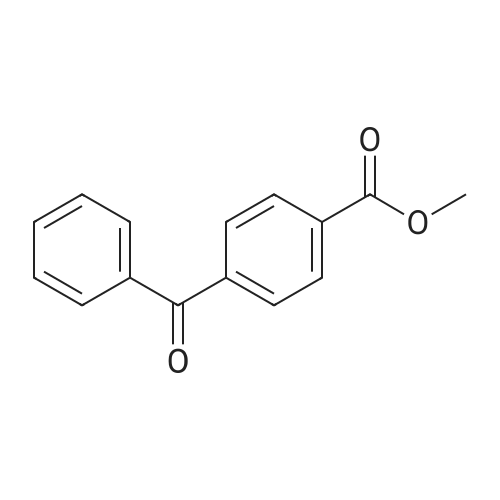
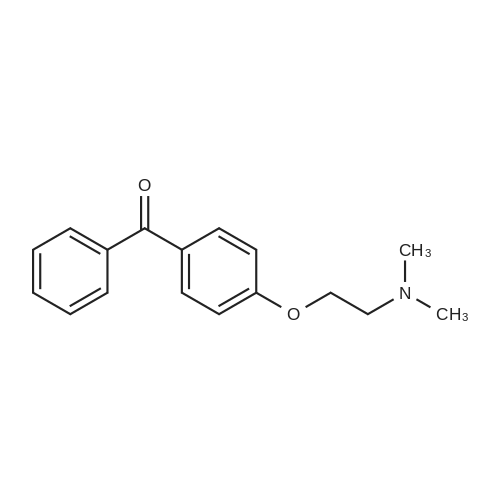
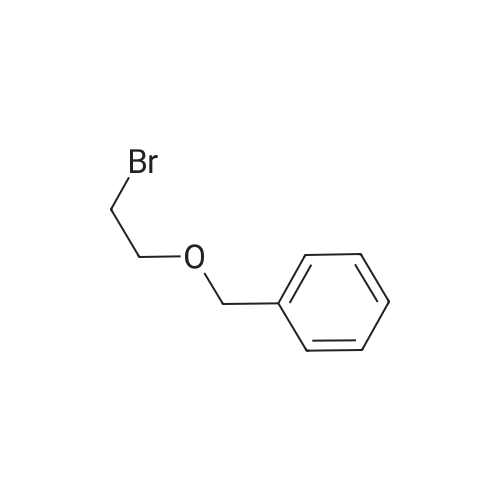
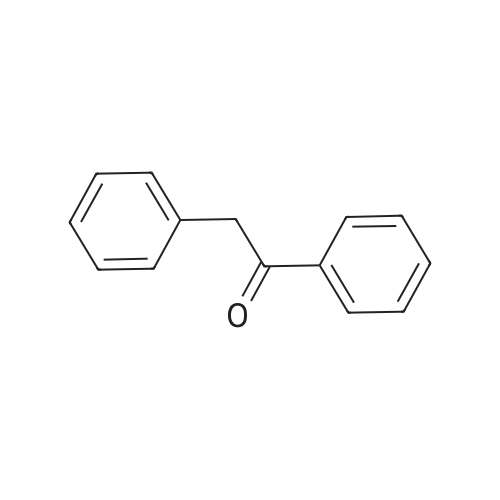
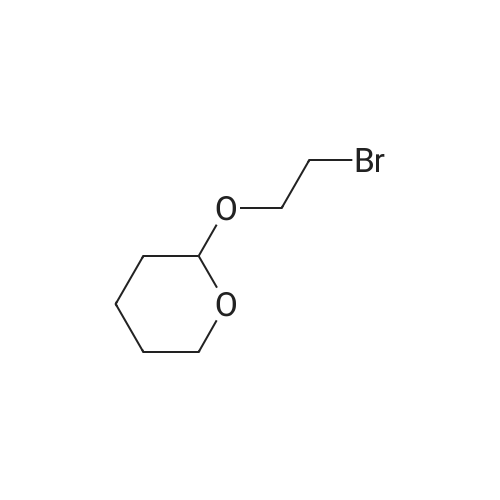
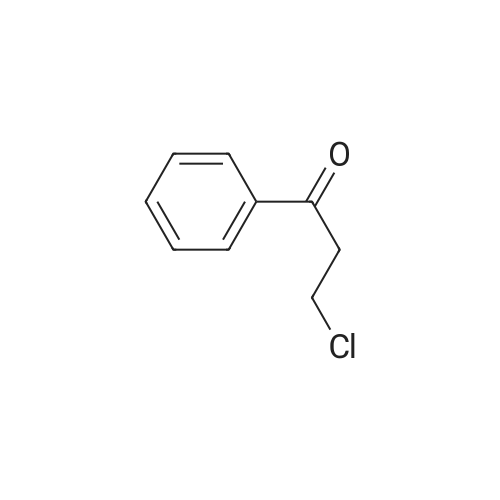
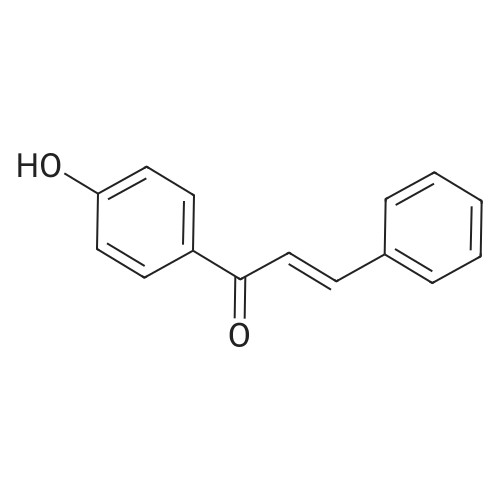
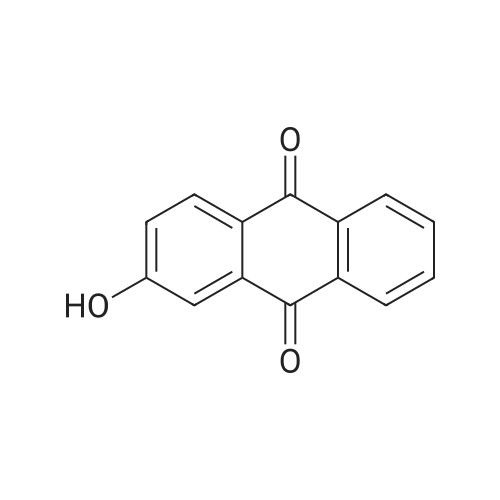
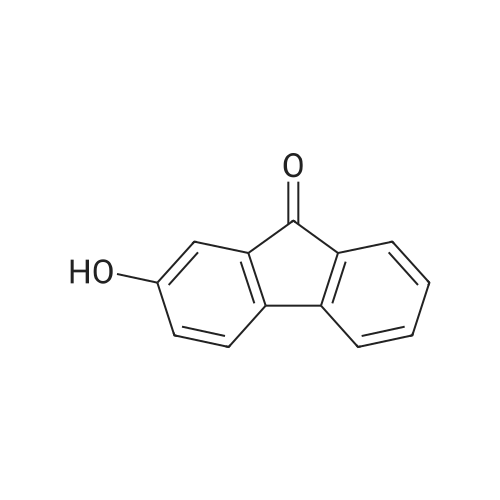
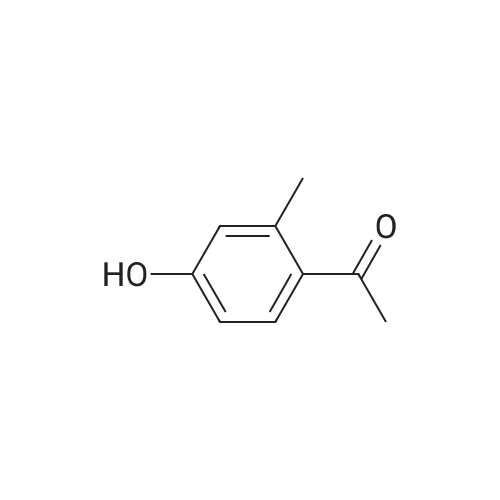
| 74.8% |
With titanium(IV) tetrachloride; zinc powder In tetrahydrofuran for 12h; Inert atmosphere; Reflux; |
|
| 74.5% |
With titanium(IV) tetrachloride; zinc powder In tetrahydrofuran at 70℃; for 24h; Inert atmosphere; Cooling with ice; |
1.1 (1) Preparation of TPE-2OH
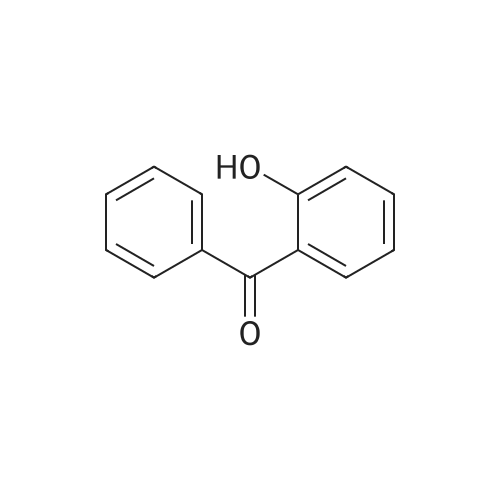
Dissolve 4-hydroxybenzophenone (1.98 g, 1 mmol) in 100 mL of anhydrous tetrahydrofuran, add zinc powder (3.45 g, 0.5 mmol), evacuate and introduce nitrogen for replacement The whole reaction process was carried out under nitrogen atmosphere for three times, and then TiCl4 (2.8 mL) was slowly added dropwise under ice-water bath conditions. After stirring for 30 min, the entire vacuum state of the device was moved to an oil bath at 70 The reaction was refluxed at for 24 hours, and then the reaction was cooled to room temperature, and K2CO3 aqueous solution (20 mL, 10%) was added under vigorous stirring to quench the reaction. The organic phase was separated by extraction with CH2Cl2, and the combined organic phase was dried with anhydrous Na2SO4. The organic layer was rotary evaporated, and the obtained crude product was purified by a silica gel column. The volume ratio of the eluent ethyl acetate to petroleum ether was 1:8, and the product was vacuum dried to obtain the product with a yield of 74.5%. |
| 56% |
With titanium(IV) tetrachloride; zinc powder In tetrahydrofuran at 85℃; for 18h; |
|
| 56% |
With titanium(IV) tetrachloride; zinc powder In tetrahydrofuran for 4h; Reflux; |
|
| 54% |
Stage #1: 4-Hydroxybenzophenone With zinc powder In tetrahydrofuran for 0.25h; Cooling with ice;
Stage #2: With titanium(IV) tetrachloride In tetrahydrofuran at 0℃; for 25h; Reflux; Inert atmosphere; |
4 2.4 Synthesis of 1,2-bis(4-hydroxyphenyl)-1,2-diphenylethene 3
A mixture of 4-hydroxybenzophenone (7.93 g, 0.04 mmol), Zn dust (15.7 g, 0.24 mmol) and 150 mL dry THF was added to a 250 mL round-bottom flask and stirred vigorously for 15 min in an ice water bath. After degasification, titanium tetrachloride (13.2 mL, 0.12 mmol) was added dropwise into above solution and stirred at 0 °C for one hour. Then the mixture was refluxed under argon atmosphere and quenched after 24 h with 10% K2CO3 aqueous solution. The precipitate was removed and the crude product was extracted with CH2Cl2 (200 * 3 mL), dried, evaporated, and purified by silica-gel column chromatography (300-400 mesh, eluent petroleum ether/ethyl acetate 2:1). White solid in 54% yield. 1H NMR (600 MHz, CDCl3), δ (TMS, ppm): 7.12-6.97 (m, 10H), 6.87 (m, 4H), 6.55 (m, 4H). |
| 54% |
With titanium(IV) tetrachloride; zinc powder In tetrahydrofuran Reflux; |
|
| 53% |
Stage #1: 4-Hydroxybenzophenone With zinc powder In tetrahydrofuran at 0℃; for 0.25h;
Stage #2: With titanium(IV) tetrachloride In tetrahydrofuran for 25h; Cooling with ice; Reflux; Inert atmosphere; |
2.2. Synthesis of TPE-2OH
To a solution of 4-hydroxybenzophenone (3.97 g, 0.02 mol) in dryTHF (80 mL) was added Zn dust (7.85 g, 0.12 mol). The mixture wasstirred vigorously for a quarter of an hour to cool to 0 °C. Into the abovemixture was dropwise added titanium tetrachloride (6.6 mL, 0.06 mol)and stirred in an ice water bath for 1 h. Then it was gradually heated toreflux under argon atmosphere for 24 h. The reaction was allowed to becooled to room temperature and quenched with 10% K2CO3 aqueoussolution and the newly formed precipitate was removed. The residuewas extracted three times with 100 mL CH2Cl2, dried, filtered. After allof the solvent CH2Cl2 has been evaporated under reduced pressure, the target product was further purified by silica-gel column chromatography(300-400 mesh, eluent ethyl acetate/petroleum ether 1:2 vol).White solid in 53% yield. 1H NMR (600 MHz, CDCl3), δ (TMS, ppm):7.13-6.98 (m, 10H), 6.91-6.85 (m, 4H), 6.59-6.54 (m, 4H). 13C NMR(150 MHz, CDCl3), δ (TMS, ppm): 154.03, 144.28, 144.17, 139.82,139.80, 136.79, 136.71, 132.87, 132.85, 131.51, 131.50, 127.82,127.72, 126.38, 114.79, 114.71. MS(ESI) m/z: [M-H]- calcd C26H20O2363.1391, found 363.1387. |
| 51.6% |
With titanium(IV) tetrachloride; zinc powder In tetrahydrofuran at -78 - 85℃; for 24h; Inert atmosphere; |
1.1 Example 1: N1,N1'-((((1,2-diphenylethylene-1,2-)bis(4,1-phenylene))bis(oxy))bis(4,1-ylidene (Phenyl)) bis(N1-(4-methoxy)benzene-1,4-diamine) preparation
The first step reaction: add 24g of 4-hydroxybenzophenone, 15.7g zinc powder, 500mL tetrahydrofuran and then cooled to -78, Under stirring and nitrogen protection, slowly add 23g of titanium tetrachloride dropwise, After returning to room temperature, it was heated to 85°C and reacted for 24 hours. After the mixture was cooled to room temperature, 500mL, 10wt% potassium carbonate solution quenches the reaction, After filtration, the organic layer was collected. The aqueous layer of the filtrate was extracted three times with ethyl acetate. After the organic phases were combined and dried over magnesium sulfate overnight, After the ethyl acetate and tetrahydrofuran were distilled off, a solid crude product was obtained. The crude product was recrystallized from methanol, After filtration and drying, 12.4 g of white crystals of 1,2-bis(4-hydroxybenzene)-1,2-stilbene were obtained, with a yield of 51.6%. |
| 46% |
With pyridine; titanium(IV) tetrachloride; zinc powder In tetrahydrofuran |
|
| 20% |
With titanium(IV) tetrachloride; zinc powder In tetrahydrofuran at 0℃; Inert atmosphere; Reflux; |
|
|
With titanium(IV) tetrachloride; zinc powder In tetrahydrofuran Reflux; |
|
|
With titanium(IV) tetrachloride; zinc powder In tetrahydrofuran for 4h; Inert atmosphere; Reflux; |
|
|
With titanium(IV) tetrachloride; zinc powder In tetrahydrofuran |
|
|
With titanium(IV) tetrachloride; zinc powder |
|
|
With titanium(IV) tetrachloride; zinc powder In tetrahydrofuran at -30 - 90℃; for 13h; |
1.1 1. Synthesis of tetrastyrene derivatives
Add 4-hydroxybenzophenone (0.2mol, 40.0g) and zinc powder (0.4mol, 25.9g) to a 1000mL four-neck flaskRepeatedly pump oxygen-nitrogen three times in a closed conditionAdd another 600 mL of refined THF,Slowly add titanium tetrachloride (0.3mol, 32.75mL) while stirring at -30 ° C. After the dropwise addition is complete, stir at room temperature for 1h.Then, the reaction was stirred under reflux at 90 ° C for 12 hours.After cooling, pour into 2L beaker,While stirring, add 10% potassium carbonate solution dropwise until the layers are separated.Filtered, the filtrate was three times with dichloromethane,The organic phase was dried over anhydrous magnesium sulfate for 4 h, and then filtered, and the filtrate was spin-dried to obtain a crude product.The crude product was dried and recrystallized with a toluene: petroleum ether = 4: 1 (v: v) mixed solvent.The product was obtained as a pale yellow solid. |
|
With titanium(IV) tetrachloride; zinc powder In tetrahydrofuran |
|
|
With titanium(IV) tetrachloride; zinc powder In tetrahydrofuran |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping