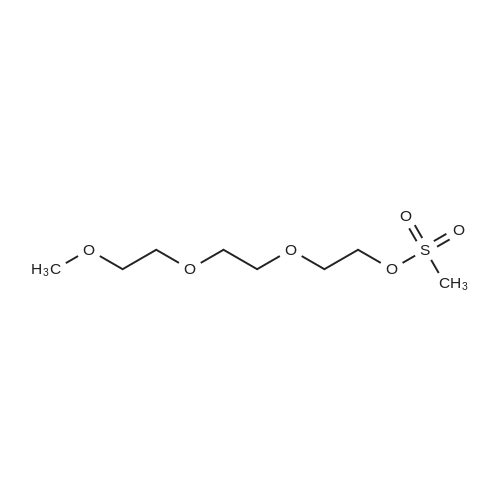
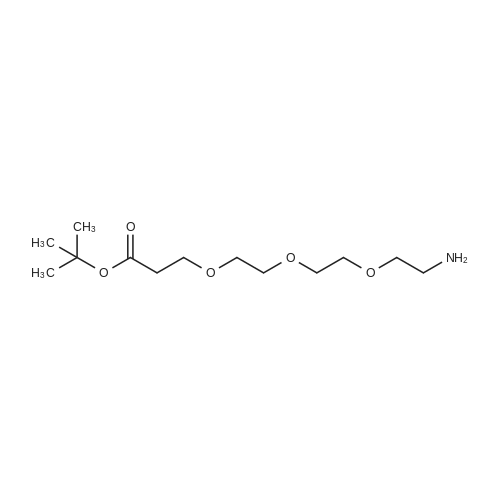
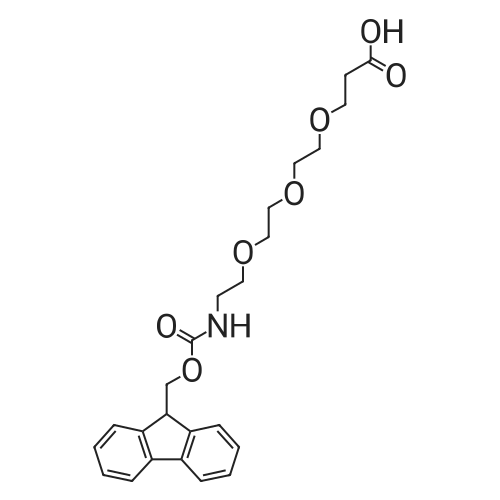
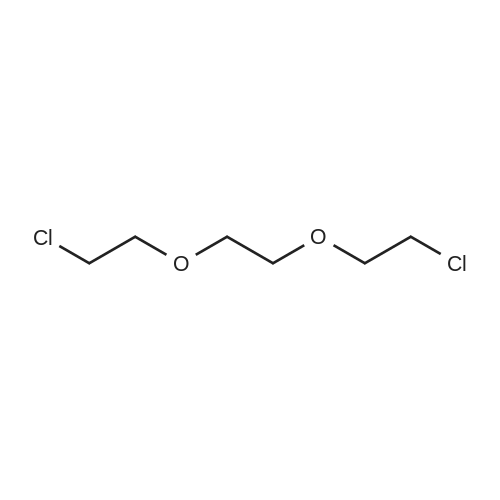
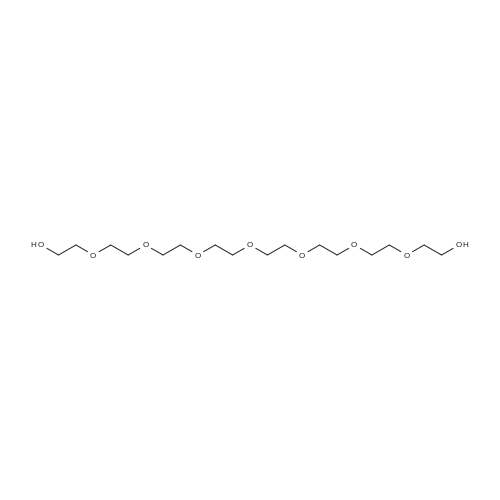
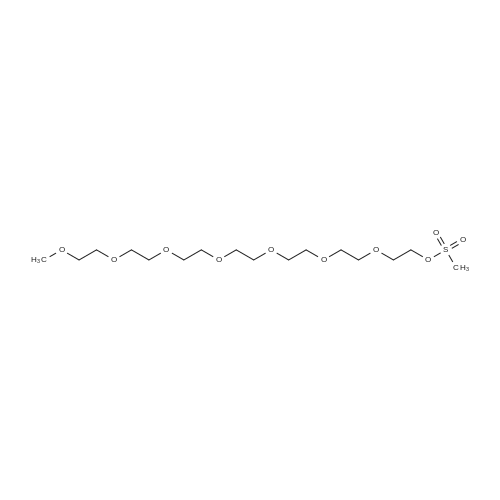
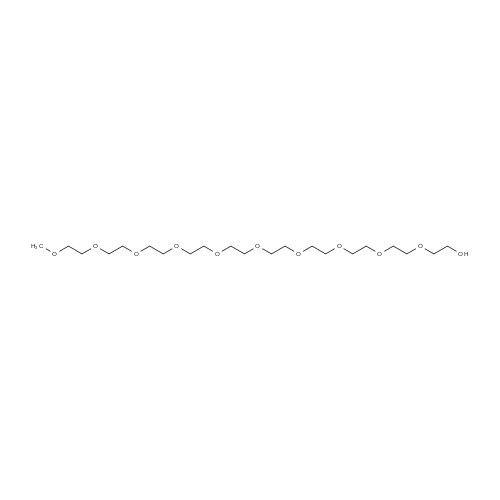
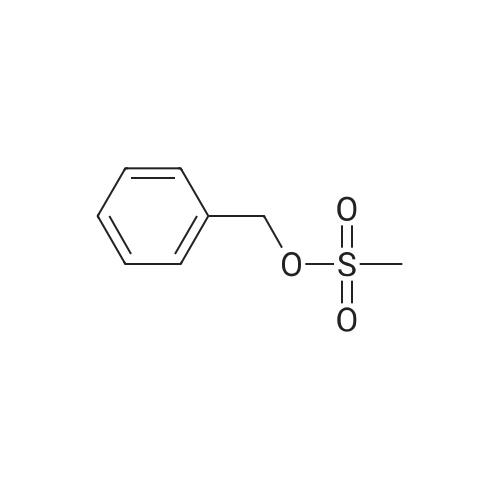
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With potassium hydroxide In dichloromethane at 0 - 10℃; for 3 h;
General procedure: Compounds 2a–c were prepared by a modified procedure known from the literature1. Oligo ethylene glycol(37.7 mmol) and p-toluenesulfonyl chloride (14.4 g, 75.4 mmol, 2 equiv) were dissolved in dichloromethane (36 mL)and this solution was cooled to 0 °C. During permanent stirring, milled potassium hydroxide (17 g, 302 mmol, 8equiv) was added and the solution was then stirred for 3 hours while the temperature was kept from 0 to 10 °C. This reaction was monitored by TLC (UV detection). After warming up to room temperature the reaction mixture was diluted by chloroform and extracted with water (3 × 1:1). Chloroform extracts were collected, dried over magnesium sulfate overnight, filtered and solvents were evaporated under reduced pressure.
96%
With potassium hydroxide In dichloromethane at 0℃; for 5 h;
Triethylene glycol (2.25 g, 15 mmol) and p-toluenesulfonyl chloride (5.72 g, 30 mmol)Dissolved in 15 ml of anhydrous dichloromethane, cooled to 0 ° C, fractionated under stirring,A small amount of KOH (6.72 g, 120 mmol) was added and the temperature was maintained at 0 ° C. After stirring at 0 ° C for 5 h,The reaction was quenched by the addition of 15 ml of dichloromethane and 30 ml of ice water. Separating the organic layer,The aqueous layer was extracted twice with 15 ml of dichloromethane, the combined organic layers were washed and washed once with 10 ml of water,After drying over anhydrous sodium sulfate, the solvent was removed by distillation under reduced pressure to give 6.6 g of the title compound as a white solid, yield 96percent.
94%
Stage #1: With triethylamine In dichloromethane at 20℃; for 0.333333 h; Stage #2: With dmap In dichloromethane at 20℃; for 16 h;
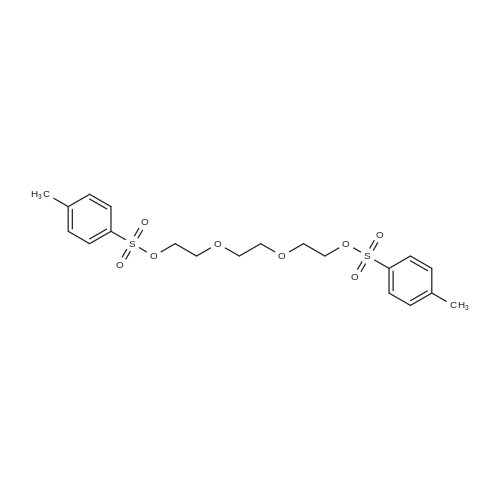
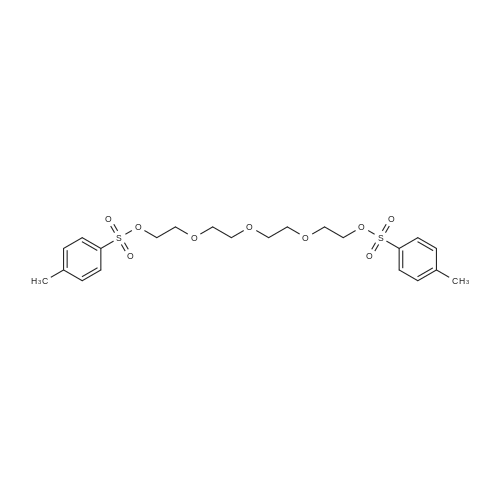
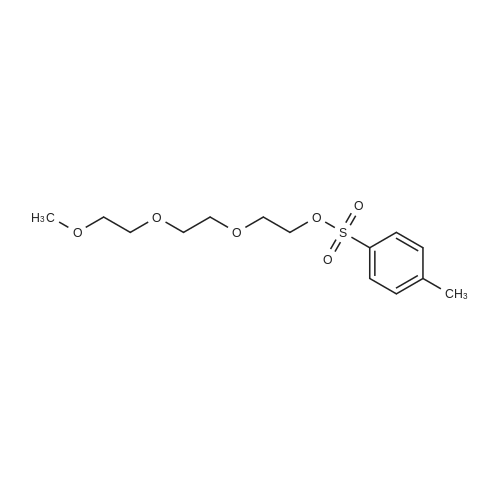
To solution of triethylene glycol (3.0 g, 20.0 mmol) in dichloromethane (20 mL) was added Et3N (6.06 g, 60.0 mmol) and reaction mixture was stirred at R.T. for 20 min. To reaction mixture were added p-toluene sulfonyl chloride (7.62 g, 40.0 mmol) and DMAP (0.12 g, 1 mmol), reaction mixture was stirred at room temperature for 16 hrs. Solvent was evaporated under vacuum, residue obtained was dissolved in ethyl acetate. Ethyl acetate layer was washed with 1N-HCl, brine and dried over Na2SO4. Organic layer was concentrated and purified on flash column to obtained (ethane-1,2-diylbis(oxy))bis(ethane-2,1-diyl) bis(4-methylbenzenesulfonate) (5A) in 94 percent yield (8.58 g). 1H NMR (400 MHz, DMSO-d6) δ 7.81 (d, J = 8.2 Hz, 4H), 7.50 (d, J = 7.8 Hz, 4H), 4.13 (t, J = 4.3 Hz, 4H), 3.57 (t, J = 4.3 Hz, 4H), 3.41 (s, 4H), 2.45 (s, 6H); 13C NMR (100 MHz, DMSO-d6) δ 145.44, 132.89, 130.67, 128.15, 70.47, 70.08, 68.40, 21.61
92%
With triethylamine In dichloromethane at 20℃; for 2 h;
To a solution of triethylene glycol (750 mg, 1.0 equiv.) in DCM (20 mL) was added triethylamine (2.1 mL, 3.0 equiv.) then tosyl chloride (2.29 g, 2.4 equiv.) portion- wise and the reaction mixture was stirred at room temperature for 2 h. The reaction mixture was washed with saturated NH4C1 solution (20 mL x 3), then by brine. The resulting mixture was subsequently dried and evaporated. The residue was purified using flash column in the eluent of ethyl acetate/hexane (1/5, then 1/1) to give a white powder (2.11 g, 92percent). 1H NMR (500 MHz, DMSO) δ 7.77 (d, J = 8.0 Hz, 4H), 7.46 (d, J = 8.0 Hz, 4H), 4.09 (t, 4H), 3.53 (t, 4H), 3.38 (t, 4H), 2.41 (s, 6H); 13C NMR (125 MHz, DMSO) δ 145.3, 132.9, 130.6, 128.1, 70.4, 70.0, 68.3, 21.5; LCMS found m/z 459.5: (M+l).
91.4%
With sodium hydroxide In tetrahydrofuran; water at 5℃; for 2 h; Large scale
16 kg (400 mol) of sodium hydroxide was dissolved in 80 L of water in a 400 L reactor, and then 18.8 L (140 mol) of triethylene glycol, 32 L of THF were added intothe reactot Afier cooling below 5° C., a solution of 47.84 kg(260 mol) of tosyl chloride and 50 L of THF was added dropwise. Following the addition, the reaction mixture waskept at this temperature for 2 hours, and it was then poured into 240 L of ice watet The precipitate was formed andfiltered, washed with a small amount of water, and dried.58.64 kg ofBPI-01 as a white crystalline powder was yielded at 91.4percent. mp: 77-80° C., HPLC: 97percent. TLC (petroleum ether:ethyl acetate=1:1) Rf=0.87.NMR data: ‘H-NMR (CDC13): ö ppm: 7.78 (d, 4H, J=10.4 Hz, benzene protons by sulfonyl group); 7.34 (d, 4H, J=1 1.6Hz, benzene protons by methyl group); 4.129 (dd, 4H, J=5.6Hz, ethylene protons by the sulfonyl group); 3.64 (dd, 4H,J=5.6 Hz, ethylene protons away from the sulfonyl group); 3.5 17 (s, 4H, ethylene protons in the middle); 2.438 (s, 6H, methyl protons on the benzene).
91.4%
With sodium hydroxide In tetrahydrofuran; water at 5℃; for 2 h; Large scale
Preparation: 16 kg (400 mol) of sodium hydroxide was dissolved in 80 L of water in a 400 L reactor, and then 18.8 L (140 mol) of triethylene glycol, 32 L of THF were added into the reactor. After cooling below 5° C., a solution of 47.84 kg (260 mol) of tosyl chloride and 50 L of THF was added dropwise. Following the addition, the reaction mixture was kept at this temperature for 2 hours, and it was then poured into 240 L of ice water. The precipitate was formed and filtered, washed with a small amount of water, and dried. 58.64 kg of BPI-01 as a white crystalline powder was yielded at 91.4percent. mp: 77-80° C., HPLC: 97percent. TLC (petroleum ether:ethyl acetate=1:1) Rf=0.87.
90%
With potassium hydroxide In dichloromethane for 8 h; Cooling with ice
The 10mL (0.075mol) of triethylene glycol,300ml dichloromethane,28.6g (0.15mol) of methyl chloride was added 500ml round-bottomed flask,Under ice-cooling,Was slowly added 33.6g (0.6mol) of potassium hydroxide,8h the reaction is substantially complete.The reaction solution was washed three times with water (3 × 100ml),The organic layer was collected,Dried over anhydrous sodium sulfate.filter,Spin dry solvent,The crude product was recrystallized from methanol,To give a white solid 30g,The yield was 90percent.
90.1%
With dmap; triethylamine In dichloromethane at 20℃; for 3.5 h; Cooling with ice
434 mg of triethylene glycol (2.9 mmol) was dried in an oven at 95 ° C for 2 hours,Was dissolved in 43.5 mL of dichloromethane,1.06 g of 4-dimethylaminopyridine (8.7 mmol) and 400 μL of triethylamine (2.9 mmol)1.55 g of p-toluenesulfonyl chloride (8.1 mmol) was added under ice-cooling,Stirring for 0.5 hours, removing the ice bath and reacting at room temperature for 3 hours;15 mL of deionized water was added, extracted, the organic phase was collected, the organic phase was washed with brine (50 g / L)Add 100mg anhydrous sodium sulfate drying, filtration, rotary evaporation to remove the organic solvent,The resulting solid was purified by silica gel chromatography (eluent: dichloromethane / methanol, V / V = 9: 1) to give solid 1,2-bis (2-toluenesulfonylethoxy) ethane(Yield 90.1percent).
88%
Stage #1: With potassium hydroxide In dichloromethane at 0 - 20℃; Stage #2: at 20℃; for 10 h;
General procedure: KOH (20 g, 356.5 mmol for 9; and 10 g, 181.5 mmol for 10–11) was added to 300 mL of a CH2Cl2 solution containing the oligoethylene glycol (10 g,94 mmol for 9, 66.6 mmol for 10, and 51.5 mmol for 11) at 0 °C. After stirring for 30 min at room temperature, TsCl (45 g, 236 mmol for 9–11) was added and the resulting solution was stirred for 10 h at room temperature. The resulting solution was filtered and then washed with aq. K2CO3. The organic layer was dried with MgSO4, filtered, and concentrated in vacuo. The resulting residue was then subjected to silicagel column chromatography (EtOAc:Hex = 1:1) to yield 35 g of 9 (90percent), 27 g of 10 (88percent), and 23 g of 11 (89percent).
87%
With triethylamine In dichloromethane at 20℃; for 12 h;
To a solution of 14 (4.5 g, 30.0 mmol) and TsCl (12.59 g, 66.0 mmol) in DCM (100 mL) was added NEt3 (6.68 g, 66.0 mmol). The reaction mixture was stirred at rt for 12 h and washed with water. The organic phase was dried over anhydrous Na2SO4. After removal of the solvent, the residue was purified by flash column chromatography (0-40percent EtOAc/hexanes) to afford 15 as a white solid (12.0 g, 87percent). 1H NMR (CDCl3, 600 MHz) δ 7.78 (d, J = 7.8 Hz, 4H), 7.34 (d, J = 7.8 Hz, 4H), 4.19-4.10 (m, 4H), 3.70-3.62 (m, 4H), 3.57-3.48 (m, 4H), 2.44 (s, 6H). HRMS (ESI+) calcd for C20H26NaO8S2 (M+Na)+ 481.0961, found 481.0963.
87%
With triethylamine In dichloromethane at 20℃; for 8 h;
p-toluenesulfonyl chloride (40.63 g, 213.09 mmol) was dissolved in methylene chloride (MC, 150 mL) in a 500 mL 1 neck round flask and, then, triethyleneglycol (16 g, 105.55 mmol) and triethylamine (29.72 mL, 213.09 mmol) were added. The reacting mixture was stirred at room temperature for 8 hr. After reaction, 200 mL of distilled water was added and extraction was carried out by using 600 mL of chloroform (CHCl3). After removing the solvents, products were separated by column chromatography (silica, hexane/ethyl acetate=1:1 (v/v)). (yield=42.59 g, 87percent)1H NMR (400 MHz, CDCl3): δ 7.80-7.78 (d, 4H), 7.35-7.33 (d, 4H), 4.15-4.12 (t, 4H), 3.66-3.64 (t, 4H), 3.52 (s, 4H), 2.44 (s, 6H). (See, FIG. 9).
76%
With triethylamine In tetrahydrofuran at 20℃; for 12 h; Inert atmosphere
To a solution of triethylene glycol (6 g, 40.00 mmol) in THF (80 mL) was added Et3N (10.12 g, 100.00mmol). Tosyl chloride (15.6 g, 80.00 mmol) was added and stirred at room temperature for 12 h. Thesolvent was removed under reduced pressure and the residue was dissolved in EtOAc. The organic layerwas washed with aqueous 1N HCl and brine. The organic layer was dried over Na2SO4, and concentratedunder reduced pressure. The crude residue was purified by flash column chromatography (EtOAc: Hexane= 10: 1) to afford compound 19 as a clear colorless oil in 76 percent yield (14 g). This compound was alreadyreported.2 1H NMR (400 MHz, DMSO-d6) δ 7.81 (d, J = 8.2 Hz, 4H), 7.50 (d, J = 7.8 Hz, 4H), 4.13 (t, J =4.3 Hz, 4H), 3.57 (t, J = 4.3 Hz, 4H), 3.41 (s, 4H), 2.45 (s, 6H); 13C NMR (100 MHz, DMSO-d6) δ 144.89,132.38, 130.13, 127.63, 69.98, 69.82, 69.70, 69.65, 69.19, 67.86, 65.53, 21.09, 15.11. ESI-MS: m/z = 459(M + H)+.
64%
With triethylamine In diethyl ether at 20℃; for 18 h; Inert atmosphere
To a solution of triethylene glycol (3 g, 0.02 mol) in diethyl ether (40 mL), triethylamine (7.8 g, 0.04 mol) was added at room temperature, followed by addition of p-toluenesulfonyl chloride (7.8 g, 0.04 mol) under argon atmosphere. The mixture was stirred at room temperature for 18 h (TLC monitoring, ethyl acetate/hexanes, 1:4). The organic solvents were removed under reduced pressure. The residue was dissolved in methylene chloride (20 mL), washed with sodium bicarbonate (20 mL, saturated), water (20 mL .x. 2), brine (30 mL), dried over magnesium sulfate and concentrated under vacuum. Colorless crystals of triethylene glycol ditosylate were obtained from ethyl acetate.Yield = 5.8 g, 64percent mp: 75-77 °C. Rf = 0.5 (ethyl acetate/hexanes, 1:4). 1H NMR (300 MHz, CDCl3): δ 7.78 and 7.34 (AA'BB', 8H), 4.12 (d, 4H), 3.65 (t, 4H), 3.53 (s, 4H), 2.45 (s, 6H). 13C NMR (75 MHz, CDCl3, δ 145.1, 133.3, 130.1, 128.2, 70.9, 69.4, 69.0, 21.8.
13.74 g
With sodium hydroxide In tetrahydrofuran; water
5 g (33.3 mmol) of triethylene glycol and 13.96 g (73.2 mmol) of 4-toluenesulfonyl chloride were dissolved in tetrahydrofuran 100ml and stirred. Then, 4.66 g (116.5 mmol) of sodium hydroxide was dissolved in the minimum amount of water," After completion of the reaction, diethyl ether and water were added to separate the layers, and the mixture was concentrated under reduced pressure,To give 13.74 g (29.97 mmol) of the titled compound.
Reference:
[1] Organic and Biomolecular Chemistry, 2006, vol. 4, # 11, p. 2082 - 2087
[2] Bioorganic and Medicinal Chemistry, 2007, vol. 15, # 14, p. 4841 - 4856
[3] Organic and Biomolecular Chemistry, 2015, vol. 13, # 30, p. 8335 - 8348
[4] Journal of Medicinal Chemistry, 2016, vol. 59, # 17, p. 7840 - 7855
[5] Journal of Organic Chemistry, 1992, vol. 57, # 24, p. 6678 - 6680
[6] Bioorganic and Medicinal Chemistry Letters, 2009, vol. 19, # 5, p. 1421 - 1424
[7] Journal of the American Chemical Society, 2012, vol. 134, # 45, p. 18809 - 18815
[8] Beilstein Journal of Organic Chemistry, 2016, vol. 12, p. 349 - 352
[9] Tetrahedron Letters, 2001, vol. 42, # 50, p. 8781 - 8783
[10] Patent: CN105218536, 2016, A, . Location in patent: Paragraph 0039-0040
[11] Reactive and Functional Polymers, 2011, vol. 71, # 9, p. 948 - 957
[12] Organic and Biomolecular Chemistry, 2017, vol. 15, # 17, p. 3681 - 3705
[13] Tetrahedron, 2007, vol. 63, # 23, p. 5083 - 5087
[14] Chinese Journal of Chemistry, 2013, vol. 31, # 1, p. 119 - 122
[15] Bioorganic and Medicinal Chemistry Letters, 2018, vol. 28, # 5, p. 915 - 921
[16] Phosphorus, Sulfur and Silicon and the Related Elements, 2006, vol. 181, # 1, p. 219 - 225
[17] Patent: WO2018/94005, 2018, A1, . Location in patent: Page/Page column 73
[18] Journal of Organic Chemistry, 1994, vol. 59, # 8, p. 2186 - 2196
[19] Patent: US9359370, 2016, B2, . Location in patent: Page/Page column 9; 10
[20] Patent: JP2015/110649, 2015, A, . Location in patent: Paragraph 0043
[21] Canadian Journal of Chemistry, 1997, vol. 75, # 11, p. 1472 - 1482
[22] Chemical Communications, 2005, # 4, p. 513 - 515
[23] Macromolecules, 2016, vol. 49, # 11, p. 4102 - 4114
[24] Patent: CN105384745, 2016, A, . Location in patent: Paragraph 0057; 0058; 0059
[25] Patent: CN106632101, 2017, A, . Location in patent: Paragraph 0036; 0038; 0043; 0049
[26] Bulletin of the Chemical Society of Japan, 1990, vol. 63, # 4, p. 1260 - 1262
[27] Journal of Organic Chemistry, 1984, vol. 49, p. 1408 - 1412
[28] Bulletin of the Chemical Society of Japan, 1990, vol. 63, # 4, p. 1260 - 1262
[29] Journal of Polymer Science, Part A: Polymer Chemistry, 2013, vol. 51, # 18, p. 3975 - 3984
[30] Organic Letters, 2015, vol. 17, # 4, p. 1018 - 1021
[31] Bulletin of the Korean Chemical Society, 2015, vol. 36, # 6, p. 1654 - 1660
[32] Chemical Communications, 2016, vol. 52, # 45, p. 7310 - 7313
[33] Chemical Biology and Drug Design, 2017, vol. 89, # 4, p. 559 - 565
[34] Journal of Organic Chemistry, 1999, vol. 64, # 18, p. 6870 - 6873
[35] Bioorganic and Medicinal Chemistry, 2016, vol. 24, # 4, p. 686 - 692
[36] Patent: US9670412, 2017, B2, . Location in patent: Page/Page column 7; 8
[37] Organic and Biomolecular Chemistry, 2005, vol. 3, # 11, p. 2114 - 2121
[38] Chemical Communications, 2012, vol. 48, # 45, p. 5650 - 5652
[39] Russian Journal of Organic Chemistry, 2012, vol. 48, # 10, p. 1345 - 1352[40] Zh. Org. Khim., 2012, vol. 48, # 10, p. 1350 - 1357,8
[41] Collection of Czechoslovak Chemical Communications, 1987, vol. 52, # 8, p. 2057 - 2060
[42] Organic and Biomolecular Chemistry, 2011, vol. 9, # 9, p. 3188 - 3198
[43] Journal of the Chemical Society - Faraday Transactions, 1996, vol. 92, # 17, p. 3173 - 3182
[44] Organic Letters, 2015, vol. 17, # 7, p. 1664 - 1667
[45] Journal of Heterocyclic Chemistry, 1981, vol. 18, p. 297 - 302
[46] Journal of the American Chemical Society, 2002, vol. 124, # 22, p. 6246 - 6247
[47] Bioorganic and Medicinal Chemistry Letters, 2018,
[48] Analytical Chemistry, 2017, vol. 89, # 13, p. 7195 - 7202
[49] Journal of Organic Chemistry, 1995, vol. 60, # 24, p. 7984 - 7996
[50] Russian Journal of Organic Chemistry, 2005, vol. 41, # 11, p. 1690 - 1693
[51] Synthesis, 2003, # 4, p. 509 - 512
[52] Journal of Medicinal Chemistry, 2012, vol. 55, # 21, p. 9283 - 9296
[53] Chemical Communications, 2017, vol. 53, # 86, p. 11822 - 11825
[54] Journal of Medicinal Chemistry, 2012, vol. 55, # 7, p. 2981 - 2993
[55] Organic and Biomolecular Chemistry, 2014, vol. 12, # 9, p. 1419 - 1429
[56] Organic Letters, 2014, vol. 16, # 9, p. 2410 - 2413
[57] Journal of Chemical Research, Miniprint, 1985, # 11, p. 3501 - 3519
[58] ACS Medicinal Chemistry Letters, 2017, vol. 8, # 2, p. 191 - 195
[59] Bioorganic and Medicinal Chemistry, 2012, vol. 20, # 12, p. 3768 - 3780
[60] Organic and Biomolecular Chemistry, 2012, vol. 10, # 42, p. 8501 - 8508
[61] Dalton Transactions, 2014, vol. 43, # 48, p. 17964 - 17970
[62] Journal of Heterocyclic Chemistry, 1998, vol. 35, # 1, p. 209 - 215
[63] Journal of the Chemical Society, Perkin Transactions 2: Physical Organic Chemistry (1972-1999), 1985, p. 607 - 624
[64] Journal of Medicinal Chemistry, 2008, vol. 51, # 10, p. 2971 - 2984
[65] Journal of Heterocyclic Chemistry, 1994, vol. 31, # 4, p. 1047 - 1052
[66] Tetrahedron, 2000, vol. 56, # 32, p. 5877 - 5883
[67] Tetrahedron, 2003, vol. 59, # 50, p. 9939 - 9950
[68] Journal of Organic Chemistry, 2002, vol. 67, # 14, p. 4847 - 4855
[69] Tetrahedron Letters, 2007, vol. 48, # 28, p. 4813 - 4815
[70] Organic Preparations and Procedures International, 2008, vol. 40, # 5, p. 499 - 504
[71] Tetrahedron Letters, 2009, vol. 50, # 43, p. 5910 - 5913
[72] Journal of Chemical Research, 2010, # 1, p. 1 - 4
[73] Journal of Chemical Research, 2009, # 12, p. 766 - 769
[74] Russian Journal of Inorganic Chemistry, 2010, vol. 55, # 4, p. 583 - 593
[75] ChemBioChem, 2010, vol. 11, # 12, p. 1769 - 1781
[76] Carbohydrate Research, 2011, vol. 346, # 6, p. 722 - 727
[77] Carbohydrate Research, 2012, vol. 352, p. 12 - 17
[78] Journal of Inorganic Biochemistry, 2011, vol. 105, # 3, p. 435 - 443
[79] Green Chemistry, 2012, vol. 14, # 2, p. 519 - 527
[80] Journal of Materials Chemistry, 2012, vol. 22, # 33, p. 16927 - 16932
[81] Bioorganic and Medicinal Chemistry, 2013, vol. 21, # 1, p. 215 - 222
[82] Journal of Medicinal Chemistry, 2013, vol. 56, # 9, p. 3478 - 3491
[83] Journal of Organic Chemistry, 2013, vol. 78, # 7, p. 3222 - 3234
[84] Journal of Materials Chemistry A, 2013, vol. 1, # 6, p. 2256 - 2266
[85] Synthesis (Germany), 2013, vol. 45, # 16, p. 2245 - 2250
[86] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 22, p. 6079 - 6082
[87] Patent: US2014/275555, 2014, A1, . Location in patent: Paragraph 0313
[88] Chinese Chemical Letters, 2014, vol. 25, # 12, p. 1643 - 1647
[89] Bioorganic and Medicinal Chemistry, 2015, vol. 23, # 3, p. 612 - 623
[90] Journal of the Iranian Chemical Society, 2015, vol. 12, # 3, p. 427 - 432
[91] Letters in Organic Chemistry, 2015, vol. 12, # 2, p. 85 - 90
[92] European Journal of Inorganic Chemistry, 2015, vol. 2015, # 28, p. 4722 - 4730
[93] Bioorganic and Medicinal Chemistry, 2016, vol. 24, # 7, p. 1488 - 1494
[94] Tetrahedron, 2014, vol. 70, # 50, p. 9545 - 9553
[95] Patent: KR101508710, 2015, B1, . Location in patent: Paragraph 0023-0032
[96] Journal of Medicinal Chemistry, 2017, vol. 60, # 7, p. 2890 - 2907
[97] Journal of Organometallic Chemistry, 2017, vol. 851, p. 52 - 56
[98] Journal of Molecular Liquids, 2017, vol. 247, p. 109 - 115
[99] Patent: CN106632571, 2017, A, . Location in patent: Paragraph 0035; 0036
[100] Patent: KR2017/125601, 2017, A, . Location in patent: Paragraph 0193-0195
[101] Analytical Chemistry, 2018, vol. 90, # 11, p. 6820 - 6826
[102] Spectrochimica Acta - Part A: Molecular and Biomolecular Spectroscopy, 2018, vol. 204, p. 225 - 231
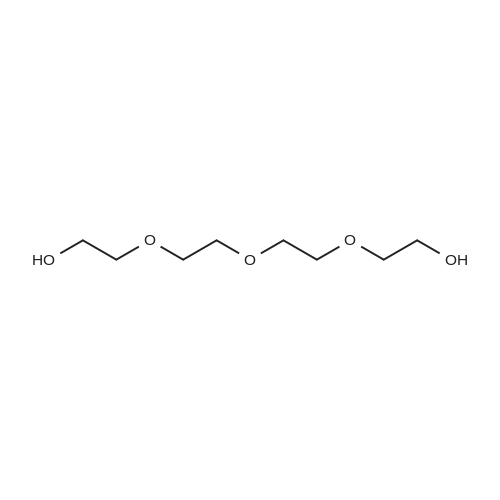
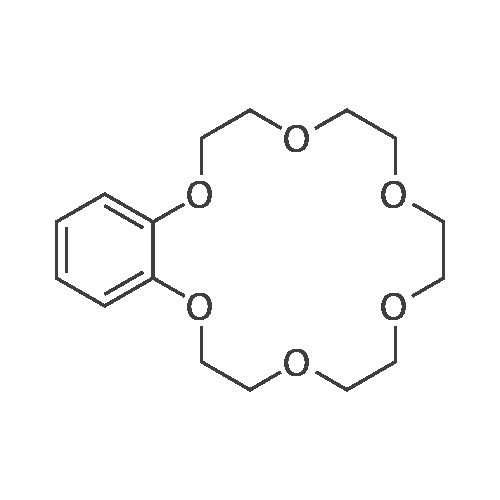
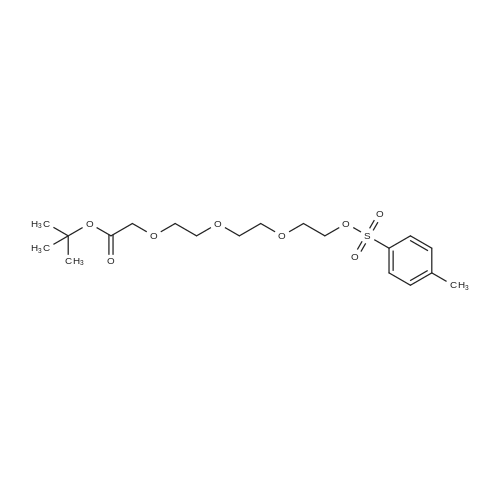
2
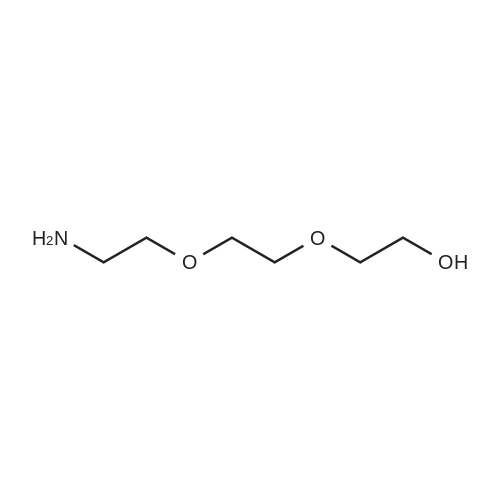
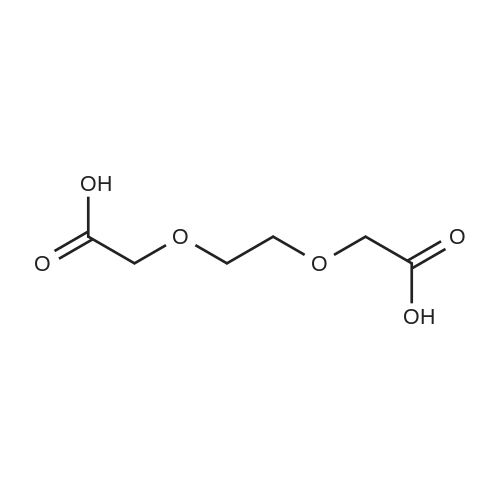
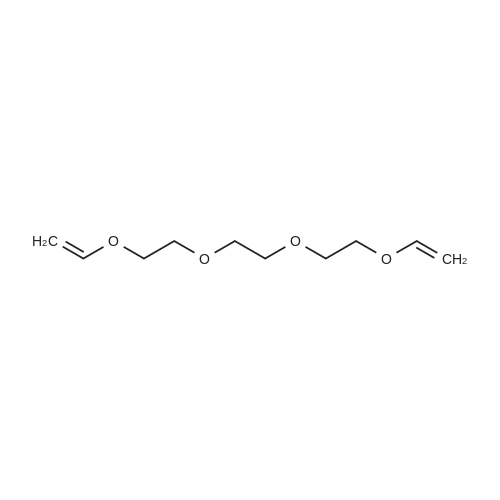
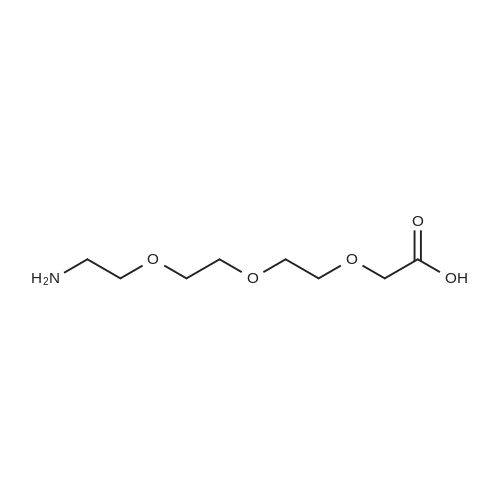
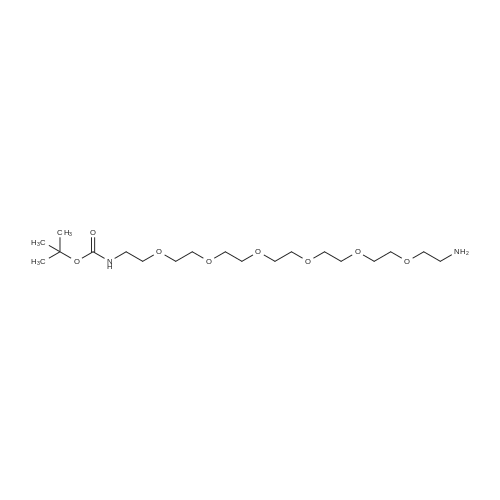
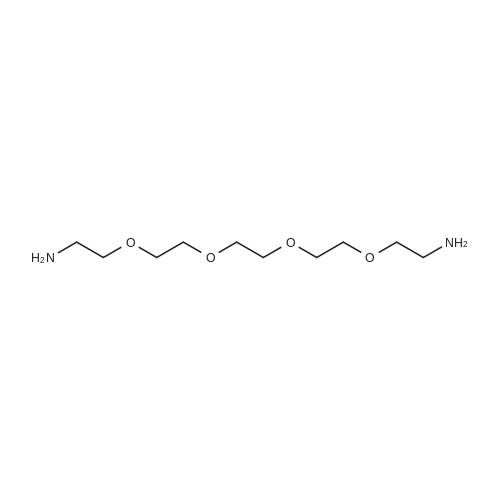
[ 98-59-9 ]
[ 112-27-6 ]
[ 77544-68-4 ]
[ 19249-03-7 ]
Reference:
[1] Organic Letters, 2002, vol. 4, # 14, p. 2329 - 2332
[2] Journal of Porphyrins and Phthalocyanines, 2013, vol. 17, # 1-2, p. 104 - 117
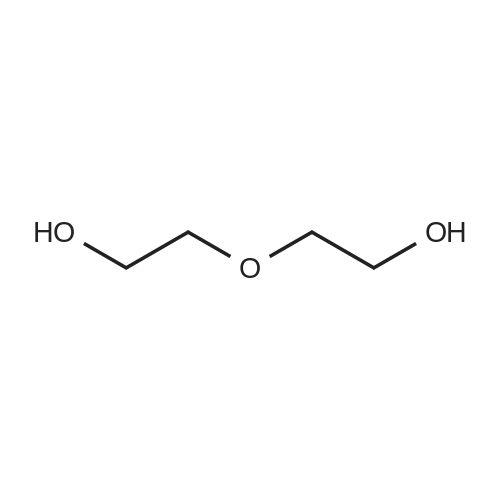
3
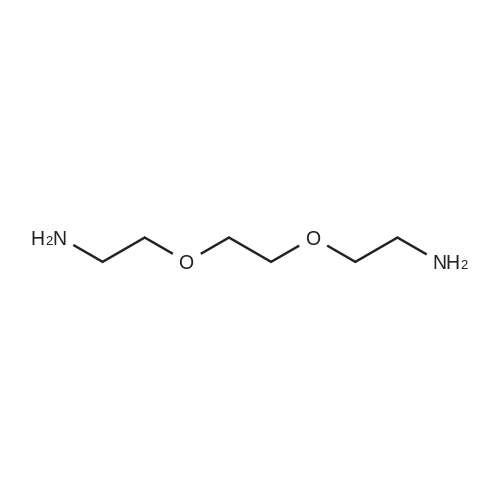
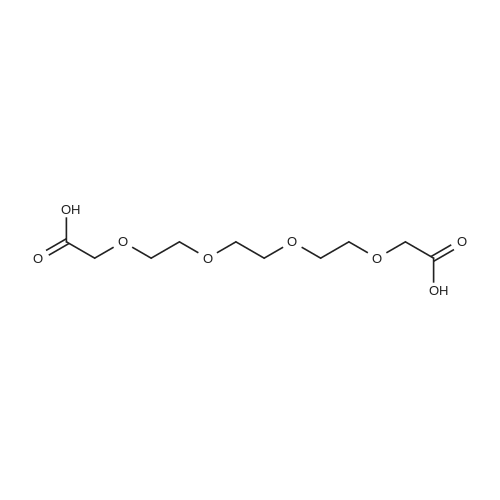
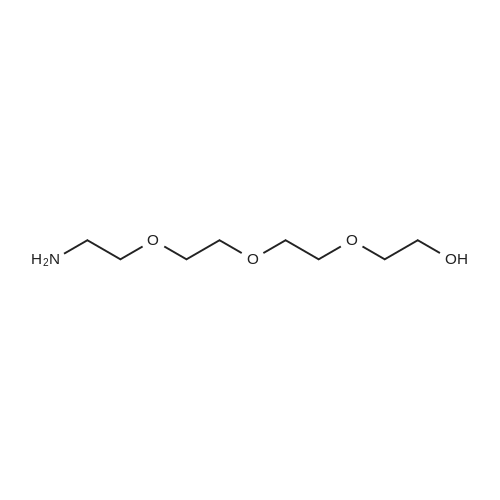
[ 104-15-4 ]
[ 112-27-6 ]
[ 19249-03-7 ]
Reference:
[1] Angewandte Chemie - International Edition, 2014, vol. 53, # 44, p. 11822 - 11827[2] Angew. Chem., 2014, vol. 126, # 44, p. 12016 - 12021,6
4
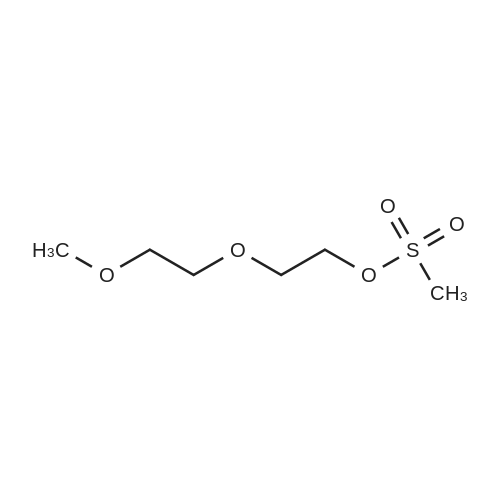
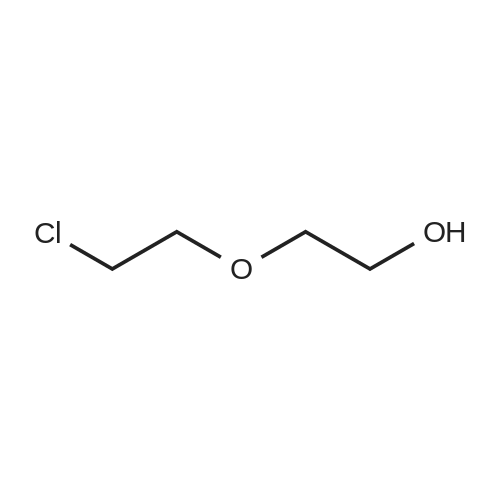
[ 112-27-6 ]
[ 19249-03-7 ]
Reference:
[1] Synlett, 2010, # 10, p. 1544 - 1548
5
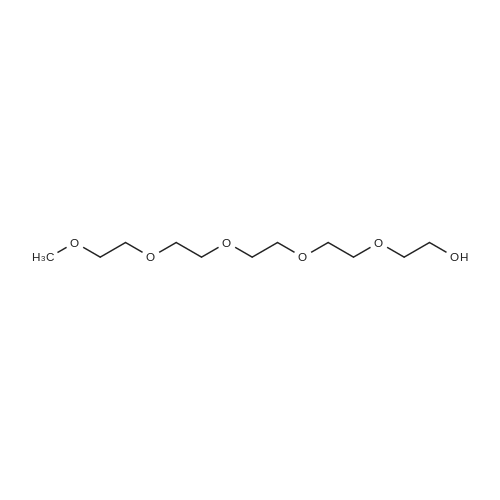
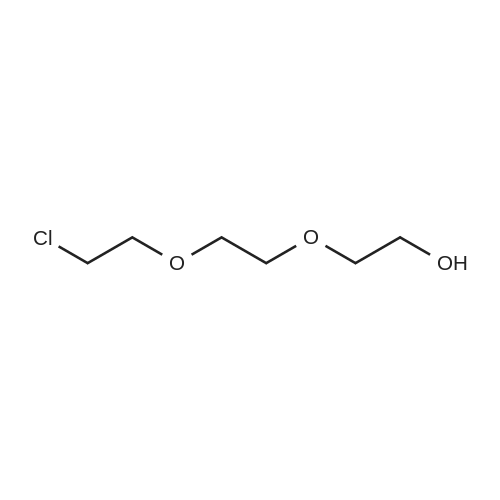
[ 98-59-9 ]
[ 112-27-6 ]
[ 37860-51-8 ]
Reference:
[1] Organic and Biomolecular Chemistry, 2014, vol. 12, # 9, p. 1430 - 1439
Reference:
[1] Bioorganic and Medicinal Chemistry, 2008, vol. 16, # 24, p. 10295 - 10300
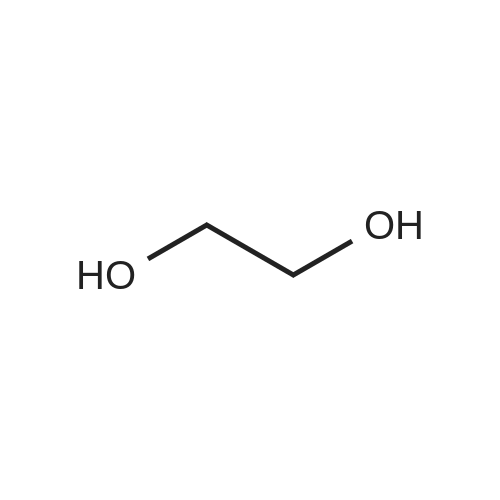
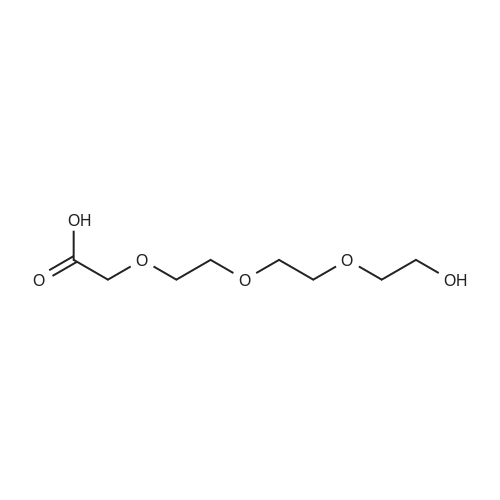
20
[ 79-11-8 ]
[ 112-27-6 ]
[ 23243-68-7 ]
Yield
Reaction Conditions
Operation in experiment
85%
at 150℃; for 24 h;
Ethylenedioxydiethanol: chloroacetic acid molar ratio of 1: 2 To a 50mL three-neck round bottom flask equipped with a magnetic stirrer, a reflux condenser and a thermometer, add ethylenedioxydiethanol (1mmol) and chloroacetic acid (2mmol). Start the stirring at 150 deg.C. React for 24 hours. After completion of the reaction, the reaction solution was cooled to room temperature, the resulting precipitate was added to 100mL of hot methanol, and stirred to dissolve, filtered, and the filtrate was slowly volatilize give a white solid, yield 85percent;
Reference:
[1] Patent: CN105669707, 2016, A, . Location in patent: Paragraph 0018; 0019
21
[ 112-27-6 ]
[ 23243-68-7 ]
Reference:
[1] Synlett, 2007, # 17, p. 2691 - 2694
[2] Tetrahedron, 2009, vol. 65, # 34, p. 7177 - 7185
[3] Russian Journal of Organic Chemistry, 2012, vol. 48, # 8, p. 1047 - 1054[4] Zh. Org. Khim., 2012, vol. 48, # 8, p. 1051 - 1058,8
[5] RSC Advances, 2015, vol. 5, # 126, p. 103782 - 103789
[6] Chemistry of Heterocyclic Compounds (New York, NY, United States), 1981, vol. 17, # 8, p. 847 - 852[7] Khimiya Geterotsiklicheskikh Soedinenii, 1981, # 8, p. 1132 - 1137
[8] Indian Journal of Chemistry, Section A: Inorganic, Physical, Theoretical & Analytical, 1982, vol. 21, # 5, p. 520 - 522
[9] Annales de Chimie (Cachan, France), 1863, vol. <3> 69, p. 321[10] Justus Liebigs Annalen der Chemie, 1862, vol. 122, p. 354
[11] Tetrahedron, 1982, vol. 38, # 14, p. 2055 - 2060
[12] Patent: WO2012/40204, 2012, A2, . Location in patent: Page/Page column 29
22
[ 62921-74-8 ]
[ 112-27-6 ]
[ 23601-40-3 ]
Yield
Reaction Conditions
Operation in experiment
69%
at 100℃; for 12 h;
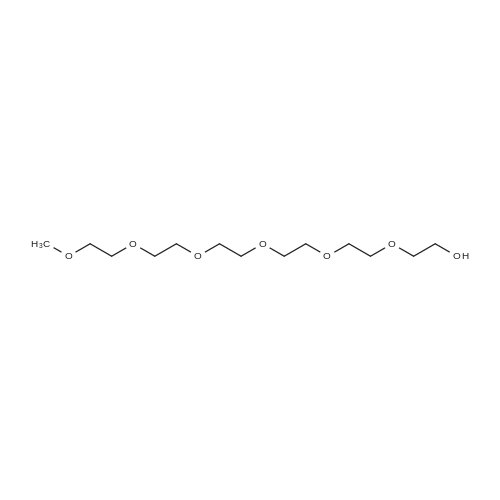
2-(2-(2-Methoxyethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (Al, 1.0 g, 3.14 mmol) was added with stir to triethylene glycol (2.10 mL, 15.7 mmol). Potassium hydroxide (510 mg, 9.42 mmol) was ground into a powder and added to the reaction and the mixture refluxed at 100°C for 12 hours, upon which the reaction was diluted with water (50 mL) and extracted with DCM. The organic layers were combined, dried over sodium sulfate, and concentrated under reduced pressure to obtain 2,5,8,1 l,14,17-hexaoxanonadecan-19-ol as a yellow oil in 69percent yield.
65%
Reflux
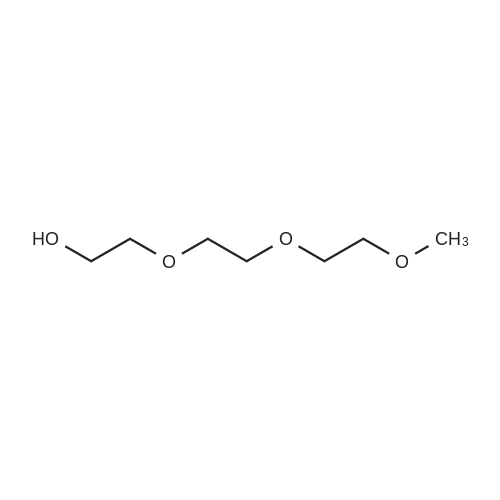
The synthesis of compound 3 is shown in Figure 5. Synthesis of compound 3 began by coupling tosylate (6) to triethylene glycol (16) under basic conditions to yield hexaethylene glycol monomethyl ether (17) in 65percent yield. Hexaethylene glycol monomethyl ether (17) was then converted to the corresponding tosylate (18) in 77percent yield, followed by conversion to the azide. The azide was reduced to the corresponding amine (19) via a hydrogenation over activated palladium on carbon in 45percent yield. The amine was coupled to cyanoacetic acid using 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) in 80percent yield to give the cyano-acetamide (20). The resulting amide (20) was coupled with previously synthesized piperidine-naphthalene (12) to give compound 3 via Knovenagel condensation in 61percent yield.
Reference:
[1] Journal of Organic Chemistry, 2006, vol. 71, # 20, p. 7499 - 7508
[2] Organic Letters, 2004, vol. 6, # 4, p. 469 - 472
[3] Patent: WO2015/143185, 2015, A1, . Location in patent: Paragraph 00819
[4] Patent: WO2016/40891, 2016, A2, . Location in patent: Paragraph 00316
[5] Bulletin of the Korean Chemical Society, 2011, vol. 32, # 7, p. 2193 - 2198
23
[ 112-35-6 ]
[ 112-27-6 ]
[ 23601-40-3 ]
Reference:
[1] Molecular Crystals and Liquid Crystals (1969-1991), 1988, vol. 160, p. 331 - 338
24
[ 74654-05-0 ]
[ 112-27-6 ]
[ 23601-40-3 ]
Reference:
[1] Bioorganic and Medicinal Chemistry, 2008, vol. 16, # 24, p. 10295 - 10300
25
[ 112-27-6 ]
[ 23601-40-3 ]
Reference:
[1] Chemistry - An Asian Journal, 2011, vol. 6, # 2, p. 452 - 458
26
[ 52995-76-3 ]
[ 112-27-6 ]
[ 23601-40-3 ]
Reference:
[1] Bulletin of the Chemical Society of Japan, 1978, vol. 51, p. 3111 - 3120
27
[ 107-13-1 ]
[ 112-27-6 ]
[ 57741-46-5 ]
Reference:
[1] Science China Chemistry, 2010, vol. 53, # 12, p. 2452 - 2460
[2] Journal of the American Chemical Society, 1943, vol. 65, p. 23,25[3] Journal of the American Chemical Society, 1948, vol. 70, p. 214,215
[4] Journal of the American Chemical Society, 1943, vol. 65, p. 23,25[5] Journal of the American Chemical Society, 1948, vol. 70, p. 214,215
[6] Journal of the American Chemical Society, 1943, vol. 65, p. 23,25[7] Journal of the American Chemical Society, 1948, vol. 70, p. 214,215
[8] Bulletin of the Chemical Society of Japan, 1988, vol. 61, # 7, p. 2443 - 2450
[9] Patent: US2437905, 1945, ,
[10] Patent: US2372808, 1941, ,
[11] Patent: US2401607, 1944, ,
[12] Patent: US2437905, 1945, ,
[13] Patent: US2372808, 1941, ,
[14] Patent: US2401607, 1944, ,
[15] Patent: US2437905, 1945, ,
[16] Patent: US2372808, 1941, ,
[17] Patent: US2401607, 1944, ,
28
[ 112-27-6 ]
[ 5617-32-3 ]
Reference:
[1] Journal of Organic Chemistry, 1999, vol. 64, # 18, p. 6870 - 6873
[2] Synthetic Communications, 1986, vol. 16, # 1, p. 19 - 26
[3] Journal of Organic Chemistry, 1992, vol. 57, # 24, p. 6678 - 6680
[4] Angewandte Chemie - International Edition, 2015, vol. 54, # 12, p. 3763 - 3767[5] Angew. Chem., 2015, vol. 127, # 12, p. 3834 - 3838,5
29
[ 112-27-6 ]
[ 31127-85-2 ]
Reference:
[1] Bulletin of the Chemical Society of Japan, 1988, vol. 61, # 7, p. 2443 - 2450
30
[ 100-44-7 ]
[ 112-27-6 ]
[ 55489-58-2 ]
Yield
Reaction Conditions
Operation in experiment
67%
Stage #1: With sodium hydroxide In water for 0.166667 h; Stage #2: at 100℃;
Triethylene glycol (30 g, 0.2 mol) was dissolved in a solution of NaOH (8 g in 8 mL of H2O) and stirred for 10 min. Then benzyl chloride (7 mL, 0.062 mol) was added and the reaction mixture was heated to 100° C. and stirred overnight. The crude reaction was diluted with sat NaCl (500 mL), washed CH2Cl2 (2.x.400 mL), organic layers dried MgSO4, and evaporated to dryness. Column chromatography (silica, ethyl acetate to ethyl acetate/MeOH, 10:1) afforded 1 a yellowish oil (9.87 g, 67percent yield).
Reference:
[1] European Journal of Organic Chemistry, 2008, # 17, p. 2900 - 2914
[2] Journal of the American Chemical Society, 2013, vol. 135, # 25, p. 9248 - 9251
[3] Synthetic Communications, 1986, vol. 16, # 1, p. 19 - 26
[4] Tetrahedron Letters, 2001, vol. 42, # 23, p. 3819 - 3822
[5] Patent: US2008/207505, 2008, A1, . Location in patent: Page/Page column 31
[6] Journal of Chemical Research, Miniprint, 1984, # 9, p. 2672 - 2691
[7] Journal of Organic Chemistry, 1994, vol. 59, # 25, p. 7695 - 7700
[8] Analytical Chemistry, 1992, vol. 64, # 15, p. 1685 - 1690
[9] Journal of Pharmaceutical Sciences, 1975, vol. 64, # 4, p. 693 - 695
[10] Arzneimittel-Forschung, 1978, vol. 28, # 11, p. 2061 - 2063
[11] Synthetic Communications, 1990, vol. 20, # 6, p. 799 - 807
[12] Journal of the Chemical Society, Chemical Communications, 1990, # 13, p. 911 - 912
[13] Liebigs Annalen der Chemie, 1992, # 10, p. 1029 - 1032
31
[ 100-39-0 ]
[ 112-27-6 ]
[ 55489-58-2 ]
Yield
Reaction Conditions
Operation in experiment
92.9%
With silver(l) oxide In dichloromethane for 2 h; Inert atmosphere
Ag2O (6.9 g, 30 mmol) was added to a solution of triethylene glycol (3.0 g, 20 mmol) in dry CH2Cl2. BnBr (2.6 mL, 22 mmol) was then added dropwise over 5 min, and the mixture was stirred for 2 h. After this time, the suspension was filtered through a pad of Celite, which was thoroughly washed with CH2Cl2. The combined filtrate was concentrated under reduced pressure, and the residue was purified by column chromatography to yield a yellow oil (4.46 g, 92.9percent). 1H NMR (300 MHz, CDCl3): δ 7.26-7.35 (m, 5H, Ph), 4.56 (s, 2H, -CH2Ph), 3.73-3.58 (m, 12H, -OCH2CH2O-), 2.80 (br, 1H, -OH); 13C NMR (75 MHz, CDCl3): δ 138.0, 128.2, 127.6, 127.5, 73.1, 72.4, 70.5, 70.4, 70.2, 69.2, 61.5.
36%
Stage #1: With sodium hydride In tetrahydrofuran; mineral oil for 1 h; Inert atmosphere Stage #2: at 0 - 20℃; for 16 h; Inert atmosphere
General procedure: Sodium hydride (60percent dispersion in mineral oil, 0.5 equiv) wasadded portion-wise to a solution of triethyleneglycol (1 equiv.) inTHF (50 mL) under nitrogen. The reaction was stirred for 1 h andthen cooled to 0 C. Benzyl bromide (0.5 equiv.) was then addeddrop-wise, and the reaction mixture was warmed to room temperatureand then stirred for 16 h. The reaction mixture was cooledin an ice bath quenched by the addition of methanol (20 mL), andthen concentrated in vacuo. The residue was dissolved in DCM(30 mL), and washed with water (30 mL). The combined organicextracts were dried over anhydrous MgSO4, filtered, and concentratedin vacuo, to afford a yellow oil, which was purified by flashchromatography.
Reference:
[1] Tetrahedron, 2012, vol. 68, # 35, p. 7148 - 7154
[2] Journal of Organic Chemistry, 2004, vol. 69, # 3, p. 639 - 647
[3] Organic and Biomolecular Chemistry, 2009, vol. 7, # 6, p. 1064 - 1067
[4] Organic and Biomolecular Chemistry, 2009, vol. 7, # 18, p. 3652 - 3656
[5] Journal of Organic Chemistry, 2012, vol. 77, # 20, p. 8879 - 8887
[6] Chemical Communications, 2007, # 47, p. 5066 - 5068
[7] Chemistry - A European Journal, 2017, vol. 23, # 56, p. 13875 - 13878
[8] European Journal of Medicinal Chemistry, 2015, vol. 102, p. 153 - 166
[9] Carbohydrate Research, 1992, vol. 230, # 1, p. 117 - 149
[10] Journal of the American Chemical Society, 1994, vol. 116, # 12, p. 5057 - 5062
[11] Journal of the Chemical Society, Perkin Transactions 2: Physical Organic Chemistry (1972-1999), 1994, # 5, p. 989 - 1002
[12] Analytical Chemistry, 1996, vol. 68, # 1, p. 208 - 215
[13] Canadian Journal of Chemistry, 2010, vol. 88, # 12, p. 1222 - 1232
32
[ 100-44-7 ]
[ 112-27-6 ]
[ 55489-58-2 ]
Reference:
[1] Journal of Organic Chemistry, 2006, vol. 71, # 26, p. 9884 - 9886
Reference:
[1] Russian Journal of General Chemistry, 2008, vol. 78, # 9, p. 1728 - 1733
35
[ 98-59-9 ]
[ 112-27-6 ]
[ 77544-68-4 ]
Yield
Reaction Conditions
Operation in experiment
89%
With sodium hydroxide In tetrahydrofuran; water at 0℃; for 2 h;
Triethylene glycol (16.81 g, 112 mmol) was dissolved in 20 mL THF, NaOH solution (6.73 g, 168 mmol in 15 mL H2O). The mixture was cooled down to 0 °C by ice/water bath, then TsCl (25.67 g, 135 mmol) in THF (150) was added dropwise to the mixture and the reaction was started simultaneously. The solution was stirred for another 1 h and the solvent was evaporated. The residue was dissolved in DCM and washed with brine (if there is a lot of salt we can wash first the organic layer with water). After that drying over MgSO4, the crude product was further purified by column chromatography with hexane/ethylacetate (2:1) as eluant, afforded the product as yellow oil; yield: 89percent. 1H NMR (CDCl3): δ = 2.44 (s, 3H, CH3), 3.51-3.70 (m, 12H, 6CH2), 7.34 (d, 2H, Ar-H), 7.80 (d, 2H, Ar-H). MS (MALDI-TOF): m/z calc, 304.1; found, 327.21 [M + Na]+.
88.3%
With sodium hydroxide In tetrahydrofuran; water at 20℃; for 6 h; Cooling with ice
(3) Take the flask, add 0.532 mοl triethylene glycol, add 50 mL of tetrahydrofuran, then slowly drop the cooled 0.2 moles of aqueous sodium hydroxide solution, stir the mixture for several minutes, Under the addition of 0.134 mol of p-methanesulfonyl chloride solution, one hour after the drop, after the completion of the drop of ice water removed, to room temperature reaction, 5 hours after the reaction to remove the solvent to get white or light yellow liquid, dissolved with ethyl acetate, saturated aqueous sodium chloride solution several times, combined with organic phase anhydrous magnesium sulfate drying. The solvent was removed by filtration to give a colorless or pale yellow oil which yielded 88.3percent yield.
83.94%
With triethylamine In dichloromethane
250mL single-neck flask was added triethylene glycol (19.69g, 131.13mmol), solution of p-toluenesulfonyl chloride (5.00g triethylamine (7.96g, 78.68mmol) and dichloromethane (50mL), ice-cooled, 26.23 mmol) was dissolved in dichloromethane (50 mL) of. After completion of the dropwise addition, the reaction system was transferred to room temperature overnight. The solvent was removed by rotary evaporation, the residue was added methylene chloride (a 500 mL) was dissolved, washed with water (a 500 mL) and saturated brine (300mL), dried over anhydrous sodium sulfate. Filtered and the solvent was removed by rotary evaporation to give a colorless oil 6.70g, yield 83.94percent.
79%
With N-ethyl-N,N-diisopropylamine In dichloromethane at 5 - 20℃; for 18 h;
FIG. 10A shows the synthesis of compound 1. Tosyl chloride (1.00 g, 5.25 mmol) was added at 5° C. to a solution of TEG (triethylene glycol, 1.57 g, 10.5 mmol) in anhydrous methylene chloride (6 mL). This was followed by drop-wise addition of DIPEA (N,N-diisopropylethylamine, 1.0 mL, 5.77 mmol). The water bath was removed and the reaction mixture stirred at room temperature for 18 hours. The resulting mixture was diluted with water (6 mL) and washed with 1 M HCl, brine (2×10 mL), and water (3×10 mL). The solution was dried over sodium sulfate and concentrated under vacuum. The resulting residue was purified by silica gel chromatography (ethyl acetate/hexane 7:3) to obtain the product as yellow oil (1.26 g, 79percent). LEN et al., “Micellar Catalysis Using a Photochromic Surfactant: Application to the Pd-Catalyzed Tsuji-Trost Reaction in Water” J. Org. Chem., 79(2): 493-500 (2014). FIG. 10B shows the 1H NMR 300 of compound 1 CDCl3.
75%
With pyridine In dichloromethane at 20℃; for 18 h;
Briefly, p-toluenesulfonyl chloride (7.50 g, 40.0 mmol) was added to a solution of triethylene glycol (60.0 g, 40.0 mmol) in pyridine (6.5 mL) and dichloromethane (400 mL) and under nitrogen. After stirring for 18 hr at room temperature, the solvent was concentrated under reduced pressure. The residue was dissolved in ethyl acetate (200 mL) and washed with aq. NaCl solution (3*200 mL). The aq. extract was back extracted with ethyl acetate (2*100 mL), and the combined ethyl acetate phases were dried over MgSO4. After filtering and evaporating to dryness the product was purified on a silica column to yield a clear oil (9.30 g, 75percent). 1H NMR (300 MHz, CDCl3, TMS): δ 7.82 (d, 3JHH=8.1 Hz, 2H), 7.36 (d, 3JHH=8.1 Hz, 2H), 7.19 (t, 3JHH=3.6 Hz, 2H), 3.59-3.78 (m, 10H), 2.47 (s, 3H). 13C NMR (75 MHz, CDCl3, TMS): δ 144.86, 132.93, 129.82, 127.96, 72.48, 70.77, 70.34, 69.13, 68.70, 61.74, 21.62. IR (NaCl): v=3421, 2886, 1594, 1358, 1169, 664 cm31 1.
75%
With potassium hydroxide In tetrahydrofuran at 20℃; for 5 h;
Compound 6-1 (235 g, 1.57 mol) was dissolved in 500 mL of tetrahydrofuran, then potassium hydroxide (41.2 g, 0.57 mmol, 82percent w/w) was added, and then p-toluenesulfonyl chloride 6-2 (100 g, 0.52 mol) was added in batches, and the mixture was stirred at room temperature for 5 hours. The reaction solution was poured into 1200 mL of water, and extracted with ethyl acetate (400 mL*3). The extract liquid was combined and washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure and purified by silica gel column chromatography, with an eluent (petroleum ether:ethyl acetate=1:1), to obtain the target product 6-3 (118 g, 75percent) as a colorless oil. LC-MS: m/z=305[M+H]+.
71%
With triethylamine In dichloromethane at 20℃; for 12 h; Inert atmosphere; Cooling with ice
The reaction process is as follows: three glycol (262.0mmol), triethylamine (40mmol) and methylene chloride (30 ml) mixed and placed in a 250 ml three flasks; will be to toluene sulfonyl chloride (26mmol) and methylene chloride (35 ml) is placed in the dropping funnel constant and is dropped in the three-mouth flask. The use of balloon under the protection of nitrogen ice salt bath stirring, all melted ice when the temperature of the into stirring at the room temperature, the total reaction time is about 12h. Using thin layer chromatography (TLC) detecting the extent of reaction. When the TLC display in the reactant material point recedes, stopping the reaction. The reaction mixture is cooled to room temperature, water washing three times, the aqueous phase dichloromethane is used for extraction, the combined organic phase with anhydrousMgSO4drying, filtering, then turns on lathe reducing the organic solvent. The final 100 - 200 mesh silica gel column chromatography separation method of purification. The ethyl acetate: dichloromethane=4:1 (V/V) as eluant. At the end of the colorless transparent oily liquid product 5.65g (18.6mmol), yield 71percent.
71.3%
With pyridine In dichloromethane at 20℃; for 18 h; Inert atmosphere
p-Toiuenesuifonyi chloride (7.50 g, 40.0 mrnoi) was added to a solution oftriethylene glycol (60.0 g, 40.0 mrnoi) in pyridine (6.5 mE) and dichiorornethane (DCM, 400 mL) and under nitrogen. After stirring at room temperature for 18 h, the solvent was removed via rotovap. The residue was dissolved in ethyl acetate (200 rnL) and washed with brine (3x100 mL). The ethyl acetate solution was dried over anhydrous Na2SO4. After filtration, the filtrate was evaporated to dryness to give crude product which was puxfied on a silicacolumn to yield 8-tosyloxy-3,6-dioxooctan-I-ol as a clear oil (868 g, 713percent). Orbitrap ESIMS Caicd. for C13H2O6S 30410 Found 305.11.
60%
With triethylamine In tetrahydrofuran at 0 - 25℃; for 18 h;
Tnethylene glycol (120 mmol, 3 eq.) was dissolved in anhydrous THF (80 ml) and triethylamine (80 mmol, 2 eq.) was added. At 0 °C p-toluenesulfonyl chloride (40 mmol, 1 eq.) in anhydrous THF (10 ml) wasadded dropwise over 45 mm. The reaction mixture was allowed to warm to room temperature and stirred overnight. The solvent was then removed in vacuo and the crude mixture purified by flash column chromatography using a gradient from 30percent-90 percent Ethyl acetate in heptane.
48%
With triethylamine In dichloromethane at 25℃; for 12 h;
Triethylene glycol (4.50 g, 30 mmol) and triethylamine (TEA) (8.0 mL) were dissolved in dichloromethane (60 mL). Then, tosylchloride (5.70 g, 30 mmol) was added in one portion. The resulting mixture was stirred for 12 h at 25 oC (Scheme 2). After washing with KHSO4 (1 mol/L, 40 mL) and NaHCO3 (5percent, 40 mL) and drying over anhydrous Na2SO4, the crude product was obtained by evaporation and subsequently purified by column chromatography over silica gel (dichloromethane/MeOH = 100:1, v/v) to obtain the target product 1 as a colorless oil (4.2 g, 14.4 mmol, 48percent).
44.4%
With dmap; triethylamine In tetrahydrofuran at 0 - 20℃; for 13 h;
To a solution of compound 1 (100 g, 665.90 mmol) in THF (500 ml), was added Et3N (46.15 ml, 332.95 mmol) and DMAP (6.51 g, 53.27 mmol) at r.t. Then PTSC (63.48 g, 332.95 mmol) in THF (200 ml) was dropped to above mixture solution for 1h and stirred at 0 °C. Next the mixture solution was stirred for about 12 h at r.t. The reaction mixture was filtered through a celite pad to remove Et3N.HCl. The solvent was evaporated under vacuum.The crude material was purified by column chromatography (DCM : MeOH = 80:1) to obtain yellow oil (90 g , yield 44.4percent). 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 8.3 Hz, 2H), 7.35 (d, J = 8.1 Hz, 2H), 4.22 –4.13 (m, 2H), 3.76 –3.67 (m,4H), 3.62 (s, 4H), 3.60 –3.56 (m, 2H), 2.45 (s, 3H), 2.05 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 144.9, 132.7, 129.8, 127.9, 72.5, 70.6, 70.1, 69.2, 68.5, 61.5, 21.6.
33%
With potassium iodide; silver(l) oxide In dichloromethane at 20℃;
Into a 250-mL round-bottom flask, was placed a solution of 2-[2-(2-hydroxyethoxy)ethoxy]-ethan-1-ol (1.5 g, 9.99 mmol, 1.00 equiv), Ag2O (3.4 g, 14.98 mmol, 1.50 equiv), KI (0.5 g, 2.99 mmol, 0.30 equiv) in dichloromethane (50 mL) at room temperature. This was followed by the addition of TsCl (2.3 g, 12.06 mmol, 1.20 equiv). The resulting solution was stirred overnight at room temperature. The reaction mixture was then quenched by the addition of water (40 mL). The insoluble solids in the reaction mixture were filtered out and the filtrate was extracted with dichloromethane (40 ml x 3). The organic layers were combined, washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was applied onto a silica gel column during with dichlorome thane/me thanol (1 : 1). This resulted in 1 g (33percent) of 14-[(4-methylbenzenesulfonyl)oxy]-3,6,9,12-tetraoxatetradecan-1-ol as yellow oil.
30%
Stage #1: With sodium hydroxide In tetrahydrofuran at 0℃; for 0.25 h; Stage #2: at 20℃; for 2 h;
j00524j A solution of triethylene glycol (30 g, 199.7 mmol) in THF (300 mL) wascooled to 0°C and charged with NaOH (11.98 g, 299.7 mmol) and stirred for 15 mm. Thissolution was charged with tosyl chloride (38.07 g, 199.7 mmol) and stirred at room temperature for 2 h. The reaction mixture was concentrated in vacuo and the residue obtained was dilutedwith water (100 mL) and extracted with ethyl acetate (3 X 200 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated in vacuo resulting in acrude compound which was purified by chromatography on silica gel, eluting with 2percentmethanol in DCM to give 18.5 g, 30percent yield of the title compound as a colorless oil. ‘H NMR(400 MHz, CDC13): ö = 7.78 (d, J= 8.3 Hz, 2H), 7.33 (d, J= 8.1 Hz, 2H), 4.25 —4.10 (m, 2H),3.75 — 3.60 (m, 6H), 3.63 — 3.52 (m, 4H), 2.43 (s, 3H).
22%
With dmap; triethylamine In dichloromethane at 0 - 20℃; for 4 h; Inert atmosphere
General procedure: p-TsCI (1.2 eq.) was added dropwise to a mixture of the appropriate di-alcohol, Et3N (1 eq.), and DMAP (cat.) in dry CH2CI2 (0.01 M) under N2 atmosphere with stirring at 0°C. The stirring was continued at room temperature for 4 hours. The reaction was quenched with saturated NH4CI aqueous solution and the whole was extracted with AcOEt. The extract was washed with brine, dried over Na2SO4, and, following solvent evaporation, the crude was purified by flash chromatography. ES154; 2-(2-(2-hydroxyethoxy)ethoxy)ethyl 4-methylbenzenesu Ifonate. It was synthesized from triethylene glycol (1 .000 g, 6.66 mmol). Elution with petroleum etherAcOEt (2:8) afforded ES154 as a waxy solid: 0.450 g (22percent).1H-NMR (CDCI3, 400 MHz)5 2.30 (s, 3H), 2.99 (br s, exchangeable with D20, IH), 3.40-3.63 (m, IOH), 4.02 (t,J=4.8 Hz, 2H), 7.21 (d, J=8.4 Hz, 2H), 7.64 (d, J=8.4 Hz, 2H). 13C-NMR (CDCI3, 100MHz) 521.48, 42.73, 61.39, 68.47, 69.24, 70.06, 70.42, 71.12, 72.46, 127.79, 129.81,132.71, 144.87.
55 g
at 0 - 20℃; Inert atmosphere
Under the protection of nitrogen, to a 1000ml three-necked flask were added 200 mL pyridine, 120 g BP103a00(1.0eq), stirred and cooled down to 0°C. 151.8g TsCl (1.0eq) was added in batches, stirred for 1h, then slowly warmed up to room temperature, and kept stirring for 3-4h. After the completion of the reaction, the reaction liquid was poured into ice-cold dilute hydrochloric acid solution, extracted with ethyl acetate. The ethyl acetate layer was washed once with dilute hydrochloric acid, washed with saturated sodium bicarbonate, washed with saturated brine, and dried over anhydrous Na2SO4. The solvents were evaporated off at reduced pressure, and chromatographed in a silica gel column to give 55g pure BP103a01.
Reference:
[1] Inorganica Chimica Acta, 2011, vol. 365, # 1, p. 38 - 48
[2] Angewandte Chemie - International Edition, 2015, vol. 54, # 35, p. 10327 - 10330[3] Angew. Chem., 2015, vol. 35, # 37, p. 10467 - 10471,5
[4] European Journal of Organic Chemistry, 2011, # 9, p. 1641 - 1644
[5] Journal of Organic Chemistry, 2004, vol. 69, # 3, p. 639 - 647
[6] Organic and Biomolecular Chemistry, 2003, vol. 1, # 15, p. 2661 - 2669
[7] Journal of the American Chemical Society, 2016, vol. 138, # 2, p. 660 - 669
[8] European Journal of Medicinal Chemistry, 2013, vol. 69, p. 848 - 854
[9] Patent: CN106749163, 2017, A, . Location in patent: Paragraph 0034; 0042
[10] Patent: CN105524053, 2016, A, . Location in patent: Paragraph 0506; 0507; 0508
[11] Chemistry Letters, 2007, vol. 36, # 10, p. 1196 - 1197
[12] Journal of the Chemical Society. Perkin Transactions 2, 1997, # 10, p. 2045 - 2053
[13] Chemical Communications, 2017, vol. 53, # 79, p. 10958 - 10961
[14] Patent: US2018/258114, 2018, A1, . Location in patent: Paragraph 0098; 0099
[15] Carbohydrate Research, 1986, vol. 150, p. 199 - 212
[16] Synthesis, 1992, # 1-2, p. 119 - 122
[17] Tetrahedron, 1993, vol. 49, # 47, p. 10931 - 10944
[18] Patent: US2009/187037, 2009, A1, . Location in patent: Page/Page column 3-4
[19] Journal of Organic Chemistry, 2014, vol. 79, # 2, p. 493 - 500
[20] Macromolecules, 2015, vol. 48, # 12, p. 3825 - 3833
[21] European Journal of Organic Chemistry, 2017, vol. 2017, # 1, p. 25 - 28
[22] Patent: US2017/2028, 2017, A1, . Location in patent: Paragraph 0106; 0107
[23] Tetrahedron Letters, 2005, vol. 46, # 28, p. 4813 - 4816
[24] Organic and Biomolecular Chemistry, 2011, vol. 9, # 24, p. 8465 - 8474
[25] Journal of Organic Chemistry, 2013, vol. 78, # 14, p. 7001 - 7012
[26] Patent: CN106045964, 2016, A, . Location in patent: Paragraph 0020; 0021; 0022; 0023; 0024
[27] Patent: WO2017/31239, 2017, A1, . Location in patent: Paragraph 0148
[28] Acta Chemica Scandinavica, 1991, vol. 45, # 6, p. 621 - 626
[29] Chemistry - A European Journal, 2008, vol. 14, # 4, p. 1155 - 1163
[30] Journal of Medicinal Chemistry, 2007, vol. 50, # 9, p. 2157 - 2165
[31] Patent: WO2018/51107, 2018, A1, . Location in patent: Page/Page column 98
[32] Journal of Medicinal Chemistry, 2013, vol. 56, # 14, p. 5797 - 5805
[33] Tetrahedron Letters, 1997, vol. 38, # 47, p. 8133 - 8136
[34] Journal of Organic Chemistry, 2001, vol. 66, # 16, p. 5449 - 5455
[35] ACS Chemical Neuroscience, 2016, vol. 7, # 7, p. 897 - 911
[36] Helvetica Chimica Acta, 1999, vol. 82, # 12, p. 2186 - 2200
[37] Journal of Organic Chemistry, 2014, vol. 79, # 13, p. 6330 - 6335
[38] Organic and Biomolecular Chemistry, 2009, vol. 7, # 24, p. 5245 - 5254
[39] Advanced Synthesis and Catalysis, 2011, vol. 353, # 16, p. 2915 - 2919
[40] Chinese Chemical Letters, 2017, vol. 28, # 4, p. 832 - 838
[41] Journal of the American Chemical Society, 2015, vol. 137, # 50, p. 15628 - 15631
[42] Chinese Chemical Letters, 2016, vol. 27, # 11, p. 1655 - 1660
[43] Chemical Communications, 2011, vol. 47, # 4, p. 1282 - 1284
[44] Bioorganic and Medicinal Chemistry Letters, 2018, vol. 28, # 4, p. 737 - 741
[45] Journal of the American Chemical Society, 2005, vol. 127, # 38, p. 13158 - 13159
[46] Patent: WO2017/11590, 2017, A1, . Location in patent: Paragraph 00390
[47] Patent: WO2015/106292, 2015, A1, . Location in patent: Paragraph 00506; 00523; 00524
[48] Journal of Medicinal Chemistry, 2012, vol. 55, # 22, p. 9708 - 9721
[49] Patent: WO2013/160728, 2013, A1, . Location in patent: Page/Page column 43; 44
[50] Synthesis, 1987, # 3, p. 280 - 281
[51] Tetrahedron Letters, 2003, vol. 44, # 8, p. 1537 - 1540
[52] Dalton Transactions, 2009, # 21, p. 4129 - 4135
[53] Journal of the American Chemical Society, 2011, vol. 133, # 18, p. 6886 - 6889
[54] Journal of the American Chemical Society, 2011, vol. 133, # 23, p. 8927 - 8933
[55] Chinese Journal of Chemistry, 2012, vol. 30, # 3, p. 577 - 584
[56] Journal of the American Chemical Society, 2012, vol. 134, # 18, p. 7632 - 7635
[57] Journal of Porphyrins and Phthalocyanines, 2013, vol. 17, # 1-2, p. 104 - 117
[58] European Journal of Organic Chemistry, 2013, # 35, p. 7952 - 7959
[59] Patent: WO2014/179620, 2014, A1, . Location in patent: Page/Page column 645-647
[60] ChemMedChem, 2015, vol. 10, # 2, p. 304 - 311
[61] ChemMedChem, 2017, vol. 12, # 18, p. 1504 - 1511
[62] Patent: WO2015/42447, 2015, A1, . Location in patent: Page/Page column 202; 203
[63] European Journal of Organic Chemistry, 2015, vol. 2015, # 13, p. 2921 - 2927
[64] Angewandte Chemie - International Edition, 2014, vol. 53, # 41, p. 10899 - 10903[65] Angew. Chem., 2014, vol. 126, # 41, p. 11079 - 11083,3
[66] Letters in Organic Chemistry, 2015, vol. 12, # 9, p. 668 - 673
[67] Journal of Materials Chemistry C, 2015, vol. 3, # 42, p. 11202 - 11211
[68] Patent: WO2015/168635, 2015, A2, . Location in patent: Page/Page column 296-300
[69] Patent: WO2015/168618, 2015, A2, . Location in patent: Page/Page column 279-283
[70] Patent: WO2015/168589, 2015, A2, . Location in patent: Page/Page column 308; 309; 310
[71] Patent: WO2016/146985, 2016, A1, . Location in patent: Page/Page column 83
[72] Patent: CN105601675, 2016, A, . Location in patent: Paragraph 0039
[73] Tetrahedron Letters, 2017, vol. 58, # 30, p. 2910 - 2914
[74] Bioconjugate Chemistry, 2017, vol. 28, # 9, p. 2284 - 2292
[75] European Journal of Organic Chemistry, 2017, vol. 2017, # 46, p. 6931 - 6941
[76] Patent: CN105061515, 2017, B, . Location in patent: Paragraph 0039
[77] Patent: EP3321279, 2018, A1, . Location in patent: Paragraph 0062
[78] Beilstein Journal of Organic Chemistry, 2018, vol. 14, p. 1281 - 1286
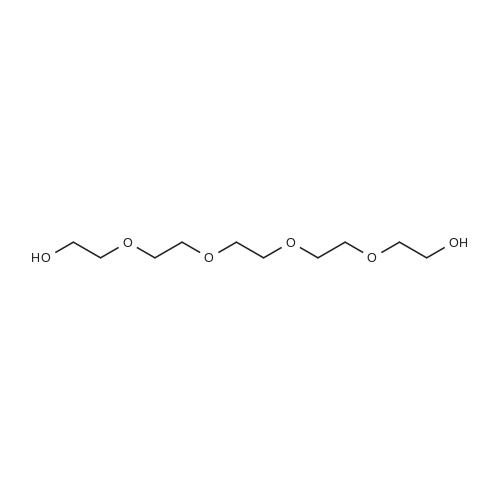
36
[ 98-59-9 ]
[ 112-27-6 ]
[ 77544-68-4 ]
[ 19249-03-7 ]
Reference:
[1] Organic Letters, 2002, vol. 4, # 14, p. 2329 - 2332
[2] Journal of Porphyrins and Phthalocyanines, 2013, vol. 17, # 1-2, p. 104 - 117
37
[ 104-15-4 ]
[ 112-27-6 ]
[ 77544-68-4 ]
Reference:
[1] Journal of Materials Chemistry B, 2017, vol. 5, # 25, p. 4973 - 4980
Reference:
[1] Journal of Organic Chemistry, 2004, vol. 69, # 3, p. 639 - 647
[2] Journal of Organic Chemistry, 2004, vol. 69, # 3, p. 639 - 647
[3] Journal of Organic Chemistry, 2004, vol. 69, # 3, p. 639 - 647
[4] Journal of Organic Chemistry, 2004, vol. 69, # 3, p. 639 - 647
[5] Journal of Organic Chemistry, 2004, vol. 69, # 3, p. 639 - 647
42
[ 1663-39-4 ]
[ 112-27-6 ]
[ 186020-66-6 ]
Yield
Reaction Conditions
Operation in experiment
96%
Stage #1: With sodium In tetrahydrofuran at 20℃; Stage #2: at 20℃; for 20 h; Stage #3: With hydrogenchloride; water In tetrahydrofuran
Synthesis of Compound 71; Step a: Sodium (0.10 g, 4.35 mml) was added to a solution of triethyleneglycol (64.0 mL, 0.48 mol) in anhydrous THF (250 mL) at room temperature. tert-Butyl acrylate (24.0 mL, 0.164 mol) was added after the sodium was dissolved. The reaction mixture was stirred at room temperature for 20 h and neutralized with 1.0 N HCl (4 mL). After removal of the solvent, the residue was suspended in brine and extracted with EtOAc (3.x.70 mL). The combined organic layers were washed with brine and dried over MgSO4. After evaporation of the solvent, the tert-butylacylate adduct (40.23 g, 0.157 mol, 96percent) was a colorless oil, which was used directly for the next reaction step without further purification. HPLC: tR=10.2 min.
95.5%
With sodium In tetrahydrofuran at 20℃; for 24 h;
250mL round bottom flask was added 2- (2- (2-hydroxyethoxy) ethoxy) ethanol (37.00mL, 276.9mmol) was dissolved in dry tetrahydrofuran (150 mL) and stirred under addition of sodium (55mg, 2.39 mmol), at room temperature, followed by dropwise addition of tert-butyl acrylate (11.30mL, 77.8mmol) to the reaction system, the reaction system was reacted at room temperature for 24 hours. Completion of the reaction system with 1mol / L dilute hydrochloric acid to the pH adjusted to 7. And (300mL × 3) and extracted with ethyl acetate, the organic layer was washed with saturated brine (100mL × 3), dried the combined organic phases were dried over anhydrous sodium sulfate, filtered and the solvent under reduced pressure to give a spin yellow oily liquid 20.7g, yield: 95.5percent.
84%
With sodium In tetrahydrofuran at 20℃;
Into a 1000-mL round-bottom flask, was placed a solution of 2,2'-(2,2'-oxybis(ethane-2,1-diyl)bis,2-diylbis(oxy))diethanol (82 g, 546.05 mmol, 3.50 equiv) in tetrahydrofuran (300 mL), sodium (110 mg, 4.78 mmol, 0.03 equiv). This was followed by the addition of tert-butyl acrylate (20 g, 156.04 mmol, 1.00 equiv) dropwise with stirring at room temperature. The resulting solution was stirred overnight at room temperature. The pH value of the solution was adjusted to 7 with 1M hydrochloric acid. The resulting solution was extracted with 3*100 mL of ethyl acetate and the organic layers combined. The resulting mixture was washed with 3*100 mL of brine. The mixture was dried over anhydrous sodium sulfate and concentrated under vacuum. This resulted in 36.6 g (84percent) of tert-butyl 3-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)propanoate as yellow oil.
79%
Stage #1: With sodium In tetrahydrofuran
To a solution of 2,2’-(ethane-1,2-diylbis(oxy))diethanol (55.0 mL, 410.75 mmol, 3.0 eq.) in anhydrous THF (200 mL) was added sodium (0.1 g). The mixture was stirred until Na disappeared and then tert-butyl acrylate (20.0 mL, 137.79 mmol, 1.0 eq.) was added dropwise. The mixture was stirred overnight and then quenched by HCl solution (20.0 mL, 1N) at 0 °C. THF was removed by rotary evaporation, brine (300 mL) was added and the resulting mixture was extracted with EtOAc (3 × 100 mL). The organic layers were washed with brine (3 × 300 mL), dried over anhydrous Na2SO4, filtered and concentrated to afford a colourless oil (30.20 g, 79.0percent yield), which was used without further purification. MS ESI m/z calcd for C13H27O6 [M + H]+ 278.1729, found 278.1730.
79%
With sodium In tetrahydrofuran
To a solution of 2,2' -(ethane- l,2-diylbis(oxy))diethanol (55.0 mL, 410.75 mmol, 3.0 eq.) in anhydrous THF (200 mL) was added sodium (0.1 g). The mixture was stirred until Na disappeared and then ieri-butyl acrylate (20.0 mL, 137.79 mmol, 1.0 eq.) was added dropwise. The mixture was stirred overnight and then quenched by HC1 solution (20.0 mL, 1 N) at 0 °C. THF was removed by rotary evaporation, brine (300 mL) was added and the resulting mixture was extracted with EtOAc (3 x 100 mL). The organic layers were washed with brine (3 x 300 mL), dried over anhydrous Na2S04, filtered and concentrated to afford a colorless oil (30.20 g, 79.0percent yield), which was used without further purification. MS ESI m/z [M + H]+ 278.17.
62%
With sodium hydride In tetrahydrofuran; mineral oil at 20℃; for 20 h;
General procedure: To a solution of tetraethylene glycol (20.0 g, 103 mol) in anhydrous THF (54.0 mL) were added NaH (60percent dispersion in mineral oil, 42 mg, 1.05 mmol) and tert-butyl acrylate (5.2 mL, 36.0 mol). The resulting solution was stirred for 20 h at room temperature. The solvent was removed under reduced pressure. The residue was purified by flash column chromatography (petroleum ether/EtOAc=1/1) to afford 16d-1 as a colorless oil (9.3 g, 80percent).
55.5%
Stage #1: With sodium In tetrahydrofuran at 20℃; for 0.2 h; Inert atmosphere Stage #2: at 20℃; Inert atmosphere
To a solution of 128ml (0.4 mol) tetraethylene glycol and 500ml of dried THF was added 340mg of freshly extruded sodium metal chips. The reaction mixture was stirred under nitrogen at room temperature until all the sodium had dissolved (approximately 2 hours). A portion of tert butyl acrylate (48ml, 0.33mol) was then added and stirred overnight under nitrogen, at room temperature. The reaction was quenched by the addition of 1M HCl and THF removed at reduced pressure. The mixture was then treated with saturated NaCl and extracted with EtOAc. The organic phase was washed with water and dried over anhydrous MgSO4, filtered and concentrated under low pressure to yield a colourless oil (61.3g, 55.5percent).
45%
Stage #1: With sodium hydride In tetrahydrofuran at 20℃; for 0.166667 h; Stage #2: at 20℃; for 15 h;
First Step tert-Butyl 3-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]propanoate 2-[2-(2-Hydroxyethoxy)ethoxy]ethanol (2 g, 13.3 mmol) was dissolved in THF (5 mL), and NaH (8 mg, 0.2 mol) was added. After the mixture was stirred at room temperature for 10 minutes, tert-butyl prop-2-enoate (0.5 g, 3.9 mmol) was slowly added, and the mixture was stirred at room temperature for 15 hours. The reaction mixture was concentrated, and the resultant residue was purified by column chromatography on silica gel (ethyl acetate) to obtain the target title compound (488 mg, 45percent) as colorless clear oil. 1H-NMR (CDCl3) δ: 3.68-3.65 (4H, m), 3.62-3.55 (10H, m), 2.47 (2H, t, J=6.4 Hz), 1.44 (9H, s).
40%
With potassium <i>tert</i>-butylate In tetrahydrofuran at 0 - 20℃; for 72 h;
[0072] To 15 grams of polyethene glycol (n=3) was added 0.45 eq of tert-butyl acrylate and 1 molpercent potassium tert-butoxide in THF at 00 C to room temperature for 3 days to yield compound 1. Compound 1 was added to 1.2 eq of methanesulfonyl chloride, 1.2 eq of triethyl amine in THF at 0° C to room temperature for 3 hours. The pH was adjusted to 8 and 1.2 eq of sodium azide was added in water. The reaction was refluxed overnight to yield compound 2. To compound 2 was added 3 eq of triphenyiphosphine in a THF:water ratio of 7:2 at room temperature for 18 hours to afford compound 3.
Reference:
[1] Patent: US2006/228325, 2006, A1, . Location in patent: Page/Page column 63
[2] Patent: CN105524053, 2016, A, . Location in patent: Paragraph 0468; 0469; 0470
[3] Journal of Organic Chemistry, 1997, vol. 62, # 4, p. 813 - 826
[4] Organic and Biomolecular Chemistry, 2015, vol. 13, # 1, p. 81 - 97
[5] Patent: US9301951, 2016, B2, . Location in patent: Page/Page column 295
[6] Synthetic Communications, 2004, vol. 34, # 13, p. 2425 - 2432
[7] Patent: WO2016/59622, 2016, A2, . Location in patent: Page/Page column 101-102
[8] Patent: WO2017/46658, 2017, A1, . Location in patent: Page/Page column 147; 148
[9] Patent: WO2018/86139, 2018, A1, . Location in patent: Page/Page column 150; 151
[10] Journal of Medicinal Chemistry, 2014, vol. 57, # 22, p. 9564 - 9577
[11] Chemistry - A European Journal, 2010, vol. 16, # 40, p. 12234 - 12243
[12] MedChemComm, 2016, vol. 7, # 4, p. 654 - 657
[13] Tetrahedron, 2011, vol. 67, # 12, p. 2251 - 2259
[14] Tetrahedron Letters, 2016, vol. 57, # 16, p. 1739 - 1742
[15] Advanced Functional Materials, 2012, vol. 22, # 5, p. 1076 - 1086
[16] Patent: US2016/2251, 2016, A1, . Location in patent: Paragraph 2235-2236
[17] Patent: WO2015/38493, 2015, A1, . Location in patent: Page/Page column 0072; 0073
[18] Synthetic Communications, 2007, vol. 37, # 11, p. 1899 - 1915
[19] Patent: US2006/205670, 2006, A1, . Location in patent: Page/Page column 104
[20] Patent: US2008/166364, 2008, A1, . Location in patent: Page/Page column 106
[21] Patent: US2009/98130, 2009, A1,
[22] Chemistry - A European Journal, 2008, vol. 14, # 19, p. 5908 - 5917
[23] Patent: WO2010/78449, 2010, A2, . Location in patent: Page/Page column 293-294
With pyridine; thionyl chloride; In 1,4-dioxane; at 80℃;
General procedure: S1, 1mol of diol and 3mol of pyridine are dissolved into 500mL of dioxane, and heated, heated to 80 C;S2, adding 2.2 mol of thionyl chloride to the product in step S1, slowly dropping, adding 2-3 h, and after the completion of the dropwise addition, reacting at 80 C for 5-6 h;S3, the product solution in step S2 is naturally cooled to room temperature, and dioxane is distilled off;S4, the residue in step S3 is added to ethyl acetate, stirred for 30 min, and the pyridinium salt is filtered off;S5. The filtrate in the step S4 was washed with water, and the ethyl acetate layer was dried over anhydrous sodium sulfate and evaporated.
60%
With pyridine; thionyl chloride; In benzene; for 19h;Reflux;
The mixture of triethylene glycol (PEG150) (0.28 mol) and pyridine (0.61 mol) in benzene (250 mL) was heated to reflux, and then thionyl chloride (0.61 mol) was added dropwise over 3 h. The mixture was kept on refluxing for another 16 h and then cooled to room temperature. Then hydrochloric acid (6.3 mL, 11.8 M) diluted with water (50 mL) was added dropwise in 15 min. The upper benzene solution was separated, and after evaporation under reduced pressure, the target compound ClPEG150Cl was obtained as a clear oil. ClPEG150Cl: colorless liquid, yield 60%, 1H NMR (CDCl3, 400 MHz) 3.77 (t, 3J = 6.0 MHz, 4 H), 3.70 (s, 4 H), 3.64 (t, 3J = 6.0 MHz, 4 H).
With pyridine; thionyl chloride; In benzene;
Step 1: Preparation of 1,8-dichloro-3,6-dioxaoctane 8 A target compound 8 (46.2 g, 0.24 mol, 91%) was obtained by reacting triethyleneglycol 7 in benzene solvent with thionylchloride melted in pyridine. 1H NMR (CDCl3): 3.55-3.57(m, 4H, OCH2), 3.60-3.69(m, 4H, OCH2), 3.76(t, 4H ClCH2)
Triethylene glycol (30 g, 0.2 mol) was dissolved in a solution of NaOH (8 g in 8 mL of H2O) and stirred for 10 min. Then benzyl chloride (7 mL, 0.062 mol) was added and the reaction mixture was heated to 100 C. and stirred overnight. The crude reaction was diluted with sat NaCl (500 mL), washed CH2Cl2 (2×400 mL), organic layers dried MgSO4, and evaporated to dryness. Column chromatography (silica, ethyl acetate to ethyl acetate/MeOH, 10:1) afforded 1 a yellowish oil (9.87 g, 67% yield).
With silver(l) oxide; In dichloromethane; for 2h;Inert atmosphere;
Ag2O (6.9 g, 30 mmol) was added to a solution of triethylene glycol (3.0 g, 20 mmol) in dry CH2Cl2. BnBr (2.6 mL, 22 mmol) was then added dropwise over 5 min, and the mixture was stirred for 2 h. After this time, the suspension was filtered through a pad of Celite, which was thoroughly washed with CH2Cl2. The combined filtrate was concentrated under reduced pressure, and the residue was purified by column chromatography to yield a yellow oil (4.46 g, 92.9%). 1H NMR (300 MHz, CDCl3): delta 7.26-7.35 (m, 5H, Ph), 4.56 (s, 2H, -CH2Ph), 3.73-3.58 (m, 12H, -OCH2CH2O-), 2.80 (br, 1H, -OH); 13C NMR (75 MHz, CDCl3): delta 138.0, 128.2, 127.6, 127.5, 73.1, 72.4, 70.5, 70.4, 70.2, 69.2, 61.5.
69%
With potassium hydroxide; at 90 - 110℃; for 16h;
Compound 1-13 (135 g, 0.9 mol) was added to a 500 mL eggplant shaped bottle, KOH (16.8 g, 0.3 mol) was added while stirring. The suspension was heated to 90 C., and stirred till KOH was completely dissolved. BnBr (35.6 mL, 0.3 mol) was added dropwise slowly. The reaction mixture was heated to 110 C. and stirred overnight. The reaction mixture was cooled to room temperature, 1000 mL water was added, then extracted with EtOAc for 3 times (200 mL×3). The organic phases were combined, washed with water for 3 times (150 mL×3) and saturated brine (150 mL), dried over anhydrous sodium sulfate, and concentrated. The crude product was purified by silica gel column chromatography (PE/EtOAc 10:1-1:1) to give 50 g product 2-13 as colorless oil, yield 69%. LCMS (ESI) m/z 240.3 (M+H)+.
43.8%
With potassium iodide; silver(l) oxide; In dichloromethane; at 20℃; for 2h;Inert atmosphere;
To a mixture of Ag2O (12.4 g, 100 mmol) and KI (4.42 g, 26.64 mmol) in DCM (200 mL) was added 2,2?-(ethane-1,2-diylbis(oxy))diethanol (10 g, 66.6 mmol) dropwise at room temperature. Then BnBr (12.5 g, 73.26 mmol) was added dropwise into the mixture over 10 min. After addition, the mixture was stirred at room temperature for 2 h. The reaction mixture was filtered. The combined filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give 2-(2-(2-(benzyloxy)ethoxy)ethoxy)ethanol (7 g, 43.8%) as a colorless oil. LC/MS (ESI, m/z): [M+1]+=241.0.
42%
To a stirred solution of 2,2?-(ethane-1,2-diylbis(oxy))bis(ethan-1-ol) (30 g, 200 mmol) in THF (125 mL) was added 60% NaH in paraffin (4 g, 100 mmol) at 10 C. The resulting reaction mixture stirred at the same temperature for 0.5 h. To this reaction mixture was then added benzyl bromide (17 g, 100 mmol) in THF (125) dropwise at 10 C. over 1.5 h. The resulting reaction mixture then stirred at rt for 16 h. The reaction mixture was then transferred into ice water and the resulting mixture was extracted using DCM (3×100 mL). The combined organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was evaporated under reduced pressure and the crude product was purified using silica gel column chromatography (4% MeOH-DCM) to give 2-(2-(2-(benzyloxy)ethoxy)ethoxy)ethan-1-ol as a yellow oil (20 g, 42%). LC-MS (ESI+) m/z 241.35 (M+H)+
36%
General procedure: Sodium hydride (60% dispersion in mineral oil, 0.5 equiv) wasadded portion-wise to a solution of triethyleneglycol (1 equiv.) inTHF (50 mL) under nitrogen. The reaction was stirred for 1 h andthen cooled to 0 C. Benzyl bromide (0.5 equiv.) was then addeddrop-wise, and the reaction mixture was warmed to room temperatureand then stirred for 16 h. The reaction mixture was cooledin an ice bath quenched by the addition of methanol (20 mL), andthen concentrated in vacuo. The residue was dissolved in DCM(30 mL), and washed with water (30 mL). The combined organicextracts were dried over anhydrous MgSO4, filtered, and concentratedin vacuo, to afford a yellow oil, which was purified by flashchromatography.
With potassium hydroxide; In tetrahydrofuran; at 70℃; for 13h;Inert atmosphere;
It includes the following steps: 1. Feeding To a 2L four-necked flask equipped with a stirrer, a thermometer, and a condenser, which is protected by nitrogen, add 225.3g of triethylene glycol (content 99.6%, 1.5mol), 750g of tetrahydrofuran, and 322g of homemade dichlorotriethylene glycol (content 95.82%) , 1.65mol), stir well and mix evenly. 2. Add potassium hydroxide The temperature was raised with an electric heating mantle, and the reflux started when the temperature reached 70C. 240g (3.96mol) of potassium hydroxide was added to the reaction solution in 12 portions, once every 1h, for a total of 12 additions. The potassium hydroxide is industrial grade with a purity of more than 90%. 3. Insulation reaction After the addition of potassium hydroxide is completed, continue to reflux for 1h. When the gas phase detection of dichlorotriethylene glycol content is less than 0.5%, the reaction can be considered as completed.If the dichlorotriethylene glycol content does not decrease during the gas phase tracking process, it is necessary to continue to add potassium hydroxide until the dichlorotriethylene glycol is consumed. 4. Distilled tetrahydrofuran and water After the heat preservation reaction is completed, the reaction liquid is cooled to below 50C, and the device is changed to a distillation device.Tetrahydrofuran is recovered by atmospheric distillation, and the temperature of the kettle is controlled at 100 at the end of one distillation. Distilled about 750g of tetrahydrofuran and water mixture, with about 3.5% water; Control the pressure of the distillation system to -0.03mpa, and raise the temperature to 100 for 0.5h, Stop the distillation and distill off about 13g of distillate; after the distillation is completed, lower the kettle temperature to below 40. 5. Add dichloromethane Add 600g of methylene chloride to the kettle liquid after distillation and continue to stir at low temperature for 10min; Filter with a Buchner funnel. After the first filtration, wash the filter cake with 300 g of methylene chloride.After washing and filtering, the potassium chloride is slightly yellow, and the salt mass is about 308g. 6. Distilled methylene chloride and water The washing liquid and the filtrate are mixed into a 2000ml four-necked flask. After stirring, 80% sulfuric acid is added dropwise, the system pH is adjusted to 7, and the acid amount is about 4-6g; Build a distillation device, transfer the neutral materials, distill methylene chloride at atmospheric pressure, stop the atmospheric distillation when the temperature of the distillation kettle is about 120C, weigh the distillate and collect it. At this time, the temperature of the system was 120C, the pressure was adjusted to -0.01mpa, and the remaining liquid was distilled under reduced pressure. At this time, the distillate is mainly water, and the vacuum is gradually increased until the pressure is -0.08mpa.The distillate does not flow out, and the temperature is raised to 130C for 0.5h and then cooled, and the distillation is stopped.Among them, about 750 g of distillate was distilled under normal pressure, and 31 g of distillate was distilled under reduced pressure. 7. Distilled crown ether Reduce the temperature of the material to below 60C, transfer to a 1L high vacuum distillation unit, and gradually pump the vacuum to a maximum of about 40pa with an Edward vacuum pump. When the kettle temperature is 150, the distillate begins to flow out. At 180-210C, the distillate flows out faster. The top temperature is stable at 168-172C. When the kettle temperature rises to 230C, the fraction hardly comes out, the top temperature starts to drop, and the distillation ends.Cool down, stop the vacuum pump after the material cools to below 100, and vent the system with nitrogen; 8. Purification of crude products (1) Add acetonitrile Weigh 250g of acetonitrile and transfer to a 1000ml four-necked flask. (2) Add crude crown ether dropwise Under the stirring state, 170g of the above distillate was added with a constant pressure dropping funnel, the dropping process was exothermic, the dropping was completed in about 0.5h, and a lot of turbidity appeared during the dropping process. (3) Heating up After the addition is completed, the temperature is increased by heating, and the temperature of the feed becomes clear after rising to 58C. (4) Cooling and crystallization Then the temperature was lowered. When the temperature dropped to 56, the feed liquid became cloudy. When the temperature continued to drop naturally to 40, the solution was placed in a cold bath at -10 (the temperature gradually decreased to -10) and the crystallization speed was 100 rpm. Stirring is stopped every 1h during the crystallization process, and after standing, the supernatant liquid is taken to detect the content of crown ether in the gas phase.Until the crown ether content no longer decreases to 0.3% stop stirring, about 4h, ready to filter. (5) Filtration and washing Filter with a Buchn...
With potassium hydroxide; In water; at 25℃; for 0.5h;
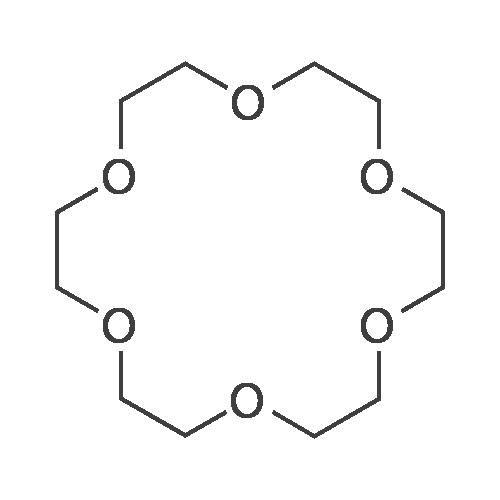
1 part by mole of triethylene glycol bis-p-toluenesulfonate, 1.1 parts by mole of triethylene glycol, 4 parts by mole of potassium hydroxide, 3 parts by mole of water, and reacted at 25 C for 30 minutes in an industrial microwave reactor. After completion of the reaction, the mixture was separated by desalting, extracted, and the solvent was evaporated, and the crude product was distilled under reduced pressure to give 18-crown ether-6, yield 54%.
With p-toluenesulfonyl chloride; potassium hydroxide; In tetrahydrofuran; at 55 - 60℃; for 3h;
A preparation method of 18-crown-6, the production steps are as follows:(1) Weigh 15g of triethylene glycol and 12g of potassium hydroxide into a 500mL reaction bottle, then add 300mL of tetrahydrofuran to the reaction bottle, and start stirring;(2) Add 19g of p-toluenesulfonyl chloride dropwise to the reaction bottle, control the reaction temperature at 55-60 , and react for 3h;(3) Add 7.7g of diethyl sulfate to the solution after the reaction to separate potassium p-toluenesulfonate, control the reaction temperature at 80 C, and react for 3h to produce diethyl p-toluenesulfonate and potassium sulfate;(4) The final solution obtained after the reaction is filtered, concentrated, and vacuum distilled, and the distillate contains 18-crown-6, diethyl p-toluenesulfonate, triethylene glycol, etc .;(5) After washing the distillate with 20 mL of distilled water and leaving it to separate into layers, the upper layer solution is diethyl p-toluenesulfonate, the lower layer solution is 18-crown-6, triethylene glycol, etc. The lower layer solution is centrifugally separated Precipitation, the precipitate is dried to obtain 18-crown-6.The mass of 18-crown-6 finally prepared was 17.2 g, and the yield was 65.2%.
Synthesis of Compound 71; Step a: Sodium (0.10 g, 4.35 mml) was added to a solution of triethyleneglycol (64.0 mL, 0.48 mol) in anhydrous THF (250 mL) at room temperature. tert-Butyl acrylate (24.0 mL, 0.164 mol) was added after the sodium was dissolved. The reaction mixture was stirred at room temperature for 20 h and neutralized with 1.0 N HCl (4 mL). After removal of the solvent, the residue was suspended in brine and extracted with EtOAc (3×70 mL). The combined organic layers were washed with brine and dried over MgSO4. After evaporation of the solvent, the tert-butylacylate adduct (40.23 g, 0.157 mol, 96%) was a colorless oil, which was used directly for the next reaction step without further purification. HPLC: tR=10.2 min.
95.5%
With sodium; In tetrahydrofuran; at 20℃; for 24h;
250mL round bottom flask was added 2- (2- (2-hydroxyethoxy) ethoxy) ethanol (37.00mL, 276.9mmol) was dissolved in dry tetrahydrofuran (150 mL) and stirred under addition of sodium (55mg, 2.39 mmol), at room temperature, followed by dropwise addition of tert-butyl acrylate (11.30mL, 77.8mmol) to the reaction system, the reaction system was reacted at room temperature for 24 hours. Completion of the reaction system with 1mol / L dilute hydrochloric acid to the pH adjusted to 7. And (300mL × 3) and extracted with ethyl acetate, the organic layer was washed with saturated brine (100mL × 3), dried the combined organic phases were dried over anhydrous sodium sulfate, filtered and the solvent under reduced pressure to give a spin yellow oily liquid 20.7g, yield: 95.5%.
84%
With sodium; In tetrahydrofuran; at 20℃;
Into a 1000-mL round-bottom flask, was placed a solution of 2,2'-(2,2'-oxybis(ethane-2,1-diyl)bis,2-diylbis(oxy))diethanol (82 g, 546.05 mmol, 3.50 equiv) in tetrahydrofuran (300 mL), sodium (110 mg, 4.78 mmol, 0.03 equiv). This was followed by the addition of tert-butyl acrylate (20 g, 156.04 mmol, 1.00 equiv) dropwise with stirring at room temperature. The resulting solution was stirred overnight at room temperature. The pH value of the solution was adjusted to 7 with 1M hydrochloric acid. The resulting solution was extracted with 3*100 mL of ethyl acetate and the organic layers combined. The resulting mixture was washed with 3*100 mL of brine. The mixture was dried over anhydrous sodium sulfate and concentrated under vacuum. This resulted in 36.6 g (84%) of tert-butyl 3-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)propanoate as yellow oil.
79.0%
To a solution of 2,2?-(ethane-1,2-diylbis(oxy))diethanol (55.0 mL, 410.75 mmol, 3.0 eq.) in anhydrous THF (200 mL) was added sodium (0.1 g). The mixture was stirred until Na disappeared and then tert-butyl acrylate (20.0 mL, 137.79 mmol, 1.0 eq.) was added dropwise. The mixture was stirred overnight and then quenched by HCl solution (20.0 mL, 1N) at 0 C. THF was removed by rotary evaporation, brine (300 mL) was added and the resulting mixture was extracted with EtOAc (3 × 100 mL). The organic layers were washed with brine (3 × 300 mL), dried over anhydrous Na2SO4, filtered and concentrated to afford a colourless oil (30.20 g, 79.0% yield), which was used without further purification. MS ESI m/z calcd for C13H27O6 [M + H]+ 278.1729, found 278.1730.
79%
With sodium; In tetrahydrofuran;
To a solution of 2,2' -(ethane- l,2-diylbis(oxy))diethanol (55.0 mL, 410.75 mmol, 3.0 eq.) in anhydrous THF (200 mL) was added sodium (0.1 g). The mixture was stirred until Na disappeared and then ieri-butyl acrylate (20.0 mL, 137.79 mmol, 1.0 eq.) was added dropwise. The mixture was stirred overnight and then quenched by HC1 solution (20.0 mL, 1 N) at 0 C. THF was removed by rotary evaporation, brine (300 mL) was added and the resulting mixture was extracted with EtOAc (3 x 100 mL). The organic layers were washed with brine (3 x 300 mL), dried over anhydrous Na2S04, filtered and concentrated to afford a colorless oil (30.20 g, 79.0% yield), which was used without further purification. MS ESI m/z [M + H]+ 278.17.
79%
With sodium; In tetrahydrofuran;
To a solution of 2, 2?- (ethane-1, 2-diylbis (oxy) ) diethanol (55.0 mL, 410.75 mmol, 3.0 eq. ) in anhydrous THF (200 mL) was added sodium (0.1 g) . The mixture was stirred until Na disappeared and then tert-butyl acrylate (20.0 mL, 137.79 mmol, 1.0 eq. ) was added dropwise. The mixture was stirred overnight and then quenched by HCl solution (20.0 mL, 1N) at 0. THF was removed by rotary evaporation, brine (300 mL) was added and the resulting mixture was extracted with EtOAc (3 × 100 mL) . The organic layers were washed with brine (3 × 300 mL) , dried over anhydrous Na2SO4, filtered and concentrated to afford a colorless oil (30.20 g, 79.0%yield) , which was used without further purification. MS ESI m/z calcd for C13H27O6[M + H]+278.1729, found 278.1730.
79%
With sodium; In tetrahydrofuran;
To 2,2 '-(ethane-1,2-diylbis (oxy)) diethanol (55.0 mL, 410.75 mmol, 3.0 eq.)To a solution of anhydrous tetrahydrofuran (200 mL) was added a block of sodium (0.1 g).Stir the mixture until the sodium mass disappears,Tert-butyl acrylate (20.0 mL, 137.79 mmol, 1.0 eq.) Was then added dropwise.The mixture was stirred overnight,It was then quenched with a hydrochloric acid solution (20.0 mL, 1N) at 0 C. Removal of tetrahydrofuran by rotary concentration,Brine (300 mL) was added and the resulting mixture was extracted with ethyl acetate (3 x 100 mL).The organic layer was washed with brine (3 × 300 mL), dried over anhydrous sodium sulfate, filtered and concentrated,Obtained as a colorless oil (30.20 g, 79.0% yield), Which was used without further
76%
To 350 mL of anhydrous THF was added 80 mg (0.0025 mol) of sodium metal and triethylene glycol 150.1 g, 1.00 mol) with stirring. After the sodium had completely dissolved, tert-butyl acrylate (24 mL, 0.33 mol) was added. The solution was stirred for 20 h at room temperature and neutralized with 8 mL of 1.0 M HCl. The solvent was removed in vacuo and the residue was suspended in brine (250 mL) and extracted with ethyl acetate (3 x 125 mL) . The combined organic layers were washed with brine (100 mL) then water (100 mL) , dried over sodium sulfate, and the solvent was removed. The resulting colorless oil was dried under vacuum to give 69.78 g (76%yields) of the title product. 1H NMR: 1.41 (s, 9H) , 2.49 (t, 2H, J=6.4 Hz) , 3.59-3.72 (m, 14H) . ESI MS m/z-C 13H 25O 6 (M-H) , cacld. 277.17, found 277.20.
69.2%
Triethylene glycol (26.7 g, 177.8 mmol) was dissolved in dry THF (125 mL) and sodium metal (50 mg, 2.18 mmol) was added. The resulting mixture was stirred for 3 hours until all the sodiu was dissolved. To the light-yellow solution was added tert-buty -aery late (12.0 mL, 82.0 mmol) and the resulting mixture was stirred at room temperature overnight. (1074) [0578] The organic solvent ws removed by evaporation and brine was added to the resulting residue and extracted with EtOAc The combined organic layers were dried over MgSOr, filtered and evaporated to dryness to give 15.8 g (69.2% yield) of a clear oil, which ws used without further purification
62%
With sodium hydride; In tetrahydrofuran; mineral oil; at 20℃; for 20h;
General procedure: To a solution of tetraethylene glycol (20.0 g, 103 mol) in anhydrous THF (54.0 mL) were added NaH (60% dispersion in mineral oil, 42 mg, 1.05 mmol) and tert-butyl acrylate (5.2 mL, 36.0 mol). The resulting solution was stirred for 20 h at room temperature. The solvent was removed under reduced pressure. The residue was purified by flash column chromatography (petroleum ether/EtOAc=1/1) to afford 16d-1 as a colorless oil (9.3 g, 80%).
55.5%
To a solution of 128ml (0.4 mol) tetraethylene glycol and 500ml of dried THF was added 340mg of freshly extruded sodium metal chips. The reaction mixture was stirred under nitrogen at room temperature until all the sodium had dissolved (approximately 2 hours). A portion of tert butyl acrylate (48ml, 0.33mol) was then added and stirred overnight under nitrogen, at room temperature. The reaction was quenched by the addition of 1M HCl and THF removed at reduced pressure. The mixture was then treated with saturated NaCl and extracted with EtOAc. The organic phase was washed with water and dried over anhydrous MgSO4, filtered and concentrated under low pressure to yield a colourless oil (61.3g, 55.5%).
45%
First Step tert-Butyl 3-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]propanoate 2-[2-(2-Hydroxyethoxy)ethoxy]ethanol (2 g, 13.3 mmol) was dissolved in THF (5 mL), and NaH (8 mg, 0.2 mol) was added. After the mixture was stirred at room temperature for 10 minutes, tert-butyl prop-2-enoate (0.5 g, 3.9 mmol) was slowly added, and the mixture was stirred at room temperature for 15 hours. The reaction mixture was concentrated, and the resultant residue was purified by column chromatography on silica gel (ethyl acetate) to obtain the target title compound (488 mg, 45%) as colorless clear oil. 1H-NMR (CDCl3) delta: 3.68-3.65 (4H, m), 3.62-3.55 (10H, m), 2.47 (2H, t, J=6.4 Hz), 1.44 (9H, s).
40%
With potassium tert-butylate; In tetrahydrofuran; at 0 - 20℃; for 72h;
[0072] To 15 grams of polyethene glycol (n=3) was added 0.45 eq of tert-butyl acrylate and 1 mol% potassium tert-butoxide in THF at 00 C to room temperature for 3 days to yield compound 1. Compound 1 was added to 1.2 eq of methanesulfonyl chloride, 1.2 eq of triethyl amine in THF at 0 C to room temperature for 3 hours. The pH was adjusted to 8 and 1.2 eq of sodium azide was added in water. The reaction was refluxed overnight to yield compound 2. To compound 2 was added 3 eq of triphenyiphosphine in a THF:water ratio of 7:2 at room temperature for 18 hours to afford compound 3.
With sodium; In tetrahydrofuran; at 0℃;
Step 1: 3-{2-[2-(2-Hydroxy-ethoxy)-ethoxy]-ethoxy}-propionic acid tert-butyl ester Na metal (catalytic) was added to a stirring solution of acrylic acid tert-butyl ester (6.7 mL, 46 mmol), and 2-[2-(2-hydroxy-ethoxy)-ethoxy]-ethanol (20.7 g, 138 mmol) in THF (100 mL) at 0 C. and the mixture was stirred overnight. Solvent was removed and the remaining oil dissolved in EtOAc (100 mL). The organic layer was washed with water (3*50 mL), and dried over Na2SO4 and the solvent removed in vacuo to give an oil which corresponds to the title compound that would be used as is for the next step. (M+1)=279.
sodium; In tetrahydrofuran; at 0℃;
Na metal (catalytic) was added to a stirring solution of acrylic acid tert-butyl ester (6.7 mL, 46 mmol), and 2-[2-(2-hydroxy-ethoxy)-ethoxy]-ethanol (20.7 g, 138 mmol) in THF (100 mL) at 0 C. and the mixture was stirred overnight. Solvent was removed and the remaining oil dissolved in EtOAc (100 mL). The organic layer was washed with water (3*50 mL), and dried over Na2SO4 and the solvent removed in vacuo to give an oil which corresponds to the title compound that would be used as is for the next step. (M+1)=279.
In tetrahydrofuran; ethyl acetate;
Step 1: 3-{2-[2-(2-Hydroxy-ethoxy)-ethoxy]-ethoxy}-propionic acid tert-butyl ester Na metal (catalytic) was added to a stirring solution of acrylic acid tert-butyl ester (6.7 mL, 46 mmol), and 2-[2-(2-hydroxy-ethoxy)-ethoxy]-ethanol (20.7 g, 138 mmol) in THF (100 mL) at 0 C. and the mixture was stirred overnight. Solvent was removed and the remaining oil dissolved in EtOAc (100 mL). The organic layer was washed with water (3*50 mL), and dried over Na2SO4 and the solvent removed in vacuo to give an oil which corresponds to the title compound that would be used as is for the next step. (M+1)=279.
To triethyleneglycol (17.6 g, 117.20 mmol, 3 00 equiv) in anhydrous THF (70 mL), was added sodium (30 mg, 1 25 mmol, 0 03 equiv) Tert-butyl acrylate (5 0 g, 39 01 mmol, 1 00 eqmv) was added after the sodium had dissolved The resulting solution was stirred overnight at room temperature and then neutralized with I O N hydrogen chloride After removal of the solvent, the residue was suspended in 50 mL of b?ne and extracted with 3x50 mL of ethyl acetate The combined organic layers were washed with saturated bnne and d?ed over anhydrous sodium sulfate After evaporation of the solvent, the tert-butyl 3-(2-(2-(2- hydroxyethoxy)ethoxy)ethoxy)propanoate (9 6 g) was isolated as a colorless oil, which was used directly for the next reaction step without further purification.
Triethylene glycol divinyl ether was obtained by base-catalyzed reaction of triethylene glycol with ethyne and distilled. The triethylene glycol divinyl ether obtained was stabilized with 100 ppm by weight of potassium hydroxide and had an APHA color number of 105. After storage for 30 days at 25 C., the triethylene glycol divinyl ether was redistilled from the still pot of the same apparatus as in Example 2 at about 0.5 kPa abs. The purified triethylene glycol divinyl ether obtained via the top had an APHA color number of only 5.
2'-({2-[2-(2-hydroxyethoxy)ethoxy]ethoxy}carbonyl)-8,12-bis[2-(methoxycarbonyl)ethyl]-2,7,13,17-tetramethyl-18-vinyl-2,21,22,23-tetrahydrobenzo[b]porphyrin-22-carboxylic acid[ No CAS ]
2'-({2-[2-(2-hydroxyethoxy)ethoxy]ethoxy}carbonyl)-13,17-bis[2-(methoxycarbonyl)ethyl]-2,7,12,18-tetramethyl-8-vinyl-2,21,22,23-tetrahydrobenzo[b]porphyrin-22-carboxylic acid[ No CAS ]
With phosphorus tribromide; at -10 - 20℃; for 4.0h;
Phosphorus tribromide (0.47 mL, 5 mmol) was added dropwise to triethylene glycol (5) (3.0 g,20.0 mmol) at -10 C. Then, the reaction was stirred at room temperature for 4 h. After the completionof the reaction, H2O was added, and the pH was adjusted to 7 using 5 M NaOH, extracted with CH2Cl2and dried over anhydrous Na2SO4. Removal of the solvent under vacuum, followed by flash columnchromatography (2% MeOH/CH2Cl2) afforded 11 as yellow oil (1.57 g, 7.36 mmol, 49%)3; 1H NMR(400 MHz, CDCl3) delta 3.83 (t, J = 6.2 Hz, 1H), 3.76 - 3.72 (m, 1H), 3.69 (s, 2H), 3.64 - 3.60 (m, 1H),3.49 (t, J = 6.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) delta 72.6, 71.2, 70.6, 70.4, 61.8, 30.3. The 1H and 13C spectroscopic data are in good agreement with those previously reported.
41%
With hydrogen bromide; In water; toluene; at 115℃; for 3.0h;
Polyethylene glycol (9 g, 60 mmol) was dissolved in 70 mL of toluene,Then, 10.2 mL of 48% HBr aqueous solution was added, heated to reflux,With sodium bicarbonate to absorb the release of gas,The reaction temperature was controlled at 115 C and the reaction was stirred for 3 days.After cooling, the reaction mixture was neutralized with saturated aqueous sodium bicarbonate and then solvent was removed. 30 mL of water was added and extracted with CH 2 Cl 2 (3 × 60 mL).The combined organic phases were dried over anhydrous sodium sulfate, and the organic solvent was stripped to give 5.2 g of compound 15 in 41% yield.
27.7%
With carbon tetrabromide; triphenylphosphine; In dichloromethane; at 0 - 20℃; for 2.0h;
Step 1: Preparation of 2-[2-(2-bromoethoxy) ethoxy] ethanol Triglycol (12.2 g, 81.3 mmol) was dissolved in dichloromethane (150 mL), stirred and cooled to 0 C., and then carbon tetrabromide (12.0 g, 27.1 mmol) and triphenylphosphine (7.8 g, 29.8 mmol) were added. The reaction mixture was stirred at room temperature for 2 h, the solvent was distilled off under reduced pressure, the residue was purified by a silica gel column to give 4.8 g of a pale yellow oil with a yield of 27.7%. NMR Detection: 1H NMR (400 MHz, CDCl3): 3.35 (t, 2H), 3.44 (t, 2H), 3.53 (m, 4H), 3.58 (t, 2H), 3.68 (t, 2H).
14%
With hydrogen bromide; In toluene; for 18.0h;Reflux;
Hydrogen bromide (86.21 g, 1.07 mmol, 115 mL) was added to a solution of 2-[2- (2-hydroxyethoxy)ethoxy]ethanol (100 g, 665.90 mmol) in toluene (1.15 L) and the resulting mixture was stirred at reflux for 18 hours, then the water layer was discarded. The organic layer was washed with aqueous NaOH solution, concentrated in vacuo, then purified by silica gel chromatography (MeOH:DCM = 1:20) to obtain 2-[2-(2-bromoethoxy)ethoxy]ethanol (20 g, 14% yield) as a liquid. 1H NMR (400 MHz, CDCl3) d 3.83 (t, J = 6.2 Hz, 2H), 3.77- 3.72 (m, 2H), 3.69 (s, 4H), 3.64- 3.61 (m, 2H), 3.49 (t, J = 6.1 Hz, 2H), 2.51 (t, J = 6.1 Hz, 1H).
titanium(IV) isopropylate; In xylene; at 160 - 180℃; for 5h;
Embodiment 2 Hexahydrophthalic anhydride of 0.5 mole, triethylene glycol of 1.05 moles, catalyst TIPT (Tetraisopropyl titanate) of 1.0 g and xylene of 34.0 g are received in a four-neck flask equipped with magnetic stir bars and a condenser for reaction at 160 C.-180 C. for 5 hours while the reactant is dehydrated during esterification. Esterification lasts until the acidity of the reactant becomes less than 3 mgKOH/g. Afterward, 0.97 mole of 2-EHA is added and esterification continues at 160 C.-230 C. for 5 hours until the acidity of the reactant becomes less than 5 mgKOH/g to finalize esterification.
Into a mixture of 17.7 g (130 mmol) of phosphorus trichloride in 15O g of toluene at ambient temperature is added dropwise a solution of 37.3 g (250 mmol) of Lambda/,Lambda/-diethyl aniline and 5 g (42 mmol) of Lambda/-methyl imidazole and 19 g (130 mmol) of methyl 3-te/f-butyl-4- hydroxyhydrocinnamate. Upon completion of the addition the temperature is increased to 6O0C and allowed to stir for 12 hours to give the desired phosphorochloridite. Into the phosphorochloridite solution at ambient temperature is added dropwise a solution 19.4 g (130 mmol) of Lambda/,Lambda/-diethyl aniline followed by a solution of 15 g (75mmol) of triethylene glycol and 3 g (30 mmol) of Lambda/-methylimidazole. This reaction mixture is allowed to stir at ambient temperature for 4 hours. The reaction mixture is then filtered and the filtrate is concentrated in vaccuo to yield a light yellow oil. 31P NMR (400MHz) (Benzene-d6) (ppm): 145
Into a mixture of 17.7 g (130 mmol) of phosphorus trichloride, 41.1 g (250 mmol) of 2-t-butyl- 6-methylphenol in 15O g of toluene at ambient temperature is added dropwise 15 g (1 deltaOmmol) of Lambda/,Lambda/-diethyl aniline. Upon completion of the addition the temperature is increased to 6O0C and allowed to stir for 12 hours to give the desired phosphorochloridite. Into the phosphorochloridite solution at ambient temperature is added dropwise a solution 1 1.3 g (75 mmol) of Lambda/-methylimidazole followed by a solution of 1 1.3 g (75 mmol) of triethylene glycol. The reaction mixture is allowed to stir at ambient temperature for 4 hours. The reaction mixture is then filtered and the filtrate is concentrated in vaccuo to afford a light yellow oil.31P NMR (400MHz) (Benzene-d6) (ppm): 161
poly(3,9-di(ethylidene)-2,4,8,10-tetraoxaspiro[5,5]undecane-co-triethylene glycol-co-triethylene glycol diglycolide)[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In tetrahydrofuran;Product distribution / selectivity;
The polyorthoester in this example was prepared from DETOSU, TEG and TEG-diGL. The molar ratio of the three components (DETOSU:TEG:TEG-diGL) was 90:80:20. Under rigorously anhydrous conditions, DETOSU (114.6 g, 540 mmol) was dissolved in a 2 L flask in 450 mL anhydrous THF and TEG (72.08 g, 480 mmol) and TEG-diGL (31.95 g, 120 mmol) was weighed into a 500 mL round bottom flask, and dissolved in anhydrous THF (50 mL). The TEG-diGL solution was added to the solution of DETOSU and TEG to initiate the polymerization. The solution came to a boil within a few minutes. The solution was allowed to cool to room temperature, then concentrated by rotary evaporation at 50 C., followed by rotary evaporation at 80 C. The material was semi-solid with a molecular weight of about 6,500. (i) Polymer A: The poly(ortho ester) in this example was prepared from DETOSU, TEG and TEG-diGL. The molar ratio of the three components (DETOSU:TEG:TEG-diGL) was 90:80:20. Under rigorously anhydrous conditions, DETOSU (114.61 g, 540 mmol) was dissolved in a 2 L flask in 450 mL anhydrous THF and TEG (72.08 g, 480 mmol) and TEG-diGL (31.95 g, 120 mmol) was weighed into a 500 mL round bottom flask, and dissolved in anhydrous THF (50 mL). The TEG-diGL solution was added to the solution of DETOSU and TEG to initiate the polymerization. The solution came to a boil within a few minutes. The solution was allowed to cool to room temperature, and then concentrated by rotary evaporation at 50 C. using a water aspirator, followed by rotary evaporation at 80 C. under high vacuum. The flask was then transferred to an oven at 70-75 C. and poured into an amber bottle over a period of about 30-60 minutes. The resulting polymer was semi-solid, viscous liquid at room temperature, with a weight average molecular weight (Mw) of approximately 6800 daltons.
poly(3,9-di(ethylidene)-2,4,8,10-tetraoxaspiro[5,5]undecane-co-triethylene glycol-co-triethylene glycol monoglycolide)[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With salicylic acid; In ethyl acetate;
The polyorthoester in this example was prepared from 3,9-di(ethylidene)-2,4,8,10-tetraoxaspiro[5,5]undecane (DETOSU), triethylene glycol (TEG), and triethyleneglycol monoglycolide (TEG-mGL). The molar ratio of the three components (DETOSU:TEG:TEG-mGL) was 65:95:5. Under rigorously anhydrous conditions, DETOSU (6.898 g, 32.5 mmol), TEG (7.133 g, 47.5 mmol) and TEG-mGL (0.521 g, 2.5 mmol) were weighed into a 250 mL round bottom flask, and the mixture dissolved in anhydrous ethyl acetate (16 mL). To this solution was added a salicylic acid solution in ethyl acetate (12 drops, 10 mg/mL) to initiate the polymerization. The solution came to a boil within a few minutes. The solution was allowed to cool to room temperature, then concentrated by rotoevaporation at 40-50 C. The flask was transferred to a vacuum oven, and dried at 40 C. for 2 hours followed by drying at 70 C. for additional 3 hours. The material was semi-solid with a molecular weight of about 4000.
General procedure: The DESs were prepared with TPAB as the organic salt. The HBD used were ethylene glycol, triethylene glycol and glycerol. The TPAB:HBD of molar ratios 1:3, 1:4, and 1:5 were used. In the case of TPAB:triethylene glycol the ratios were 1:2.5, 1:3, and 1:4. DES samples synthesized in different TPAB/HBD molar ratios are shown in Table 1. An incubator shaker (Brunswick Scientific Model INNOVA 40R) was used to mix the salt and the HBD. Each DES mixture was mixed at 400 rpm and 353.15 K for 2 h until a homogeneous transparent colorless liquid was formed. DES samples were synthesized at atmospheric pressure and under tight control of moisture content. Samples were kept in well sealed glass vials after preparation and fresh samples were used for analysis to avoid structural change or environmental effects on their physical properties. The properties measured were density, viscosity, surface tension, conductivity, reflective index and pH. The temperature range considered for all measured physical properties was 293.15-353.15 K.
General procedure: The DESs were prepared with TPAB as the organic salt. The HBD used were ethylene glycol, triethylene glycol and glycerol. The TPAB:HBD of molar ratios 1:3, 1:4, and 1:5 were used. In the case of TPAB:triethylene glycol the ratios were 1:2.5, 1:3, and 1:4. DES samples synthesized in different TPAB/HBD molar ratios are shown in Table 1. An incubator shaker (Brunswick Scientific Model INNOVA 40R) was used to mix the salt and the HBD. Each DES mixture was mixed at 400 rpm and 353.15 K for 2 h until a homogeneous transparent colorless liquid was formed. DES samples were synthesized at atmospheric pressure and under tight control of moisture content. Samples were kept in well sealed glass vials after preparation and fresh samples were used for analysis to avoid structural change or environmental effects on their physical properties. The properties measured were density, viscosity, surface tension, conductivity, reflective index and pH. The temperature range considered for all measured physical properties was 293.15-353.15 K.
General procedure: The DESs were prepared with TPAB as the organic salt. The HBD used were ethylene glycol, triethylene glycol and glycerol. The TPAB:HBD of molar ratios 1:3, 1:4, and 1:5 were used. In the case of TPAB:triethylene glycol the ratios were 1:2.5, 1:3, and 1:4. DES samples synthesized in different TPAB/HBD molar ratios are shown in Table 1. An incubator shaker (Brunswick Scientific Model INNOVA 40R) was used to mix the salt and the HBD. Each DES mixture was mixed at 400 rpm and 353.15 K for 2 h until a homogeneous transparent colorless liquid was formed. DES samples were synthesized at atmospheric pressure and under tight control of moisture content. Samples were kept in well sealed glass vials after preparation and fresh samples were used for analysis to avoid structural change or environmental effects on their physical properties. The properties measured were density, viscosity, surface tension, conductivity, reflective index and pH. The temperature range considered for all measured physical properties was 293.15-353.15 K.
2-(2-(2-hydroxyethoxy)ethoxy)ethyl 2-(2-chlorophenoxy)acetate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
67%
With iodine; In toluene;Reflux;
General procedure: A mixture of phenoxy carboxylic acid 1 (2 mmol), ethylene glycol (10 mmol), I2 (0.025g, 0.1 mmol) in toluene (10 mL) was heated under reflux for 2-3 h. When the reaction was completed (TLC), the mixture was washed with 10% Na2S2O3 (10 mL × 3) and H2O (10 mL × 2). The organic layer was dried over anhydrous Na2SO4. The crude was purified by column chromatography to afford the desired products 3a-3p.
2-(2-(2-hydroxyethoxy)ethoxy)ethyl 2-(2-chloro-4-methylphenoxy)acetate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
65%
With iodine; In toluene;Reflux;
General procedure: A mixture of phenoxy carboxylic acid 1 (2 mmol), ethylene glycol (10 mmol), I2 (0.025g, 0.1 mmol) in toluene (10 mL) was heated under reflux for 2-3 h. When the reaction was completed (TLC), the mixture was washed with 10% Na2S2O3 (10 mL × 3) and H2O (10 mL × 2). The organic layer was dried over anhydrous Na2SO4. The crude was purified by column chromatography to afford the desired products 3a-3p.
KOH (3.66 g, 65.2 mmol) was added to a stirred DMSO solution (100 mL) of triethylene glycol (9.79 g, 65.2 mmol). After 1 hour at room temperature, <strong>[4199-89-7]5-chloro-1,10-phenanthroline</strong> (2.80 g, 13.0 mmol) was introduced into the above stirred solution at 60 [deg.] C for an additional 40 hours . The reaction mixture was poured onto 200 mL EtOAc and washed thoroughly with water (300 mL x 5 times) to remove DMSO. The organic layer was recovered, dried over anhydrous MgSO4, and concentrated under vacuum. Silica gel column purification was carried out with increasing the polarity of the effluent from 100% CH2Cl2 to CH2Cl2: CH3OH = 9: 1 by volume to give a yellow liquid in 67% yield.
With dmap; N-(3-dimethylaminopropyl)-N-ethylcarbodiimide; In dichloromethane; at 20℃; for 168h;Inert atmosphere; Cooling with ice;
<strong>[82419-36-1]Ofloxacin</strong> (2.1 mol) and DMAP (1.05 eqv.) were weighed in a flask and stirred in anhydrous dichloromethane (900 mL) under N2 at room temperature until dissolved. Triethylene glycol (1 mol eqv.) was added into reaction flask. The reaction flask was then placed in an ice bath and solution was stirred under N2. EDC (8.4 mol eqv.) was weighed and quickly added into the reaction flask. The reaction was allowed to proceed for 1 week at room temperature under N2. The resulting solution was washed with water (2×2 L). The organic layer was dried over sodium sulphate. Mixture solution was filtered and filtrate was collected. Dichloromethane was removed to approximately 20% of the original volume by rotary evaporator. Acetone was added into the flask at 1:1 (v/v) ratio and placed in -20 C. freezer overnight to precipitate. Precipitate was then filtered, collected and dried. Compound 12: HPLC (mobile phase H2O/TFA and MeCN/TFA) 19.678 min and 19.868 min. Mass spectroscopy (m/z) 836.4. 1H NMR (300 MHz, CDCl3) delta (ppm) 1.56 (CH3-CH, <strong>[82419-36-1]ofloxacin</strong>), 2.37 (CH3-N, <strong>[82419-36-1]ofloxacin</strong>), 2.56 (-O-CH2-CH (CH3), <strong>[82419-36-1]ofloxacin</strong>), 3.34 (-N-CH2-CH2-N-, <strong>[82419-36-1]ofloxacin</strong>), 3.77 (O-CH2-CH2-O, TEG), 3.85 (-CH2-O, TEG), 4.38 (CH-C(CH3), <strong>[82419-36-1]ofloxacin</strong>), 4.84 (CH2-OOC, TEG), 7.21 (HC?C-F, <strong>[82419-36-1]ofloxacin</strong>), 8.22 (N-C(H)?C(CO)-COO-, <strong>[82419-36-1]ofloxacin</strong>).
2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)-3-nitrobenzoic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
82%
With caesium carbonate; at 60℃; for 16h;
Example 12 2-(2-(2-(2-Hydroxyethoxy)ethoxy)ethoxy)-3-nitrobenzoic acid (20) A heterogeneous mixture of triethyleneglycol (183 mL, 1375 mmol), (<strong>[317-46-4]2-fluoro-3-nitrobenzoic acid</strong> 19) (10.18 g, 55 mmol), and cesium carbonate (Cs2CO3) (39.4 g, 121 mmol) was warmed to 60 C. to give a homogenous solution. After 16 hours, heating was discontinued and the mixture was cooled to room temperature. The cooled reaction mixture was diluted with water (400 mL) and, with cooling and swirling, slowly acidified with 12N HCl to pH 2-3 (16 mL of 12 N HCl; 190 mmol). The resulting acidic solution was then extracted with Et2O (?75 mL). The layers were separated and the aqueous phase was washed with CHCl3 (3*; 400 mL, 300 mL and 150 mL respectively). The CHCl3 layers were combined and washed with water (2*; 100 mL each), and then with brine (100 mL). The CHCl3 organic phase was then dried over Na2SO4, filtered, concentrated under reduced pressure and then under high vacuum overnight to yield 14.11 g (82%) of a yellow tinted oil. LC/MS: >95% (M+23=338.4, consistent with desired product). The isolated 2-(2-(2-(2-Hydroxyethoxy)ethoxy)ethoxy)-3-nitrobenzoic acid was used for the subsequent step without further manipulation.
62%
With caesium carbonate; at 60℃; for 5h;
A heterogenous mixture of triethyleneglycol (33 mL, 250 mmol), <strong>[317-46-4]2-fluoro-3-nitrobenzoic acid</strong> (1.85 g, 10 mmol), and cesium carbonate (7.2 g, 22 mmol) was warmed to 60 C. to give a homogenous solution. After 5 hours, heating was discontinued and the mixture was cooled to room temperature. The cooled reaction mixture was diluted with water (100 mL), and with swirling slowly acidified with 12N HCl to pH 2 (3.5 mL of 12 N HCl; 42 mmol). The resulting acidic solution was then extracted with Et2O (15 mL). The layers were separated and the aqueous phase was washed with CHCl3 (2×; 100 mL and 75 mL respectively). The CHCl3 layers were combined and washed with water (2×; 30 mL each), and then with brine (50 mL). The CHCl3 organic phase was then dried over Na2SO4, filtered, and concentrated under reduced pressure to give 1.95 g (62%) of 2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)-3-nitrobenzoic acid as a yellow tinted oil. TLC: 10:1:0.5 EtOAc:MeOH:NH4OH Rf=0.05, with smallest trace impurity at Rf=0.15; LC/MS: Virtually clean and consistent with desired product (M+H=315.9). The isolated 2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)-3-nitrobenzoic acid was used for subsequent steps without further manipulation.
With potassium carbonate; at 155℃; under 750.075 Torr; for 6h;
Example 8 This embodiment serves to illustrate the present invention prepartion method of triethylene glycol dicyclohexanoateIn a 100ml round-bottomed flask with condenser, liquid separator and stirrer was added 11.53g (0.077 mol) of triethylene glycol (determined by gas chromatography analysis of chromatographic purity 99.2%), 40.43g (0.028 mol) cyclohexanoic acid methyl ester, 2.07g (0.015mol) of anhydrous potassium carbonate, round bottom flask was contacted in an oil bath heated to 155C, the reaction 0.1MPa, during reaction, simultaneously go through distillation to separate and remove the generated distilled alcohol. After 6h of reaction, add toluene to the round-bottomed flask. Before and after it was added a total of 7 times , was added in two adjacent intervals of 2 hours, each time adding 6 mL, for each time toluene is added, it was reacted for 1.4 hours and at the same time distill off the toluene and azeotropes of the round-bottomed flask. After the reaction mixture was cooled for approximately 6 minutes, again add the next toluene. After completion of reaction, cool down the round-bottomed flask and filter to remove anhydrous potassium carbonate. Again, vacuum distill the filtrate at 350Pa at 48C to remove unreacted cyclohexanoic acid methyl ester and other impurities to obtain triethylene glycol dicyclohexyl carboxylate in 88% yield, by the gas chromatography - mass spectrometry (GC-MS) analysis, the purity of 99.1%. The results are shown in Table 1.
With potassium carbonate; In dimethyl sulfoxide; at 0 - 60℃;Inert atmosphere;
Using <strong>[51762-67-5]3-nitrophthalonitrile</strong> (20 mmol) and triethylene glycol (20-70 mmol, preferably 60 mmol) as reactants and dimethyl sulfoxide (40-200 mL, preferably 140 mL) as solvent in potassium carbonate ( 30 to 90 mmol, preferably 80 mmol) are present and the reaction is stirred at room temperature to 60 C. (preferably 60 C.) for 24 to 72 hours under nitrogen protection and monitored by thin layer chromatography. After the reaction was completed, the reaction solution was poured into ice water to precipitate a light yellow precipitate, which was filtered to collect the residue. The filtrate was extracted with CHCl 3 , and the extract was back-extracted with water. The CHCl 3 extract was evaporated and vacuum dried at room temperature to give a yellow solid. The filter residue was combined with the product obtained by extraction in a yield of 65%.
65%
With potassium carbonate; In dimethyl sulfoxide; at 20 - 60℃;Inert atmosphere;
Using <strong>[51762-67-5]3-nitrophthalonitrile</strong> (20 mmol) and triethylene glycol (20-70 mmol, preferably 60 mmol) as reactants and dimethyl sulfoxide (40-200 mL, preferably 140 mL) as solvent in potassium carbonate ( 30-90 mmol, preferably 80 mmol) are present and incubated under nitrogen for 24 to 72 hours at room temperature to 60C (preferably 60C) and monitored by thin layer chromatography. After the reaction was completed, the reaction solution was poured into ice water to precipitate a light yellow precipitate, which was filtered to collect the residue. The filtrate was extracted with CHCl 3 , and the extract was back-extracted with water. The CHCl 3 extract was evaporated and vacuum dried at room temperature to give a yellow solid. The filter residue was combined with the product obtained by extraction in a yield of 65%.
58%
In acetonitrile; at 90℃; for 18h;Inert atmosphere;
(. 3. 18 g, 21 17 mmol), anhydrous potassium carbonate (. 15.20 g, 0 11 mol) was added to the IJ IOOml round-bottomed flask, dissolved in acetonitrile and heated to 90 C under nitrogen; 3- nitro-phthalonitrile (. 1.83 g, 10 58 mmol) dissolved in acetonitrile after transfer to constant pressure dropping funnel and slowly added dropwise to the flask, continued after the addition was complete the reaction at 90 C at 18 h; After completion of the reaction, acetonitrile was removed by rotary evaporation under reduced pressure, the residue was extracted with methylene chloride; the resulting crude product with dichloromethane - methanol 30: 1 (v / v) as eluent, separated by silica gel column chromatography to give a pale after yellow oily liquid Technology Xhttp://www.technology-x.net/A61K/201410486382_2.html
To a solution of Compound 15 (1.2 eq) in THF/DMF (1 : 5, 0.1 M) was added NaH (1.2 eq) at 0 C. The mixture was stirred for 5 minutes and Compound 14 (1.0 eq) and Nal (0.1 eq) was added into the mixture at 0 C. The mixture was warmed to room temperature and stirred for 14 hours. After the reaction, the mixture was cooled to 0 C, quenched with ice-water, and then extracted with EtOAc (x3). The combined organic layer was washed with brine, dried over MgS04 and then concentrated under reduced pressure. The crude residue was purified by flash column chromatography (CH2CI2 to 1 : 10 MeOH : CH2CI2) to afford Compound 16.
With di-isopropyl azodicarboxylate; triphenylphosphine; In tetrahydrofuran; at 20℃; for 4h;
2-Chloroquinolin-6-ol ((253 mg, 1.1 mmol) was dissolved in dry THF, and then PPh3 (393 mg, 1.5 mmol) was added thereto in succession.Triethylene glycol (68.3 muL, 0.5 mmol)And DIAD (diisopropylazodicarboxylate, 294 muL, 1.5 mmol).The reaction was kept at room temperature for 4 h, TLC traced It should be until the reaction is complete. After completion of the reaction, the mixture was concentrated under reduced pressure and purified by column chromatography to obtain Compound 6 (red solid, 95.8%; ethyl acetate:dichloromethane=1:1).
The protected version of this tether was obtained through standard transformations involving monoprotection of triethyleneglycol (45-0) followed by conversion of the remaining alcohol to a mesylate, displacement with azide and catalytic reduction in the presence of di-t-butyl dicarbonate.
With titanium(IV) isopropylate; at 40 - 110℃; for 6h;
100 G of methyl 3-(3,5-di-tert-butyl-4-hydroxyphenyl)propionate and 450 G of triethylene glycol were added to the reaction flask. The temperature was raised to 40 C, stirring was started, and 0.75 G of tetraisopropyl titanate was added. The reaction temperature was raised to 75 C and the distillation of methanol was started. The reaction temperature was raised up to 110 C until there was no methanol distillate and the reaction was carried out for 6 hours. After the reaction was stopped, 350 mL of toluene was added to the reaction mixture, and the lower excess triethylene glycol was separated under heating (temperature: 50 C). The organic phase was washed once more with 50 mL of pure water, and the collected organic phase was concentrated by distillation under reduced pressure to remove all volatile materials to give a pale yellow viscous material (ie, compound MPCP) 145G.
Decaethylene glycol monomethyl ether (32). Monodispersed compound 32 was prepared from compound 31 and monodispersed triethylene glycol using the procedure described above in Example 17. Oil; Rf 0.41 (methanol:chloroform=6:10); MS m/z calc'd for C21H44O11 472.29 (M++1), found 472.29.
General procedure: A mixture of triethylene glycol (2.0 equiv) and sodium hydride (1.0 equiv) was stirred at RT in tetrahydrofuran (THF) (10 mL) for 2 h in Schlenk flask. Then, 3a (400 mg, 2.4 mmol) was added dropwise, and the mixture was stirred at 60 C for 12 h. After cooling at RT, distilled water was added and the mixture was stirred in air for 5 min. After extracting the mixture with CH2Cl2, the organic layer was dried with anhydrous MgSO4. The crude product was purified by column chromatography, leading to the desired product 4a (480 mg, 83% yield): 1H NMR (300 MHz, CDCl3, ppm): delta 3.48-3.82 (m, 12H), 4.57 (s, 2H), 7.23-7.36 (m, 4H). 13C NMR (75 MHz, CDCl3, ppm): delta 61.5, 70.0, 70.3, 70.4, 73.1, 127.6, 127.7, 128.4, 138.1.
(21E,23E,25E,26E,36R,37S,38R,39R,41S,43S,46S,47R,48R,57R)-47,57-dihydroxy-45-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]-46-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxy-cyclohexyl]-1-methyl-ethyl]-48-methoxy-36,37,38,39,49,50-hexamethyl-68,69-dioxa-58-azatricyclohexatriaconta-21,23,25(49),26(50)-tetraene-51,52,53,54,55-pentone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
28%
With toluene-4-sulfonic acid; In tetrahydrofuran; water; at 20℃; for 2h;
A mixture of <strong>[159351-69-6]everolimus</strong> (3.92 g, 26.09 mmol) and p-toluenesulfonic acid hydrate (0.45 g, 2.61 mmol) in THF (10 mL) was stirred at 20 C for 2 hours. The mixture was poured into ice cold saturated NaHCO3 (30 mL) and extracted with EtOAc (50 mL × 3). The organic layers were combined and then washed with water and brine, then concentrated in vacuo. The residue was purified by reversed phase chromatography (CH3CN/pure water = 1:1) to afford (21E,23E,25E,26E,36R,37S,38R,39R,40S,43S,45S,47R,48R,57R)-47,57-dihydroxy-44-[2-[2- (2-hydroxyethoxy)ethoxy]ethoxy]-45-[(1R)-2-[(1S,2R,3R)-3-(2-hydroxyethoxy)-2-methoxy- cyclohexyl]-1-methyl-ethyl]-48-methoxy-36,37,38,39,49,50-hexamethyl-68,69-dioxa-58- azatricyclohexatriaconta-21,23,25(49),26(50)-tetraene-51,52,53,54,55-pentone (I-32: 0.155 g, 28% yield) as a white solid. ESI-MS (EI+, m/z): 1098.4 [M+Na]+. 1H NMR (500 MHz, CDCl3) d 6.49- 5.83 (m, 4H), 5.67- 5.35 (m, 2H), 5.33- 5.01 (m, 2H), 4.92- 4.08 (m, 2H), 4.05- 3.51 (m, 17H), 3.52- 3.23 (m, 11H), 3.23- 3.01 (m, 3H), 2.66 (m, 4H), 2.40- 1.95 (m, 5H), 1.95- 1.55 (m, 17H), 1.52- 1.13 (m, 9H), 1.13- 0.79 (m, 16H), 0.71 (dd, J = 23.8, 11.9 Hz, 1H).
(21E,23E,25E,26E,36R,37S,38R,39R,41S,43S,45R,46S,47R,48R,57R)-47,57-dihydroxy-45-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]-46-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxy-cyclohexyl]-1-methyl-ethyl]-48-methoxy-36,37,38,39,49,50-hexamethyl-68,69-dioxa-58-azatricyclohexatriaconta-21,23,25(49),26(50)-tetraene-51,52,53,54,55-pentone[ No CAS ]
(21E,23E,25E,26E,36R,37S,38R,39R,41S,43S,45S,46S,47R,48R,57R)-47,57-dihydroxy-45-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]-46-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxy-cyclohexyl]-1-methyl-ethyl]-48-methoxy-36,37,38,39,49,50-hexamethyl-68,69-dioxa-58-azatricyclohexatriaconta-21,23,25(49),26(50)-tetraene-51,52,53,54,55-pentone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
33 mg; 37 mg
With toluene-4-sulfonic acid; In tetrahydrofuran; water; for 2h;
A mixture of <strong>[159351-69-6]everolimus</strong> (0.5 g, 0.522 mmol), 2-[2-(2- hydroxyethoxy)ethoxy]ethanol (3.92 g, 26.09 mmol) and p-toluenesulfonic acid monohydrate (0.45 g, 2.61 mmol) in THF (10 mL) was stirred at 20 C for 2 h. The mixture was then poured into ice cold sat. NaHCO3 (30 mL) and extracted with EtOAc (50 mL × 3). The combined organic layers were washed with water and brine then dried over anhydrous Na2SO4, filtered and concentrated. The reslting residue was purified via reverse phase chromatography (C18, CH3CN:H2O= 6.5: 3.5) to afford (21E,23E,25E,26E,36R,37S,38R,39R,41S,43S,46S,47R,48R,57R)-47,57-dihydroxy-45-[2-[2- (2-hydroxyethoxy)ethoxy]ethoxy]-46-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxy- cyclohexyl]-1-methyl-ethyl]-48-methoxy-36,37,38,39,49,50-hexamethyl-68,69-dioxa-58- azatricyclohexatriaconta-21,23,25(49),26(50)-tetraene-51,52,53,54,55-pentone (0.155 g, 28%) as white solid. ESI-MS (EI+, m/z):1098.4 [M+Na]+. 1H NMR (500 MHz, CDCl3) d 6.42- 5.87 (m, 4H), 5.63- 5.34 (m, 2H), 5.32- 5.00 (m, 2H), 4.85 (s, 1H), 4.35- 4.09 (m, 1H), 4.05- 3.49 (m, 18H), 3.49- 3.01 (m, 14H), 2.66 (dddd, J = 31.3, 24.8, 21.2, 13.0 Hz, 4H), 2.33 (d, J = 12.0 Hz, 2H), 2.06 (dd, J = 39.9, 10.6 Hz, 3H), 1.77- 1.53 (m, 13H), 1.51- 1.14 (m, 10H), 1.14- 0.81 (m, 18H), 0.71 (dd, J = 23.8, 11.9 Hz, 1H). 170 mg of (21E,23E,25E,26E,36R,37S,38R,39R,41S,43S,46S,47R,48R,57R)- 47,57-dihydroxy-45-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]-46-[(1R)-2-[(1S,3R,4R)-4-(2- hydroxyethoxy)-3-methoxy-cyclohexyl]-1-methyl-ethyl]-48-methoxy-36,37,38,39,49,50- hexamethyl-68,69-dioxa-58-azatricyclohexatriaconta-21,23,25(49),26(50)-tetraene- 51,52,53,54,55-pentone purified via prep chiral HPLC and the resulting epimers purified via silica gel chromatography (hexane:DCM:EtOAc:MeOH = 3:3:1:0.8) to obtain (21E,23E,25E,26E,36R,37S,38R,39R,41S,43S,45S,46S,47R,48R,57R)-47,57-dihydroxy-45- [2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]-46-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3- methoxy-cyclohexyl]-1-methyl-ethyl]-48-methoxy-36,37,38,39,49,50-hexamethyl-68,69- dioxa-58-azatricyclohexatriaconta-21,23,25(49),26(50)-tetraene-51,52,53,54,55-pentone (I- 110: 37 mg, 21% yield) and (21E,23E,25E,26E,36R,37S,38R,39R,41S,43S,45R,46S,47R,48R,57R)-47,57-dihydroxy-45- [2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]-46-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3- methoxy-cyclohexyl]-1-methyl-ethyl]-48-methoxy-36,37,38,39,49,50-hexamethyl-68,69- dioxa-58-azatricyclohexatriaconta-21,23,25(49),26(50)-tetraene-51,52,53,54,55-pentone (I- 109: 33 mg, 19% yield), both as white solids. Chiral analysis method: C I-110: ESI-MS (EI+, m/z):1098.0 [M+Na]+. 1H NMR (500 MHz, CDCl3) d 6.41- 6.18 (m, 2H), 6.12 (dd, J = 15.0, 10.3 Hz, 1H), 5.93 (dd, J = 33.7, 10.6 Hz, 1H), 5.47 (ddd, J = 33.0, 20.8, 9.5 Hz, 2H), 5.26 (d, J = 5.6 Hz, 1H), 5.13 (dt, J = 21.8, 10.8 Hz, 1H), 4.88 (s, 1H), 4.19 (t, J = 9.3 Hz, 1H), 3.94- 3.52 (m, 19H), 3.49- 3.25 (m, 12H), 3.24- 3.02 (m, 3H), 2.76 (ddd, J = 26.2, 16.6, 10.3 Hz, 3H), 2.57 (dd, J = 17.0, 6.3 Hz, 1H), 2.29 (t, J = 26.2 Hz, 2H), 2.16- 1.85 (m, 6H), 1.74- 1.53 (m, 10H), 1.53- 1.16 (m, 9H), 1.15- 1.01 (m, 8H), 1.01- 0.82 (m, 10H), 0.71 (dd, J = 23.9, 12.0 Hz, 1H). I-109: ESI-MS (EI+, m/z):1098.0 [M+Na]+. 1H NMR (500 MHz, CDCl3) d 6.43- 5.90 (m, 4H), 5.62- 5.02 (m, 5H), 4.24 (d, J = 63.6 Hz, 1H), 3.97 (dd, J = 21.5, 6.8 Hz, 1H), 3.86- 3.50 (m, 18H), 3.45- 3.01 (m, 14H), 2.73- 2.46 (m, 3H), 2.39- 1.94 (m, 6H), 1.91- 1.69 (m, 10H), 1.50- 1.31 (m, 12H), 1.16- 0.85 (m, 18H), 0.69 (d, J = 11.7 Hz, 1H).
3'-O-(tert-butyl)dimethylsilyl-5-[(4-decyl-1,2,3-triazol-1-yl)methyl]-2'-deoxyuridine[ No CAS ]
[ 112-27-6 ]
3'-O-(tert-butyl)dimethylsilyl-5'-O-carbonyltriethylene glycol-5-[(4-decyl-1,2,3-triazol-1-yl)methyl]-2'-deoxyuridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
60%
General procedure: A nucleoside (0.28mmol) was dissolved in dry dimethylformamide(1ml) and CDT was added (137mg, 0.84mmol).The mixture was heated for 24 h at 37 C. Then the anhydrous tri- or tetraethylene glycol (1.65 g for triethylene glycol, 2.1 gfor tetraethylene glycol, 11mol) and dioxane (0.5ml) wereadded. The mixture was heated for 24 h at 37 C and thenevaporated to obtain crude oil with constant volume. Theproduct was extracted in the chloroform-water system, the organic layer was evaporated. The product was isolated by column chromatography in the chloroform:ethyl acetate:ethanol (5:5:0.1 v/v) eluting system.
3'-O-(tert-butyl)dimethylsilyl-5-[(4-dodecyl-1,2,3-triazol-1-yl)methyl]-2'-deoxyuridine[ No CAS ]
[ 112-27-6 ]
3'-O-(tert-butyl)dimethylsilyl-5'-O-carbonyltriethylene glycol-5-[(4-dodecyl-1,2,3-triazol-1-yl)methyl]-2'-deoxyuridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
62%
General procedure: A nucleoside (0.28mmol) was dissolved in dry dimethylformamide(1ml) and CDT was added (137mg, 0.84mmol).The mixture was heated for 24 h at 37 C. Then the anhydroustri- or tetraethylene glycol (1.65 g for triethylene glycol, 2.1 gfor tetraethylene glycol, 11mol) and dioxane (0.5ml) wereadded. The mixture was heated for 24 h at 37 C and thenevaporated to obtain crude oil with constant volume. Theproduct was extracted in the chloroform-water system, theorganic layer was evaporated. The product was isolated bycolumn chromatography in the chloroform:ethyl acetate:ethanol (5:5:0.1 v/v) eluting system.
5'-O-(tert-butyl)dimethylsilyl-5-azidomethyl-2'-deoxyuridine[ No CAS ]
[ 112-27-6 ]
3'-O-carbonyltriethylene glycol-5'-O-(tert-butyl)dimethylsilyl-5-azidomethyl-2'-deoxyuridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
70%
General procedure: A nucleoside (0.28mmol) was dissolved in dry dimethylformamide(1ml) and CDT was added (137mg, 0.84mmol).The mixture was heated for 24 h at 37 C. Then the anhydroustri- or tetraethylene glycol (1.65 g for triethylene glycol, 2.1 gfor tetraethylene glycol, 11mol) and dioxane (0.5ml) wereadded. The mixture was heated for 24 h at 37 C and thenevaporated to obtain crude oil with constant volume. Theproduct was extracted in the chloroform-water system, theorganic layer was evaporated. The product was isolated bycolumn chromatography in the chloroform:ethyl acetate:ethanol (5:5:0.1 v/v) eluting system.
5'-O-(tert-butyl)dimethylsilyl-5-[(4-decyl-1,2,3-triazol-1-yl)methyl]-2'-deoxyuridine[ No CAS ]
[ 112-27-6 ]
3'-O-carbonyltriethylene glycol-5'-O-(tert-butyl)dimethylsilyl-5-[(4-decyl-1,2,3-triazol-1-yl)methyl]-2'-deoxyuridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
67%
General procedure: A nucleoside (0.28mmol) was dissolved in dry dimethylformamide(1ml) and CDT was added (137mg, 0.84mmol).The mixture was heated for 24 h at 37 C. Then the anhydroustri- or tetraethylene glycol (1.65 g for triethylene glycol, 2.1 gfor tetraethylene glycol, 11mol) and dioxane (0.5ml) wereadded. The mixture was heated for 24 h at 37 C and thenevaporated to obtain crude oil with constant volume. Theproduct was extracted in the chloroform-water system, theorganic layer was evaporated. The product was isolated bycolumn chromatography in the chloroform:ethyl acetate:ethanol (5:5:0.1 v/v) eluting system.
5'-O-(tert-butyl)dimethylsilyl-5-[(4-dodecyl-1,2,3-triazol-1-yl)methyl]-2'-deoxyuridine[ No CAS ]
[ 112-27-6 ]
3'-O-carbonyltriethylene glycol-5'-O-(tert-butyl)dimethylsilyl-5-[(4-dodecyl-1,2,3-triazol-1-yl)methyl]-2'-deoxyuridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
63%
General procedure: A nucleoside (0.28mmol) was dissolved in dry dimethylformamide(1ml) and CDT was added (137mg, 0.84mmol).The mixture was heated for 24 h at 37 C. Then the anhydroustri- or tetraethylene glycol (1.65 g for triethylene glycol, 2.1 gfor tetraethylene glycol, 11mol) and dioxane (0.5ml) wereadded. The mixture was heated for 24 h at 37 C and thenevaporated to obtain crude oil with constant volume. Theproduct was extracted in the chloroform-water system, theorganic layer was evaporated. The product was isolated bycolumn chromatography in the chloroform:ethyl acetate:ethanol (5:5:0.1 v/v) eluting system.
(1) Weigh 0.447g of sodium hydroxide (11mmol) into a 50mL three-necked flask, dissolve it with 2mL of water, add 10g of triethylene glycol (66mmol), mix well, and then weigh 1.278g of 4-toluenesulfonyl chloride (TsCl, 6mmol), put into a 50mL beaker, add 17mL of THF to dissolve, put it into a constant pressure dropping funnel, slowly add in an ice bath, dropwise after 2.5 hours, continue the reaction for 10 minutes, TLC monitors the reaction is complete After the reaction was stopped, 20 mL of water was added to the reaction solution, followed by extraction with dichloromethane. The organic layers were combined, the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, filtered with suction, and 1.763 g of a crude intermediate product was obtained by rotary evaporation.(2) In a 25 mL round-bottomed flask, 89.4 mg of the crude intermediate obtained in step (1), 87 mg of tertiary acetic acid (0.2 mmol), 53 mg of dicyclohexylcarbodiimide (DCC, 0.26 mmol), and 15 mg of 4 were sequentially added. -Dimethylaminopyridine (DMAP, 0.12 mmol) was dissolved in 2 mL of DCM, reacted at room temperature for 24 h, the reaction was stopped, the solution was cloudy, and 265 mg of a pale yellow solid was obtained after suction filtration. 120 mg of intermediate product was purified by column chromatography. The elution system was petroleum ether: ethanol = 3: 2.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






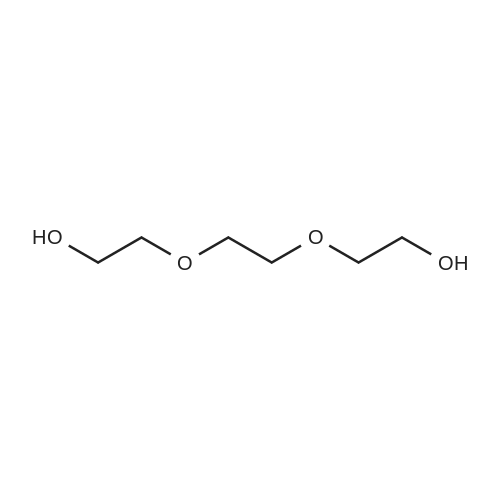

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping