* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Synthesis, 2008, # 11, p. 1793 - 1797
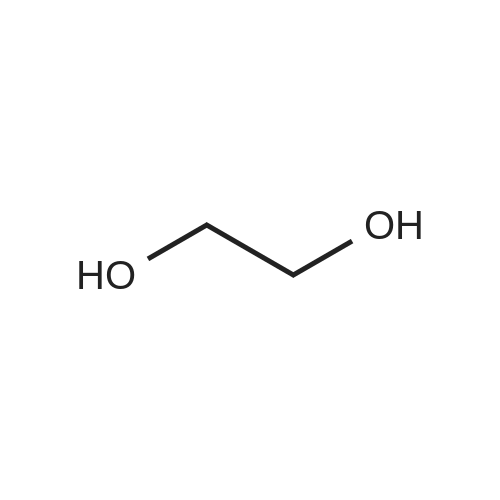
2
[ 872-31-1 ]
[ 1072-53-3 ]
[ 13781-67-4 ]
Reference:
[1] Synthesis, 2008, # 11, p. 1793 - 1797
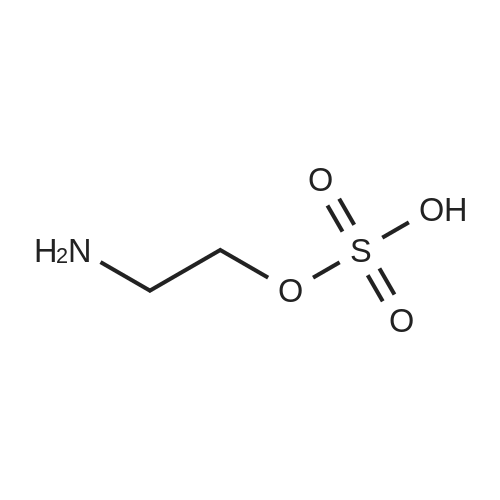
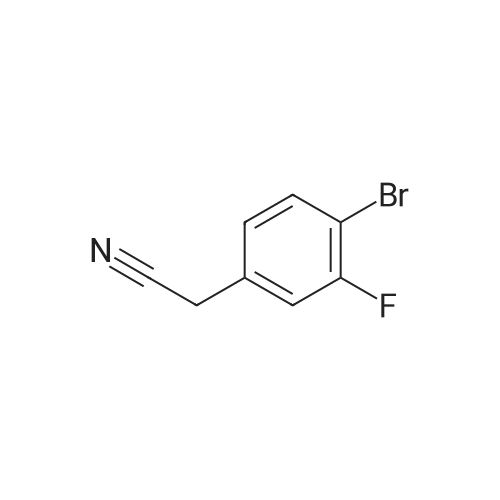
3
[ 3741-38-6 ]
[ 1072-53-3 ]
Yield
Reaction Conditions
Operation in experiment
95.84%
With manganese(IV) oxide In water at 5℃; for 0.75 h;
Taking 216.3 g (2 mol) of ethylene sulfite, 200 ml of distilled water and 0.4 g of a particulate catalyst (by mass ratio, Cu:NiO:MnO2 = 1:1.2:0.8),Stir slowly and control the temperature at 5 °C.Ozone was added at 0.05 L/min (ozone concentration was 20 mL/L) and the reaction was carried out for 45 min.After the reaction is completed, the layers are allowed to stand, and the organic layer is taken.The organic layer was washed twice with distilled water and dried to give a vinyl sulfate.The yield was 95.84percent. The product was analyzed by liquid chromatography and the results showed thatThe content of ethylene sulfate was 95.63percent, and the content of ethylene sulfite was 4.28percent.
90.66%
With sodium hydrogencarbonate; iron(II) sulfate In dichloromethane
(2) oxidation reaction: first of all to the 2nd reaction kettle is added as a solvent of dichloromethane 450 g and as the quality of the catalyst concentration is 10percent ferric sulfate solution 49.38 g, then by adding sodium bicarbonate to adjust the pH of the solution in the 2nd reaction value is 7 - 8, then the 2nd reactor cooling to 4 °C, then to the 2nd reaction in the cauldron adds by drops quality concentration is 10percent sodium percarbonate solution 7756.29 g, dropping time is 2h, keeping the heat after dropping 1.5h, to obtain the aqueous phase and organic phase coexistence of the reaction solution, then the reaction fluid settlement, layered separating the aqueous phase, get the organic phase, then the weight of the aqueous phase such as methylene chloride to the separated aqueous phase extraction three times, the residual sulfuric acid in the aqueous phase is extracted to vinyl acetate, thereby obtaining the vinyl acetate dichloromethane extract containing sulfuric acid, then the dichloromethane extract combined with the organic phase, then combined to form the solution is distilled under reduced pressure at room temperature to remove the methylene chloride, thereby obtaining the sulfuric acid ethylene the ester is thick product 247.38g (the purity is 95.66percent), molar yield is 90.88percent;(3) refining: use with sulfuric acid and ethylene the ester is thick product of the weight of the dichloromethane as solvent to sulfuric acid ethylene the ester is thick product of re-crystallization processes, thus obtaining high-purity sulfuric acid vinyl 236.29 g (purity of 99.91percent, the water content is 39 ppm, an acid value of 45 ppm), molar yield is 90.66percent.
Reference:
[1] Patent: CN108822073, 2018, A, . Location in patent: Paragraph 0017-0028
[2] Patent: CN106187989, 2016, A, . Location in patent: Paragraph 0016
[3] Acta Chemica Scandinavica, 1996, vol. 50, # 2, p. 170 - 177
[4] Journal of the Chemical Society, Chemical Communications, 1983, # 23, p. 1392 - 1394
[5] Molecules, 2005, vol. 10, # 9, p. 1169 - 1178
[6] Organic Letters, 2015, vol. 17, # 23, p. 5898 - 5901
[7] Tetrahedron Asymmetry, 1994, vol. 5, # 1, p. 83 - 92
[8] Tetrahedron, 1995, vol. 51, # 17, p. 5169 - 5180
[9] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 7, p. 2151 - 2155
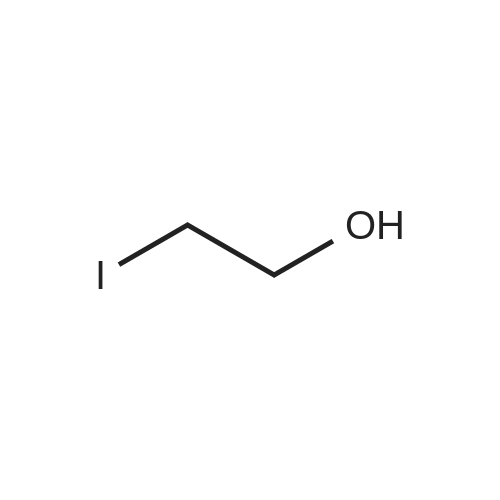
4
[ 107-21-1 ]
[ 1072-53-3 ]
Yield
Reaction Conditions
Operation in experiment
81.8%
Stage #1: With fluorosulfonyl fluoride; sodium sulfate; tetra(n-butyl)ammonium hydrogensulfate; sodium hydroxide In tert-butyl methyl ether at 5 - 10℃; for 1 h; Autoclave Stage #2: With 15-crown-5; 18-crown-6 ether In dichloromethane
In a 1000 mL autoclave, 40 g of ethylene glycol, 250 mL of methyl tert-butyl ether, and 59 g of solid sodium hydroxide were added.Sodium sulfate 20g,Tetra-n-butylammonium hydrogensulfate (4.3 g) was kept at an internal temperature of 5 to 10°C, 72 g of sulfuryl fluoride gas was slowly introduced under stirring, and the reaction was sealed for 1 hour. At the end of the reaction, nitrogen was blown for 1 hour, filtered, and the filtrate was desolvated under reduced pressure to give a crude solid which was added to dichloromethane (100 mL), 15-crown-5 0.05 g, and 18-crown-6 0.05 g. The mixture was heated to reflux and dissolved, and then slowly cooled to room temperature. 65.5 g of the product was obtained by filtration and drying. The yield was 81.8percent
106.3 g
Stage #1: With potassium hydroxide In 5,5-dimethyl-1,3-cyclohexadiene at 130 - 140℃; for 20 h; Stage #2: With fluorosulfonyl fluoride In acetonitrile at -15 - -5℃; for 1 h; Sealed tube; Inert atmosphere
200 ml of xylene was placed in the reaction flask.81.2 g of ethylene glycol and 110 g of potassium hydroxide,Stir to a reflux state of about 130-140 ° C,After 20 hours, the solvent was evaporated to dryness, and 400 ml of anhydrous acetonitrile was added.Stir into a uniform suspension. Sealed system after nitrogen replacement,The suspension was continuously added to a mixed solution of 130 g of sulfuryl fluoride and 400 ml of acetonitrile, and the temperature of the reaction system was maintained at -15 to -5 ° C, and the addition was completed for about 1 hour, and the temperature was maintained for 1 hour.At the end of the reaction, the reaction system was blown with nitrogen, and the insoluble matter was removed by filtration.The filtrate was decomposed to dryness under reduced pressure at 60 ° C.The obtained solid was stirred at room temperature for 1 hour in a solution of 0.6 g of 15-crown-5 and 0.5 g of 18-crown-6 in 250 ml of n-hexane, and filtered.Drying white powder 106.3g;
Reference:
[1] Patent: CN107629032, 2018, A, . Location in patent: Paragraph 0071; 0072; 0073; 0074; 0075; 0076; 0077; 0091
[2] Tetrahedron Letters, 1997, vol. 38, # 27, p. 4841 - 4844
[3] Patent: CN108409708, 2018, A, . Location in patent: Paragraph 0072; 0073; 0084; 0085; 0086; 0087
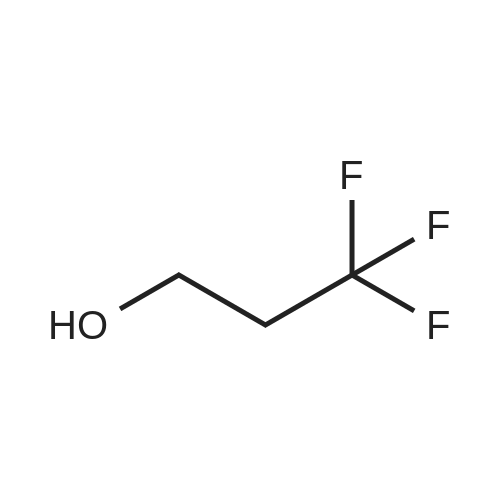
5
[ 75-21-8 ]
[ 1072-53-3 ]
Yield
Reaction Conditions
Operation in experiment
97.8%
at 140℃; for 2 h; Inert atmosphere; Autoclave
(1) In a 200 ml autoclave with a solenoid valve stirring, 8.0 g of a composite catalyst (composite catalyst consisting of iron trichloride, antimony trichloride and cerium oxide in a molar ratio of 3:2:1) was added. After replacing the air with nitrogen, 100 g of ethylene oxide was added, and then the reaction solution was heated and heated, but when the reaction temperature was raised to 140 ° C, sulfur dioxide gas was introduced to 6 MPa, and the reaction was carried out for 2 h while maintaining the temperature and pressure in the autoclave. The cooling water is passed through, cooled to 40-50 ° C, and the product in the kettle is discharged to obtain a crude product of vinyl sulfite. The obtained crude product is subjected to vacuum distillation under vacuum to obtain 242.5 g of refined sulfite, which is chromatographed. The purity was 99.4percent and the yield was 98.8percent (relative to ethylene oxide).(2) Adding an aqueous solution of sodium hydrogencarbonate to the vinyl sulfite obtained in the step (1), adjusting the pH to 8-9, cooling to -3 to 5 ° C, and dropping it at a temperature of -3 to 5 ° C. a mixture of sodium iodate and antimony trichloride (wherein the amount of sodium periodate is 106.9 g, the molar ratio of sodium periodate to antimony trichloride is 1:0.000005), the duration of the addition is 2.5 h, and the dropwise addition is carried out. After the completion of the heat for 1 h, the reaction liquid in which the aqueous phase and the organic phase coexist is obtained, and the layer is allowed to stand, and the aqueous phase is separated, and the obtained organic phase is a crude product of vinyl sulfate;(3) The crude vinyl sulphate obtained in the step (2) is subjected to molecular distillation at 70-80 ° C to obtain 242.44 g of vinyl sulfate, and the purity is 99.1percent, and the yield is 97.8percent (relatively high). Sodium iodate.
Reference:
[1] Patent: CN108658928, 2018, A, . Location in patent: Paragraph 0023; 0024; 0025; 0026; 0027; 0028; 0029; 0031
[2] Patent: US5274123, 1993, A,
6
[ 7719-09-7 ]
[ 107-21-1 ]
[ 1072-53-3 ]
Yield
Reaction Conditions
Operation in experiment
74%
Stage #1: at 25 - 30℃; for 3 h; Inert atmosphere Stage #2: at 0 - 10℃; for 3 h;
Weigh 31.0g of ethylene glycol was added to the industrial 1L three bottles, which leads to dry steady stream of nitrogen, sealed and exhaust system connected through the water absorbed into the bottle.Solution of 65.5g of thionyl chloride, dropping the control temperature of the system does not exceed 30 , 1h drops completion.Dropping bubbles emerge.Bi drops to warm to 25-30 , keep stirring 3h, TLC monitoring no starting material remaining glycol, insulation stop, this time the body system as a light brown clear solution.The cooling system to 0-10 deg.] C, 8g of sodium bicarbonate and weighed 112g formulated into aqueous sodium bicarbonate solution was added to the system manipulation system pH between 7-8, and then added 0.3g of ruthenium trichloride trihydrate , the system as a dark brown turbid liquid.0-10 temperature and stirred solution of 313g mass concentration of 13percent sodium hypochlorite solution until the reaction mixture turns a pale yellow turbid solution was dropped at 2h, dropping Bi 0-10 insulation 1h, starch -KI test strip aqueous phase has oxidation, TLC monitoring of the reaction mixture remaining after no DTO, continue to the next step operation.Aqueous sodium hydrogen sulfite, 120g of 0-10 holding temperature of the system was added 10percent by mass concentration of potassium iodide starch test paper non-oxidizing.After stirring for 10min temperature 0-5 filtered to give 49.8g of yellow solid particles, GC> 96percent, yield: 80percent.The crude toluene with 50g hot melt, hot filtered and the filtrate temperature drop precipitated solid was filtered to give 46g of white crystalline powder, GC> 99.9percent, the total yield of 74percent.
Reference:
[1] Patent: CN105481826, 2016, A, . Location in patent: Paragraph 0022; 0038; 0039; 0040; 0041
7
[ 23248-21-7 ]
[ 1072-53-3 ]
Yield
Reaction Conditions
Operation in experiment
37.9 g
Stage #1: With sulfuryl dichloride In dichloromethane at -10 - 10℃; for 2 h; Inert atmosphere Stage #2: With 18-crown-6 ether In toluene at 20℃; for 1 h;
100 ml of xylene, 40.5 g of ethylene glycol and 55.3 g of potassium hydroxide were placed in the reaction flask.Stir to reflux at about 130-140 ° C, and evaporate the solvent after 15 hours.200 ml of anhydrous dichloromethane was added and stirred to form a uniform suspension. After nitrogen replacement,The suspension was continuously added to a mixed solution of 86 g of sulfuryl chloride and 200 ml of dichloromethane.The temperature of the reaction system was kept at -10 to 10 ° C, and the addition was completed in about 1 hour, and the temperature was kept for 1 hour.At the end of the reaction, add 100 ml of water and stir to separate the layers.The organic phase is desolvated under reduced pressure at 40 ° C until the solid precipitates.Add 200 ml of toluene and 0.6 g of 18-crown-6 and stir at room temperature for 1 hour, filter and dry to obtain a white powder.37.9 g; yield 61.0percent; GC content > 99.5percent, potassium ion 7 ppm
Reference:
[1] Patent: CN108373460, 2018, A, . Location in patent: Paragraph 0069-0077
8
[ 74-85-1 ]
[ 1072-53-3 ]
Reference:
[1] Tetrahedron Letters, 1986, vol. 27, # 34, p. 3971 - 3974
[2] Journal of Organic Chemistry USSR (English Translation), 1986, vol. 22, p. 398 - 399[3] Zhurnal Organicheskoi Khimii, 1986, vol. 22, # 2, p. 450 - 452
9
[ 3741-38-6 ]
[ 1072-53-3 ]
Reference:
[1] Patent: US4960904, 1990, A,
10
[ 106-93-4 ]
[ 1072-53-3 ]
Reference:
[1] Journal of the Chemical Society, 1932, p. 86,87
[2] Australian Journal of Chemistry, 1977, vol. 30, p. 569 - 578
[3] Journal of the Chemical Society, 1960, p. 201 - 211
11
[ 1072-53-3 ]
[ 926-39-6 ]
Reference:
[1] Journal of Heterocyclic Chemistry, 1972, vol. 9, p. 891 - 894
With manganese(IV) oxide; In water; at 5℃; for 0.75h;
Taking 216.3 g (2 mol) of ethylene sulfite, 200 ml of distilled water and 0.4 g of a particulate catalyst (by mass ratio, Cu:NiO:MnO2 = 1:1.2:0.8),Stir slowly and control the temperature at 5 C.Ozone was added at 0.05 L/min (ozone concentration was 20 mL/L) and the reaction was carried out for 45 min.After the reaction is completed, the layers are allowed to stand, and the organic layer is taken.The organic layer was washed twice with distilled water and dried to give a vinyl sulfate.The yield was 95.84%. The product was analyzed by liquid chromatography and the results showed thatThe content of ethylene sulfate was 95.63%, and the content of ethylene sulfite was 4.28%.
90.66%
With sodium hydrogencarbonate; iron(II) sulfate; In dichloromethane;pH 7 - 8;
(2) oxidation reaction: first of all to the 2nd reaction kettle is added as a solvent of dichloromethane 450 g and as the quality of the catalyst concentration is 10% ferric sulfate solution 49.38 g, then by adding sodium bicarbonate to adjust the pH of the solution in the 2nd reaction value is 7 - 8, then the 2nd reactor cooling to 4 C, then to the 2nd reaction in the cauldron adds by drops quality concentration is 10% sodium percarbonate solution 7756.29 g, dropping time is 2h, keeping the heat after dropping 1.5h, to obtain the aqueous phase and organic phase coexistence of the reaction solution, then the reaction fluid settlement, layered separating the aqueous phase, get the organic phase, then the weight of the aqueous phase such as methylene chloride to the separated aqueous phase extraction three times, the residual sulfuric acid in the aqueous phase is extracted to vinyl acetate, thereby obtaining the vinyl acetate dichloromethane extract containing sulfuric acid, then the dichloromethane extract combined with the organic phase, then combined to form the solution is distilled under reduced pressure at room temperature to remove the methylene chloride, thereby obtaining the sulfuric acid ethylene the ester is thick product 247.38g (the purity is 95.66%), molar yield is 90.88%;(3) refining: use with sulfuric acid and ethylene the ester is thick product of the weight of the dichloromethane as solvent to sulfuric acid ethylene the ester is thick product of re-crystallization processes, thus obtaining high-purity sulfuric acid vinyl 236.29 g (purity of 99.91%, the water content is 39 ppm, an acid value of 45 ppm), molar yield is 90.66%.
82.4%
With sodium hypochlorite; water; at 0 - 5℃; for 0.5h;Large scale;
(2) Oxidation reaction: first, 2413 g of glycol sulfite is added to the oxidation vessel. Then add 1 kg of ice, 2 kg of buffer solution, 1.8 g of catalyst, and mix well.Further, sodium hypochlorite was added dropwise thereto, and the reaction temperature was controlled to be 0 to 5 C by adding ice. Observe the color change and stop the drop when the solution turns milky white. The reaction was completed and the entire reaction time was controlled within 30 min.The mixture is filtered to obtain 3253 g of crude glycol sulfite with a purity of about 90.88%; (3) Refining: The obtained crude vinyl sulphate product is dissolved in 6 times by weight of dichloroethane solvent, and then washed, dehydrated, Extraction and drying treatment. Control drying conditions, vacuum: -0.08 ~ -0.09MPa; temperature control: 60 C; Drying time: 2 hours. Thus, 2145 g of high-purity glycol acetate (purity of 99.8207%, moisture content of 35 ppm) was obtained. The molar yield was 82.48%.
68.1%
With potassium sulfate; rhenium(III) chloride; Marshall's acid; potassium carbonate; In dichloromethane; water; at 0 - 4℃; for 1h;
Add 1,200 g of pure water to the reaction bottle,600 g of dichloromethane,510g of potassium carbonate,Potassium sulfate 190g,The structural formula is 54 g of vinyl sulfite,Samarium trichloride 1.35g,The reaction system was stirred and cooled to 0 C. with an ice-water bath, and then 460 g of the peroxysulfuric acid solution prepared in Example 1 was added dropwise to the reaction system within half an hour, and the temperature was maintained at 2 to 4 C. during the dropwise addition. After the dropwise addition, stirring was continued for 30 min. After filtration, the organic layer was separated. The organic layer was dehydrated through molecular sieves, and then evaporated to dryness to remove dichloromethane to obtain 46.3 g of a crude vinyl sulfate having a structural formula of 97.6% purity. The crude product was recrystallized twice from dichloromethane to obtain 42.2 g of a finished product of vinyl sulfate with a purity of 99.7% and a yield of 68.1%.
In a 1000 mL autoclave, 40 g of ethylene glycol, 250 mL of methyl tert-butyl ether, and 59 g of solid sodium hydroxide were added.Sodium sulfate 20g,Tetra-n-butylammonium hydrogensulfate (4.3 g) was kept at an internal temperature of 5 to 10C, 72 g of sulfuryl fluoride gas was slowly introduced under stirring, and the reaction was sealed for 1 hour. At the end of the reaction, nitrogen was blown for 1 hour, filtered, and the filtrate was desolvated under reduced pressure to give a crude solid which was added to dichloromethane (100 mL), 15-crown-5 0.05 g, and 18-crown-6 0.05 g. The mixture was heated to reflux and dissolved, and then slowly cooled to room temperature. 65.5 g of the product was obtained by filtration and drying. The yield was 81.8%
106.3 g
200 ml of xylene was placed in the reaction flask.81.2 g of ethylene glycol and 110 g of potassium hydroxide,Stir to a reflux state of about 130-140 C,After 20 hours, the solvent was evaporated to dryness, and 400 ml of anhydrous acetonitrile was added.Stir into a uniform suspension. Sealed system after nitrogen replacement,The suspension was continuously added to a mixed solution of 130 g of sulfuryl fluoride and 400 ml of acetonitrile, and the temperature of the reaction system was maintained at -15 to -5 C, and the addition was completed for about 1 hour, and the temperature was maintained for 1 hour.At the end of the reaction, the reaction system was blown with nitrogen, and the insoluble matter was removed by filtration.The filtrate was decomposed to dryness under reduced pressure at 60 C.The obtained solid was stirred at room temperature for 1 hour in a solution of 0.6 g of 15-crown-5 and 0.5 g of 18-crown-6 in 250 ml of n-hexane, and filtered.Drying white powder 106.3g;
375 g
With sulfuryl dichloride; In chloroform; at 5 - 60℃; for 3h;
(1) Synthesis of ethylene sulfate200 g of ethylene glycol was added to the four-necked bottle, stirring was started, and 1000 mL of chloroform was added to the reaction flask.Slowly add 457 g of sulfuryl chloride (molar ratio of ethylene glycol to sulfonyl chloride is 1:1.05) at room temperature.The dropping rate of the sulfonyl chloride is controlled so that the reaction temperature in the reactor does not exceed 30 C, and the temperature of the reaction liquid rises immediately when the dropping is started.When the amount of sulfuryl chloride added is half of the total amount,The temperature of the reaction solution began to decrease, and after the addition of the sulfuryl chloride was completed, the temperature of the reaction liquid was lowered to 5 C.The sulfuryl chloride addition time is 1.5 hours.The color of the reaction liquid changed from colorless to pale yellow during the reaction.The temperature was slowly raised to 60 C, and the reaction solution was stirred at 60 C for 1.5 hours, and the color of the reaction liquid changed from pale yellow to yellow during the reaction.During the whole reaction process, white hydrogen chloride gas is continuously emitted, and the generated hydrogen chloride gas is absorbed by the tap water.ownedThe concentration of the aqueous hydrochloric acid solution is 15-20%.When the TLC test confirmed that the raw material ethylene glycol reaction was complete, the stirring was stopped.Reaction solution in iceAdd 200 mL of water to the bath.Adding saturated sodium bicarbonate solution to neutralize,The pH of the solution was adjusted to 7-8, the reaction solution was allowed to stand for stratification, and the aqueous phase was extracted with chloroform (200 mL×2). The organic phase was combined and the organic phase was washed with saturated sodium chloride solution.Polyester, liquid separation,5.0 g of activated carbon was added to the organic phase, and the mixture was decolorized by stirring at 10-15 C for 15 min.The filtrate was filtered and dried over anhydrous sodium sulfate.(2) Purification of ethylene sulfate. 401 g of crude ethylene sulfate was poured into a three-necked bottle.Add 480mL chloroform dissolved in a nitrogen flow seal In the system, slowly heat up to 60 C,Cooled back to reflux, the ethylene sulfate was completely dissolved, slowly cooled to 20 C, and allowed to stand for white solid,The filter cake was concentrated under reduced pressure in a water bath of 30 C to give 375 g of pure ethylene sulfate (total yield: 93.76%).GC > 99.92%, moisture 33 ppm, acidity 56 ppm.
Stage #1: di-isopropylphosphine With n-butyllithium In tetrahydrofuran at -70 - 20℃;
Stage #2: ethyleneglycol sulfate In tetrahydrofuran at -70 - 20℃;
Stage #3: lithium diphenylphosphide In tetrahydrofuran at -70 - 60℃;
2-(cis-4-(5-bromo-2-(2,5-dimethyl-1H-pyrrol-1-yl)-6-methylpyrimidin-4-ylamino)cyclohexyloxy)ethanol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
45%
Example 95. 2-c/s-4-(5-Bromo-2-(2 ,5-dimethvl-1 H-pvrrol-1 -vl)-6-methvlDvrimidin-4- vlamino)cvclohexvloxv)ethanol; To a cooled (O0C) solution of c/s-4-(5-bromo-2-(2,5-dimethyl-1 H-pyrrol-1-yl)-6-methylpyrimidin-4- ylamino)cyclohexanol (2.50 g, 6.59 mmol) in dimethylformamide (17.0 ml.) was added sodium hydride (60% dispersion in oil, 527 mg,13.2 mmol). After 2.5 hr at O0C a solution of 1 ,3,2-dioxathiolane 2,2 dioxane (1.23 g, 9.89 mmol) in dimethylformamide (7.0 mi_) was added drop wise over 1 hr. After stirring 00C overnight an additional 4 eq sodium hydride was added followed by 1 ,3,2-dioxathiolane 2,2 dioxane in 0.25 eq portions every 15 min up to 2.25 eq. The reaction was quenched with methanol and concentrated. The residue was then diluted with 1 ,4 dioxane (200 ml) and water (5.0 ml), p- Toluenesulfonic acid (g mmol) was added and the mixture was heated to 4O0C for 1.5 hour. The solution cooled to O0C and saturated with solid sodium bicarbonate. Diluted with water (100 mL) and extracted with dichloromethane (3 x 500 mL). Combined organics were washed with brine (150 mL), dried (MgSO4), filtered, and concentrated. The crude product was purified by flash chromatography eluting with hexanes/methyl tert-butylether (15-75%) to afford the title compound (1.27 g, 45%). 1 H NMR (400 MHz, DMSO-cfe) delta ppm 1.40 - 1.50 (m, 2 H) 1.51 - 1.61 (m, 2 H) 1.72 - 1.80 (m, 2 H) 1.80 - 1.88 (m, 2 H) 2.26 (s, 6 H) 2.41 (s, 3 H) 3.39 (t, J=5.43 Hz, 2 H) 3.48 - 3.57 (m, 3 H) 3.94 - 4.03 (m, 1 H) 4.50 (t, J=5.56 Hz, 1 H) 5.75 (s, 2 H) 6.80 (d, J=8.08 Hz, 1 H) (MH-H)+ 424
Butyl lithium (2.5M in hexanes, 61 mL, 0.152mol) was added dropwise to an ice-cold solution of cyclobutanol (10g, 0.139mol) in tetrahydrofuran (250mL) so as to maintain the reaction temperature below10 C. The mixture was then stirred for a further 2 hours at5-10 C, and a solution of 1,3, 2-dioxathiolane 2,2-dioxide(18. 90g, 0.152mol) in tetrahydrofuran (50mL) was added dropwise so as to maintain the reaction temperature below15 C. Once addition was complete the reaction was stirred for a further 3 hours at room temperature, water (3mL) followed by concentrated sulphuric acid (7.5mL) then added and the reaction stirred for an additional 18 hours. The reaction was carefully neutralised by the addition of solid sodium carbonate and sodium bicarbonate, and the mixture concentrated under reduced pressure at room temperature. The residue was diluted with water, saturated with sodium chloride added until saturation was achieved and the solution then extracted with ethyl acetate(4x100mL). The combined organic extracts were dried over magnesium sulphate and evaporated under reduced pressure at room temperature. The residual orange oil was purified by Kugelrohr distillation to afford the title compound, 7.7g. bp70-80 C at 10mmHg. 1H NMR (CDCI3 400 MHz) : 1. 38-1. 57 (m, 1 H), 1.63 (m, 1 H), 1.80-1. 98 (m, 2H), 2.06-2. 15 (m, 2H), 3.40 (t, 2H), 3.65 (t, 2H), 3.95 (m, 1H).
n-Butyl lithium (39mL, 2.5M in hexanes, 97. 5mmol) was added dropwise to an ice- cooled solution of3, 3,3-trifluoropropan-1-ol(10g, 87.7mmol) in tetrahydrofuran(130mL), so as to maintain the temperaturebelow 5 C, and once addition was complete the reaction was stirred for a further hour at 0 C. A solution of 1,3, 2-dioxathiolane 2,2-dioxide(11. 97g, 96.5mmol) in tetrahydrofuran (35mL) was then added dropwise so as to maintain the internal temperature below5 C, and once addition was complete the reaction was stirred at room temperature for 18 hours. Water (2mL) followed by concentrated sulphuric acid (5mL) were added and the reaction stirred for a further 6 hours at room temperature. The mixture was neutralised by the addition of sodium carbonate, then diluted with water (20mL) and the resulting solid filtered off and washed with ethyl acetate. The filtrate was concentrated under reduced pressure and the residue suspended in brine and extracted with ethyl acetate (3x). The combined organic extracts were dried over magnesium sulphate and evaporated under reduced pressure. The residual gum was distilled under high vacuum to afford the title compound as a colourless liquid, 6.75g (b. p.57-80 C). 'H NMR (CDCI3 400 MHz) 8 : 2.38 (m, 2H), 2.57 (m, 2H), 3.69 (m, 4H).
c) tert-Butvl 3-(2-hydroxyethoxy)-4-(4-methoxyphenyl)piperidine-1-carboxylate A solution of 1.01 g of tert-butyl 3-hydroxy-4- (4-methoxyphenyl) piperidine-1-carboxylate in 40 ml of N, N-dimethylformamide is added at 0C to the suspension of 0.26 g of sodium hydride dispersion (60%) in 40 mi of N, N-dimethylformamide. After 30 minutes, the reaction mixture is admixed with 0.83 g of [1,3, 2] dioxathiolane 2,2-dioxide and stirred at room temperature over 18 hours. The reaction mixture is cooled to 0C, admixed with water and concentrated by evaporation. The residue is stirred in a 1: 10 mixture of 0.1 M H2SO4/dioxane at 50C over 60 hours, neutralized with saturated aqueous sodium hydrogencarbonate solution and concentrated by evaporation. The residue is dissolved in ethyl acetate, washed with water and brine, dried over sodium sulphate and concentrated by evaporation. The title compound is obtained as a yellow oil from the residue by means of flash chromatography (Si02 60F). Rf = 0.20 (1: 1 EtOAc-heptane); Rt = 4.24.
With cerium(IV) oxide; iron(III) chloride; sulfur dioxide; antimony(III) chloride; at 140℃; under 45004.5 Torr; for 2h;Inert atmosphere; Autoclave;
(1) In a 200 ml autoclave with a solenoid valve stirring, 8.0 g of a composite catalyst (composite catalyst consisting of iron trichloride, antimony trichloride and cerium oxide in a molar ratio of 3:2:1) was added. After replacing the air with nitrogen, 100 g of ethylene oxide was added, and then the reaction solution was heated and heated, but when the reaction temperature was raised to 140 C, sulfur dioxide gas was introduced to 6 MPa, and the reaction was carried out for 2 h while maintaining the temperature and pressure in the autoclave. The cooling water is passed through, cooled to 40-50 C, and the product in the kettle is discharged to obtain a crude product of vinyl sulfite. The obtained crude product is subjected to vacuum distillation under vacuum to obtain 242.5 g of refined sulfite, which is chromatographed. The purity was 99.4% and the yield was 98.8% (relative to ethylene oxide).(2) Adding an aqueous solution of sodium hydrogencarbonate to the vinyl sulfite obtained in the step (1), adjusting the pH to 8-9, cooling to -3 to 5 C, and dropping it at a temperature of -3 to 5 C. a mixture of sodium iodate and antimony trichloride (wherein the amount of sodium periodate is 106.9 g, the molar ratio of sodium periodate to antimony trichloride is 1:0.000005), the duration of the addition is 2.5 h, and the dropwise addition is carried out. After the completion of the heat for 1 h, the reaction liquid in which the aqueous phase and the organic phase coexist is obtained, and the layer is allowed to stand, and the aqueous phase is separated, and the obtained organic phase is a crude product of vinyl sulfate;(3) The crude vinyl sulphate obtained in the step (2) is subjected to molecular distillation at 70-80 C to obtain 242.44 g of vinyl sulfate, and the purity is 99.1%, and the yield is 97.8% (relatively high). Sodium iodate.
After cooling, the determination of the reaction mixture: - by HPLC shows that the yield of ethylene sulphate is 95% relative to the ethylene oxide used, - by GPC the degree of conversion of ethylene oxide is 100%. After removal of the dioxane by distillation at reduced pressure (20 mm Hg; 2.6 kPa), the crude ethylene sulphate, which is 88% pure, is extracted with 500 g of dichloromethane. The chloromethylene solution is washed with concentrated sulphuric acid and then with water until neutral and is finally dried over sodium sulphate. After filtering and removing the solvents, 103.5 g of ethylene sulphate are obtained in the form of a white powder melting at 99 C., its purity being 97%.
With sodium hypochlorite; In dichloromethane; isopropyl alcohol;
EXAMPLE 1 Ethylene sulphite (27 g, 0.25 mole), methylene chloride (175 cc) and ruthenium (IV) oxide dihydrate (18 mg, 0.106*10-3 mole) are placed in a 250 cc round-bottomed flask. The mixture is cooled to 5 C., then a solution of sodium hypochlorite (2 moles per liter; 125 cc, 0.25 mole) is added with vigorous stirring in the course of 40 minutes. Stirring is continued for 10 minutes at 5 C. After decantaion, the organic phase is stirred with isopropanol (1 cc) for 10 minutes at 10 C., then it is washed with water (40 cc) at 5 C. After concentrating the organic phase at 30 C. under reduced pressure (100 mm Hg; 13.3 kPa), ethylene sulphate (25.4 g, 0.205 mole) is obtained in the form of white crystals melting at 99 C. The yield is 82%.
Stage #1: ethyleneglycol sulfate; 3,5-dimethyl-1-hexyn-3-ol With n-butyllithium In tetrahydrofuran; hexane at -78 - 40℃; for 49h;
Stage #2: With sulfuric acid In diethyl ether; water at 20℃; for 23h; Further stages.;
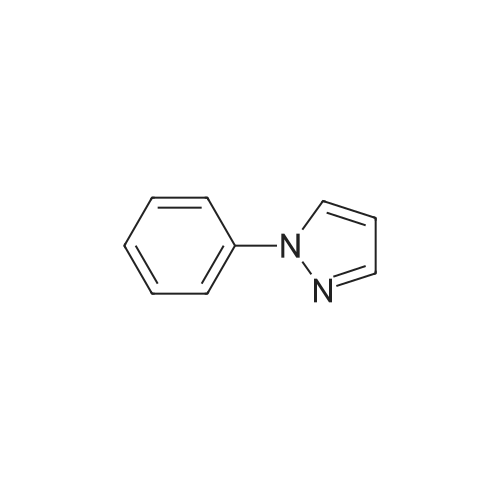
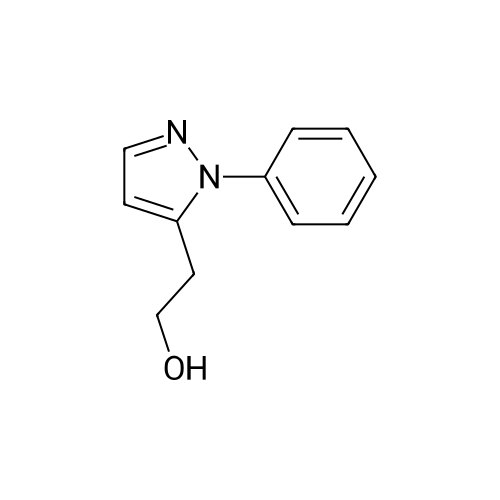
Under N2-atmosphere phenyl-pyrazole (1.0 g, 6.94 mmol) was dissolved in abs. THF (70 mL) and cooled to -78 C. Following that n-buthyl lithium in hexane (1.6 M, 4.3 mL, 6.94 mmol) was added dropwise and thereafter the mixture was stirred for 2 h at -78 C. Following that <strong>[1072-53-3]ethylene sulfate</strong> (1.03 g, 8.32 mmol), dissolved in abs. THF (10 mL) was slowly added. After a further hour of stirring at -78 C the reaction mixture was heated to room temperature and stirred for a further 71 h. After addition of water (30 mL) and H2SO4 conc. (5 mL) it was heated for 40 h under reflux and strong stirring, before it was neutralized with NaOH (5 N, 60 mL) and extracted 3 times with CH2Cl2-The pooled organic phases were dried over K2CO3, filtered and the solvent removed in vacuum. The crude product (1.31 g) was prified using flash-chromatography (Ø=6 cm, h = 15 cm, n-hexane:ethyl acetate = 1:1, 40 mL, Rf = 0.13). Slightly yellow oil, Yield: 981 mg (75%) C11H12N2O (188.3) C H N Calc. 70.2 6.43 14.9 Found 70.2 6.55 14.9MS (EI): m/z (rel. Int.) = 189 [MH+, 42], 188 [M+, 52], 158 [MH - CH2OH, 41], 157 [M - CH2OH, 100], 77 [Phenyl, 18]. IR (neat): v (cm-1) = 3328 (O-H), 3067 (C-H aromat.), 2930, 2876 (C-H aliphat.), 1598, 1534, 1501 (C=C), 764, 695 (C-H). 1H-NMR (CDCl3): delta (ppm) = 2.11 (s breit, 1H, CH2CH2OH), 2.91 (t, J = 6.5 Hz, 2H, CH2CH2OH), 3.79 (t, J = 6.7 Hz, 2H, CH2CH2OH), 6.29 (d, J = 1.6 Hz, 1 H, Pyrazol-4-CH), 7.38 - 7.48 (m, 5H, Phenyl-CH), 7.60 (d, J = 1.6 Hz, 1H, Pyrazol-3-CH). 13C-NMR (CDCl3): delta (ppm) = 29.7 (1C, ArCH2CH2OH), 61.4 (1C, ArCH2CH2OH), 106.1 (1C, Pyrazol-4-CH), 125.9 (2C, Phenyl-CH, ortho), 128.4 (1C, Phenyl-CH, para), 129.4 (2C, Phenyl-CH, meta), 139.7 (1C, Phenyl-C, quartaer), 140.2 (1C, Pyrazol-3-CH), 140.5 (1C, Pyrazol-5-C).
step 30.3 Preparation of 2-(3-Methoxyphenyl)ethanol3-Bromanisole (300 muL, 2.39 mmol) was dissolved under nitrogen atmosphere in abs. THF (35 mL) and cooled to -78 0C. Afterwards n-butyllithium in n-hexane (1.48 M, 1.62 mL 2.39 mmol) was added dropwise during 30 min and subsequently stirred for 2 h at -78 0C. In the following step ethylensulfat (355.3 mg, 2.87 mmol) was dissolved in abs. THF (5 mL) und transferred dropwise to the solution for 20 min. The reaction mixture was allowed to come to room temperature and strirred for 16 h. After the addition Of H2O (10 mL) and cone. H2Stheta4 (1.8 mL) the suspension was heated at reflux for 47 h. Finally the solution was neutralized by 5M NaOH (20 mL) and extracted three times with CH2Cl2 (20 mL). The combined organic layers were dried with Na2Stheta4 and concentrated in vacuum. The residue was purified by flash chromatography (n-hexane : ethyl acetate 8 : 2, 0 4 cm, fraction size 30 mL, Rf = 0.16). The titled compound was obtained as colourless oil. step
step 30.3 Preparation of 2-(3-Methoxyphenyl)ethanol [202] 3-Bromanisole (300 L, 2.39 mmol) was dissolved under nitrogen atmosphere in abs. 1500 THF (35 mL) and cooled to -78 C. Afterwards n-butyllithium in n-hexane (1.48 M, 1 .62 mL 2.39 mmol) was added dropwise during 30 min and subsequently stirred for 2 h at -78 C. In the following step ethylensulfat (355.3 mg, 2.87 mmol) was dissolved in abs. THF (5 mL) und transferred dropwise to the solution for 20 min. The reaction mixture was allowed to come to room temperature and stirred for 16 h. After the addition Of H20 (10 mL) and cone. H2S04 1505 (1.8 mL) the suspension was heated at reflux for 47 h. Finally the solution was neutralized by 5M NaOH (20 mL) and extracted three times with CH2CI2 (20 mL). The combined organic layers were dried with Na2S04 and concentrated in vacuum. The residue was purified by flash chromatography (n-hexane : ethyl acetate 8 : 2, 0 4 cm, fraction size 30 mL, Rf = 0.16). The titled compound was obtained as colourless oil.
Sodium hydride (0.12 g, 3 mmol, 60% dispersion in mineral oil) was dissolved in N,N-dimethylformamide (1.5 mL), and 4-methyl -N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide (0.47 g, 1.1 mmol) was added thereto, and then, the resulting mixture was stirred at room temperature for 30 minutes. Then, 1,3,2-dioxathiolane-2,2-dioxide (0.14 g, 1.2 mmol) was added thereto, and the resulting mixture was stirred at room temperature. After 1 hour, sodium hydride (40 mg, 1.0 mmol, oily, 60%) was added thereto again, and the resulting mixture was stirred for 30 minutes. Then, 1,3,2-dioxathiolane-2,2-dioxide (12 mg, 0.11 mmol) was added thereto, and the resulting mixture was stirred at room temperature for 1 hour. After the mixture was concentrated under reduced pressure, methanol (5 mL) was added to the residue and insoluble substances were removed by filtration, and the filtrate was concentrated again. To the residue, tetrahydrofuran (2 mL) and 6 M hydrochloric acid (2 mL) were added, and the resulting mixture was stirred at 60C for 16 hours. The reaction was cooled to room temperature, and then dissolved in ethyl acetate, and washed with water and saturated saline. The organic layer was dried over anhydrous sodium sulfate and filtered. Then, the filtrate was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (ethyl acetate), whereby the objective compound (0.25 g, 48%) was obtained. 1H-NMR (400 MHz, CDCl3) delta: 7.89-7.79 (m, 6H), 7.66-7.58 (m, 2H), 7.49 (s, 1H), 7.36 (d, 1H, J = 7.4Hz), 3.81-3.63 (m, 4H), 3.05 (s, 3H), 2.08 (s, 3H). HR-MS (ESI) calcd for C22H22F3N2O4S [M+H]+, required m/z: 467.1252, found: 467.1246. Anal. calcd for C22H21F3N2O4S: C, 56.65; H, 4.54; N, 6.01; F, 12.22; S, 6.87. found: C, 56.39; H, 4.58; N, 5.99; F, 12.72; S, 6.92.
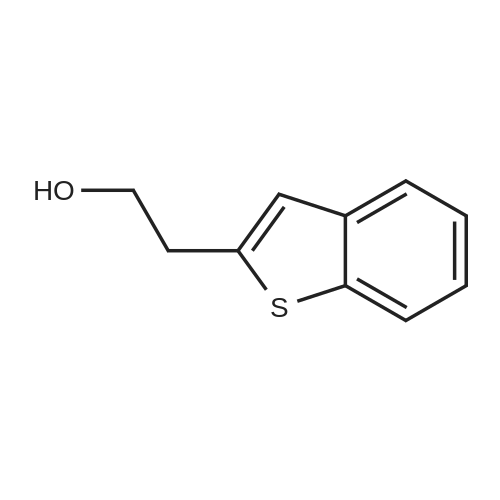
EXAMPLE 1B; SYNTHESIS OF REAGENT (2-BENZO[B]THIOPHEN-2-YL-ETHYL)-DIMETHYL-AMINE, ROUTE 2; An alternative synthesis for compound 1a follows: Benzo[b]thiophene (20 g, 149 mmol) was dissolved in anhydrous THF (600 mL) and cooled to -50 C. N-BuLi (85 mL, 135 mmol, 1.6M in hexanes) was added and the mixture was stirred for 45 min at -40 C. A solution of [1,3,2]Dioxathiolane 2,2-dioxide (16.8 g, 135 mmol) in THF (100 mL) was added dropwise and the temperature was allowed to slowly reach room temperature, while being stirred overnight. Water and ether were added and the product was extracted with water. The combined water layers were concentrated in vacuo, MeOH and dichloromethane were added and the mixture was again concentrated in vacuo. In a separate vessel AcCl (70 mL) was slowly added to cooled (0 C.) MeOH (700 mL) and stirred for 15 min. This mixture was cannulated to the residue obtained as described above and stirred for 4 hrs at room temperature. After concentration, EtOAc was added and the mixture was washed with sat. NaHCO3 and brine, dried (Na2SO4), filtered and concentrated in vacuo to generate 19.1 g of 2-Benzo[b]thiophen-2-yl-ethanol as a white solid in 79% yield.
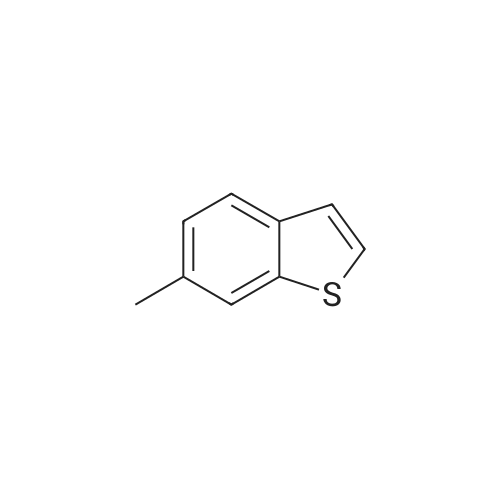
EXAMPLE 4 <strong>[16587-47-6]6-Methyl-benzo[b]thiophene</strong> was dissolved in anhydrous THF (75 mL) and cooled to -60 C. N-BuLi (11.4 mL, 18.2 mmol, 1.6M in hexanes) was added and the mixture was stirred for 45 min at -60 C. A solution of [1,3,2]Dioxathiolane 2,2-dioxide (2.26 g, 1.82 mmol) in THF (15 mL) was added dropwise and the temperature was allowed to slowly reach room temperature, while being stirred overnight. MeOH was added and the mixture was concentrated in vacuo. In a separate vessel AcCl (15 mL) was slowly added to cooled (0 C.) MeOH (150 mL) and stirred for 15 min. This mixture was cannulated to the residue obtained as described above and stirred for 4 hrs at room temperature. After concentration, EtOAc was added and the mixture was washed with sat. NaHCO3 and brine, dried (Na2SO4), filtered and concentrated in vacuo. Silica gel column chromatography (eluent: hexanes with gradient up to hexanes/EtOAc=4/1) afforded 2.83 g of 2-(6-methyl-benzo[b]thiophen-2-yl)-ethanol as a white solid in 89% yield.
disodium 5,5-bis(octyloxymethyl)-3,7-dioxa-1,9-nonadiyl disulfate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
70.2%
Example 23A; Sodium 5,5-bis(octyloxymethyl)-3,7-dioxa-1,9-nonanediol disulfate (23a); 23a[00227] To a stirred ice-bath cooled solution of 2,2-bis(octyloxymethyl)-1,3-propanediol 5 2a (Example 2A) (5.40 g, 0.015 mol) in THF (100 mL) is added a hexanes-washed 60% oil dispersion of sodium hydride (2.60 g, 0.065 mol, 4.3 eq). The cooled reaction mixture is stirred for 30 min, then a solution of <strong>[1072-53-3]ethylene sulfate</strong> (prepared by the method of Baker, W.; Field, F. B. J. Chem. Soc. 1932, 86-91 and/or Brimacombe, J. S.; Foster, A. B.; Hancock, E. B.; Overend, W. G.; Stacey, M. J.Chem. Soc. 1960, 201-211) (4.0 g,10 0.032 mol, 2.2 eq) in THF (50 mL) is added dropwise to the reaction mixture over a 20 min period. The reaction mixture is stirred for 19 h at rt, then MeOH is added carefully dropwise until foaming ceased. 10% HCI is added to neutralize the reaction (pH paper). Concentration gives a white paste that is purified using column chromatography with a solvent gradient changing from pure EtOAc to MeOH : EtOAc 2:3. The product is15 obtained as a white solid (6.87 g, 70.2%): mp 143.0-145.50C, 1H NMR (500.13 MHz, MeOD: CDCI3, 50:50) delta 0.89 (t, J = 6.8 Hz, 6H, 2 x CH3), 1.31 (br m, 2OH, alkyl protons), 1.54 (quintet, J = 7.0 Hz, 4H, 2 x OCH2CH2), 3.38 (s, 4H, CH2CH2CH2OCH2C), 3.39 (t, J = 6.5Hz, 4H, 2 x OCH2CH2), 3.49 (s, 4H, O3SOCH2CH2OCH2C), 3.68 (XX' part of AA1XX' pattern, JAX + JAX = 10.0 Hz, 4H, 2 x O3SOCH2CH2), 4.14 (AA' part of AA'XX' pattern, JAX 20 + JA-X = 10.0 Hz, 4H, 2 x O3SOCH2CH2); 13C NMR (125.77 MHz, MeOD) delta 14.3 (2 x CH3), 23.2 (2 x CH2CH3), 26.8 ( 2 x OCH2CH2CH2), 30.0, 30.1 , 30.2 (2 x OCH2CH2 and alkyl chain carbons), 32.5 (2 x CH2CH2CH3), 46.3 (q C), 67.6 (2 x OSO3CH2), 70.1 (2 x CH2OCH2CH2CH2), 70.7 (2 x OSO3CH2CH2), 71.1 (2 x O3SOCH2CH2OCH2), 72.4 (2 x OCH2CH2CH2); ESI MS (neg ion mode): calc for C25H50O12S2Na (M-Na) 629.26, found25 629.3; calc for (M-2Na)/2 303.14, found 303.3, calc for M-2Na+H 607.28, found 607.1; calc for 2M-Na 1281.52, found 1281.1. Anal. Calc. for C25H50O12S2Na2 : C 46.00, H 7.72. Found: C 46.11 , H 7.47.
2,2-bis((dodecyloxy)methyl)propane-1,3-diol[ No CAS ]
[ 1072-53-3 ]
disodium 5,5-bis(dodecyloxymethyl)-3,7-dioxa-1,9-nonadiyl disulfate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83.3%
Example 23C; Sodium 5,5-bis(dodecyloxymethyl)-3,7-dioxa-1,9-nonanediol disulfate (23c); 23c[00229] To an ice-bath cooled solution of 2,2-bis(dodecyloxymethyl)-1 ,3-propanediol 2c (Example 2C) (7.08 g, 0.015 mol) in THF (100 ml_) is added a hexanes-washed 60% oil dispersion of NaH (2.20 g, 0.055 mol, 3.6 eq). The cooled reaction mixture is stirred for 30 min then a solution of <strong>[1072-53-3]ethylene sulfate</strong> (prepared by the method of Baker, W.; Field, F. B. J. Chem. Soc. 1932, 86-91 and/or Brimacombe, J. S.; Foster, A. B.; Hancock, E. B.; Overend, W. G.; Stacey, M. J.Chem. Soc. 1960, 201-211) (4.0 g, 0.032 mol, 2.2 eq) in THF (50 mL) is added dropwise over a 20 min period. After the reaction mixture has been stirred for 21 h at rt, not all starting material has been consumed (TLC). More <strong>[1072-53-3]ethylene sulfate</strong> (0.1 g, 0.8 mmol) is added and the reaction mixture is stirred for another 3 h. MeOH is added carefully dropwise until foaming ceases, then 10% HCI is added until the reaction mixture is neutral (pH paper). Concentration yields a white paste that is taken up in a 1 :1 CH2CI2/Me0H. The mixture is heated to a boil then filtered. The filtrate is then heated until the mixture became clear. The solution is allowed to cool to rt before being refrigerated. The product is collected by filtration (9.53 g, 0.0125 mol, 83.3%): mp 144.5 - 146.00C; 1H NMR (500.13 MHz, MeOD) delta 0.90 (t, J = 7.1 Hz, 6H, 2 x CH3), 1.29 (br m, 36H, alkyl protons), 1.54 (quintet, J = 7.2 Hz, 4H, 2 x OCH2CH2), 3.38 (s, 4H, 2 x CH2CH2CH2OCH2C), 3.39 (t, J = 6.6 Hz, 4H, 2 x OCH2CH2CH2), 3.47 (s, 4H,03SOCH2CH2OCH2C), 3.64 (AA' part of AA'XX1 pattern, JAX + JAX = 10.0 Hz, 4H, 2 x " O3SOCH2CH2), 4.09 (XX' part of AA'XX' pattern, JAX + JAX = 10.0 Hz, 4H, 2 x " O3SOCH2CH2); 13C NMR (125.77 MHz, MeOD) delta 14.6 (2 x CH3), 23.9 (2 x CH2CH3), 27.6 (2 x OCH2CH2CH2), 30.6, 30.8, 30.9, 30.9, 31.0 (2 x OCH2CH2 and 12 alkyl chain carbons), 33.2 (2 x CH2CH2CH3), 47.0 (q C), 68.3 (2 x OSO3CH2), 70.7(CH2OCH2CH2CH2), 71.3 (2 x OSO3CH2CH2), 71.4 (2 x OSO3CH2CH2OCH2), 72.7 (2 x OCH2CH2CH2); ESI MS (neg ion mode): calc for (M+Na) m/z C33H66O12S2Na 741.39, found 741.5; calc for (M)/2, 359.20, found 359.3; calc for M +H, 719.40, found 719.2; calc for 2M+Na, 1505.77, found 1506.3. Anal. Calc. for C33H64O12S2Na2: C 51.81 , H 8.70. Found: C 52.04, H 8.86.
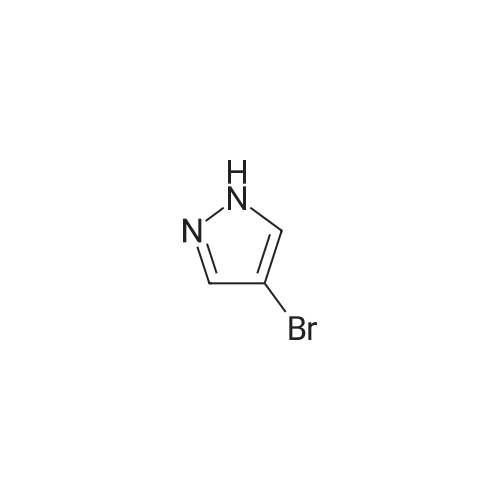
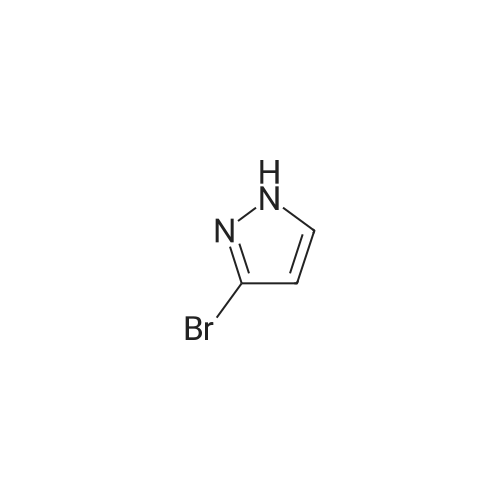
Example 84A (Ena?tiomer 1) Step 1: 2-(3-Bromo-lH-pyrazol-l-yl)ethanol; Br'Potassium tert-butoxide in THF(0.510 mL, 0.510 mmol) was added to a solution of 3-bromo-l H- pyrazole (50.0 mg, 0.340 mmol) in DMF (3 mL) and stirred at r.t. for 10 min, and then 1,3,2- dioxathiolane 2,2-dioxide (0.0633 g, 0.510 mmol) was added and the stirred at r.t. for 1 h. At this time, cone. HCI (0.3 mL) was added to the above reaction mixture and then stirred at r.t. overnight. The mixture was diluted with ethyl acetate, washed with saturated NaHCC>3, water and brine, dried over Na2SO4, filtered, and concentrated to give the crude product which was directly used in the next reaction without further purification. LCMS (M+H)+: m/z = 191.0 .
880 mg
To a solution of <strong>[14521-80-3]3-bromo-1H-pyrazole</strong> obtained in Step C (1.20 g) in N,N-dimethylformamide (82 mL) was added potassium tert-butoxide tetrahydrofuran solution (1 M, 12.3 mL) at room temperature. The reaction mixture was stirred at room temperature for 10 min, 1,3,2-dioxathiolane 2,2-dioxide (1.52 g) was added thereto at room temperature, and the mixture was stirred for 1 hr. conc. Hydrochloric acid (7.5 mL) was added thereto at room temperature, and the reaction mixture was stirred at room temperature for 18 hr. To the reaction mixture was added ethyl acetate, and the mixture was neutralized with saturated aqueous sodium hydrogen carbonate solution, and washed with saturated brine. The organic layer was dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate/hexane) to give the title compound (880 mg). 1H NMR (400 MHz, CDCl3) delta 3.99 (2H, t, J = 4.8 Hz), 4.21 (2H, t, J = 4.8 Hz), 6.27 (1H, d, J = 2.4 Hz), 7.36 (1H, d, J = 2.4 Hz)
Comparative Examples 1 to 7 and Examples 8 to 12 NEODOL 23, a C12/C13 primary alcohol composition, is commercially available from The Shell Chemical Company. Hexadecanol and 2-undecanol are available from Aldrich and 4-tridecanol is available from Chemical Samples Co, Columbus, Ohio, USA. Comparative Examples 1 to 7 and Examples 8 to 12 of the present invention are carried out according to the following process unless otherwise indicated in Table 1. Under a nitrogen atmosphere the alcohol, solvent-1 and the base were reacted for 1 h under the conditions detailed in Table 1, in order to form a sodium alkoxylate mixture. Ethylene sulfate (available from Eastar Chemical Corporation, Sacramento, Ca, USA) dissolved in solvent-2 was added to the stirred sodium alkoxylate mixture at such a rate that the designated temperature could be maintained. The reaction mixture was then stirred at the indicated temperature overnight. At the times indicated in Table 1, small samples were taken. These samples were hydrolysed by treatment with 6N H2SO4 at 90C for less than 1h and subsequently analysed by gas chromatography (GC). GC was carried out on a Hewlett-Packard HP6890 apparatus with the following column: Varian-Chrompack capillary column CP-SM 5CB (low-bleed), length 50 m, internal diameter 0.25 mm, film thickness 0.4 mum and with the following temperature program: 125 C (5 min); 125 - 325 C (10 C/min); 325 C (5 min). Flame ionization detection and an internal normalization method of quantification were employed.; Table 1: Preparation of Alcohol Ethoxysulfates Example Alcohol (mmol) Base (eq) Solvent-1Ethylene sulfatea (eq) Solvent-2 Temp (C) Time (h)Conversionb (%) Remarks 1* Neodol 23 KOH (0.05) toluene 1.0 toluene 90 <½12c prepared according to WO 96/035663 (50)Na2CO3 (1.0) water toluene 40 ½ 2* hexadecanol (20) NaOH (1.0) toluene 1.0 toluene 20 24 10 water removal with Dean-Stark setup at 130C 3* Neodol 23 Na (1.0) p-dioxane 1.0 p-dioxane 20 ½ 59 deprotonation at (50) 20 24 63 120C for 20h 4* 4-tridecanol (50) Na (1.15) p-dioxane 1.0 p-dioxane 20 2 54 deprotonation at 120C for 18h 5* Neodol 23 (50)NaHCO3 (1.0) water 1.0 p-dioxane 90 1622d 6* 4-tridecanol NaOH (1.5) p-dioxane 1.3CH2Cl2 <25 ½ 0 (50) 24 0 7* 4-tridecanol (50) NaOH (1.5) p-dioxane 1.3 p-dioxane <25 ½ 0 8 Neodol 23 NaOH (5.0) DMSO 1.0CH2Cl2 20-40 ½ 65 (50) 40 20 66 +0.5 40 6 76 +0.5 <25 ½ 84 9 Neodol 23 NaOH (1.5) sulfolane 1.0CH2Cl2 <25 ½ 49 minimum amount (50) 20 49CH2Cl2 to lower the viscosity. 10 2-undecanol (50) NaOH (1.5) DMSO 1.3CH2Cl2 <25 ½ 48 11 2-undecanol (50) NaOH (5.0) DMSO 1.3CH2Cl2 <25 ½ 57 12 Neodol 23 (50) NaOH (5.0) DMSO 1.3CH2Cl2 <0 0-20 24 69 <0C during addition; then slowly heated to 20C 13 Neodol 67 (300) NaOH (5.0) DMSO 1.3CH2Cl2 <25 1 -69 * Comparative Example. a. Ethylene sulfate is available from Eastar Chemical Corporation, Sacramento, Ca, USA and has been used without purification. b. Measured by GC after hydrolysis in 6N sulphuric acid at 90C for <1h (wt% of 1EO-adduct on alcohol intake). c. Trace (<1%) of 2EO derivative also present. d. 2EO and 3EO derivatives also observed in GC after hydrolysis.
Stage #1: undecan-2-ol With sodium hydroxide In dimethyl sulfoxide at 25℃; for 1h; Inert atmosphere;
Stage #2: ethyleneglycol sulfate In dichloromethane; dimethyl sulfoxide at 25℃; for 0.5h;
11
Comparative Examples 1 to 7 and Examples 8 to 12 NEODOL 23, a C12/C13 primary alcohol composition, is commercially available from The Shell Chemical Company. Hexadecanol and 2-undecanol are available from Aldrich and 4-tridecanol is available from Chemical Samples Co, Columbus, Ohio, USA. Comparative Examples 1 to 7 and Examples 8 to 12 of the present invention are carried out according to the following process unless otherwise indicated in Table 1. Under a nitrogen atmosphere the alcohol, solvent-1 and the base were reacted for 1 h under the conditions detailed in Table 1, in order to form a sodium alkoxylate mixture. Ethylene sulfate (available from Eastar Chemical Corporation, Sacramento, Ca, USA) dissolved in solvent-2 was added to the stirred sodium alkoxylate mixture at such a rate that the designated temperature could be maintained. The reaction mixture was then stirred at the indicated temperature overnight. At the times indicated in Table 1, small samples were taken. These samples were hydrolysed by treatment with 6N H2SO4 at 90°C for less than 1h and subsequently analysed by gas chromatography (GC). GC was carried out on a Hewlett-Packard HP6890 apparatus with the following column: Varian-Chrompack capillary column CP-SM 5CB (low-bleed), length 50 m, internal diameter 0.25 mm, film thickness 0.4 µm and with the following temperature program: 125 °C (5 min); 125 - 325 °C (10 °C/min); 325 °C (5 min). Flame ionization detection and an internal normalization method of quantification were employed.; Table 1: Preparation of Alcohol Ethoxysulfates Example Alcohol (mmol) Base (eq) Solvent-1Ethylene sulfatea (eq) Solvent-2 Temp (°C) Time (h)Conversionb (%) Remarks 1* Neodol 23 KOH (0.05) toluene 1.0 toluene 90 c prepared according to WO 96/035663 (50)Na2CO3 (1.0) water toluene 40 ½ 2* hexadecanol (20) NaOH (1.0) toluene 1.0 toluene 20 24 10 water removal with Dean-Stark setup at 130°C 3* Neodol 23 Na (1.0) p-dioxane 1.0 p-dioxane 20 ½ 59 deprotonation at (50) 20 24 63 120°C for 20h 4* 4-tridecanol (50) Na (1.15) p-dioxane 1.0 p-dioxane 20 2 54 deprotonation at 120°C for 18h 5* Neodol 23 (50)NaHCO3 (1.0) water 1.0 p-dioxane 90 1622d 6* 4-tridecanol NaOH (1.5) p-dioxane 1.3CH2Cl2 <25 ½ 0 (50) 24 0 7* 4-tridecanol (50) NaOH (1.5) p-dioxane 1.3 p-dioxane <25 ½ 0 8 Neodol 23 NaOH (5.0) DMSO 1.0CH2Cl2 20-40 ½ 65 (50) 40 20 66 +0.5 40 6 76 +0.5 <25 ½ 84 9 Neodol 23 NaOH (1.5) sulfolane 1.0CH2Cl2 <25 ½ 49 minimum amount (50) 20 49CH2Cl2 to lower the viscosity. 10 2-undecanol (50) NaOH (1.5) DMSO 1.3CH2Cl2 <25 ½ 48 11 2-undecanol (50) NaOH (5.0) DMSO 1.3CH2Cl2 <25 ½ 57 12 Neodol 23 (50) NaOH (5.0) DMSO 1.3CH2Cl2 <0 0-20 24 69 <0°C during addition; then slowly heated to 20°C 13 Neodol 67 (300) NaOH (5.0) DMSO 1.3CH2Cl2 <25 1 -69 * Comparative Example. a. Ethylene sulfate is available from Eastar Chemical Corporation, Sacramento, Ca, USA and has been used without purification. b. Measured by GC after hydrolysis in 6N sulphuric acid at 90°C for <1h (wt% of 1EO-adduct on alcohol intake). c. Trace (<1%) of 2EO derivative also present. d. 2EO and 3EO derivatives also observed in GC after hydrolysis.
Step 2: 2-(benzyloxy)-5-(2-hydroxyethyl)-4-methoxynicotinonitrile2.5 M of n-Butyllithium in hexane (4.85 mL, 12.1 mmol) was added drop-wise to a solution of 2-(benzyloxy)-5-iodo-4-methoxynicotinonitrile (3.7 g, 10 mmol) in THF (150 mL) at -78 C. The reaction was held at -78 C. for 1.5 h, at which time 1,3,2-dioxathiolane 2,2-dioxide (1.25 g, 10.1 mmol, Aldrich) in THF (5.0 mL) was introduced drop-wise. The mixture was allowed to warm to RT and stir for 16 h. Conc. HCl was added (1.85 mL) and the mixture was stirred for 30 min. Saturated NaHCO3 solution was added to adjust the pH to 7. Some water was added and the product was extracted with three portions of EtOAc. The extracts were combined, dried over sodium sulfate and concentrated. Flash column chromatography, eluting with a gradient from 0-70% ethyl acetate in hexanes afforded product as a white solid (1.62 g, 56%). 1H NMR (300 MHz, CDCl3): delta 8.00 (s, 1H), 7.51-7.45 (m, 2H), 7.41-7.30 (m, 3H), 5.47 (s, 2H), 4.33 (s, 3H), 3.77 (dd, 2H), 2.77 (t, 2H); LCMS (M+H)+: 285.1.
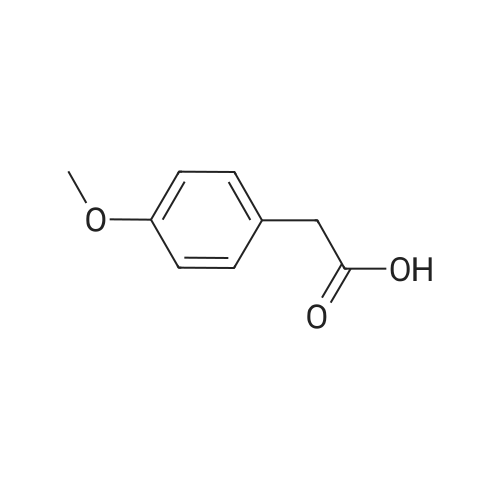
[00263] Step 1 : To a -78 C solution of diisopropylamine (5.84 mL, 41.7 mmol) in THF (12 mL) was added a solution of 2.5 M butyllithium (16.7 mL, 41.7 mmol) in hexanes via a syringe. The mixture was stirred at -78 C for 10 minutes, warmed to 0 C for 5 minutes and cooled to -78 C for 10 minutes. A solution of 2-(4-methoxyphenyl)acetic acid (3.30 g, 19.9 mmol) in THF (12 mL) was added to the lithium diisopropylamide ("LDA") solution at - 78 C using a cannula. The reaction mixture was stirred at -78 C for 20 minutes, warmed to ambient temperature and stirred for 45 minutes. A solution of 1,3,2-dioxathiolane 2,2-dioxide (2.46 g, 19.9 mmol) in THF (12 mL) was added via a syringe. Dimethyl ether ("DME") (10 mL) was added, and the reaction mixture was refluxed for 16 hours. Cool to ambient temperature and concentrated. The residue was partitioned between EtOAc and water. The aqueous layer was extracted with EtOAc (3 X). The combined organics were washed with saturated NaHC03, dried, filtered and concentrated to give 3-(4- methoxyphenyl)dihydrofuran-2(3H)-one (2.22 g, 58.2%).
2-[2-chloro-6-(trifluoromethyl)pyridin-4-yl]ethanol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
2-Chloro-4-iodo-6-(trifluoromethyl)pyridine (0.50 g, 1.6 mmol, European Journal of Organic Chemistry, (18), 3793-3798, 2004) was dissolved in tetrahydrofuran (9.0 mL) and cooled to -78 C. 2.5 M n-Butyllithium in hexanes (0.98 mL, 2.4 mmol) was added dropwise. The reaction was stirred at -78 C for 45 minutes, at which time 1,3,2-dioxathiolane 2,2-dioxide (0.24 g, 2.0 mmol, Aldrich) in Tetrahydrofuran (2.2 mL) was added. The mixture was then allowed to warm to ambient temperature and stir over 1.5 hours. 12.0 M hydrogen chloride in water (0.81 mL, 9.8 mmol) was added to the mixture and the reaction was stirred overnight IN NaOH was then added to achieve a pH between 8 and 9, and brine was also added. The product was extracted with EtOAc, and the organic layer was dried over sodium sulfate, filtered and concentrated. Flash chromatography using a 40 g silica gel cartridge, eluting with a gradient from 0-40% EtOAc in hexanes afforded product as a mixture of isomers, which contained about 50% of the desired isomer (0.09 g, 24%). LCMS (M+H)+: 226.0/228.1.
tert-butyl 4-[6-(trifluoromethyl)pyridin-2-yl]oxy}piperidine-1-carboxylate[ No CAS ]
2-[2-(piperidin-4-yloxy)-6-(trifluoromethyl)pyridin-4-yl]ethanol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
2.5 M n-Butyllithium in hexanes (1.2 mL, 2.9 mmol) was added dropwise to a solution of tert-butyl 4-[6-(trifluoromethyl)pyridin-2-yl]oxy}piperidine-l- carboxylate (0.50 g, 1.4 mmol, from Step 1) in tetrahydrofuran (8.0 mL) at -78 C. The reaction was stirred at this temperature for 1.5 hours, and 1,3,2-dioxathiolane 2,2- dioxide (0.36 g, 2.9 mmol, Aldrich) in Tetrahydrofuran (2.0 mL) was added. The mixture was allowed to warm to room temperature and stir overnight. 12 N Hydrogen chloride in water (0.72 mL, 8.7 mmol) was added to the mixture. The mixture was stirred at room temperature for 4 hours. The mixture was then treated with saturated sodium bicarbonate to pH between 7 and 8, and brine was added. The mixture was then extracted with DCM four times. The combined extracts were dried over sodium sulfate, filtered and concentrated. Preparative HPLC-MS (CI 8 eluting with a gradient of MeCN/0 containing 0.15% NH40H) was used to purify the product. It was the second peak to elute having the desired mass. NMR (300 MHz, CD3OD) delta 7.24 (s, 1H), 6.86 (s, 1 H), 5.16 (tt, J= 8.4, 3.9 Hz, 1H), 3.80 (t, J= 6.3 Hz, 2H), 3.06 (dt, J= 12.6, 4.6 Hz, 2H), 2.85 (t, = 6.3 Hz, 2H), 2.73 (ddd, J= 12.7, 9.5, 3.2 Hz, 2H), 2.13 - 1.93 (m, 2H), 1.68 (dtd, J= 13.0, 9.1 , 3.9 Hz, 2H). I 9F NMR (282 MHz, CD3OD) 8 -70.29 (s). LCMS (M+H)+: 291.1
To a cooled (0 C.) solution of trans-4-((5-bromo-2-(2,5-dimethyl-1H-pyrrol-1-yl)-6-methylpyrimidin-4-yl)amino)cyclohexanol (1.0 g, 2.65 mmol) in dimethylacetamide (15 mL) was added sodium hydride (60% dispersion in oil, 0.19 g, 7.96 mmol) portion-wise. After 2.5 hours, 1,3,2-dioxathiolane-2,2-dioxane (0.48 g, 3.98 mmol) was added portion-wise (0.25 eq. each time). The mixture was stirred at room temperature overnight, quenched with methanol and concentrated. The residue was diluted with 1, 4 dioxane (100 mL) and water (3 mL). p-Toluenesulfonicacid (0.685 g, 3.98 mmol) was added and the mixture was heated at 40 C. for 3 hours. Additional p-toluenesulfonic acid (0.685 g, 3.98 mmol) was added to the mixture and the resulting mixture was heated at 40 C. for another 3 hours. More p-toluenesulfonic acid (0.91 g, 5.3 mmol) was added and the mixture was heated at 40 C. overnight. The reaction mixture was cooled to room temperature and slowly quenched with a solution of sodium bicarbonate (4 g) in water (20 mL). The mixture was concentrated and the residue was extracted with ethylacetate. The combined organic layers were dried with Na2SO4, filtered and concentrated. The crude product was purified by flash silica gel chromatography (20/1 to 10/1 ethyl acetate/hexane) to afford 2-((trans-4-((5-bromo-2-(2,5-dimethyl-1H-pyrrol-1-yl)-6-methylpyrimidin-4-yl)amino) cyclohexyl)oxy)ethanol. [0185] 1H NMR (500 MHz, CDCl3): ppm 5.862 (s, 2H) 5.32 (d, 1H) 4.03 (t, 1H) 3.72 (t, 2H) 3.59 (t, 2H) 3.34 (t, 1H) 2.51 (s, 3H) 2.36 (s, 6H) 2.05-2.15 (m. 5H) 1.21-1.45 (m. 6H); [0186] M+H: 424
To a solution of compound 4a (3.78 g, 10 mmol) in DMF (38 mL) was added NaH (70% dispersion in oil, 1.715 g, 50 mmol) at 0 C, and the resulting mixture was stirred at 0 C for 2.5 hrs. 1,3,2-dioxathiolane 2,2-dioxane (1.86 g, 15 mmol) was added in 0.25 eq portions every 15 minutes. The resulting mixture was stirred at 0 C for 2 hrs. The reaction was quenched with methanol (5mL) and concentrated in vacuo. The residue was diluted with 1,4-dioxane (150 mL) and water (3.8 ml). P-toluenesulfonic acid (3.80 g, 20 mmol) was added and the mixture was stirred 40 C for 3 hours. After removing the volatiles in vacuo, the residue was diluted with water (100 mL) and extracted with dichloromethane (50 mL×3). The combined organic layers were washed with water (50 mL×2) and brine (50 mL×2), dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate 3:1, v/v) to give the title compound 4b (1.52 g, 36% yield) as ayellow oil.
General procedure: To a solution of compound 4a (3.78 g, 10 mmol) in DMF (38 mL) was added NaH (70% dispersion in oil, 1.715 g, 50 mmol) at 0 C, and the resulting mixture was stirred at 0 C for 2.5 hrs. 1,3,2-dioxathiolane 2,2-dioxane (1.86 g, 15 mmol) was added in 0.25 eq portions every 15 minutes. The resulting mixture was stirred at 0 C for 2 hrs. The reaction was quenched with methanol (5mL) and concentrated in vacuo. The residue was diluted with 1,4-dioxane (150 mL) and water (3.8 ml). P-toluenesulfonic acid (3.80 g, 20 mmol) was added and the mixture was stirred 40 C for 3 hours. After removing the volatiles in vacuo, the residue was diluted with water (100 mL) and extracted with dichloromethane (50 mL×3). The combined organic layers were washed with water (50 mL×2) and brine (50 mL×2), dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate 3:1, v/v) to give the title compound 4b (1.52 g, 36% yield) as ayellow oil.
General procedure: To a solution of compound 4a (3.78 g, 10 mmol) in DMF (38 mL) was added NaH (70% dispersion in oil, 1.715 g, 50 mmol) at 0 C, and the resulting mixture was stirred at 0 C for 2.5 hrs. 1,3,2-dioxathiolane 2,2-dioxane (1.86 g, 15 mmol) was added in 0.25 eq portions every 15 minutes. The resulting mixture was stirred at 0 C for 2 hrs. The reaction was quenched with methanol (5mL) and concentrated in vacuo. The residue was diluted with 1,4-dioxane (150 mL) and water (3.8 ml). P-toluenesulfonic acid (3.80 g, 20 mmol) was added and the mixture was stirred 40 C for 3 hours. After removing the volatiles in vacuo, the residue was diluted with water (100 mL) and extracted with dichloromethane (50 mL×3). The combined organic layers were washed with water (50 mL×2) and brine (50 mL×2), dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate 3:1, v/v) to give the title compound 4b (1.52 g, 36% yield) as ayellow oil.
To a solution of 2-fluoro-3-iodopyridine (Ark Pharm, 2.989 g, 13.40 mmol) in THF (50 mL) at -78 C., a solution of lithium diisopropylanide in heptane/THF/ethylbenzene (2.0 M; 8.10 mL, 16.2 mmol) was added. The mixture was stirred at -78 C. for 90 min. A solution of 1,3,2-dioxathiolane 2,2-dioxide (2.206 g, 17.77 mmol) in THF (30 mL) was then added slowly to the mixture at -78 C., over a period of 20 min. After stirring at -78 C. for 20 min., the reaction mixture was allowed to warm to room temperature and stirred for 2 h. The mixture was then cooled to 0 C., and 12.0 M HCl in water (5.0 mL, 60 mmol) was added. The reaction was allowed to warm to room temperature and stirred for 3 h. Saturated aq. NaHCO3 (250 mL) was added. The mixture was extracted with EtOAc (3*150 mL). The combined extracts were washed with brine (250 mL), dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (0-100% EtOAc in hexanes) to give the sub-title compound as a white solid (3.13 g, 87%). LCMS calc. for C7H8FINO (M+H)+: m/z=268.0. found 268.0
87%
A solution of 2.0 M lithium diisopropylamide in heptane/THF/ethylbenzene (8.10 mL, 16.2 mmol) was added to a solution of 2-fluoro-3-iodopyridine (Ark Pharm, 2.989 g, 13.40 mmol) in THF (50 mL) at -78 C., then the mixture was stirred at -78 C. for 90 min. With the temperature maintained at -78 C., a solution of 1,3,2-dioxathiolane 2,2-dioxide (2.206 g, 17.77 mmol) in THF (30 mL) was added slowly over a period of 20 min., the solution was stirred at -78 C. for a further 20 min., then allowed to warm to room temperature and stirred for 2 h. The mixture was then cooled to 0 C., and 12.0 M aq. HCl (5.0 mL, 60. mmol) was added. The reaction mixture was allowed to warm to room temperature and stirred for 3 h. Saturated aq. NaHCO3 (250 mL) was added, then the mixture was extracted with EtOAc (3×150 mL). The combined extracts were washed with brine (250 mL), dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by chromatography on silica gel (0-100% EtOAc in hexanes) to give the sub-title compound as a white solid (3.13 g, 87%). LCMS calc. for C7H8FINO (M+H)+: m/z=268.0. found 268.0.
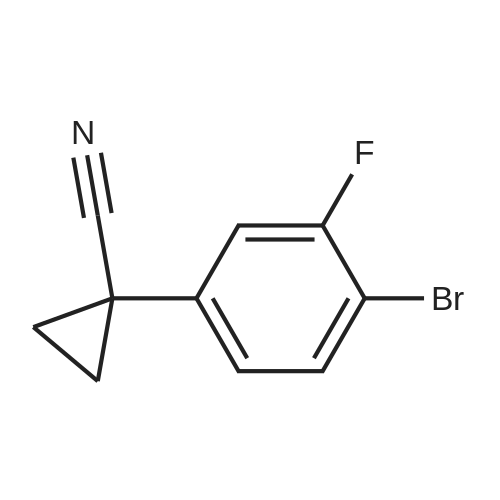
4-bromo-3-fluorophenyl-cyclopropane carboxylic acid ethyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With lithium diisopropyl amide; In tetrahydrofuran; n-heptane; ethylbenzene; at -20℃; for 8.2h;Inert atmosphere; Reflux;
6.6 ml of lithium diisopropylamide (2M in THF/heptane/ethylbenzene, 13.2 mmol, 2.2 eq.) were loaded into a 50 ml dried reactor at T= -20C under nitrogen. 1.71 g of 3-fluoro-4-bromophenylacetate (6.0 mmol, 1.0 eq.), dissolved in 8 ml of dry THF, and 0.82 g of <strong>[1072-53-3]ethylene sulfate</strong> (6.6 mmol, 1.1 eq.), dissolved in 8 ml of dry THF, were added dropwise in 10 minutes. The mixture was kept under stirring at T = -20C for 3h then heated to 20C and maintained under reflux for 5h. The mixture was cooled to room temperature. The reaction was quenched by adding NH4Cl (saturated solution) and extracted with toluene. The organic layer was concentrated to dryness at reduced pressure to yield 1.15 g of crude product (purity = 72.4%).
1.15 g
With lithium diisopropyl amide; In tetrahydrofuran; n-heptane; ethylbenzene; at -20 - 20℃; for 5h;Inert atmosphere;
6.6 ml of lithium diisopropylamide (2M in THF/heptane/ethylbenzene, 13.2 mmol, 2.2 eq.) were loaded into a 50 ml dried reactor at T= -20C under nitrogen. 1.71 g of 3-fluoro-4-bromophenylacetate (6.0 mmol, 1.0 equiv.), dissolved in 8 ml of dry THF, and 0.82 g of <strong>[1072-53-3]ethylene sulfate</strong> (6.6 mmol, 1.1 equiv.), dissolved in 8 ml of dry THF, were added dropwise in 10 minutes. The mixture was kept under stirring at T = -20C for 3h then heated to 20C and maintained under reflux for 5h. The mixture was cooled . to room temperature. The reaction was quenched by adding NH4C1 (saturated solution) and extracted with toluene. The organic layer was concentrated to dryness at reduced pressure to yield 1.15 g of crude product (purity = 72,4%).
With lithium hexamethyldisilazane; In tetrahydrofuran; at -20℃; for 4h;Inert atmosphere;
24 ml of lithium bis(trimethylsilyl)amide (1M in THF, 24 mmol, 2.2 eq.) were loaded at T= -20C in a 50 ml dried reactor under nitrogen. 2.34 g of <strong>[499983-13-0]4-bromo-3-fluorophenylacetonitrile</strong> (10.92 mmol, 1.0 eq.), dissolved in 5 ml of dry THF, and 1.49 g of ethylene sulfate (12.0 mmol, 1.1 eq.), dissolved in 5 ml of dry THF, were added in the reactor. The mixture was kept under stirring at T = -20C for 4h and then heated to 20C. The reaction was quenched by adding NH4Cl (saturated solution) and extracted with toluene. The organic layer was concentrated to dryness at reduced pressure to yield 3.01 g of crude product (assay = 69.4%; molar yield = 79.7%).
With lithium hexamethyldisilazane; In tetrahydrofuran; at -20 - 20℃; for 4h;Inert atmosphere;
EXAMPLE 2Cyclopropanation of <strong>[499983-13-0]4-bromo-3-fluorophenylacetonitrile</strong> (II) with ethylene sulfate (III) to give 4-bromo-3-fluorophenyl-cyclopropanenitrile (IV)24 ml of lithium bis(trimethylsilyl)amide (1M in THF, 24 mmol, 2.2 eq.) were loaded at T=-20 C in a 50 ml dried reactor under nitrogen. 2.34 g of <strong>[499983-13-0]4-bromo-3-fluorophenylacetonitrile</strong> (10.92 mmol, 1.0 eq.), dissolved in 5 ml of dry THF, and 1.49 g of ethylene sulfate (12.0 mmol, 1.1 eq.), dissolved in 5 ml of dry THF, were added in the reactor. The mixture was kept under stirring at T=-20 C. for 4 h and then heated to 20 C. The reaction was quenched by adding NH4Cl (saturated solution) and extracted with toluene. The organic layer was concentrated to dryness at reduced pressure to yield 3.01 g of crude product (assay=69.4%; molar yield=79.7%).
4-bromo-3-fluorophenyl-cyclopropane carboxylic acid ethyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With lithium diisopropyl amide; In tetrahydrofuran; n-heptane; ethylbenzene; at -20 - 20℃; for 8h;Inert atmosphere;
EXAMPLE 4Cyclopropanation of ethyl 4-bromo-3-fluorophenylacetate (II) with ethylene sulfate (III) to give the ethyl ester of 4-bromo-3-fluorophenyl-cyclopropane carboxylic acid. (IV)6.6 ml of lithium diisopropylamide (2M in THF/ heptane/ethylbenzene, 13.2 mmol, 2.2 eq.) were loaded into a 50 ml dried reactor at T=-20 C. under nitrogen. 1.71 g of <strong>[942282-40-8]3-fluoro-4-bromophenylacetate</strong> (6.0 mmol, 1.0 equiv.), dissolved in 8 ml of dry THF, and 0.82 g of ethylene sulfate (6.6 mmol, 1.1 equiv.), dissolved in 8 ml of dry THF, were added dropwise in 10 minutes. The mixture was kept under stirring at T=-20 C for 3 h then heated to 20 C and maintained under reflux for 5 h. The mixture was cooled to room temperature. The reaction was quenched by adding NH4Cl (saturated solution) and extracted with toluene. The organic layer was concentrated to dryness at reduced pressure to yield 1.15 g of crude product (purity=72.4%).
13e (1.02 g, 8.9 mmol) was dissolved in THF (70 mL) and cooled down to -15 C (acetone/dry ice). 1 M LiHMDS solution (in THF, 10.5 mL) was added and the mixture was stirred at -15 C for 30 min. Cyclic sulfate (2.88 g, 23.2 mmol) was dissolved in THF (36 mL) and cooled to -15 C simultaneously. Deprotonated 13e was then transferred to the solution of cyclic sulfate in THF via canula and the mixture was stirred for 2 h at -15 C. After warming to rt, THF was removed in vacuum and ethanolic NaOH (76 mL ethanol/water 2:1, 1.75 g NaOH) was added. The mixture was heated to reflux overnight. The pH value of the reaction mixture was adjusted to pH 3 by addition of 1/4 concd H2SO4 and the aqueous layer was extracted with CH2Cl2 (3*). The combined organic layers were dried (Na2SO4), filtered and concentrated under reduced pressure. The crude product was immediately (.) used in the next reaction step without further purification. Brown oil, yield 665.5 mg (59%). C7H10O2 (126.2 g/mol). 1H NMR (CDCl3): delta [ppm] = 0.21-0.34 (m, 1H, CH2cycloprop), 0.37-0.47 (m, 1H, CH2cycloprop), 0.50-0.56 (m, 1H, CH2cycloprop), 0.64-0.72 (m, 1H, CH2cycloprop), 1.17-1.28 (m, 1H, CHcycloprop), 2.01-2.08 (m, 1H, 4-H), 2.10-2.21 (m, 1H, 4-H), 2.24-2.33 (m, 1H, 3-H), 4.14-4.23 (m, 1H, 5-H), 4.29-4.40 (m, 1H, 5-H). 13C NMR (CDCl3): delta [ppm] = 2.3 (1C, CH2cycloprop), 3.6 (1C, CH2cycloprop), 11.3 (1C, CHcycloprop), 28.4 (1C, C-4), 61.9 (1C, C-5), 66.4 (1C, C-3), 178.6 (1C, C=O). MS (APCI): m/z = 127.0766 (calcd 127.0754 for C7H11O2 [MH+]). IR: [cm-1] = 2970 (C-H), 1709 (C=O), 1184 (C-O).
Ester 13C (2.72 g, 16.2 mmol) was dissolved in THF (146 mL) and the solution was cooled down to -15 C (acetone/dry ice). 0.1 M LiHMDS (in THF, 18.4 mL) was added and the mixture was stirred at -15 C for 30 min. Cyclic sulfate (5.06 g, 40.7 mmol) was dissolved in THF (57 mL) and cooled to -15 C simultaneously. The solution of the enolate of 13C was transferred to the THF solution of cyclic sulfate via canula and the mixture was stirred for 2 h at -15 C. The mixture was warmed to rt, the solvent was removed in vacuo and ethanolic NaOH (146 mL ethanol/water 2:1, 3.27 g NaOH) was added. The resulting solution was heated to reflux overnight. The pH value of the reaction mixture was then adjusted to pH 3 with 1/4 concd H2SO4 and the aqueous layer was extracted with CH2Cl2 (3*). The combined organic layers were dried (Na2SO4), filtered and concentrated under reduced pressure. The crude product was purified by flash column chromatography (Ø 4.5 cm, length 14 cm, CH2Cl2/EtOAc 90:10, fraction size 30 mL, Rf = 0.26 (CH2Cl2/EtOAc 40:60)). Brown oil, yield 1.51 g (52%). C10H9FO2 (180.2 g/mol). 1H NMR (CDCl3): delta [ppm] = 2.41 (dddd, J = 12.8/10.7/9.6/8.3 Hz, 1H, 4-H), 2.72 (dddd, J = 12.8/9.0/6.6/3.0 Hz, 1H, 4-H), 3.79 (dd, J = 10.6/8.8 Hz, 1H, 3-H), 4.34 (td, J = 9.4/6.5 Hz, 1H, 5-H), 4.48 (td, J = 8.7/3.0 Hz, 1H, 5-H), 7.02-7.07 (m, 2H, 3-Harom, 5-Harom), 7.24-7.28 (m, 2H, 2-Harom, 6-Harom). 13C NMR (CDCl3): delta [ppm] = 31.6 (1C, C-4), 44.7 (1C, C-3), 66.4 (1C, C-5), 115.5 and 115.8 (d, each J = 21.5 Hz, 2C, C-3arom, C-5arom), 129.5 and 130.9 (d, each J = 8.1 Hz, 2C, C-2arom, C-6arom), 132.2 (d, J = 3.4 Hz, 1C, C-1arom), 162.2 (d, J = 246.7 Hz, 1C, C-4arom), 177.1 (1C, C=O). MS (APCI): m/z = 181.0653 (calcd 181.0659 for C10H10FO2 [MH+]). IR: [cm-1] = 1762 (C=O), 1219 (C-Farom), 1150 (C-O), 814 (C-Harom).
5-chloro-1-(phenylsulfonyl)-1H-pyrrolo[3,2-b]pyridine[ No CAS ]
C15H13ClN2O3S[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Step 1. 2-(2-[tert-butyl(dimethyl)silyl]oxy}ethyl)-5-chloro-1-(phenylsulfonyl)-1H-pyrrolo[3,2-b]pyridine 1.6 M n-Butyllithium in hexanes (12 mL, 19 mmol) was added dropwise to a solution of N,N-diisopropylamine (2.9 mL, 20. mmol) in THF (41 mL) at -78 C. After complete addition, the reaction temperature was raised to 0 C. for 30 minutes, followed by re-cooling to -78 C. A solution of 5-chloro-1-(phenylsulfonyl)-1H-pyrrolo[3,2-b]pyridine (3.74 g, 12.8 mmol, prepared as in Example 2, Step 1) in THF (19 mL) was added dropwise. After stirring for 1 h at -78 C., 1,3,2-dioxathiolane 2,2-dioxide (3.2 g, 26 mmol, Aldrich) in THF (9.4 mL) was added. The cold bath was then removed and the mixture was allowed to warm to room temperature over 1 hour. The reaction was cooled to 0 C. and 12.0 M hydrogen chloride in water (6.4 mL, 77 mmol), then was allowed to warm to room temperature and stir overnight. The mixture was then neutralized with NaHCO3 and was extracted with two portions of EtOAc. The combined extracts were dried over sodium sulfate, filtered and concentrated to afford the crude alcohol, which was dissolved in DCM (100. mL), and tert-butyldimethylsilyl chloride (2.9 g, 19 mmol, Aldrich) and 1H-imidazole (1.4 g, 20. mmol, Aldrich) were added. After 15 minutes, the mixture was washed with water, brine, dried over sodium sulfate, filtered and concentrated. Flash chromatography, eluting with a gradient from 0-10% EtOAc in hexanes afforded product as a light yellow oil (5.02 g, 87%). 1H NMR (300 MHz, CDCl3) delta 8.37 (dd, J=8.7, 0.7 Hz, 1H), 7.75-7.65 (m, 2H), 7.63-7.53 (m, 1H), 7.49-7.41 (m, 2H), 7.20 (d, J=8.7 Hz, 1H), 6.68-6.56 (m, 1H), 3.97 (t, J=6.2 Hz, 2H), 3.21 (t, J=6.2 Hz, 2H), 0.84 (s, 9H), -0.00 (s, 6H). LCMS (M+H)+: 450.9.
(S)-3',3'-dimethyldihydro-1'H-spiro[cyclopropane-1,6'-pyrrolo[1,2-c]oxazol]-5'(3'H)-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
44%
With lithium diisopropyl amide; In tetrahydrofuran; at -78 - 25℃; for 8.75h;Inert atmosphere;
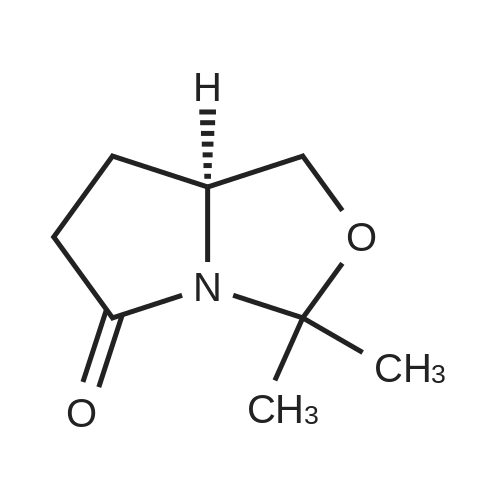
A solution of (S)-3,3-dimethyltetra-hydropyrrolo[1,2-c]oxazol-5(1H)-one (CAS 99208-71-6, 233 mg, 1.5 mmol) in THF (10 mL) was treated with LDA (2.0 M, 1.6 mL) at about -78 C. A solution of 1,3,2-dioxathiolane 2,2-dioxide (CAS 1072-53-3, 242 mg, 1.9 mmol) in THF (10 mL) was added at a rate to maintain the internal temp at about less than -65 C. The mixture was stirred at about -78 C. for about 10 min then warmed to about -20 C. The mixture was stirred for about 45 min and gradually warmed to about -3 C. before being re-cooled to about -78 C. LDA (2.0 M, 1.95 mmol) was added and the mixture was stirred for about 10 min at about -78 C., then slowly warmed to about 25 C. and kept for about 8 h. The mixture was treated with half-saturated aqueous NH4Cl and extracted with EtOAc. The EtOAc extracts were washed with saturated aqueous NH4Cl, dried over MgSO4 filtered and concentrated. The residue was purified by chromatography to provide the title compound C42. Yield: 120 mg (44%). 1H NMR (400 MHz, CDCl3) delta 4.29-4.21 (m, 1H), 4.08-4.05 (m, 1H), 3.43-3.38 (m, 1H), 2.04-1.99 (m, 1H), 1.94-1.90 (m, 1H), 1.61 (s, 3H), 1.45 (s, 3H), 1.19-1.14 (m, 1H), 1.25-1.17 (m, 1H), 1.16-1.14 (m, 1H), 0.95-0.94 (m, 1H), 0.93-0.90 (m, 1H).
2-{4,6-dichloro-2-[(3S)-3-methylmorpholin-4-yl]pyrimidin-5-yl}ethanol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85%
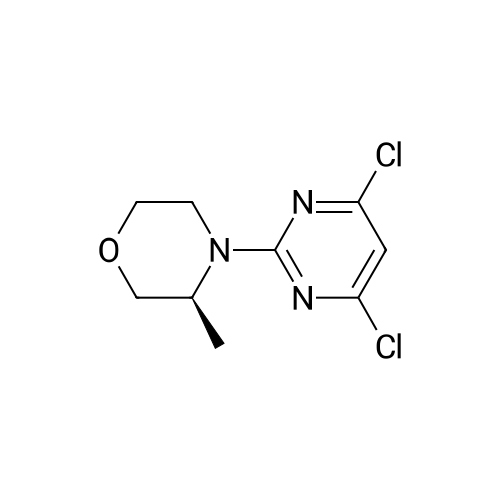
To a solution of (3S)-4-(4,6-dichloropyrim idin-2-yl)-3-methylmorpholine (992 mg,4.0 mmol) in THE (20 mL) was added n-butyllithium (1.56 mL, 1.6 M) dropwise at -78 Cand the mixture was stirred for 30 mm. i ,3,2-dioxathiolane 2,2-dioxide (672 mg, 5.41 mmol) was added, and stirring was continued for 30 mm, whereupon 6 N HCI (13.3 mL) was added. The reaction mixture was stirred for 18 h and then heated at 40 C for 4 h. The reaction mixture was extracted with ethyl acetate, and the organic layer waswashed with brine, dried with Mg504, filtered and concentrated by rotary evaporation.The resulting residue was purified by silica gel chromatography using a gradient ofEtOAc/heptane (0-50%) to give the title compound (991 mg, 85%). 1H NMR (400 MHz,CDCI3) O 4.62 (qd, J = 6.7, 3.4 Hz, 1 H), 4.26 (dd, J = 13.7, 2.8 Hz, 1 H), 3.97 (dd, J =11.4, 3.7 Hz, 1 H), 3.87 - 3.82 (m, 2 H), 3.78 - 3.74 (m, 1 H), 3.68 - 3.63 (m, 1 H), 3.50(td, J= 11.9, 3.0 Hz, 1 H), 3.28 (ddd, J= 13.5, 12.4, 3.8 Hz, 1 H), 3.03 (t, J= 7.0 Hz, 2 H), 1.31 (d, J = 6.8 Hz, 3 H). m/z (APCI+) for C11H15C12N302 291.9 (M+H).
2-[4,6-dichloro-2-(methylsulfanyl)pyrimidin-5-yl]ethanol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
76%
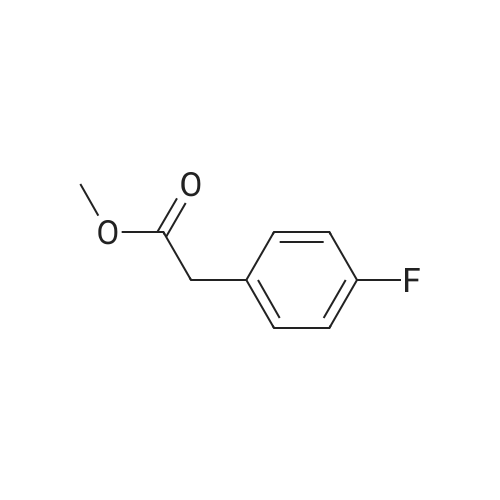
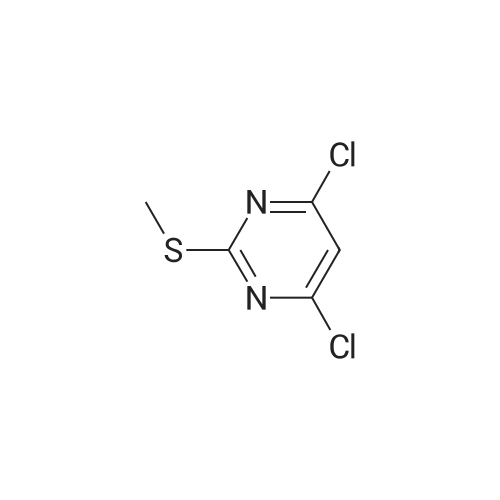
To a vial containing 4,6-dichloro-2-(methylsulfanyl)pyrimidine (200 mg, 0.102 mmol) in THE (10 mL) was added n-butyllithium (0.554 mL, 1.38 mmol, 2.5 M inhexane) at -78 C and the resulting mixture was stirred for 30 mm. i ,3,2-dioxathiolane2,2-dioxide (172 mg, 1.39 mmol) was added at -78 C and stirring was continued for 2h. The reaction vial was removed from the dry ice bath, 6 N HCI (3.5 mL, 21 mmol) wasadded, and the mixture was stirred at room temperature for 18 h. 2-Methyltetrahydrofuran (20 mL) was added and the solution was washed with a 1:1mixture of brine/water and then with saturated aqueous NaHCO3. The organic layer wasdried over Na2504, filtered, concentrated and purified via silica gel chromatography using a gradient of EtOAC/heptane (30-50%) to give the title compound (185 mg, 76% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) O 4.84 (t, J = 5.4 Hz, 1 H), 3.61 (q, J = 6.3 Hz, 2 H), 2.93 (t, J = 6.8 Hz, 2 H), 2.52 (5, 3 H). m/z (APCI+) for C7H8C12N205239.0 (M+H).
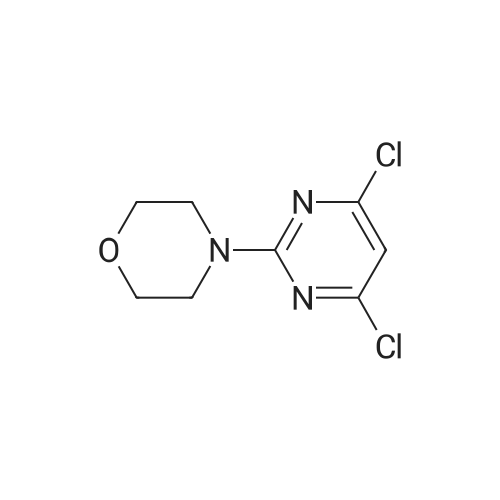
2-[4,6-dichloro-2-(morpholin-4-yl)pyrimidin-5-yl]ethanol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
42%
To a solution of <strong>[10397-13-4]4-(4,6-dichloropyrimidin-2-yl)morpholine</strong> (468 mg, 2.0 mmol) in THE (20 mL) was added n-butyllithium (1.56 mL, 1.6 M) dropwise at -78 C. After stirring for 30 mm, 1 ,3,2-dioxathiolane 2,2-dioxide (336 mg, 2.71 mmol) was added, and afterstirring for an additional 40 mm, 6 N HCI (6.67 mL) was added. The reaction was stirred at room temperature for 18 h and then at 40C for 4 h. The reaction mixture was extracted with ethyl acetate and the organic layer was washed with brine, dried with MgSO4, filtered and concentrated by rotary evaporation. The resulting residue was purified by silica gel chromatography using a gradient of EtOAc/heptane (25-75%) togive the title compound (231 mg, 42%). 1H NMR (400 MHz, CDCI3) O 3.84 (t, J = 7.0 Hz,2 H), 3.80 - 3.76 (m, 4 H), 3.76 - 3.72 (m, 4 H), 3.04 (t, J = 7.0 Hz, 2 H). m/z (APCI+) for C10H13C12N302 277.9 (M+H).
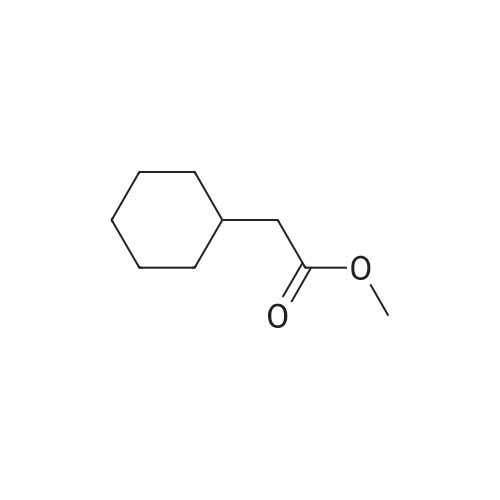
Stage #1: methyl cyclohexylacetate With lithium diisopropyl amide In tetrahydrofuran at -78℃; for 0.333333h; Inert atmosphere;
Stage #2: ethyleneglycol sulfate In tetrahydrofuran at -78 - 20℃; for 5h; Inert atmosphere;
Stage #1: methyl 3,3-dimethylbutyrate With lithium diisopropyl amide In tetrahydrofuran at -78℃; for 0.333333h; Inert atmosphere;
Stage #2: ethyleneglycol sulfate In tetrahydrofuran at -78 - 20℃; for 5h; Inert atmosphere;
2-(2-[tert-butyl(dimethyl)silyl]oxy}ethyl)thiophene-3-carbaldehyde[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
3.102 g
Stage #1: ethyleneglycol sulfate; 2-(thiophen-3-yl)-1,3-dioxolane With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 0.5h;
Stage #2: With sulfuric acid In tetrahydrofuran; hexane; water at 75℃;
Stage #3: tert-butyldimethylsilyl chloride With 1H-imidazole In tetrahydrofuran; hexane; water for 0.5h;
15.2 Step 2: 2-(2-[tert-Butyl(dimethyl)silyl]oxy}ethyl)thiophene-3-carbaldehyde
Step 2: 2-(2-[tert-Butyl(dimethyl)silyl]oxy}ethyl)thiophene-3-carbaldehyde [00333]A solution of 3-thiophenecarboxaldehyde ethylene acetal (4.33 g, 27.7 mmol) in THF (130.0 mL, 1603 mmol) was cooled to -78 °C, at which point 2.50 M of -BuLi in hexane ( 17.74 mL, 44.35 mmol) was added. A solution of ethylenesulfate (3.7846 g, 30.492 mmol) in THF ( 10.0 mL, 123 mmol) was added solution and the reaction was stirred for 30 min at -78 °C. The reaction was concentrated to -20% of the original volume in vacuo, and then a solution of 6 ml 98% H2S04 in 30ml water was added to the mixture. This mixture was then stirred at 75 °C overnight. The solution was added slowly to 150 ml saturated NaHC03 aqueous solution to neutralize and then extracted with 3 x 50 ml DCM. The combined organic layers were concentrated to dryness, and the residue was dissolved into DCM ( 100 mL, 2000 mmol), to which were added IH-imidazole (3.774 g, 55.44 mmol) and tert-butyldimethylsilyl chloride (5.013 g, 33.26 mmol). The reaction was stirred for 30 min, and poured into 60ml water. The aqueous was extracted with 2 x 40 mL DCM. The combined organic layers were concentrated in vacuo and purified by flash column (80g, eluent was 0-25% EtOAc in hexane over 15min) to afford 3.102g (41 %) of the title compound. NMR (400 MHz, Chloroform-d) δ 10.04 (s, 1H), 7.43 (d, J = 5.4 Hz, 1H), 7.15 (dd, J = 5.4, 0.5 Hz, 1 H), 3.89 (t, J = 6.0 Hz, 2H), 3.40 (t, J = 6.0 Hz, 2H), 0.88 (s, 9H), -0.00 (s, 6H).
A suspension of 2-(l -(2-fluorobenzyl)-5-(isoxazol-3-yl)-lH-pyrazol-3-yl)pyrimidine- -4,5-diamine (described in previous patents: WO2012/3405 Al, WO2013/101830 Ai) (1.0 equiv.) in anhydrous THF was treated with triethylamine (2.0 equiv.) followed by l ,3,2-dioxathiolane-2,2-dioxide (1.2 equiv.). After 18 h, additional amount of l ,3,2-dioxathiolane-2,2-dioxide (0.30 equiv.) was added and the contents were stirred for 5 h. The reaction mixture was then concentrated in vacuo, re-suspended in aqueous 6 N HC1/THF (3:1 v/v) and heated at 60 C for 18 h. After cooling to 23 C, the contents were poured into half-saturated sodium bicarbonate solution and extracted with dichloromethane/iPrOH (4:1). The organic layers were dried over sodium sulfate, filtered and the solvent was removed in vacuo. The crude material was purified via silica gel chromatography utilizing 70-100% acetonitrile-methanol (7:1) in dichloromethane to deliver the desired compound (31 mg, 62% yield) as a beige-colored solid. 'H-NMR (500 MHz, DMSO-ife) delta ppm 9.19 (d, 1 H), 8.83 (br. s, 1 H), 8.10 (br. s, 1 H), 7.58 (s, 1 H), 7.46 (s, 1 H), 7.39 (m, 1 H), 7.26 (d, 1 H), 7.24 (m, 1 H), 7.19-7.10 (m, 2 H), 5.99 (s, 1 H), 5.89 (s, 2 H), 5.07 (t, 1 H), 4.50 (m, 2 H), 3.62 (m, 2 H).
Weigh 31.0g of ethylene glycol was added to the industrial 1L three bottles, which leads to dry steady stream of nitrogen, sealed and exhaust system connected through the water absorbed into the bottle.Solution of 65.5g of thionyl chloride, dropping the control temperature of the system does not exceed 30 , 1h drops completion.Dropping bubbles emerge.Bi drops to warm to 25-30 , keep stirring 3h, TLC monitoring no starting material remaining glycol, insulation stop, this time the body system as a light brown clear solution.The cooling system to 0-10 deg.] C, 8g of sodium bicarbonate and weighed 112g formulated into aqueous sodium bicarbonate solution was added to the system manipulation system pH between 7-8, and then added 0.3g of ruthenium trichloride trihydrate , the system as a dark brown turbid liquid.0-10 temperature and stirred solution of 313g mass concentration of 13% sodium hypochlorite solution until the reaction mixture turns a pale yellow turbid solution was dropped at 2h, dropping Bi 0-10 insulation 1h, starch -KI test strip aqueous phase has oxidation, TLC monitoring of the reaction mixture remaining after no DTO, continue to the next step operation.Aqueous sodium hydrogen sulfite, 120g of 0-10 holding temperature of the system was added 10% by mass concentration of potassium iodide starch test paper non-oxidizing.After stirring for 10min temperature 0-5 filtered to give 49.8g of yellow solid particles, GC> 96%, yield: 80%.The crude toluene with 50g hot melt, hot filtered and the filtrate temperature drop precipitated solid was filtered to give 46g of white crystalline powder, GC> 99.9%, the total yield of 74%.
With N-Bromosuccinimide; In dichloromethane; at 20℃; for 6h;
Using vinyl sulfate as a raw material, 1 mol of vinyl sulfate was dissolved in 800 mL of methylene chloride, and the mixture was added portionwise at 20 CWas added to 1.1 mol of NBS for 6 h to give the intermediate 4-bromo-vinylsulfate. The resulting intermediate was then placed in dichloromethane with sodium fluoride in the presence of dodecyltrimethylammonium chloride Exchange reaction to obtain 4-fluoro-vinyl sulfate, wherein the molar ratio of the intermediate to the sodium fluoride is 1: 1.3 and the solvent methylene chloride is used in an amount of 600 times the mass of the vinyl sulfate. The calculated yield was 85.4%, the purity was 99.7%, and the water content was 17PPM
With N-chloro-succinimide; In dichloromethane; at 41℃; for 6h;
Using vinyl sulfate as a raw material, 1 mol of vinyl sulfate was dissolved in 500 mL of methylene chloride, and the mixture was added portionwise at 41 C1.28 mol of NCS was added and reacted for 6 h to give the intermediate 4-chloro-vinylsulfate. The resulting intermediate was then subjected to an exchange reaction with potassium cyanide in dichloromethane in the presence of tetrabutylammonium bromide to give 4-cyano-vinylsulfate, wherein the molar ratio of the intermediate to potassium cyanide is 1: 1 and the amount of the solvent methylene chloride is 600 times the mass of the vinyl sulfate. The calculated yield was 85.5%, the purity was 99.7%, and the water content was 17PPM.
A solution of 2.0 M lithium diisopropylamide in heptane/THF/ethylbenzene (8.10 mL, 16.2 mmol) was added to a solution of 2-fluoro-4-iodopyridine (Ark Pharm, 2.989 g, 13.40 mmol) in THF (50 mL) at -78 C, then the mixture was stirred at -78 C for 90 min. With the temperature maintained at -78 C, a solution of 1,3,2-dioxathiolane 2,2-dioxide (2.206 g, 17.77 mmol) in THF (30 mL) was added slowly over a period of 20 min., the solution was stirred at -78 C for a further 20 min., then allowed to warm to room temperature and stirred for 2 h at that temperature. The mixture was then cooled to 0 C, and 12.0 M aqueous HC1 (5.0 mL, 60. mmol) was added. The reaction mixture was allowed to warm to room temperature and stirred at that temperature for 3 h. Saturated aqueous NaHCC (250 mL) was added, then the reaction mixture was extracted with EtOAc (3 x 150 mL). The combined extracts were washed with brine (250 mL), dried over Na2S04 and concentrated under reduced pressure. The residue was purified by chromatography on silica gel (gradient elution with 0-100% EtOAc in hexanes) to give the sub-title compound as a white solid (3.13 g, 87%). LCMS calc. for CvHsFINO (M+H)+: m/z = 268.0; found 268.0.
1950 mg
A solution of LDA, 2M in n-heptane / THF / ethylbenzene (5.3 mL; 10.6 mmol) was added to a solution of 2-fluoro-4-iodopyridine (2000 mg; 8.79 mmol) in THF (40 mL) at -78 C. The reaction mixture was stirred at -78 C for 1.5 h. With the temperature maintained at -78 C, a solution of 1,3,2-dioxathiolane 2,2-dioxide (1450 mg; 11.4 mmol) in THF (20 mL) was added slowly over a period of 20 min. The solution was further stirred at -78 C for 20 min, then allowed to warm to rt and stirred for additional 2 h. The mixture was then cooled to 0 C, and 12 M aqueous HC1 (3.3 mL) was added. The reaction mixture was allowed to warm to rt and stirred for 3 h. Saturated aqueous solution of NaHCC>3 (200 mL) was added and the product was extracted with EA (3 x 150 mL). The combined extracts were washed with brine (250 mL), dried over Na2S04, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (silica gel; n-Hex : EA; 1 :0 to 0: 1 ; v/v) to afford 2-(2-fluoro-4-iodo- 3-pyridyl)ethanol (1950 mg) as a white solid. (0758) MS m/z (+ESI): 268.0 [M+H]+. (0759) 'H-NMR (400 MHz, DMSO-/ + D20) delta ppm: 7.77 (d, J= 5.2 Hz, 1H), 7.72 (d, J= 5.2 Hz, 1H), 3.52 (t, J= 7.2 Hz, 2H), 2.88 (t, J= 7.2 Hz, 2H).
General procedure: To a stirred solution of diisopropylamine (1.86 g, 18.4 mmol)in dried THF (50 mL) cooled at -78 C in N2 atmospherewas dropwise added via syringe a 1.6 M solution of n-BuLi inn-hexane (11 mL, 17.6 mmol), and after addition the resultingsolution was stirred for another 0.5 h followed by dropwiseaddition via syringe of 18a-18k (8 mmol) dissolved in driedTHF (5 mL). The mixture thus obtained was stirred for another1 h, and 1,3,2-dioxathiolane-2,2-dioxide (19a, 1.49 g, 12 mmol)or 19b-19f dissolved in dried THF (10 mL) and DMPU (1.13 g,8.8 mmol) dissolved in dried THF (5 mL) were dropwise added successively via syringe. The reaction mixture was stirred atroom temperature for 10 h. The reaction mixture was pouredinto ice-water (200 mL), and the mixture thus obtained wasextracted with CH2Cl2 (3 × 100 mL). The combined extractswere washed with 5% brine (5 × 100 mL), dried over anhydrousNa2SO4 and evaporated on a rotary evaporator to aord aresidue, which was purifed by column chromatography to yield17a-18j after trituration with EtOAc/n-hexane if the productwas a solid.Methyl 1-(phenylthio)cyclopropanecarboxylate (17a). YieldsareshowninTable2.Physicalpropertieswereinagreementwiththose for the sample from synthetic approach A.
methyl 1-[(4-methoxyphenyl)thio]cyclopropanecarboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
41%
General procedure: To a stirred solution of diisopropylamine (1.86 g, 18.4 mmol)in dried THF (50 mL) cooled at -78 C in N2 atmospherewas dropwise added via syringe a 1.6 M solution of n-BuLi inn-hexane (11 mL, 17.6 mmol), and after addition the resultingsolution was stirred for another 0.5 h followed by dropwiseaddition via syringe of 18a-18k (8 mmol) dissolved in driedTHF (5 mL). The mixture thus obtained was stirred for another1 h, and 1,3,2-dioxathiolane-2,2-dioxide (19a, 1.49 g, 12 mmol)or 19b-19f dissolved in dried THF (10 mL) and DMPU (1.13 g,8.8 mmol) dissolved in dried THF (5 mL) were dropwise added successively via syringe. The reaction mixture was stirred atroom temperature for 10 h. The reaction mixture was pouredinto ice-water (200 mL), and the mixture thus obtained wasextracted with CH2Cl2 (3 × 100 mL). The combined extractswere washed with 5% brine (5 × 100 mL), dried over anhydrousNa2SO4 and evaporated on a rotary evaporator to aord aresidue, which was purifed by column chromatography to yield17a-18j after trituration with EtOAc/n-hexane if the productwas a solid.
methyl 1-[(4-nitrophenyl)thio]cyclopropanecarboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
37%
General procedure: To a stirred solution of diisopropylamine (1.86 g, 18.4 mmol)in dried THF (50 mL) cooled at -78 C in N2 atmospherewas dropwise added via syringe a 1.6 M solution of n-BuLi inn-hexane (11 mL, 17.6 mmol), and after addition the resultingsolution was stirred for another 0.5 h followed by dropwiseaddition via syringe of 18a-18k (8 mmol) dissolved in driedTHF (5 mL). The mixture thus obtained was stirred for another1 h, and 1,3,2-dioxathiolane-2,2-dioxide (19a, 1.49 g, 12 mmol)or 19b-19f dissolved in dried THF (10 mL) and DMPU (1.13 g,8.8 mmol) dissolved in dried THF (5 mL) were dropwise added successively via syringe. The reaction mixture was stirred atroom temperature for 10 h. The reaction mixture was pouredinto ice-water (200 mL), and the mixture thus obtained wasextracted with CH2Cl2 (3 × 100 mL). The combined extractswere washed with 5% brine (5 × 100 mL), dried over anhydrousNa2SO4 and evaporated on a rotary evaporator to aord aresidue, which was purifed by column chromatography to yield17a-18j after trituration with EtOAc/n-hexane if the productwas a solid.
methyl 1-(n-hexylthio)cyclopropanecarboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
36%
General procedure: To a stirred solution of diisopropylamine (1.86 g, 18.4 mmol)in dried THF (50 mL) cooled at -78 C in N2 atmospherewas dropwise added via syringe a 1.6 M solution of n-BuLi inn-hexane (11 mL, 17.6 mmol), and after addition the resultingsolution was stirred for another 0.5 h followed by dropwiseaddition via syringe of 18a-18k (8 mmol) dissolved in driedTHF (5 mL). The mixture thus obtained was stirred for another1 h, and 1,3,2-dioxathiolane-2,2-dioxide (19a, 1.49 g, 12 mmol)or 19b-19f dissolved in dried THF (10 mL) and DMPU (1.13 g,8.8 mmol) dissolved in dried THF (5 mL) were dropwise added successively via syringe. The reaction mixture was stirred atroom temperature for 10 h. The reaction mixture was pouredinto ice-water (200 mL), and the mixture thus obtained wasextracted with CH2Cl2 (3 × 100 mL). The combined extractswere washed with 5% brine (5 × 100 mL), dried over anhydrousNa2SO4 and evaporated on a rotary evaporator to aord aresidue, which was purifed by column chromatography to yield17a-18j after trituration with EtOAc/n-hexane if the productwas a solid.
methyl 1-(t-butylthio)cyclopropanecarboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
39%
General procedure: To a stirred solution of diisopropylamine (1.86 g, 18.4 mmol)in dried THF (50 mL) cooled at -78 C in N2 atmospherewas dropwise added via syringe a 1.6 M solution of n-BuLi inn-hexane (11 mL, 17.6 mmol), and after addition the resultingsolution was stirred for another 0.5 h followed by dropwiseaddition via syringe of 18a-18k (8 mmol) dissolved in driedTHF (5 mL). The mixture thus obtained was stirred for another1 h, and 1,3,2-dioxathiolane-2,2-dioxide (19a, 1.49 g, 12 mmol)or 19b-19f dissolved in dried THF (10 mL) and DMPU (1.13 g,8.8 mmol) dissolved in dried THF (5 mL) were dropwise added successively via syringe. The reaction mixture was stirred atroom temperature for 10 h. The reaction mixture was pouredinto ice-water (200 mL), and the mixture thus obtained wasextracted with CH2Cl2 (3 × 100 mL). The combined extractswere washed with 5% brine (5 × 100 mL), dried over anhydrousNa2SO4 and evaporated on a rotary evaporator to aord aresidue, which was purifed by column chromatography to yield17a-18j after trituration with EtOAc/n-hexane if the productwas a solid.
methyl 1-(cyclohexylthio)cyclopropanecarboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
40%
General procedure: To a stirred solution of diisopropylamine (1.86 g, 18.4 mmol)in dried THF (50 mL) cooled at -78 C in N2 atmospherewas dropwise added via syringe a 1.6 M solution of n-BuLi inn-hexane (11 mL, 17.6 mmol), and after addition the resultingsolution was stirred for another 0.5 h followed by dropwiseaddition via syringe of 18a-18k (8 mmol) dissolved in driedTHF (5 mL). The mixture thus obtained was stirred for another1 h, and 1,3,2-dioxathiolane-2,2-dioxide (19a, 1.49 g, 12 mmol)or 19b-19f dissolved in dried THF (10 mL) and DMPU (1.13 g,8.8 mmol) dissolved in dried THF (5 mL) were dropwise added successively via syringe. The reaction mixture was stirred atroom temperature for 10 h. The reaction mixture was pouredinto ice-water (200 mL), and the mixture thus obtained wasextracted with CH2Cl2 (3 × 100 mL). The combined extractswere washed with 5% brine (5 × 100 mL), dried over anhydrousNa2SO4 and evaporated on a rotary evaporator to aord aresidue, which was purifed by column chromatography to yield17a-18j after trituration with EtOAc/n-hexane if the productwas a solid.
methyl 1-[(naphth-2-yl)thio]cyclopropanecarboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
43%
General procedure: To a stirred solution of diisopropylamine (1.86 g, 18.4 mmol)in dried THF (50 mL) cooled at -78 C in N2 atmospherewas dropwise added via syringe a 1.6 M solution of n-BuLi inn-hexane (11 mL, 17.6 mmol), and after addition the resultingsolution was stirred for another 0.5 h followed by dropwiseaddition via syringe of 18a-18k (8 mmol) dissolved in driedTHF (5 mL). The mixture thus obtained was stirred for another1 h, and 1,3,2-dioxathiolane-2,2-dioxide (19a, 1.49 g, 12 mmol)or 19b-19f dissolved in dried THF (10 mL) and DMPU (1.13 g,8.8 mmol) dissolved in dried THF (5 mL) were dropwise added successively via syringe. The reaction mixture was stirred atroom temperature for 10 h. The reaction mixture was pouredinto ice-water (200 mL), and the mixture thus obtained wasextracted with CH2Cl2 (3 × 100 mL). The combined extractswere washed with 5% brine (5 × 100 mL), dried over anhydrousNa2SO4 and evaporated on a rotary evaporator to aord aresidue, which was purifed by column chromatography to yield17a-18j after trituration with EtOAc/n-hexane if the productwas a solid.
methyl 2-[(3-methylfuran-2-yl)thio]acetate[ No CAS ]
methyl 1-[(3-methylfuran-2-yl)thio]cyclopropanecarboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
38%
General procedure: To a stirred solution of diisopropylamine (1.86 g, 18.4 mmol)in dried THF (50 mL) cooled at -78 C in N2 atmospherewas dropwise added via syringe a 1.6 M solution of n-BuLi inn-hexane (11 mL, 17.6 mmol), and after addition the resultingsolution was stirred for another 0.5 h followed by dropwiseaddition via syringe of 18a-18k (8 mmol) dissolved in driedTHF (5 mL). The mixture thus obtained was stirred for another1 h, and 1,3,2-dioxathiolane-2,2-dioxide (19a, 1.49 g, 12 mmol)or 19b-19f dissolved in dried THF (10 mL) and DMPU (1.13 g,8.8 mmol) dissolved in dried THF (5 mL) were dropwise added successively via syringe. The reaction mixture was stirred atroom temperature for 10 h. The reaction mixture was pouredinto ice-water (200 mL), and the mixture thus obtained wasextracted with CH2Cl2 (3 × 100 mL). The combined extractswere washed with 5% brine (5 × 100 mL), dried over anhydrousNa2SO4 and evaporated on a rotary evaporator to aord aresidue, which was purifed by column chromatography to yield17a-18j after trituration with EtOAc/n-hexane if the productwas a solid.
methyl 1-[(thiophen-2-yl)thio]cyclopropanecarboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
35%
General procedure: To a stirred solution of diisopropylamine (1.86 g, 18.4 mmol)in dried THF (50 mL) cooled at -78 C in N2 atmospherewas dropwise added via syringe a 1.6 M solution of n-BuLi inn-hexane (11 mL, 17.6 mmol), and after addition the resultingsolution was stirred for another 0.5 h followed by dropwiseaddition via syringe of 18a-18k (8 mmol) dissolved in driedTHF (5 mL). The mixture thus obtained was stirred for another1 h, and 1,3,2-dioxathiolane-2,2-dioxide (19a, 1.49 g, 12 mmol)or 19b-19f dissolved in dried THF (10 mL) and DMPU (1.13 g,8.8 mmol) dissolved in dried THF (5 mL) were dropwise added successively via syringe. The reaction mixture was stirred atroom temperature for 10 h. The reaction mixture was pouredinto ice-water (200 mL), and the mixture thus obtained wasextracted with CH2Cl2 (3 × 100 mL). The combined extractswere washed with 5% brine (5 × 100 mL), dried over anhydrousNa2SO4 and evaporated on a rotary evaporator to aord aresidue, which was purifed by column chromatography to yield17a-18j after trituration with EtOAc/n-hexane if the productwas a solid.Methyl 1-(phenylthio)cyclopropanecarboxylate (17a). Yieldsare shown in Table 2. Physical properties were in agreement withthose for the sample from synthetic approach A.
methyl 2-[(6-bromoquinolin-4-yl)thio]acetate[ No CAS ]
methyl 1-[(6-bromoquinolin-4-yl)thio]cyclopropanecarboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
40%
General procedure: To a stirred solution of diisopropylamine (1.86 g, 18.4 mmol)in dried THF (50 mL) cooled at -78 C in N2 atmospherewas dropwise added via syringe a 1.6 M solution of n-BuLi inn-hexane (11 mL, 17.6 mmol), and after addition the resultingsolution was stirred for another 0.5 h followed by dropwiseaddition via syringe of 18a-18k (8 mmol) dissolved in driedTHF (5 mL). The mixture thus obtained was stirred for another1 h, and 1,3,2-dioxathiolane-2,2-dioxide (19a, 1.49 g, 12 mmol)or 19b-19f dissolved in dried THF (10 mL) and DMPU (1.13 g,8.8 mmol) dissolved in dried THF (5 mL) were dropwise added successively via syringe. The reaction mixture was stirred atroom temperature for 10 h. The reaction mixture was pouredinto ice-water (200 mL), and the mixture thus obtained wasextracted with CH2Cl2 (3 × 100 mL). The combined extractswere washed with 5% brine (5 × 100 mL), dried over anhydrousNa2SO4 and evaporated on a rotary evaporator to aord aresidue, which was purifed by column chromatography to yield17a-18j after trituration with EtOAc/n-hexane if the productwas a solid.Methyl 1-(phenylthio)cyclopropanecarboxylate (17a). Yieldsare shown in Table 2. Physical properties were in agreement withthose for the sample from synthetic approach A.
methyl 1-(pyrimidin-5- yl)cyclopropane-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
LiHMDS (1 M solution in THF, 21 1 mL, 21 1 mmol) was added drop wise at -70 C to a stirred solution of methyl 2-(pyrimidin-5-yl)acetate (26.73 g, 175.7 mmol) in THF (200 mL) and the resulting mixture was stirred at this temperature for 1 hr. A solution of 1 ,3,2-dioxathiolane 2,2-dioxide (26.2 g, 21 1 mmol) in THF (200 mL) was added drop wise so as to maintain the temperature at -70 C and on complete addition, the reaction was brought slowly to -20 C and stirred for 1 .5 hr. The reaction mixture was again cooled to -72 C and an additional portion of LiHMDS (1 M in THF, 21 1 mL, 21 1 mmol) added. The reaction mixture was then allowed to warm to room temperature and stirred for 15 hrs. The reaction was quenched with saturated NH4CI solution (300 mL) and the mixture extracted with EtOAc (3 x 500 mL). The combined organic extracts were washed with brine, dried (Na2S04) and concentrated in vacuo. The crude product was purified by column chromatography eluting with pet. Ether: EtOAc (100:0 to 65:35) to afford the title compound as a yellow oil, 25.0 g, 80 %. 1H NMR (400MHz, CDCI3): delta 1 .20-1 .25 (m, 2H), 1 .67-1.74 (m, 2H), 3.63 (s, 3H), 8.69 (s, 2H), 9.03 (s, 1 H). LCMS m/z = 179 [M+H]+
100 ml of xylene, 40.5 g of ethylene glycol and 55.3 g of potassium hydroxide were placed in the reaction flask.Stir to reflux at about 130-140 C, and evaporate the solvent after 15 hours.200 ml of anhydrous dichloromethane was added and stirred to form a uniform suspension. After nitrogen replacement,The suspension was continuously added to a mixed solution of 86 g of sulfuryl chloride and 200 ml of dichloromethane.The temperature of the reaction system was kept at -10 to 10 C, and the addition was completed in about 1 hour, and the temperature was kept for 1 hour.At the end of the reaction, add 100 ml of water and stir to separate the layers.The organic phase is desolvated under reduced pressure at 40 C until the solid precipitates.Add 200 ml of toluene and 0.6 g of 18-crown-6 and stir at room temperature for 1 hour, filter and dry to obtain a white powder.37.9 g; yield 61.0%; GC content > 99.5%, potassium ion 7 ppm
benzyl N-[2-[2-(cyanomethyl)phenoxy]ethyl]-N-methylcarbamate[ No CAS ]
benzyl N-[2-[2-(1-cyanocyclopropyl)phenoxy]ethyl]-N-methylcarbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
84.6%
To benzyl N-[2-[2-(cyanomethyl)phenoxy]ethyl]-N-methyl-carbamate (2 g, 0.006 mol,1.0 equivalent) in dimethyl sulfoxide (20 ml, 10 relative volumes)The solution in the solution was added with powdered sodium hydroxide (0.59 g, 0.014 mol,2.5 equivalents), and the resulting material was stirred at 20 C to 25 C for 10 min.Then at 20 C -25 C,1,3,2-dioxathiolane-2,2-dioxide (1.09 g, 0.009 mol, 1.5 eq.) in tetrahydrofuran (4 ml, 4 relative volumes)The solution in the solution is added dropwise to the reaction substance,The resulting material was stirred for 1 h.Charge another batch of powdered sodium hydroxide (0.24 g, 0.006 mol, 1.0 eq.)The reaction mass was then stirred for an additional 15 h.The completion of the reaction was monitored by HPLC (N-[2-[2-(cyanomethyl)phenoxy]ethyl]-N-methyl-carbamate below 4%).The reaction mass was quenched with methyl-tert-butyl ether (20 mL, 10 vol.), then water (20 mL, 10 vol.) and allowed to settle.The two layers were separated and the resulting organic layer was washed with aqueous sodium chloride (5% w/v, 20 ml, 10 vol.).The organic layer was then concentrated under reduced pressure to dryness.To obtain a pale yellow oilBenzyl N-[2-[2-(1-cyanocyclopropyl)phenoxy]ethyl]-N-methyl-carbamate (1.7 g, 91.2% purity, 84.6% yield).
2-[2-[2-[benzyl(methyl)amino]ethoxy]phenyl]acetonitrile[ No CAS ]
1-[2-[2-[benzyl(methyl)amino]ethoxy]phenyl]cyclopropanenitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
75%
With sodium hydroxide; In sulfolane; dimethyl sulfoxide; at 17 - 28℃; for 16h;
A solution of 2-[2-[2-[benzyl(methyl)amino]ethoxy]phenyl]acetonitrile in DMSO (138.9 g, 10.8% w/w, 15.0g at 100% w/w, 0.49 moles, 1.0 eqv) was further diluted with DMSO (60 mL), followed by addition of sodium hydroxide powder (5.35 g, 2.5 eqv). The resulting suspension was stirred before a solution of ethylene sulphate (8.66g, 1.3 eqv) in sulfolane (37.5 mL, 2.5 rel vol) was added dropwise over a period of 2 h at 20-25 C. The mixture was stirred and further sodium hydroxide powder was added (2.2 lg, 1.0 mol eqv). The mixture was stirred at ambient temperature for 16 h, followed by addition of MTBE (135mL, 9.0 rel vol) and water (150mL, 10.0 rel vol). The two-phase mixture was then allowed to settle and the lower aqueous phase was separated and re-extracted with more MTBE. The combined MTBE extracts were washed twice with 10%> w/v aqueous sodium chloride solution. l-[2-[2- [benzyl(methyl)amino) ethoxy]phenyl]cyclopropanecarbonitrile was isolated as a 6.9% w/w solution in methyl tert-butyl ether (12.3 g at 100% strength) with 75% yield.
ethyl 2-(6-(8-fluoronaphthalen-2-yl)-2-methoxypyridin-3-yl)acetate[ No CAS ]
ethyl 1-(6-(8-fluoronaphthalen-2-yl)-2-methoxypyridin-3-yl)cyclopropane carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Into a 40-mL vial maintained with an inert atmosphere of nitrogen, were placed THF (5 mL). To the mixture were then added LDA (0.33 mL, 0.64 mmol, 2.2 equiv.), and ethyl 2-(6-(8-fluoronaphthalen-2-yl)-2-methoxypyridin-3-yl)acetate (100 mg, 0.29 mmol, 1 equiv.), with stirring at 0 C. The mixture was stirred at 0 C. for 20 min. Ethylene sulfate (40 mg, 0.32 mmol, 1.1 equiv.) was added and the resulting solution was stirred 16 h at 30 C. The mixture was quenched by the addition of H2O, extracted with EA, the organic layers was concentrated under vacuum. The residue was purified by silica cal column with ethyl acetate/petroleum ether (30:70) to ethyl 1-(6-(8-fluoronaphthalen-2-yl)-2-methoxypyridin-3-yl)cyclopropanecarboxylate as yellow oil. Mass spectrum (ESI, m/z): Calculated for C22H20FNO3, 366.1 (M+H), found 365.8.
Into a 40-mL vial maintained with an inert atmosphere of nitrogen, were placed THF (5 mL). To the mixture were then added LDA (0.33 mL, 0.64 mmol, 2.2 equiv.), and ethyl 2-(6-(8-fluoronaphthalen-2-yl)-2-methoxypyridin-3-yl)acetate (100 mg, 0.29 mmol, 1 equiv.), with stirring at 0 C. The mixture was stirred at 0 C. for 20 min. Ethylene sulfate (40 mg, 0.32 mmol, 1.1 equiv.) was added and the resulting solution was stirred 16 h at 30 C. The mixture was quenched by the addition of H2O, extracted with EA, the organic layers was concentrated under vacuum. The residue was purified by silica cal column with ethyl acetate/petroleum ether (30:70) to ethyl 1-(6-(8-fluoronaphthalen-2-yl)-2-methoxypyridin-3-yl)cyclopropanecarboxylate as yellow oil. Mass spectrum (ESI, m/z): Calculated for C22H20FNO3, 366.1 (M+H), found 365.8.
Into a 40-mL vial maintained with an inert atmosphere of nitrogen, were placed THF (5 mL). To the mixture were then added LDA (0.33 mL, 0.64 mmol, 2.2 equiv.), and ethyl 2-(6-(8-fluoronaphthalen-2-yl)-2-methoxypyridin-3-yl)acetate (100 mg, 0.29 mmol, 1 equiv.), with stirring at 0 C. The mixture was stirred at 0 C. for 20 min. Ethylene sulfate (40 mg, 0.32 mmol, 1.1 equiv.) was added and the resulting solution was stirred 16 h at 30 C. The mixture was quenched by the addition of H2O, extracted with EA, the organic layers was concentrated under vacuum. The residue was purified by silica cal column with ethyl acetate/petroleum ether (30:70) to ethyl 1-(6-(8-fluoronaphthalen-2-yl)-2-methoxypyridin-3-yl)cyclopropanecarboxylate as yellow oil. Mass spectrum (ESI, m/z): Calculated for C22H20FNO3, 366.1 (M+H), found 365.8.
2-(4-chloro-1-(phenylsulfonyl)-1H-pyrrolo[2,3-b]pyridin-2-yl)ethan-1-ol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
64%
A stirred solution of 1-(benzenesulfonyl)-4-chloro-pyrrolo[2,3-b]pyridine (4.39 g, 15.0 mmol)(which can be prepared as described in U52014/200227) in dry THF (42 mL) at -78 00 wastreated dropwise with a freshly prepared solution of LDA (18.0 mL, 1.OM in THF/hexane,18.0 mmol) over 10 mm. The resulting orange solution was stirred at -78 00 for 1 h. Then1 ,3,2-dioxathiolane 2,2-dioxide (2.79 g, 22.5 mmol) was added in one portion at -78 00. The resulting solution was stirred at -78 00 for 2 h, then warmed to room temperature over 30 mm and stirred for an additional 1 h. The reaction was quenched by the addition of water, then 12M HCI was added and the resulting solution stirred at 30 00 overnight. The solution was concentrated under reduced pressure, then slowly diluted with a saturated aqueoussolution of NaHCO3 until pH = 9. The aqueous phase was extracted with EtOAc and. the combined organic layers were washed with brine, dried (Mg504), and concentrated in vacuo. The residue was purified by FCC eluting with 30-50% EtOAc in petrol to give the title compound (3.22 g, 64%) as a pale yellow solid.LCMS (Method 4): Rt 2.63 mi mlz 337 I 339 [MH].
A solution of lithium diisopropylamide (1M in THF) (13.00 mL, 26.0 mmol) was added to tetrahydrofuran (THF) (25 mL) in the reaction flask at -78 C.,Stir for 5 minutes. Methyl butyrate (2.84 mL,25 mmol) in THF (12.5 mL),Add slowly at -78 C,Stir for 30 minutes.1,3,2-dioxathiolane 2,2-dioxide (3.23 g, 26.0 mmol) was added slowly in THF (12.5 mL),The reaction was then slowly warmed to room temperature over 2 hours.The reaction was diluted with methanol (15 mL) and then evaporated to remove volatiles.The crude residue was dissolved in 20% aqueous H2SO4 (6 mL) and toluene (50 mL),The biphasic mixture was then heated to reflux with vigorous stirring for 6 hours. The organic phase is separated,The aqueous phase was extracted with ethyl acetate (3 × 50 mL). Combine the combined organic phases with sodium bicarbonate,Washed with brine, dried over MgSO4,Concentration to dryness gave a crude residue.The residue was purified by silica gel chromatography eluting with 0-50% ethyl acetate / heptane to give the product (1.3 g, 46% yield).
2-[4-(thiadiazol-4-yl)pyridin-1-ium-1-yl]ethyl sulfate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In 1,2-dichloro-ethane at 85℃; for 18h;
7 Example 7: Preparation of 2-[4-(thiadiazol-4-yl')pyridin-1 -ium-1 -yl1ethyl sulfate A65
A mixture of 4-(4-pyridyl)thiadiazole (0.1 g), 1 ,3, 2-dioxathiolane 2,2-dioxide (0.087 g) and 1 ,2- dichloroethane (6 ml_) was heated at 85°C for 18 hours. The resulting precipitate was filtered off and air dried to give 2-[4-(thiadiazol-4-yl)pyridin-1 -ium-1 -yl]ethyl sulfate as a white solid. NMR (400 MHz, DMSO-d6) 10.23 - 10.36 (m, 1 H), 9.09 - 9.24 (m, 2H), 8.73 - 8.96 (m, 2H), 4.73 - 4.94 (m, 2H), 4.18 - 4.34 (m, 2H)
Stage #1: ethyl 2-(1H-imidazol-1-yl)acetate With lithium hexamethyldisilazane In tetrahydrofuran at -78℃; for 0.666667h;
Stage #2: ethyleneglycol sulfate In tetrahydrofuran at -78℃; for 0.333333h;






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






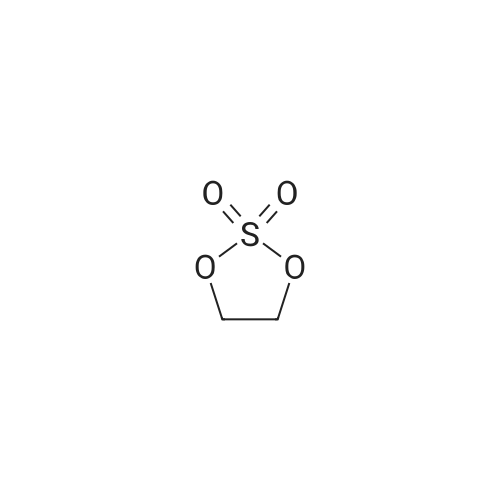

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping