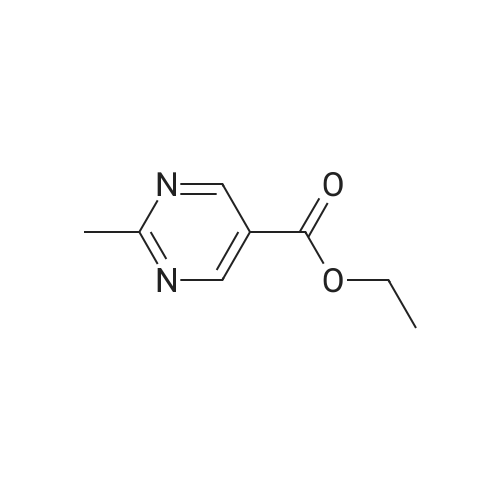
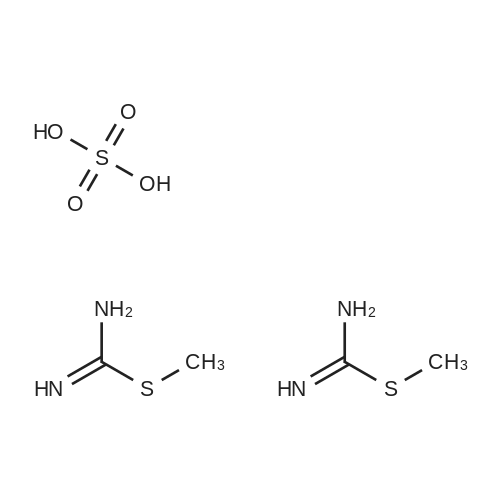
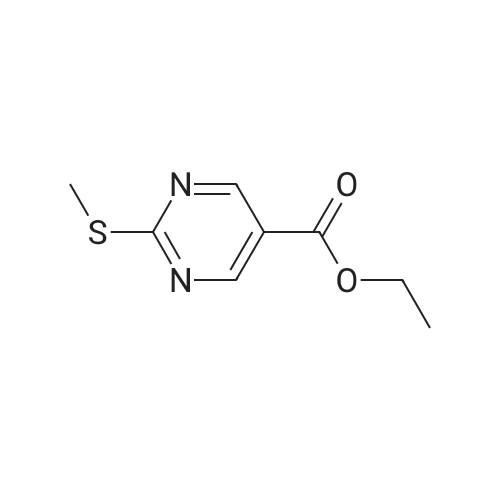
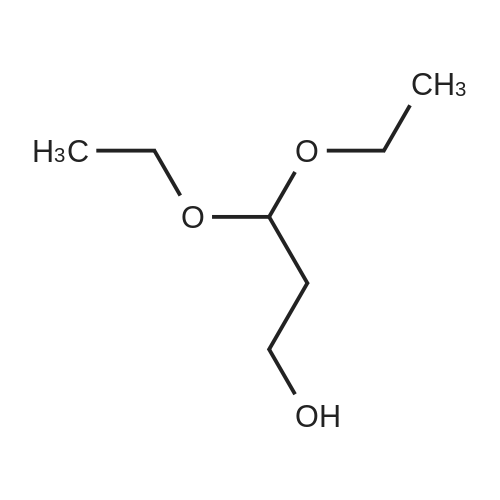
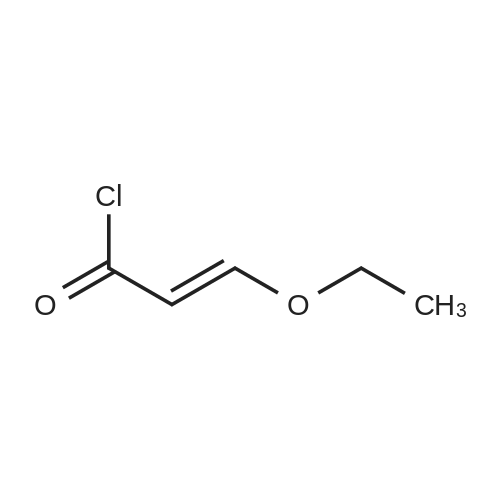
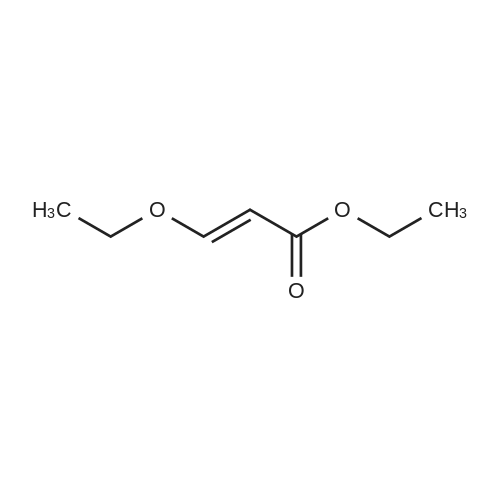
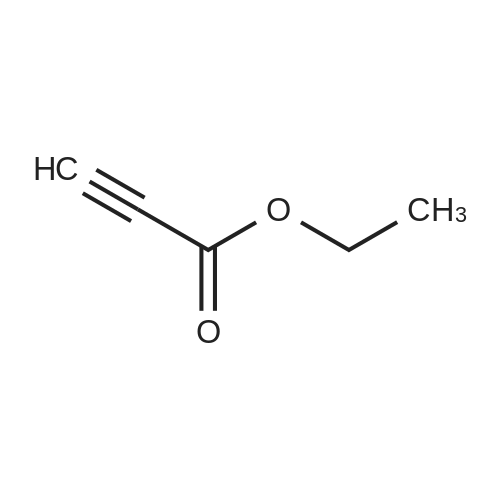
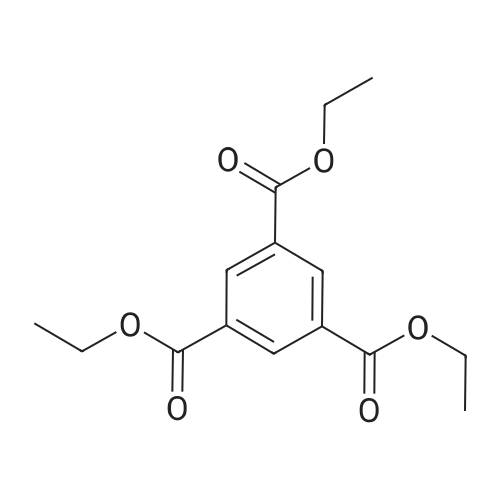
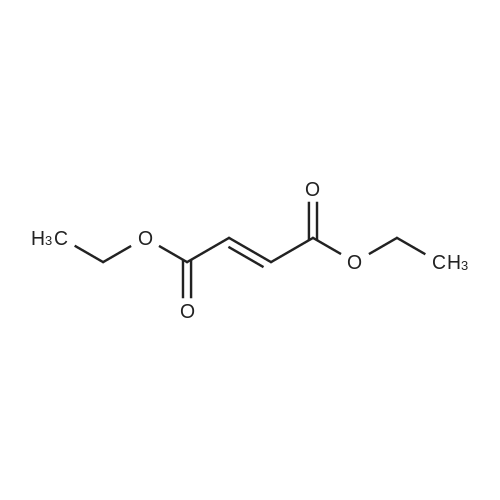
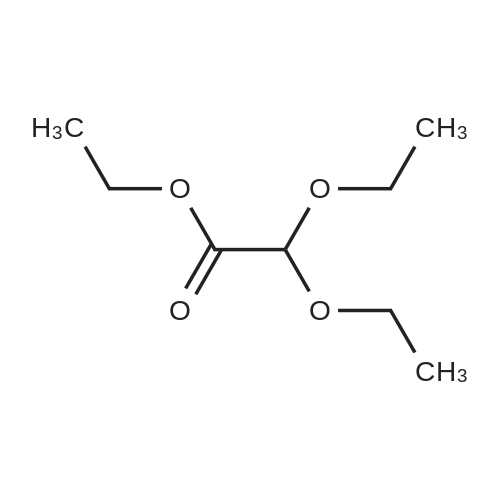
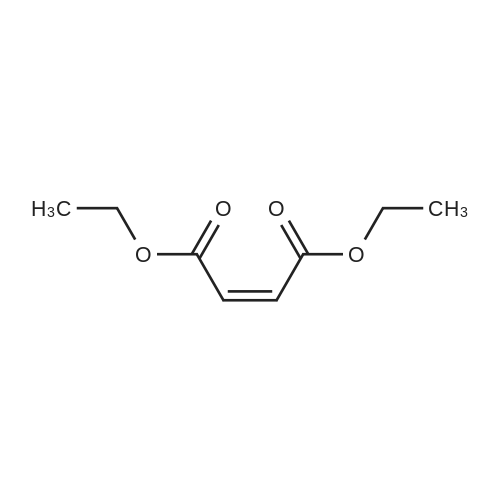
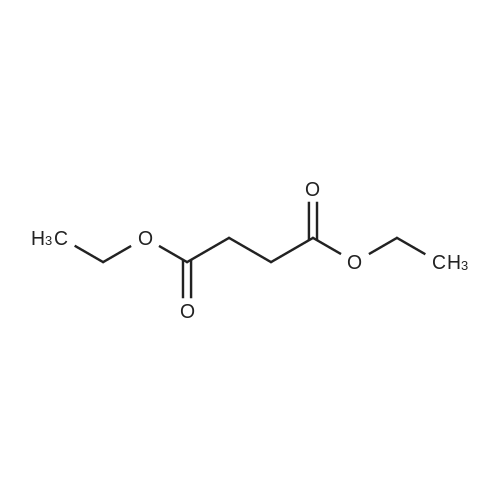
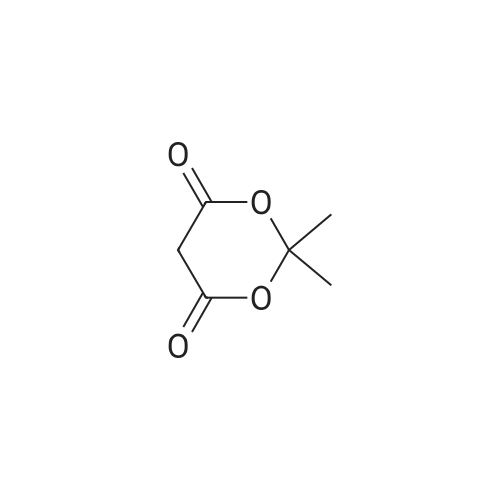
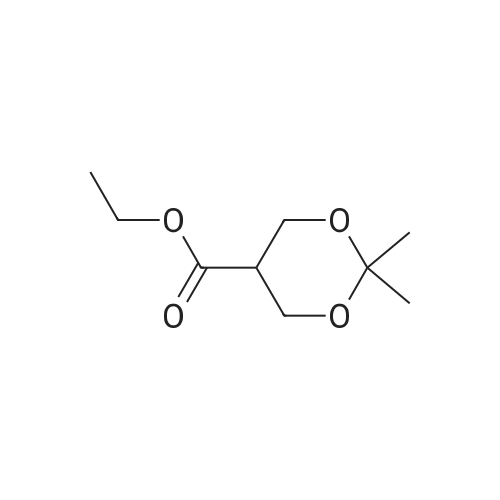
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
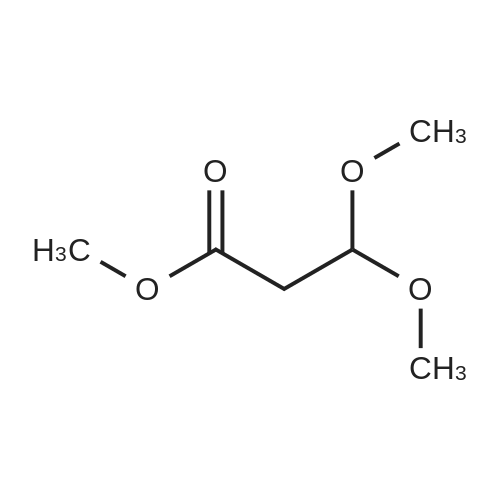
Stage #1: With lithium aluminium tetrahydride In tetrahydrofuran at 10 - 20℃; for 1 h; Stage #2: With water In tetrahydrofuran for 2 h;
In the following, tetrahydrofuran (THF) to be used was dehydrated on molecular sieves of 4 angstrom overnight. Inside of a 5 L four-neck flask was substituted with nitrogen gas and 2.2 kg of THF was placed therein, followed by cooling to 10° C. or lower. Then, 50 g of lithium aluminum hydride was placed therein and 250 g of ethyl 3,3-diethoxypropionate was added dropwise so that inner temperature did not exceed 10° C. After completion of the dropwise addition, temperature was elevated to 20° C. and reaction was carried out for 1 hour. After the reaction, 420 g of distilled water was added and the whole was stirred for 2 hours, then filtration was performed through No. 2 filtration paper, and subsequently a cake was washed with 1.1 kg of THF. The filtrate was concentrated at 40° C. to obtain 213 g of crude 3,3-diethoxy-1-propanol (hereinafter 33DEP).Then, 200 g of crude 33DEP was placed in a 5 L four-neck flask and 1 kg of hexane was added thereto, followed by stirring for 1 hour (step (A)). After 1 kg of a 0.1M phosphate buffer (pH 7.5) was added and the whole was stirred for 20 minutes, it was transferred to a 5 L separatory funnel and allowed to stand until it was separated into layers. After an aqueous layer was taken out, a hexane layer was returned to the 5 L four-neck flask and 1 kg of the 0.1M phosphate buffer (pH 7.5) was again added. After stirring for 20 minutes, the whole was again transferred to the 5 L separatory funnel and allowed to stand until it was separated into layers and then an aqueous layer was recovered. The aqueous layer was recovered into the 5 L four-neck flask (step (B)). Then, 2 kg of chloroform was added thereto and, after stirring for 30 minutes, the whole was transferred to the 5 L separatory funnel and allowed to stand for 15 minutes to separate it into layers. A chloroform layer was recovered and an aqueous layer was returned to the 5 L four-neck flask and the same operations were performed with 2 kg of chloroform to recover a chloroform layer (step (C)). Then, the recovered chloroform layer was concentrated at 40° C. using an evaporator and, after evaporation ceased, the solvent was evaporated by bubbling with a minute amount of nitrogen. Thereafter, the product was stirred with 20 g of molecular sieves of 4 angstrom for 5 hours and then filtration was performed to obtain 188 g of 33DEP.Then, 150 g of 33DEP obtained by the above operations was placed in a 300 ml four-neck flask and distillation was started under reduced pressure. A first fraction begun to be distilled at a degree of vacuum of 0.4 kPa, a bath temperature of 38° C., an inner temperature of 36° C., and a column top temperature of 33° C. After the distillation ceased, the bath temperature was gradually elevated and a main fraction begun to be distilled at a degree of vacuum of 0.2 kPa, a bath temperature of 58° C., an inner temperature of 55° C., and a column top temperature of 54° C. Thereafter, distillation was performed so that the column top temperature did not exceed 60° C. to obtain 138 g of the main fraction (step (D)). GC analysis was performed for the obtained main fraction. Results are shown in Table 1.
Reference:
[1] Patent: US2012/277477, 2012, A1, . Location in patent: Page/Page column 5
[2] Acta Chemica Scandinavica (1947-1973), 1973, vol. 27, p. 239 - 250
[3] Patent: US4634773, 1987, A,
Reference:
[1] Journal of Chemical Research, Miniprint, 1985, # 3, p. 987 - 996
[2] Journal of Chemical Research, Miniprint, 1985, # 3, p. 987 - 996
[3] Journal of Chemical Research, Miniprint, 1985, # 3, p. 987 - 996
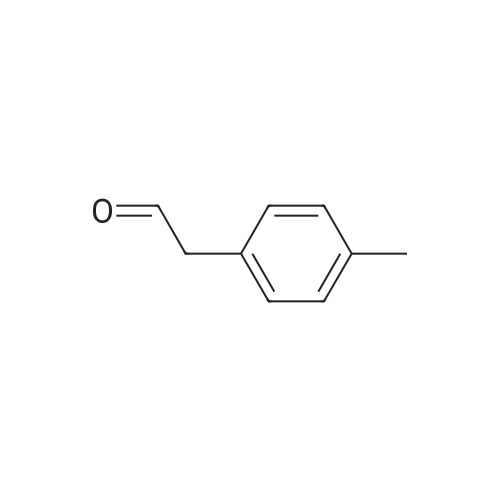
11
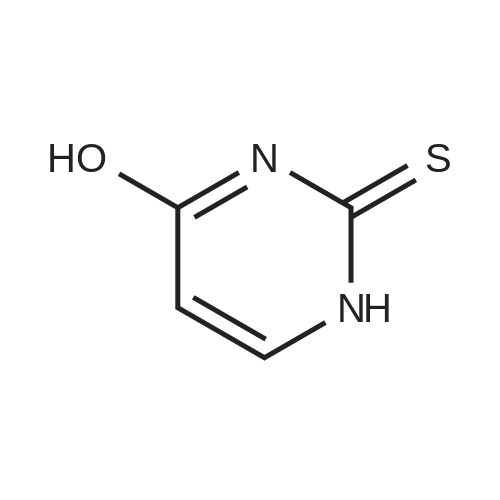
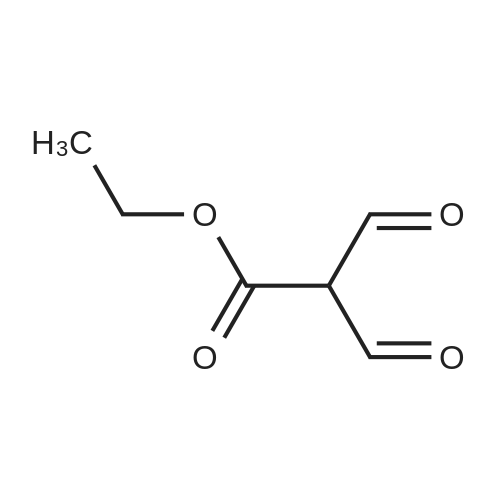
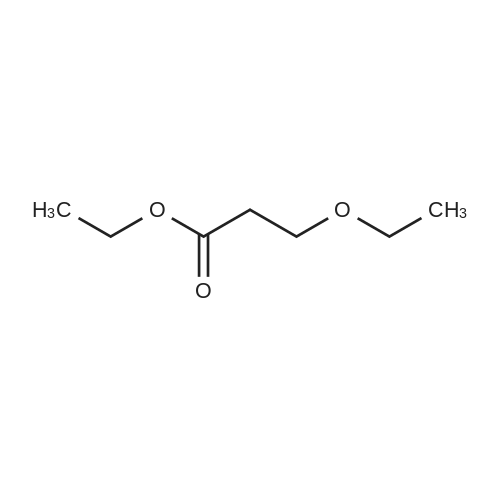
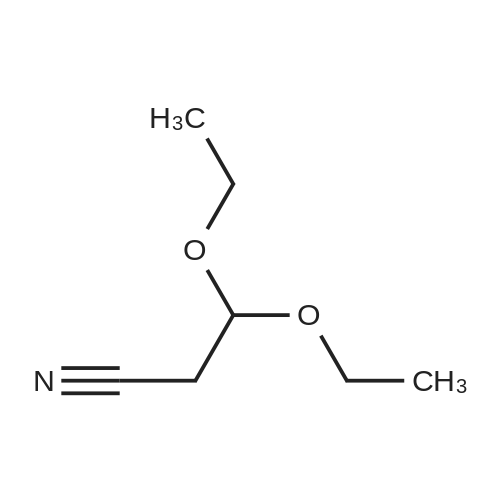
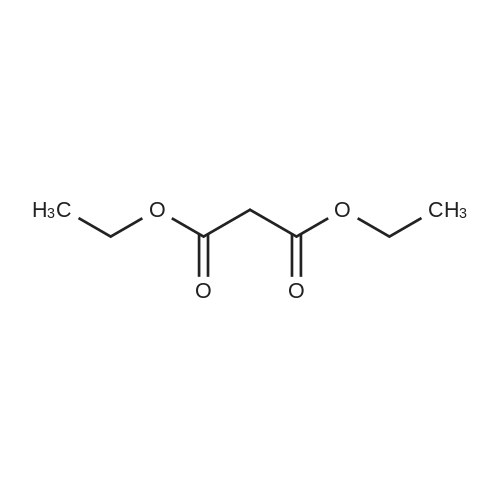
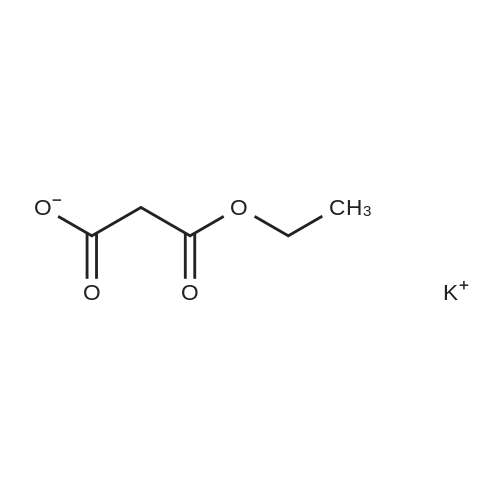
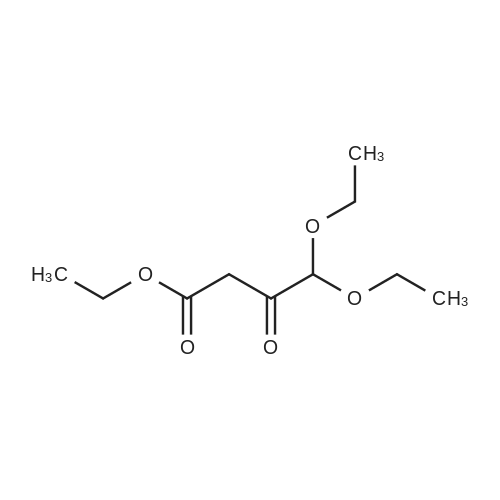
[ 10601-80-6 ]
[ 109-94-4 ]
[ 80370-42-9 ]
Yield
Reaction Conditions
Operation in experiment
100%
With sodium hydride In diethyl ether at 0 - 20℃; for 15 h;
12.6 g of sodium hydride (purity: 60percent, 525.0 mmoles) was washed with diethyl ether by decantation several times and then made into a solution in 500 ml of diethyl ether. Thereto were added, in a nitrogen current at 0 to 10°C, 194 g (2.6 moles) of ethyl formate and 50 g (262.0 mmoles) of ethyl 3,3-diethoxy-propionate. The resulting mixture was stirred at room temperature for 15 hours to give rise to a reaction. After confirmation of the completion of the reaction, the reaction mixture was poured into water, followed by washing with diethyl ether. The resulting aqueous layer was allowed to have a pH of 1 with hydrochloric acid, followed by extraction with dichloromethane. The resulting organic layer was washed with an aqueous sodium chloride solution and then dried over anhydrous magnesium sulfate. The resulting solution was subjected to vacuum distillation to remove the solvent contained therein, to obtain 37.6 g (yield: 100percent) of crude (ethoxycarbonyl)malondialdehyde as a dark red oily substance. 1H-NMR [CDCl3/TMS, δ (ppm)]:
With sodium hydride In diethyl ether at 0 - 20℃; for 15 h;
REFERENCE EXAMPLE 28 Production of (ethoxycarbonyl)malondialdehyde; 12.6 g of sodium hydride (purity: 60percent, 525.0 mmoles) was washed with diethyl ether by decantation several times and then made into a solution in 500 ml of diethyl ether. Thereto were added, in a nitrogen current at 0 to 10° C., 194 g (2.6 moles) of ethyl formate and 50 g (262.0 mmoles) of ethyl 3,3-diethoxy-propionate. The resulting mixture was stirred at room temperature for 15 hours to give rise to a reaction. After confirmation of the completion of the reaction, the reaction mixture was poured into water, followed by washing with diethyl ether. The resulting aqueous layer was allowed to have a pH of 1 with hydrochloric acid, followed by extraction with dichloromethane. The resulting organic layer was washed with an aqueous sodium chloride solution and then dried over anhydrous magnesium sulfate. The resulting solution was subjected to vacuum distillation to remove the solvent contained therein, to obtain 37.6 g (yield: 100percent) of crude (ethoxycarbonyl)malondialdehyde as a dark red oily substance. 1H-NMR [CDCl3/TMS, δ (ppm)]: 9.09 (2H,s), 5.26 (1H,s), 4.27 (2H,q), 1.28 (3H,t)
99.7%
With sodium hydride In tetrahydrofuran at 0 - 20℃;
[00442] To a stirred suspension of sodium hydride (60percent, 1.68 g, 42.1 mmol) in THF (20 mL) was added ethyl formate (8.5 mL, 105.7 mmol). The solution was cooled to 0 °C and a solution of ethyl 3,3-diethoxypropanoate (4 g, 21.0 mmol) in THF (10 mL) was added dropwise over 30 mins and the reaction mixture stirred at r.t. overnight. 2M aq HCl (30 mL) was added under whilst cooling with ice and the reaction stirred at r.t. for 30 mins. The reaction mixture was extracted with diethyl ether (2 x 50 mL) and the combined organic extracts were dried over MgS04 and concentrated in vacuo to afford the title compound (3.02 g, 99.7percent) as an amber liquid. [00443] 1H NMR (250 MHz, Chloroform-d) δ 9.13 (s, 2H), 4.29 (q, J = 7.1 Hz, 2H), 1.37 - 1.28 (m, 3H).
74.2%
With sodium hydride In diethyl ether; hexane at 20℃; for 15 h; Cooling with ice
Weighed out sodium hydride (2.46 g, 61.5 mmol) in a dry 100-mL pear flask. Washed with hexanes then with diethyl ether. Suspended in ether (100 mL), cooled in ice bath then ethyl formate (24.84 ml, 308 mmol) was added then ethyl 3,3-diethoxypropanoate (5.98 ml, 30.8 mmol). The resulting mixture was stirred at room temperature for 15 hours to give rise to a reaction. After confirmation of the completion of the reaction, the reaction mixture was poured into water, followed by washing with diethyl ether. The resulting aqueous layer was allowed to have a pH of 1 with hydrochloric acid, followed by extraction with dichloromethane. The resulting organic layer was washed with an aqueous sodium chloride solution and then dried over anhydrous magnesium sulfate, filtered and concentrated to give ethyl 2-formyl-3-oxopropanoate (3.29 g, 22.83 mmol, 74.2 percent yield) as a golden syrup
70%
Stage #1: With sodium hydride In tetrahydrofuran at 0 - 20℃; Stage #2: With hydrogenchloride In water
EXAMPLE 1Preparation of Ethyl-2-formyl-3-oxopropionate[0059] A three- or four-neck round bottom flask equipped with magnetic stir bar, thermocouple, digital thermometer, gas inlet and outlet and addition funnel was flushed <n="19"/>with argon. Ethyl 3,3-diethoxypropionate (64.5 g) in tetrahydrofuran were charged to the addition funnel. Sodium hydride (21.2 g of a 60percent dispersion) was charged to the reaction flask followed by tetrahydrofuran. The contents of the flask were cooled to 0- 5°C in an ice-bath, and ethyl formate (257 g) was added. The mixture was cooled to 0- 50C and the contents of the addition funnel added drop wise, maintaining an internal temperature of less than 5°C. The ice-bath was removed and the contents allowed to warm to ambient temperature. Consumption of ethyl 3,3-diethoxypropionate was monitored by TLC analysis. The reaction was quenched by addition of ice-water (10.6 vol), and extracted three times with methyl t-butyl ether (5.4 vol each), and the organic layers discarded. The aqueous phase was acidified with cone, hydrochloric acid to a pH of 1 to 1.5. The acidified aqueous layer was extracted three times with dichloromethane and the combined organic layers dried over sodium sulfate. The solvent was removed under reduced pressure, and the residue distilled under vacuum, to provide ethyl 2-formyl-3-oxopropionate, 27.92 g, 70percent yield.
70%
With sodium hydride In tetrahydrofuran at 0 - 20℃;
[0066] A three- or four-neck round bottom flask equipped with magnetic stir bar, thermocouple, digital thermometer, gas inlet and outlet and addition funnel was flushed with argon. Ethyl 3,3- diethoxypropionate (64.5 g) in tetrahydrofuran were charged to the addition funnel. Sodium hydride (21.2 g of a 60percent dispersion) was charged to the reaction flask followed by tetrahydrofuran. The contents of the flask were cooled to 0-50C in an ice-bath, and ethyl formate (257 g) was added. The mixture was cooled to 0-50C and the contents of the addition funnel added dropwise, maintaining an internal temperature of less than 5°C. The ice-bath was removed and the contents allowed to warm to ambient temperature. Consumption of ethyl 3,3- diethoxypropionate was monitored by TLC analysis. The reaction was quenched by addition of ice-water (10.6 vol), and extracted three times with methyl t-butyl ether (5.4 vol each), and the organic layers discarded. The aqueous phase was acidified with cone, hydrochloric acid to a pH of 1 to 1.5. The acidified aqueous layer was extracted three times with dichloromethane and the combined organic layers dried over sodium sulfate. The solvent was removed under reduced pressure, and the residue distilled under vacuum, to provide ethyl 2-formyl-3 -oxopropionate, 27.92 g, 70percent yield.
59%
With sodium hydride In tetrahydrofuran at 0 - 30℃; for 24 h;
4) In a 500 ml four-necked flask equipped with magnetic stirring, 160 ml of tetrahydrofuran and9.84g NaH, mass fraction of NaH in tetrahydrofuran solution is 60percent, the solution is cooled in an ice bath to 0 °C to 5 °C, and then 119g of ethyl formate is added to slowly increase the temperature, when the temperature stabilizes to 0 °C At 5 °C, 80 ml of a tetrahydrofuran solution containing D was continuously added dropwise into a 500 ml four-necked flask, wherein the mass fraction of D was 5percent. After the dropwise addition, the water bath was removed and heated to room temperature. At this time, a large amount of hydrogen was evolved. It will first rise to 30 °C and then gradually decrease to 25 °C. After 24 hours of reaction, the reaction solution was poured into ice water and quenched. 5) The resulting product is first extracted with 500 ml of tert-butyl methyl ether three times to remove the organic phase, and then with concentrated hydrochloric acid to adjust the pH of the aqueous phase to 1~1.5; then extract the aqueous phase with 300 ml of dichloromethane, twice The combined organic phases were combined; the combined organic phases were then dried over anhydrous sodium sulfate and evaporated to give a brown-red liquid which was evaporated in vacuo. GC yield was 99.3percent. The yield was 85.9percent. Finally, the 9 mm Hg diaphragm was distilled under reduced pressure. The colorless transparent liquid E, ethyl 2-formyl-3-oxopropanoate, was obtained in a yield of 59percent.
Reference:
[1] Patent: EP1364946, 2003, A1, . Location in patent: Page/Page column 196
[2] Patent: US2005/256004, 2005, A1, . Location in patent: Page/Page column 31-32
[3] Patent: WO2017/59191, 2017, A1, . Location in patent: Paragraph 00441-00443
[4] Journal of Organic Chemistry, 1982, vol. 47, # 11, p. 2216 - 2217
[5] Tetrahedron, 2008, vol. 64, # 33, p. 7745 - 7758
[6] Patent: WO2014/160203, 2014, A2, . Location in patent: Page/Page column 67
[7] Patent: WO2007/92372, 2007, A1, . Location in patent: Page/Page column 17-18
[8] Patent: WO2008/143667, 2008, A1, . Location in patent: Page/Page column 20
[9] Patent: CN106928060, 2017, A, . Location in patent: Paragraph 0007; 0022; 0023
[10] Patent: US2007/225280, 2007, A1, . Location in patent: Page/Page column 30-31
[11] European Journal of Medicinal Chemistry, 2017, vol. 137, p. 96 - 107
Reference:
[1] European Journal of Organic Chemistry, 2014, vol. 2014, # 24, p. 5281 - 5301
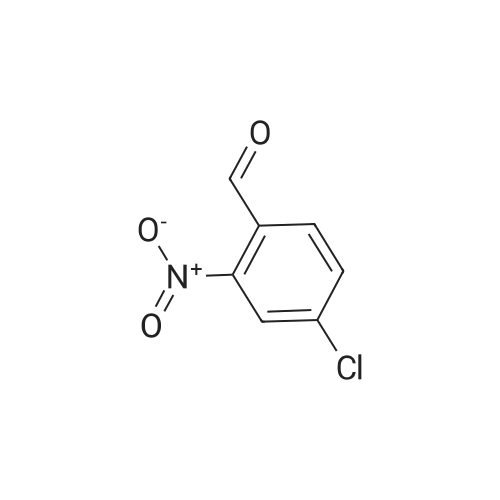
14
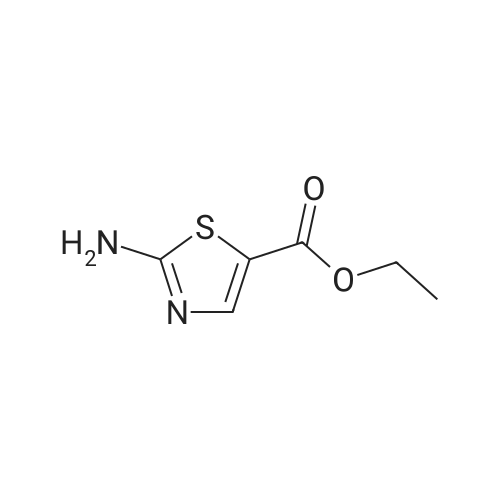
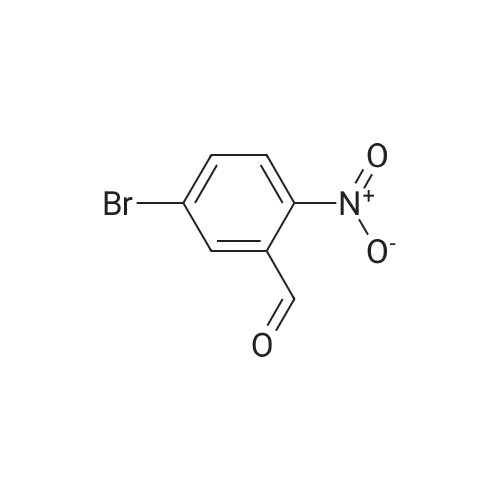
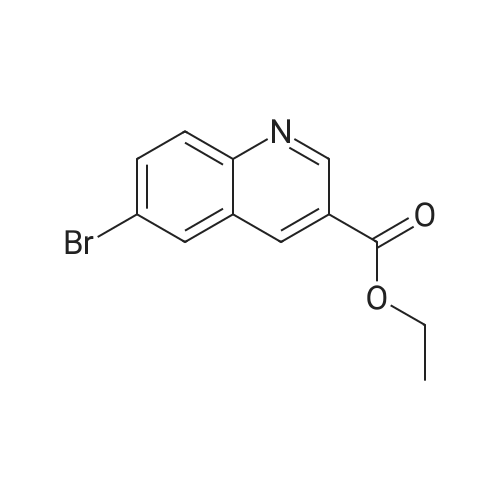
[ 10601-80-6 ]
[ 20357-20-4 ]
[ 481054-89-1 ]
Yield
Reaction Conditions
Operation in experiment
60%
With tin(II) chloride dihdyrate In ethanol at 20 - 90℃;
Step 1: ethyl 6-bromoquinoline-3-carboxylate To a solution of 5-bromo-2-nitrobenzaldehyde (2 g, 9 mmol) in ethanol (46 mL) was added tin(II) chloride dihydrate (7.95 g, 35.2 mmol) and 3,3-diethoxypropionic acid ethyl ester (4.2 mL, 22 mmol). The reaction was heated to 90° C. for 16 hours. The reaction was then allowed to cool to room temperature and stir overnight. The reaction mixture was concentrated and the residue was dissolved in ethyl acetate. The mixture was poured into saturated aqueous sodium bicarbonate. The resulting emulsion was filtered through Celite, rinsing with ethyl acetate. The layers were separated and the aqueous was extracted with ethyl acetate. The combined organics were washed with brine, dried over magnesium sulfate, filtered, and concentrated. Purification by flash column chromatography (0-50percent ethyl acetate/heptanes) gave the title compound (1.41 g, 60percent) as a solid.
Reference:
[1] Journal of Organic Chemistry, 2010, vol. 75, # 10, p. 3488 - 3491
[2] Patent: US2012/270893, 2012, A1, . Location in patent: Page/Page column 32
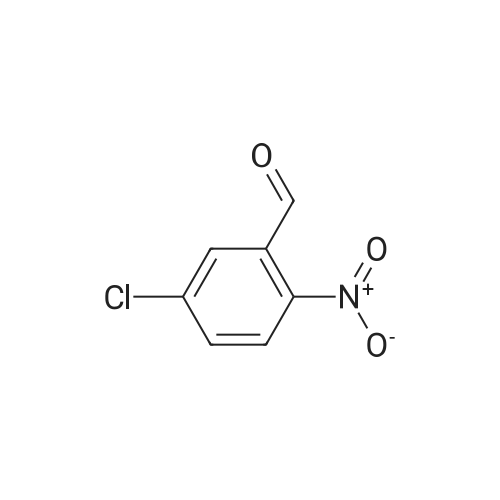
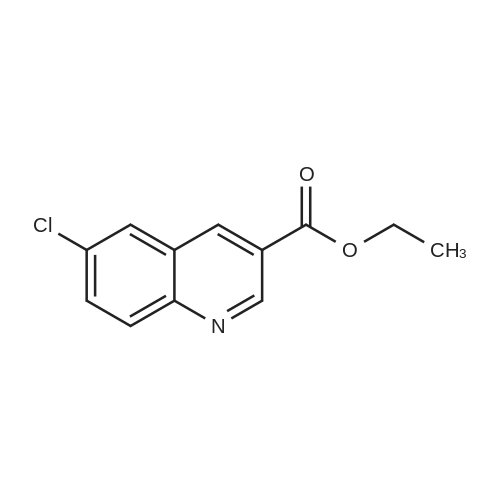
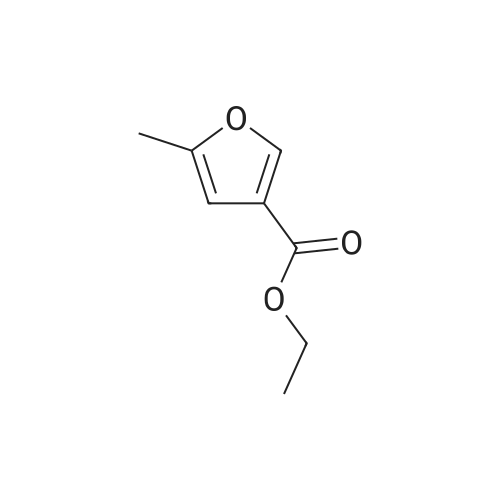
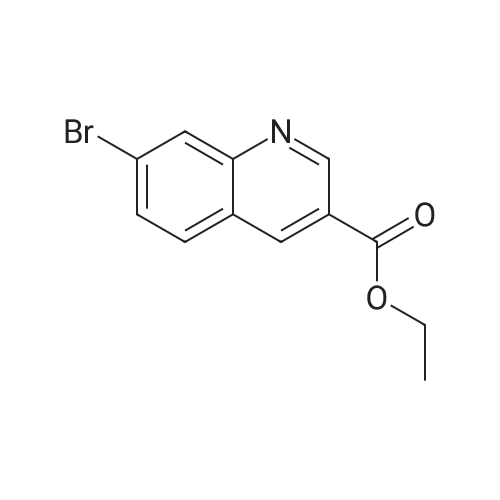
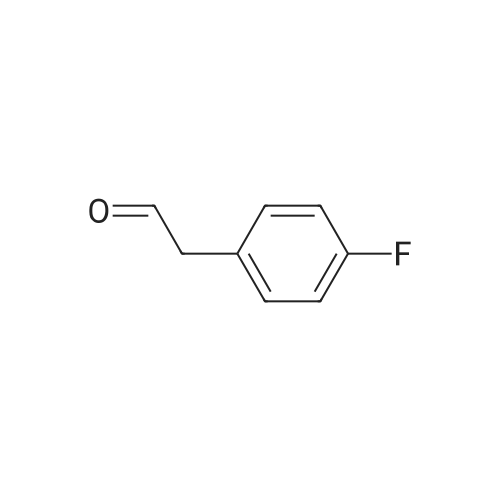
15
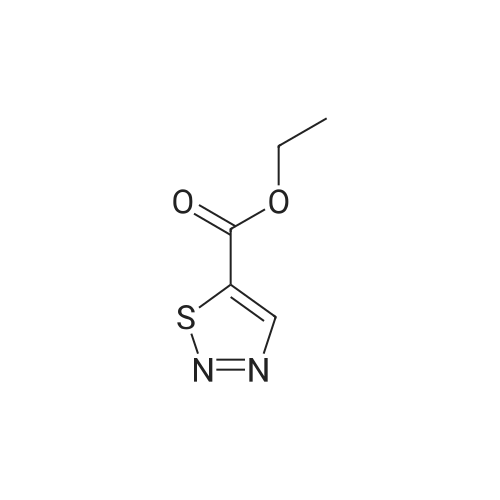
[ 10601-80-6 ]
[ 6628-86-0 ]
[ 375854-57-2 ]
Reference:
[1] Journal of Organic Chemistry, 2010, vol. 75, # 10, p. 3488 - 3491
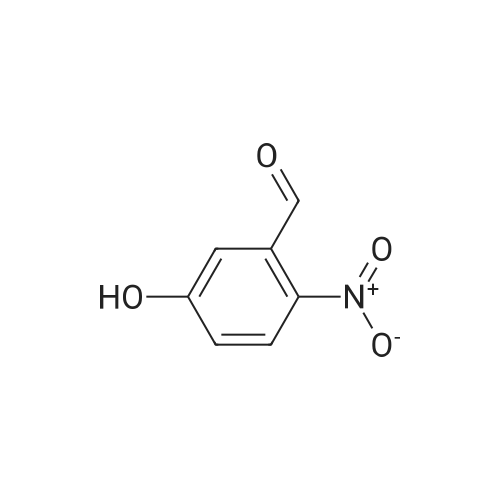
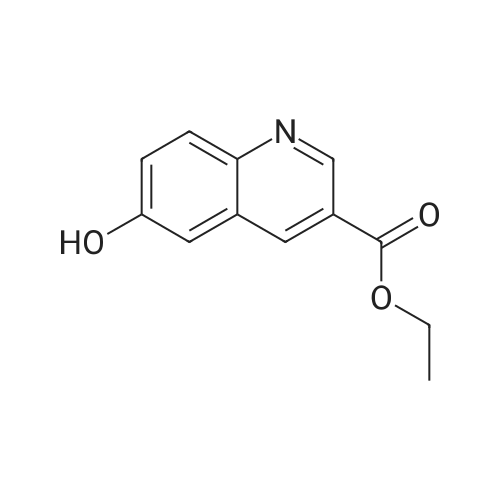
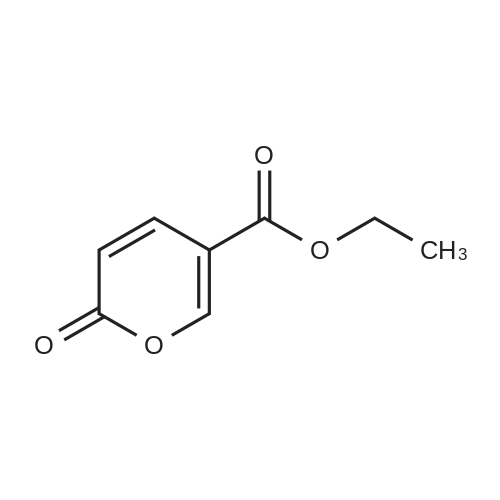
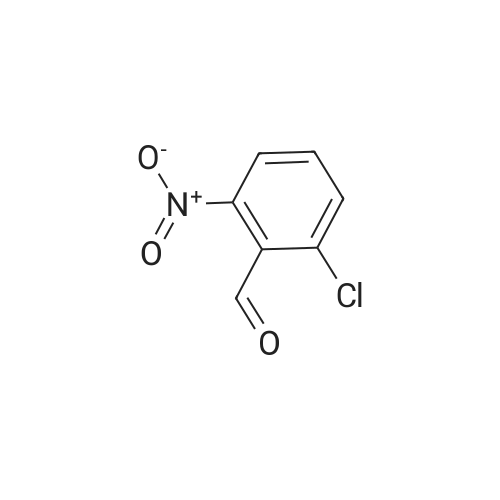
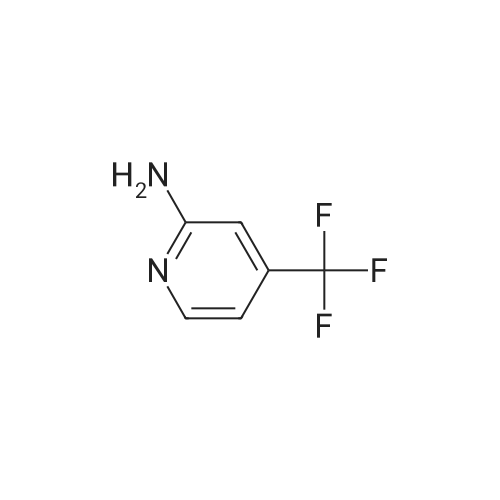
16
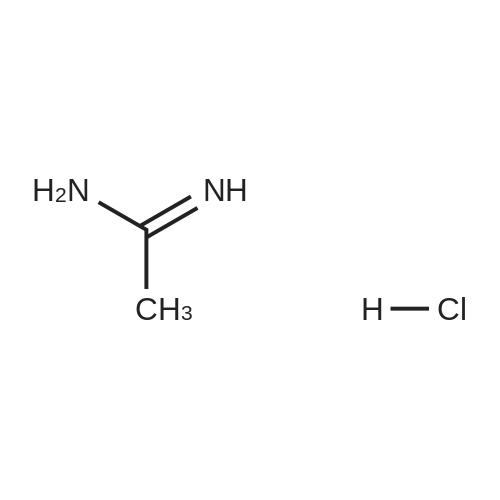
[ 10601-80-6 ]
[ 42454-06-8 ]
[ 6972-86-7 ]
Reference:
[1] Journal of Organic Chemistry, 2010, vol. 75, # 10, p. 3488 - 3491
In the following, tetrahydrofuran (THF) to be used was dehydrated on molecular sieves of 4 angstrom overnight. Inside of a 5 L four-neck flask was substituted with nitrogen gas and 2.2 kg of THF was placed therein, followed by cooling to 10 C. or lower. Then, 50 g of lithium aluminum hydride was placed therein and 250 g of ethyl 3,3-diethoxypropionate was added dropwise so that inner temperature did not exceed 10 C. After completion of the dropwise addition, temperature was elevated to 20 C. and reaction was carried out for 1 hour. After the reaction, 420 g of distilled water was added and the whole was stirred for 2 hours, then filtration was performed through No. 2 filtration paper, and subsequently a cake was washed with 1.1 kg of THF. The filtrate was concentrated at 40 C. to obtain 213 g of crude 3,3-diethoxy-1-propanol (hereinafter 33DEP).Then, 200 g of crude 33DEP was placed in a 5 L four-neck flask and 1 kg of hexane was added thereto, followed by stirring for 1 hour (step (A)). After 1 kg of a 0.1M phosphate buffer (pH 7.5) was added and the whole was stirred for 20 minutes, it was transferred to a 5 L separatory funnel and allowed to stand until it was separated into layers. After an aqueous layer was taken out, a hexane layer was returned to the 5 L four-neck flask and 1 kg of the 0.1M phosphate buffer (pH 7.5) was again added. After stirring for 20 minutes, the whole was again transferred to the 5 L separatory funnel and allowed to stand until it was separated into layers and then an aqueous layer was recovered. The aqueous layer was recovered into the 5 L four-neck flask (step (B)). Then, 2 kg of chloroform was added thereto and, after stirring for 30 minutes, the whole was transferred to the 5 L separatory funnel and allowed to stand for 15 minutes to separate it into layers. A chloroform layer was recovered and an aqueous layer was returned to the 5 L four-neck flask and the same operations were performed with 2 kg of chloroform to recover a chloroform layer (step (C)). Then, the recovered chloroform layer was concentrated at 40 C. using an evaporator and, after evaporation ceased, the solvent was evaporated by bubbling with a minute amount of nitrogen. Thereafter, the product was stirred with 20 g of molecular sieves of 4 angstrom for 5 hours and then filtration was performed to obtain 188 g of 33DEP.Then, 150 g of 33DEP obtained by the above operations was placed in a 300 ml four-neck flask and distillation was started under reduced pressure. A first fraction begun to be distilled at a degree of vacuum of 0.4 kPa, a bath temperature of 38 C., an inner temperature of 36 C., and a column top temperature of 33 C. After the distillation ceased, the bath temperature was gradually elevated and a main fraction begun to be distilled at a degree of vacuum of 0.2 kPa, a bath temperature of 58 C., an inner temperature of 55 C., and a column top temperature of 54 C. Thereafter, distillation was performed so that the column top temperature did not exceed 60 C. to obtain 138 g of the main fraction (step (D)). GC analysis was performed for the obtained main fraction. Results are shown in Table 1.
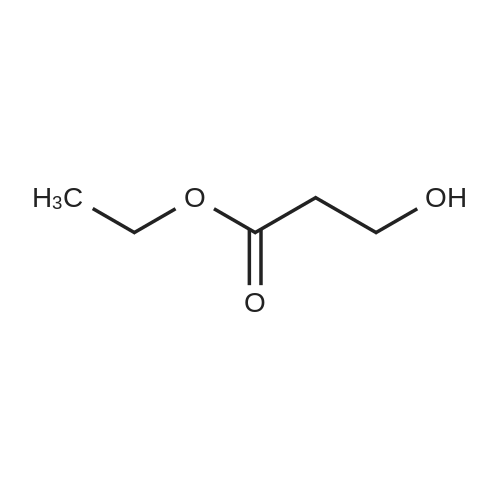
In water;
(2) A three liter three-necked flask, condenser and addition funnel were dried in an oven at 140-150 C. overnight, assembled hot and cooled under a flow of dry nitrogen. Lithium aluminum hydride (7.2 g; 0.19 mole) and 800-900 ml of ether were placed in the flask with a magnetic stirring bar and the slurry was stirred vigorously. A solution of ethyl-3,3-diethoxypropionate (35 g; 0.184 mole) dissolved in 100-200 ml of ether was added to the lithium aluminum hydride slurry at a rate sufficient to keep the stirred slurry boiling. When addition was complete, the reaction mixture was refluxed for twelve hours. After cooling, 200 ml of water was added very cautiously with stirring and then carbon dioxide was bubbled into the mixture for two hours to hydrolyze the salts. The reaction mixture was filtered and the solids were washed several times with ether. The ether layer was separated, dried over sodium sulfate and evaporated to a colorless oil. Boiling range 85-87 C. at 10 mm (Lit. 90-92 C. at 10 mm) yield of 3-hydroxypropanal diethyl acetal 22 g; 85% by weight.
40 g of powder Zetaeta were washed with dilute hydrochloric acid once, H20 washed 3 times, washed once in methanol, acetone and once with 20 Mm Hg and 100 degrees Celsius for 10 minutes after drying, remove the nitrogen 25 g was added to 100 ml three Zetaeta Round-bottomed flask containing a catalytic amount of iodine was added 6 ml of anhydrous benzene was heated and stirred at reflux, was added over 45 minutes containing 8.35 grams, 0.05 mole of ethyl bromoacetate and 8.89 g XI, 0 06 mol triethylorthoformate XII. After the dropwise addition, the treated again take Zetaeta 6.25 g flour added to the system, after the reaction was refluxed for 6 hours, the reaction solution was transferred to a 100 ml ice-water and 50 g, excess acetic acid was added, the ether layer was separated, with washed with saturated sodium bicarbonate solution, dried over anhydrous sodium sulfate, and concentrated under 1.5 mm Hg vacuum distillation, 79-81 ° C was collected as a colorless oil distillate 3, 3-ethoxy-propionate esters XIII2 8 g, yield 29.4percent; collected above to give the 2.8 g 3, 3-ethoxypropionate XIII at 190-200 degrees Celsius was heated under reflux for 2 hours with a delicate smell ., colorless liquid of ethyl acrylate, 3- XIV2 2 g, yield 100percent;. the 2.2 g, 152 mmol of ethyl acrylate, 3- XIV added to 7.5 ml of water and 7.5 ml two six ring oxygen mixed solvent, -10 degrees centigrade to cool large, slowly added 2.97 g after 16.72 mmol of NBS, the reaction 1 hour at room temperature, then 1.15 g, 15.2 mmol) was added thiourea after 1 hour at 80 ° C, ice-cooling, after addition of an excess of ammonia brown solid appeared, suction filtered to give a solid, after washing with water and drying give 1.4 g 2-aminothiazol-5-carboxylate XV of sitting Jie, The yield was 53.8percent
62.1 g
With potassium hydrogensulfate; at 100℃; for 5h;Inert atmosphere; Green chemistry;
In a three-necked flask, 91 g (0.5 mol) of trichloroacetyl chloride was added, and 54 g (0.75 mol) of vinyl ether was slowly added dropwise, and the mixture was dropped for about 1.5 hours. The temperature was controlled at 30 ° C for 5 hours, and then steamed under 40 ° C. The low-boiling by-product is obtained; after steaming, 66 g (0.65 mol) of triethylamine and 100 g of ethanol are added, and the reaction is kept at 35 ° C for 5 h, filtered, and the cake is recovered. The filtrate is evaporated under reduced pressure at 50 ° C or less; Add 6.8 g (0.05 mol) of potassium hydrogen sulfate, raise the temperature to 100 ° C, pass nitrogen gas, the flow rate of nitrogen is 300 mL / min, keep the reaction for 5 h, the reaction is finished, and distilled under reduced pressure to obtain 62.1 g of ethyl 3-ethoxyacrylate. The rate was 86.1percent, and the purity by gas phase normalization was 98.8percent
With sodium hydride; In diethyl ether; at 0 - 20℃; for 15h;
12.6 g of sodium hydride (purity: 60percent, 525.0 mmoles) was washed with diethyl ether by decantation several times and then made into a solution in 500 ml of diethyl ether. Thereto were added, in a nitrogen current at 0 to 10°C, 194 g (2.6 moles) of ethyl formate and 50 g (262.0 mmoles) of ethyl 3,3-diethoxy-propionate. The resulting mixture was stirred at room temperature for 15 hours to give rise to a reaction. After confirmation of the completion of the reaction, the reaction mixture was poured into water, followed by washing with diethyl ether. The resulting aqueous layer was allowed to have a pH of 1 with hydrochloric acid, followed by extraction with dichloromethane. The resulting organic layer was washed with an aqueous sodium chloride solution and then dried over anhydrous magnesium sulfate. The resulting solution was subjected to vacuum distillation to remove the solvent contained therein, to obtain 37.6 g (yield: 100percent) of crude (ethoxycarbonyl)malondialdehyde as a dark red oily substance. 1H-NMR [CDCl3/TMS, delta (ppm)]: 9.09 (2H,s), 5.26 (1H,s), 4.27 (2H,q), 1.28 (3H,t)
100%
With sodium hydride; In diethyl ether; at 0 - 20℃; for 15h;
REFERENCE EXAMPLE 28 Production of (ethoxycarbonyl)malondialdehyde; 12.6 g of sodium hydride (purity: 60percent, 525.0 mmoles) was washed with diethyl ether by decantation several times and then made into a solution in 500 ml of diethyl ether. Thereto were added, in a nitrogen current at 0 to 10° C., 194 g (2.6 moles) of ethyl formate and 50 g (262.0 mmoles) of ethyl 3,3-diethoxy-propionate. The resulting mixture was stirred at room temperature for 15 hours to give rise to a reaction. After confirmation of the completion of the reaction, the reaction mixture was poured into water, followed by washing with diethyl ether. The resulting aqueous layer was allowed to have a pH of 1 with hydrochloric acid, followed by extraction with dichloromethane. The resulting organic layer was washed with an aqueous sodium chloride solution and then dried over anhydrous magnesium sulfate. The resulting solution was subjected to vacuum distillation to remove the solvent contained therein, to obtain 37.6 g (yield: 100percent) of crude (ethoxycarbonyl)malondialdehyde as a dark red oily substance. 1H-NMR [CDCl3/TMS, delta (ppm)]: 9.09 (2H,s), 5.26 (1H,s), 4.27 (2H,q), 1.28 (3H,t)
99.7%
With sodium hydride; In tetrahydrofuran; at 0 - 20℃;
[00442] To a stirred suspension of sodium hydride (60percent, 1.68 g, 42.1 mmol) in THF (20 mL) was added ethyl formate (8.5 mL, 105.7 mmol). The solution was cooled to 0 °C and a solution of ethyl 3,3-diethoxypropanoate (4 g, 21.0 mmol) in THF (10 mL) was added dropwise over 30 mins and the reaction mixture stirred at r.t. overnight. 2M aq HCl (30 mL) was added under whilst cooling with ice and the reaction stirred at r.t. for 30 mins. The reaction mixture was extracted with diethyl ether (2 x 50 mL) and the combined organic extracts were dried over MgS04 and concentrated in vacuo to afford the title compound (3.02 g, 99.7percent) as an amber liquid. [00443] 1H NMR (250 MHz, Chloroform-d) delta 9.13 (s, 2H), 4.29 (q, J = 7.1 Hz, 2H), 1.37 - 1.28 (m, 3H).
74.2%
With sodium hydride; In diethyl ether; hexane; at 20℃; for 15h;Cooling with ice;
Weighed out sodium hydride (2.46 g, 61.5 mmol) in a dry 100-mL pear flask. Washed with hexanes then with diethyl ether. Suspended in ether (100 mL), cooled in ice bath then ethyl formate (24.84 ml, 308 mmol) was added then ethyl 3,3-diethoxypropanoate (5.98 ml, 30.8 mmol). The resulting mixture was stirred at room temperature for 15 hours to give rise to a reaction. After confirmation of the completion of the reaction, the reaction mixture was poured into water, followed by washing with diethyl ether. The resulting aqueous layer was allowed to have a pH of 1 with hydrochloric acid, followed by extraction with dichloromethane. The resulting organic layer was washed with an aqueous sodium chloride solution and then dried over anhydrous magnesium sulfate, filtered and concentrated to give ethyl 2-formyl-3-oxopropanoate (3.29 g, 22.83 mmol, 74.2 percent yield) as a golden syrup
70%
EXAMPLE 1Preparation of Ethyl-2-formyl-3-oxopropionate[0059] A three- or four-neck round bottom flask equipped with magnetic stir bar, thermocouple, digital thermometer, gas inlet and outlet and addition funnel was flushed <n="19"/>with argon. Ethyl 3,3-diethoxypropionate (64.5 g) in tetrahydrofuran were charged to the addition funnel. Sodium hydride (21.2 g of a 60percent dispersion) was charged to the reaction flask followed by tetrahydrofuran. The contents of the flask were cooled to 0- 5°C in an ice-bath, and ethyl formate (257 g) was added. The mixture was cooled to 0- 50C and the contents of the addition funnel added drop wise, maintaining an internal temperature of less than 5°C. The ice-bath was removed and the contents allowed to warm to ambient temperature. Consumption of ethyl 3,3-diethoxypropionate was monitored by TLC analysis. The reaction was quenched by addition of ice-water (10.6 vol), and extracted three times with methyl t-butyl ether (5.4 vol each), and the organic layers discarded. The aqueous phase was acidified with cone, hydrochloric acid to a pH of 1 to 1.5. The acidified aqueous layer was extracted three times with dichloromethane and the combined organic layers dried over sodium sulfate. The solvent was removed under reduced pressure, and the residue distilled under vacuum, to provide ethyl 2-formyl-3-oxopropionate, 27.92 g, 70percent yield.
70%
With sodium hydride; In tetrahydrofuran; at 0 - 20℃;
[0066] A three- or four-neck round bottom flask equipped with magnetic stir bar, thermocouple, digital thermometer, gas inlet and outlet and addition funnel was flushed with argon. Ethyl 3,3- diethoxypropionate (64.5 g) in tetrahydrofuran were charged to the addition funnel. Sodium hydride (21.2 g of a 60percent dispersion) was charged to the reaction flask followed by tetrahydrofuran. The contents of the flask were cooled to 0-50C in an ice-bath, and ethyl formate (257 g) was added. The mixture was cooled to 0-50C and the contents of the addition funnel added dropwise, maintaining an internal temperature of less than 5°C. The ice-bath was removed and the contents allowed to warm to ambient temperature. Consumption of ethyl 3,3- diethoxypropionate was monitored by TLC analysis. The reaction was quenched by addition of ice-water (10.6 vol), and extracted three times with methyl t-butyl ether (5.4 vol each), and the organic layers discarded. The aqueous phase was acidified with cone, hydrochloric acid to a pH of 1 to 1.5. The acidified aqueous layer was extracted three times with dichloromethane and the combined organic layers dried over sodium sulfate. The solvent was removed under reduced pressure, and the residue distilled under vacuum, to provide ethyl 2-formyl-3 -oxopropionate, 27.92 g, 70percent yield.
59%
With sodium hydride; In tetrahydrofuran; at 0 - 30℃; for 24h;
4) In a 500 ml four-necked flask equipped with magnetic stirring, 160 ml of tetrahydrofuran and9.84g NaH, mass fraction of NaH in tetrahydrofuran solution is 60percent, the solution is cooled in an ice bath to 0 °C to 5 °C, and then 119g of ethyl formate is added to slowly increase the temperature, when the temperature stabilizes to 0 °C At 5 °C, 80 ml of a tetrahydrofuran solution containing D was continuously added dropwise into a 500 ml four-necked flask, wherein the mass fraction of D was 5percent. After the dropwise addition, the water bath was removed and heated to room temperature. At this time, a large amount of hydrogen was evolved. It will first rise to 30 °C and then gradually decrease to 25 °C. After 24 hours of reaction, the reaction solution was poured into ice water and quenched. 5) The resulting product is first extracted with 500 ml of tert-butyl methyl ether three times to remove the organic phase, and then with concentrated hydrochloric acid to adjust the pH of the aqueous phase to 1~1.5; then extract the aqueous phase with 300 ml of dichloromethane, twice The combined organic phases were combined; the combined organic phases were then dried over anhydrous sodium sulfate and evaporated to give a brown-red liquid which was evaporated in vacuo. GC yield was 99.3percent. The yield was 85.9percent. Finally, the 9 mm Hg diaphragm was distilled under reduced pressure. The colorless transparent liquid E, ethyl 2-formyl-3-oxopropanoate, was obtained in a yield of 59percent.
Step 1: 2-Formyl-3-oxo-propionic acid ethyl ester Ethyl 3,3-diethoxypropionate (100 g, 525.7 mmol) was dissolved in THF (360 ml) at room temp. Ethyl formate (175.1 ml, 2.1 mol) was added at room temp. The solution was cooled in an ice-bath to 0° C. and tBuOK (1M solution in THF, 1,156 ml, 1.156 mol) was added via an addition funnel slowly over 30 min, maintaining internal temperature below 5° C. The color changed instantly from colorless to dark orange. The reaction mixture was allowed to warm up to room temp. and stirred for 2 h. The reaction was allowed to stir at room temp. for 18 h. The reaction mixture was concentrated in vacuo and 1 L of solvent was removed. The remaining brownish solution with white solid in it was cooled in an ice-bath and hydrochloric acid (6N, 200 ml) was added to adjust pH=3, maintaining internal temperature below 20° C. The resulting bright yellow suspension was then warmed up to room temp. and stirred for 1 h. Additional 700 ml solvent was removed in vacuo at room temp. Water (400 ml) was added to dissolve all the white solid and ethyl acetate (500 ml) was added and the mixture transferred to a separatory funnel. The aqueous was extracted once with ethyl acetate (200 ml). The combined organic extracts was washed once with brine (100 ml). After drying over MgSO4 and concentrating in vacuo, 2-formyl-3-oxo-propionic acid ethyl ester (75.85 g) was obtained as yellow oil.
With sodium hydride; In tetrahydrofuran; at 0 - 20℃; for 24h;
Sodium hydride (2.00 g, 50.6 mmol, 60percent) was added to a solution of ethyl formate (10.13 g, 168.7 mmol) in tetrahydrofuran while maintaining the internal temperature below 0 °C. Then, ethyl 3,3-diethoxy propionate (5.00 g, 33.75 mmol) was added dropwise to the reaction mixture and stirred for 24 h at ambient temperature. After confirming that the reaction was complete by using TLC analysis, the reaction was quenched with 20 mL water and the tetrahydrofuran was removed under reduced pressure. The mixture was extracted with ethyl acetate and then brine. The organic phase was dried over Na2SO4 overnight and the solvent was removed in vacuo to give the title compound 14.
With hydrogenchloride;titanium tetrachloride; In nitromethane; dichloromethane;
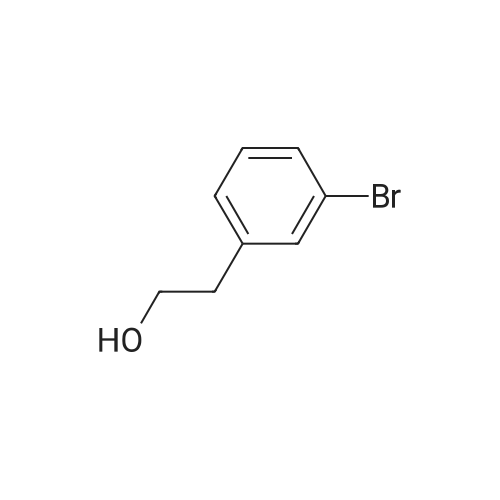
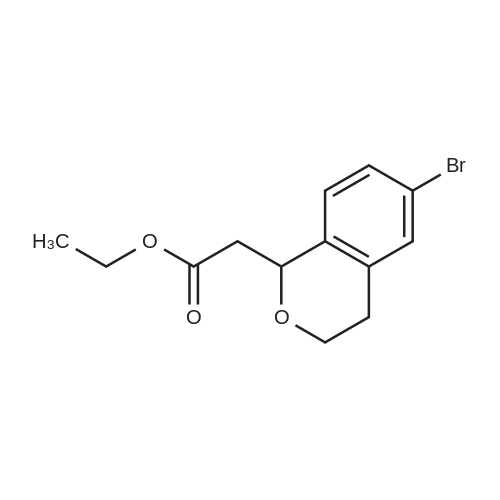
Titanium tetrachloride (1M in methylene chloride, 51 ml) is added over a period of 10 min to an ice-cooled mixture of <strong>[28229-69-8]2-(3-bromophenyl)ethanol</strong> (XLV, 4.34 g, 21.6 mmol) and ethyl 3,3-diethoxypropionate (LXXXV, 4.93 g, 25.9 mmol) in nitromethane (5 ml). After stirring for 10 min, the ice bath is removed and the mixture is allowed to stir at 20-250 for 5 hr, at which time it is poured onto ice/aqueous hydrochloric acid (~1N). The mixture is extracted with dichloromethane and backwashed with hydrochloric acid (0.5N)/saline and saline. The organic phase is separated dried over sodium sulfate, concentrated, and the residue chromatographed on silica gel eluding with ethyl acetate/hexane (5/95). The appropriate fractions are pooled and concentrated to give ethyl (6-bromoisochroman-1-yl)acetate (LXXVII), NMR (CDCl3) 1.28, 2.66-2.87, 2.96, 3.768, 3.807, 4.11, 4.21, 5.19, 6.93 and 7.28 δ.
15 Example 15
Example 15 A reaction was carried out by the procedure of Example 1, except for using ethyl 3,3-diethoxypropionate instead of methyl acrylate. After the reaction, the reaction mixture was analyzed by gas chromatography to find that ethyl 5-methylfuran-3-carboxylate was produced in a yield of 56%.
Into 25 ml of ether was suspended 762 mg of 60% NaH, and then 20 g of ethyl formate was added dropwise under ice-cooling. Then, 12 ml of an ether solution of 5.0 g of ethyl 3.3-diethoxypropanoate was added dropwise thereto. After 2 days of stirring at the same temperature, 2.50 g of acetamidine hydrochloride was added thereto, followed by 1 day of stirring at room temperature. Then, 5 ml of acetic acid and water were added to the reaction solution, followed by extraction with ethyl acetate. After the organic layer was dried over anhydrous sodium sulfate, the solvent was removed by evaporation and the resulting residue was subjected to silica gel column chromatography to obtain 2.93 g of the title compound from an eluate eluted with ethyl acetate-hexane (3:7, v/v).1H-NMR (DMSO-d6) delta: 1.34 (3H, t), 2.71 (3H, s), 4.36 (2H, q), 9.12 (2H, s)
With tin(ll) chloride; In ethanol; at 90℃; for 4h;
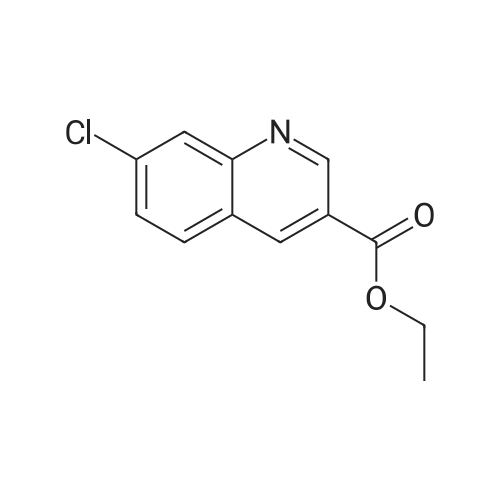
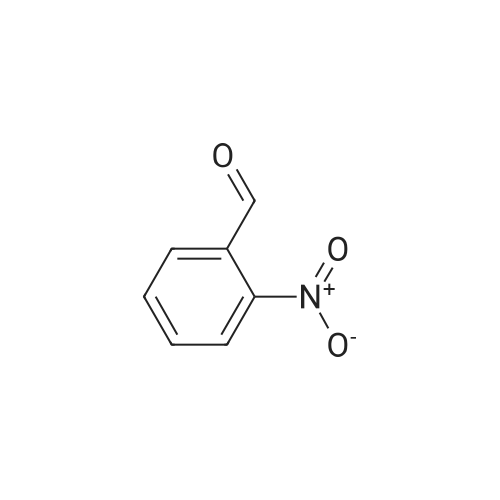
<strong>[5551-11-1]4-Chloro-<strong>[5551-11-1]2-nitrobenzaldehyde</strong></strong> (3.71 g, 0.02 mol) was dissolved in ethanol (40 ml).Further adding stannous chloride (18.04g, 0.08mol) and ethyl 3,3-diethoxypropionate (9.5g, 0.05mol), and reacting at 90 C for 4 hours, the reaction of the raw materials of the plate is completed; After the solvent was added, ethyl acetate was dissolved, and neutralized to a weak basic with saturated sodium carbonate; filtered, the organic layer was separated, and the aqueous layer was extracted with ethyl acetate, and then the organic layer was combined, and then evaporated to dryness. Ethyl acetate = 10:1, then petroleum ether: ethyl acetate = 5:1) After washing, ethyl 7-chloro-3-quinolinecarboxylate (3.478 g, 74%).
With tin(II) chloride dihdyrate; In ethanol; at 90℃; for 12h;
To a solution of compound 1 (20 g, 0.087 mol) and ethyl 3,3-diethoxypropionate (33 g, 0.17 mol) in EtOH (500 mE) was added SnCl2-2H20 (87 g, 0.39 mol), the mixture was stirred at 90 C. for 12 hours. TEC monitored completion. Afier cooled, evaporated to obtain the crude product. Then put into EA (600 mE), H20 (300 mE), Na2CO3 (32 g) and stirred for 1 hour. The organic layer was separated, dried, filtered and concentrated to obtain compound 2 (15 g, 92%) as a yellow solid.
21.5%
With tin(II) chloride dihdyrate; In ethanol; at 90℃; for 4h;
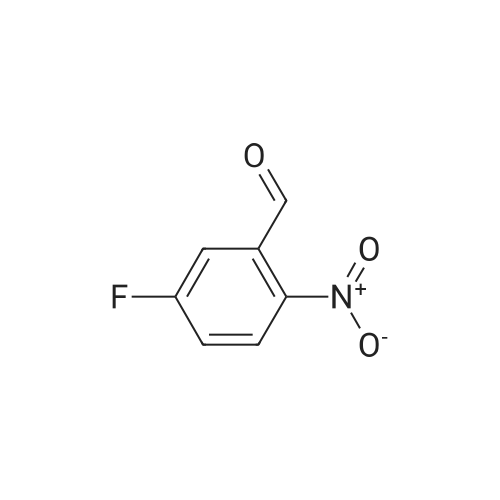
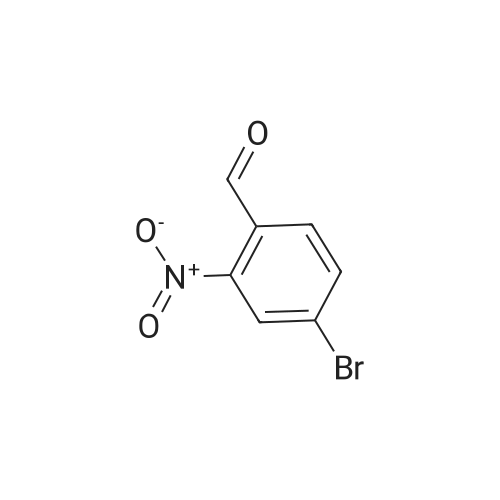
Tin (II) chloride dihydrate (9.026 g, 40.0 mmol) was added to a solution of <strong>[5551-12-2]4-bromo-2-nitrobenzaldehyde</strong> (2.3 g, 10.0 mmol) and EtOH (40 ml) followed by 3,3-diethoxypropionic acid ethyl ester (4.75 g, 25.0 mmol ). The reaction was heated to 90 oc for4 hr upon the cooling, the reaction mixture was concentrated and the residue was dissolved inEtOAc and quenched with sat. NaHC03. The resulting emulsion was filtered through a pad ofcelite and rinsed well with EtOAc. The remaining aqueous layer was extracted with EtOAcand the combined organic layer were washed with brine, dried over Na2S04 andconcentrated. The crude product was purified by silica gel column chromatography with 10-30% ofEtOAc in hexanes to give ethyl 7-bromoquinoline-3-carboxylate (0.6 g, 21.5%). OH(400 MHz; CDCh) 1.47 (3H, t, J= 8.0 Hz), 4.50 (2H, q, J= 8.0 Hz), 7.72 (1H, t, J= 4.0 Hz),7.80 (1H, s), 8.36 (1H,s), 8.82 (1H, s), 9.45 (1H, s). 8c (100 MHz; CDCh) 14.3, 61.7, 123.5,125.4,126.3, 130.2, 131.1, 138.5, 150.2, 151.0, 165.0.
With tin(II) chloride dihdyrate; In ethanol; at 90℃; for 4h;
4-Bromo-2-nitrobenzaldehyde (2.3 g), ethyl 3,3-diethoxypropionate (4.85 ml)Stannous chloride dihydrate (9.05 g),Ethanol (50 ml) was added to a three-necked flask, the oil bath was 90 C,After stirring for 4 hours (thin plate chromatography test), the reaction solution was concentrated to a viscous shape by a rotary evaporator. The viscous substance was dissolved in ethyl acetate, stirred with an excess of sodium carbonate solution,The organic layer was separated by filtration and washed repeatedly with brine, and then dried over anhydrous magnesium sulfate,The organic phase was collected by filtration and evaporated to dryness, recrystallized from ethyl acetate,(7- bromo)quinolinyl-3-carboxylate
With tin(II) chloride dihdyrate; In ethanol; at 20 - 90℃;
Step 1: ethyl 6-bromoquinoline-3-carboxylate To a solution of 5-bromo-2-nitrobenzaldehyde (2 g, 9 mmol) in ethanol (46 mL) was added tin(II) chloride dihydrate (7.95 g, 35.2 mmol) and 3,3-diethoxypropionic acid ethyl ester (4.2 mL, 22 mmol). The reaction was heated to 90 C. for 16 hours. The reaction was then allowed to cool to room temperature and stir overnight. The reaction mixture was concentrated and the residue was dissolved in ethyl acetate. The mixture was poured into saturated aqueous sodium bicarbonate. The resulting emulsion was filtered through Celite, rinsing with ethyl acetate. The layers were separated and the aqueous was extracted with ethyl acetate. The combined organics were washed with brine, dried over magnesium sulfate, filtered, and concentrated. Purification by flash column chromatography (0-50% ethyl acetate/heptanes) gave the title compound (1.41 g, 60%) as a solid.
With ytterbium(III) triflate; In 1,4-dioxane; at 90℃;
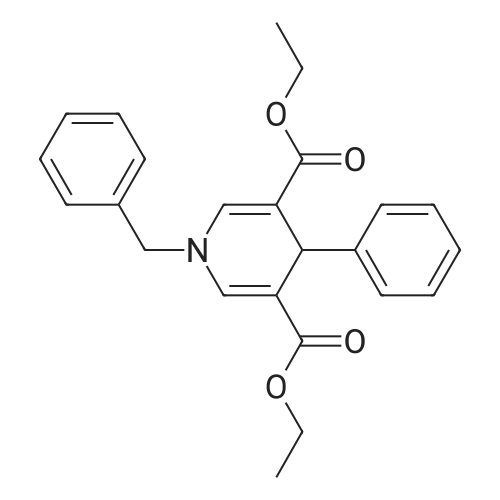
General procedure: A mixture of Yb(OTf)3 (2.5 mol%), amine (1.0 eq.), ethyl 3,3-diethoxypropionate (2.5 eq.) and aldehyde (1.1 eq.) in 1,4-dioxane (1.5 mL / 1 eq.) was stirred at 90 C for 6-17 h. After the reaction, the resulting mixture was washed with PBS buffer solution (25 mL) and extracted with CH2Cl2 (15 mL×3). The organic layer was dried over MgSO4. Removal of the solvent in vacuo, followed by chromatography on silica gel column (AcOEt/Hexane or CHCl3/MeOH), afforded dihydropyridine 4.
General procedure: To a solution of ethyl propiolate (98mg, 1.0mmol) in methanol (2mL), catalytic PtCl2 (5mg, 0.02mmol) was added. The mixture was stirred at 60C for 12h. The reaction solution was cooled to room temperature, and CH2Cl2 (20mL) was added. The mixture was then filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel with hexane-ethyl acetate (20:1) as the eluent to give a mixture of (E)-ethyl 3-methoxyacrylate (4) and ethyl 3,3-dimethoxypropanoate (5) (112mg, 85% yield, 4:5=13:1, the ratio was determined by 1H NMR) as a colorless oil.
(E)-ethyl 3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)acrylate[ No CAS ]
(E)-3-(p-methylstyryl)-4H-pyrido[1,2-a]pyrimidin-4-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
1: 39%
2: 13%
With toluene-4-sulfonic acid In toluene at 80℃; for 6h;
Typical procedure for the synthesis of 5a
General procedure: 2-aminopyridine 1a (47 mg, 0.5 mmol), ethyl 3,3-diethoxypropanoate 2 (143 mg, 0.75 mmol), 2-phenylacetaldehyde 4a (108mg, 0.90 mmol) and p-TsOH (8.6 mg, 10 mol %) were stirred in 2 mL of toluene for 6h at 80 °C. After completion of the reaction (monitored by TLC), the water (10 mL) was added. The aqueous solution was extracted with ethyl acetate (3×8 mL) and the combined extract was dried with anhydrous MgSO4. The solvent was removed and the crude product was separated by column chromatography (eluted with petroleum ether/ethyl acetate = 3:1) to give a pure sample of 5a (Yellow crystal, 57 mg, 46%).
(E)-ethyl 3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)acrylate[ No CAS ]
(E)-3-(4-fluorostyryl)-4H-pyrido[1,2-a]pyrimidin-4-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
1: 49%
2: 5%
With toluene-4-sulfonic acid In toluene at 80℃; for 6h;
Typical procedure for the synthesis of 5a
General procedure: 2-aminopyridine 1a (47 mg, 0.5 mmol), ethyl 3,3-diethoxypropanoate 2 (143 mg, 0.75 mmol), 2-phenylacetaldehyde 4a (108mg, 0.90 mmol) and p-TsOH (8.6 mg, 10 mol %) were stirred in 2 mL of toluene for 6h at 80 °C. After completion of the reaction (monitored by TLC), the water (10 mL) was added. The aqueous solution was extracted with ethyl acetate (3×8 mL) and the combined extract was dried with anhydrous MgSO4. The solvent was removed and the crude product was separated by column chromatography (eluted with petroleum ether/ethyl acetate = 3:1) to give a pure sample of 5a (Yellow crystal, 57 mg, 46%).
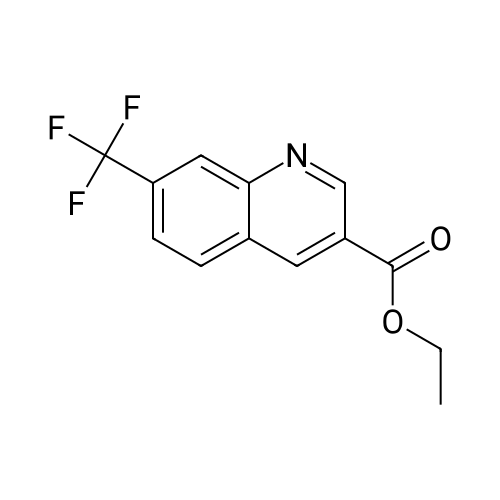
ethyl (E)-3-(8-(trifluoromethyl)-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)acrylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
With toluene-4-sulfonic acid; In toluene; at 80℃; for 6h;
General procedure: 2-aminopyridine 1a (47 mg, 0.5 mmol), ethyl 3,3-diethoxypropanoate 2 (285 mg, 1.5 mmol) and p-TsOH (8.6 mg, 10 mol %) were stirred in 2 mL of toluene for 6 h at 80 C. After completion of the reaction (monitored by TLC), the water (10 mL) was added. The aqueous solution was extracted with ethyl acetate (3×8 mL) and the combined extract was dried with anhydrous MgSO4. The solvent was removed and the crude product was separated by column chromatography (eluted with petroleum ether/ethyl acetate = 2:1) to give a pure sample of 3a (Yellow crystal, 100 mg, 82%).
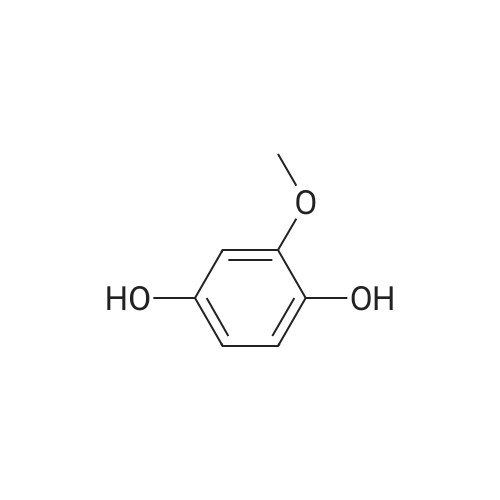
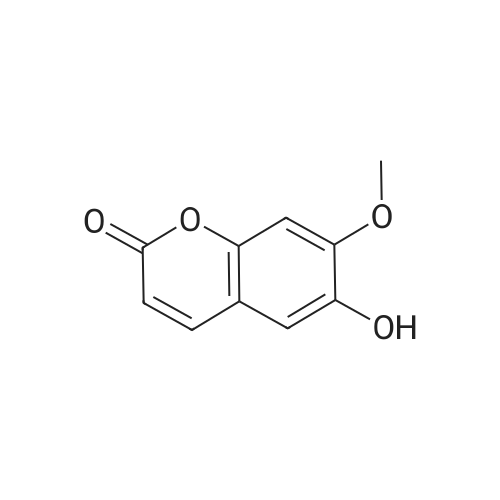
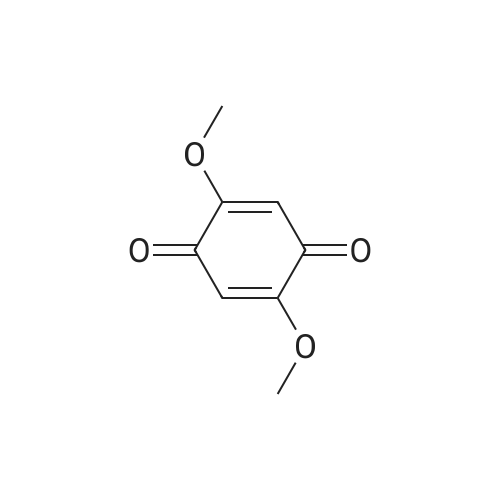
3.1.4. Compound 10 (6-hydroxy-7-methoxy-2H-chromen-2-one)
A solution of 2-methoxyhydroquinone (8) and 3,3-diethoxyethylpropionate (9) in conc. H3PO4 was well stirred for 2 h at 100 °C. The resulting mixture was then poured into icy water and the resulting yellow solid formed filtered under vacuum. The desired product was obtained (m.p. 230-232 °C) in 62% yield. Analytical data were identical to those obtained for a commercial sample used for comparison.
With boron trifluoride diethyl etherate at 60℃; for 4h; Inert atmosphere;
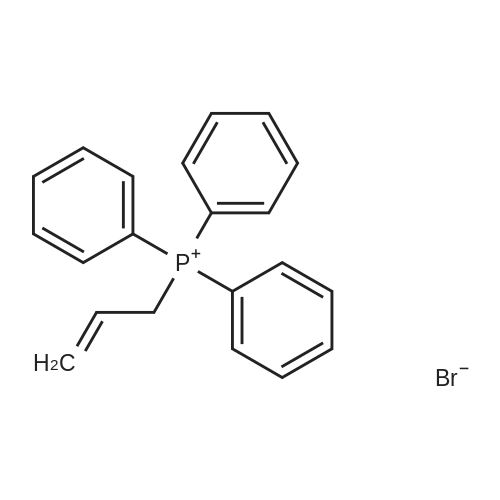
1 Preparation of ethyl ((4R, 6S) -6- triphenylphosphine alkenyl ylidene-methyl-2-ethoxycarbonyl-1,3-dioxane-4-yl) acetate
To 1000 ml reaction flask were added 150 g of tetrahydrofuran, 300 g of toluene, 136.2 g (0.52 mol) of triphenylphosphine was stirred at room temperature Was dissolved, was added dropwise 60.5 g (0.5 mol) of 3-bromo-propene, dropping was completed, the reaction at room temperature for 12 hours. Was added 209 g (1.1 mol) of 3,3-ethoxypropionate, 6.0 g (0.035 mole) of 40% solution of boron trifluoride etherate, nitrogen gas, 3 changes, warmed to 60 ° C for 4 hours (recovery ether). Completion of the reaction, 70 ° C or less under reduced recovery tetrahydrofuran, cooled to 20 ° C, 50 g of water was added, separated and the aqueous layer was extracted three times (each with toluene Times 30 g), and the combined organic phases. Transferred to a flask, azeotropic removal of water, cooled to minus 10 ° C, was added in portions 40.8 g (0.6 mol) of sodium solid ethanol, plus complete, 10 ° C Anti For 4 hours, filtered, and the filtrate was recovered toluene, the residue was treated with 600 g of methyl tert-butyl ether, 120 g of ethanol and 5 g of activated carbon and recrystallized to give the class White solid powder, 2 - ((4R, 6S) -6- triphenylphosphine alkenyl ylidene-methyl-2-ethoxycarbonyl-1,3-dioxane-4-yl) acetate 212.5 g, purity 99.5% (HPLC), yield 79.6%.
With [methyl-3-(butyl-4-sulfonate) imidazolium]CuPW12O40; oxygen; at 159.84℃; under 6000.6 Torr; for 5h;
General procedure: In a typical process, 0.25 g lignin, 0.9 mmol POM-IL catalyst and 20 mL 100% ethanol werecharged into a 100 mL stainless autoclave (Andorra MED1220, Premex Co. Ltd.). After airpurging with pure oxygen five times and pressurizing to 0.8 MPa, the reactor was heated to thedesignated temperature and maintained for the desired time. Once the latter elapsed, the autoclavewas cooled rapidly to room temperature in an ice water bath. The reaction mixture was removedand the reactor was washed with anhydrous ethanol (3 5.0 mL). The IL catalyst was precipitatedat room temperature and used for the next run after drying (extra fresh catalyst was added tooffset transfer losses). The liquid mixture was then diluted by ethanol to 50 mL for qualitative andquantitative analysis, while dimethyl phthalate was used as the internal standard. When aqueoussolutions of ethanol were used, the spent mixture was rotary evaporated under reduced pressurefor solvent recovery. The concentrated liquor was esterified with 10 mL anhydrous ethanol at 373K for 2 h and then diluted to 50 mL with ethanol. Volatile products were qualitatively andquantitatively analyzed via gas chromatography-mass spectrometry (GC-MS) and gaschromatography-flame ionization detection (GC-FID). Residual lignin can be obtained throughsimple precipitation processes. Organosolv lignin was recovered as follows: 60 mL deionized water was added into 20 mL of the above reaction mixture causing precipitation. The mixture wasthen separated using centrifugation and was dried until a constant weight was obtained. For therecovery of dealkaline lignin the mixture obtained after reaction was acidified to pH=2 with 1.0mol L-1 HCl solution and the same procedure described for organosolv lignin was conducted.In the atmosphere investigation, a mixture of nitrogen and oxygen with various molar ratioswas used, while depolymerization of lignin was conducted at 433 K for 5.0 h in the single stageexperiments. For a typical two-stage process, the lignin was first depolymerized employing theaforementioned conditions. When the mixture was cooled to room temperature, an extra 0.8 MPanitrogen or oxygen was purged into the reactor and the reaction was heated to 433 K for 1.0 or 2.0h. The product separation and analysis procedure remained unchanged to that describedpreviously. In comparative and control experiments, a series of model compounds (monolignolsand potential intermediate products) were tested under the same procedures as that for lignin (i.e.,0.25 g model compound, 0.9 mmol POM-IL catalyst and 20 mL 100% ethanol solvent). Triplicateexperiments were conducted and the data shown in this study is the average.
With [methyl-3-(butyl-4-sulfonate) imidazolium]CuPW12O40; oxygen at 159.84℃; for 5h;
General procedure for catalytic oxidation of lignin and model compound.
General procedure: In a typical process, 0.25 g lignin, 0.9 mmol POM-IL catalyst and 20 mL 100% ethanol werecharged into a 100 mL stainless autoclave (Andorra MED1220, Premex Co. Ltd.). After airpurging with pure oxygen five times and pressurizing to 0.8 MPa, the reactor was heated to thedesignated temperature and maintained for the desired time. Once the latter elapsed, the autoclavewas cooled rapidly to room temperature in an ice water bath. The reaction mixture was removedand the reactor was washed with anhydrous ethanol (3 5.0 mL). The IL catalyst was precipitatedat room temperature and used for the next run after drying (extra fresh catalyst was added tooffset transfer losses). The liquid mixture was then diluted by ethanol to 50 mL for qualitative andquantitative analysis, while dimethyl phthalate was used as the internal standard. When aqueoussolutions of ethanol were used, the spent mixture was rotary evaporated under reduced pressurefor solvent recovery. The concentrated liquor was esterified with 10 mL anhydrous ethanol at 373K for 2 h and then diluted to 50 mL with ethanol. Volatile products were qualitatively andquantitatively analyzed via gas chromatography-mass spectrometry (GC-MS) and gaschromatography-flame ionization detection (GC-FID). Residual lignin can be obtained throughsimple precipitation processes. Organosolv lignin was recovered as follows: 60 mL deionized water was added into 20 mL of the above reaction mixture causing precipitation. The mixture wasthen separated using centrifugation and was dried until a constant weight was obtained. For therecovery of dealkaline lignin the mixture obtained after reaction was acidified to pH=2 with 1.0mol L-1 HCl solution and the same procedure described for organosolv lignin was conducted.In the atmosphere investigation, a mixture of nitrogen and oxygen with various molar ratioswas used, while depolymerization of lignin was conducted at 433 K for 5.0 h in the single stageexperiments. For a typical two-stage process, the lignin was first depolymerized employing theaforementioned conditions. When the mixture was cooled to room temperature, an extra 0.8 MPanitrogen or oxygen was purged into the reactor and the reaction was heated to 433 K for 1.0 or 2.0h. The product separation and analysis procedure remained unchanged to that describedpreviously. In comparative and control experiments, a series of model compounds (monolignolsand potential intermediate products) were tested under the same procedures as that for lignin (i.e.,0.25 g model compound, 0.9 mmol POM-IL catalyst and 20 mL 100% ethanol solvent). Triplicateexperiments were conducted and the data shown in this study is the average.
With [methyl-3-(butyl-4-sulfonate) imidazolium]CuPW12O40; oxygen at 159.84℃; for 5h;
General procedure for catalytic oxidation of lignin and model compound.
General procedure: In a typical process, 0.25 g lignin, 0.9 mmol POM-IL catalyst and 20 mL 100% ethanol werecharged into a 100 mL stainless autoclave (Andorra MED1220, Premex Co. Ltd.). After airpurging with pure oxygen five times and pressurizing to 0.8 MPa, the reactor was heated to thedesignated temperature and maintained for the desired time. Once the latter elapsed, the autoclavewas cooled rapidly to room temperature in an ice water bath. The reaction mixture was removedand the reactor was washed with anhydrous ethanol (3 5.0 mL). The IL catalyst was precipitatedat room temperature and used for the next run after drying (extra fresh catalyst was added tooffset transfer losses). The liquid mixture was then diluted by ethanol to 50 mL for qualitative andquantitative analysis, while dimethyl phthalate was used as the internal standard. When aqueoussolutions of ethanol were used, the spent mixture was rotary evaporated under reduced pressurefor solvent recovery. The concentrated liquor was esterified with 10 mL anhydrous ethanol at 373K for 2 h and then diluted to 50 mL with ethanol. Volatile products were qualitatively andquantitatively analyzed via gas chromatography-mass spectrometry (GC-MS) and gaschromatography-flame ionization detection (GC-FID). Residual lignin can be obtained throughsimple precipitation processes. Organosolv lignin was recovered as follows: 60 mL deionized water was added into 20 mL of the above reaction mixture causing precipitation. The mixture wasthen separated using centrifugation and was dried until a constant weight was obtained. For therecovery of dealkaline lignin the mixture obtained after reaction was acidified to pH=2 with 1.0mol L-1 HCl solution and the same procedure described for organosolv lignin was conducted.In the atmosphere investigation, a mixture of nitrogen and oxygen with various molar ratioswas used, while depolymerization of lignin was conducted at 433 K for 5.0 h in the single stageexperiments. For a typical two-stage process, the lignin was first depolymerized employing theaforementioned conditions. When the mixture was cooled to room temperature, an extra 0.8 MPanitrogen or oxygen was purged into the reactor and the reaction was heated to 433 K for 1.0 or 2.0h. The product separation and analysis procedure remained unchanged to that describedpreviously. In comparative and control experiments, a series of model compounds (monolignolsand potential intermediate products) were tested under the same procedures as that for lignin (i.e.,0.25 g model compound, 0.9 mmol POM-IL catalyst and 20 mL 100% ethanol solvent). Triplicateexperiments were conducted and the data shown in this study is the average.
With [methyl-3-(butyl-4-sulfonate) imidazolium]CuPW12O40; oxygen; at 159.84℃; under 6000.6 Torr; for 5h;
General procedure: In a typical process, 0.25 g lignin, 0.9 mmol POM-IL catalyst and 20 mL 100% ethanol werecharged into a 100 mL stainless autoclave (Andorra MED1220, Premex Co. Ltd.). After airpurging with pure oxygen five times and pressurizing to 0.8 MPa, the reactor was heated to thedesignated temperature and maintained for the desired time. Once the latter elapsed, the autoclavewas cooled rapidly to room temperature in an ice water bath. The reaction mixture was removedand the reactor was washed with anhydrous ethanol (3 5.0 mL). The IL catalyst was precipitatedat room temperature and used for the next run after drying (extra fresh catalyst was added tooffset transfer losses). The liquid mixture was then diluted by ethanol to 50 mL for qualitative andquantitative analysis, while dimethyl phthalate was used as the internal standard. When aqueoussolutions of ethanol were used, the spent mixture was rotary evaporated under reduced pressurefor solvent recovery. The concentrated liquor was esterified with 10 mL anhydrous ethanol at 373K for 2 h and then diluted to 50 mL with ethanol. Volatile products were qualitatively andquantitatively analyzed via gas chromatography-mass spectrometry (GC-MS) and gaschromatography-flame ionization detection (GC-FID). Residual lignin can be obtained throughsimple precipitation processes. Organosolv lignin was recovered as follows: 60 mL deionized water was added into 20 mL of the above reaction mixture causing precipitation. The mixture wasthen separated using centrifugation and was dried until a constant weight was obtained. For therecovery of dealkaline lignin the mixture obtained after reaction was acidified to pH=2 with 1.0mol L-1 HCl solution and the same procedure described for organosolv lignin was conducted.In the atmosphere investigation, a mixture of nitrogen and oxygen with various molar ratioswas used, while depolymerization of lignin was conducted at 433 K for 5.0 h in the single stageexperiments. For a typical two-stage process, the lignin was first depolymerized employing theaforementioned conditions. When the mixture was cooled to room temperature, an extra 0.8 MPanitrogen or oxygen was purged into the reactor and the reaction was heated to 433 K for 1.0 or 2.0h. The product separation and analysis procedure remained unchanged to that describedpreviously. In comparative and control experiments, a series of model compounds (monolignolsand potential intermediate products) were tested under the same procedures as that for lignin (i.e.,0.25 g model compound, 0.9 mmol POM-IL catalyst and 20 mL 100% ethanol solvent). Triplicateexperiments were conducted and the data shown in this study is the average.
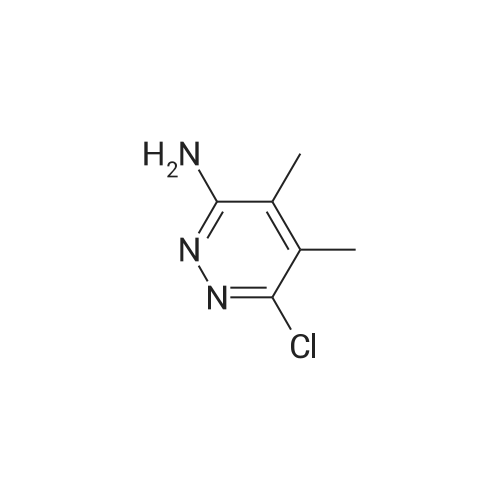
7-chloro-8,9-dimethylpyrimido[1,2-b]pyridazin-4-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
50%
With polyphosphoric acid; at 85℃; for 23.0h;Sealed tube;
A mixture of 6-chIoro-3-amino-4,5-dimethylpyridazine (550 mg), ethyl 3,3-dietboxypropiQnate (1.02 mL), and poyphosphoric acid (5 mL) was added to a sealed tube. The mixture was stirred at 85 C for 5 hours then cooled to room temperature where additional ethyl 3,3-diethoxypropionate (1.02 L) was added to the reaction mixture. The vial was sealed and the mixture heated to 85 C for 18 hours. The reaction mixture was then slowly added to a stirred solution of 150 L of saturated aqueous NaHCCb and 100 mL of DCM. After complete addition, the mixture was allowed to stir for 20 minutes, the organic layer was separated, and the aqueous layer was further extracted with chioroform/PA (4:1) (x3). The organic layers were pooled, dried over MgSG4, filtered, and concentrated. The crude product was purified using Teledyne ISCO Combi- Flash system (liquid loading with DCM, 40G column, 0 - 70% EtGAc/Hex, 35 min run) to give the title compound as a solid (366 mg, 50% yield). XH MMR (400 MHz, CDC ) d 8.22-8.20 (d, J = 6.5 Hz, 1H), 6.64 - 6.62 (d, J = 6.5 Hz, 1H), 2.62 (d, J = 0.6 Hz, 3H), 2.51 (d, J = 0.6 Hz, 3H). ES-MS [M+l]+: 210
50%
With polyphosphoric acid; at 85℃; for 23.0h;Sealed tube;
A mixture of 6-chIoro-3-amino-4,5-dimethylpyridazine (550 mg), ethyl 3,3-dietboxypropiQnate (1.02 mL), and poyphosphoric acid (5 mL) was added to a sealed tube. The mixture was stirred at 85 C for 5 hours then cooled to room temperature where additional ethyl 3,3-diethoxypropionate (1.02 L) was added to the reaction mixture. The vial was sealed and the mixture heated to 85 C for 18 hours. The reaction mixture was then slowly added to a stirred solution of 150 L of saturated aqueous NaHCCb and 100 mL of DCM. After complete addition, the mixture was allowed to stir for 20 minutes, the organic layer was separated, and the aqueous layer was further extracted with chioroform/PA (4:1) (x3). The organic layers were pooled, dried over MgSG4, filtered, and concentrated. The crude product was purified using Teledyne ISCO Combi- Flash system (liquid loading with DCM, 40G column, 0 - 70% EtGAc/Hex, 35 min run) to give the title compound as a solid (366 mg, 50% yield). XH MMR (400 MHz, CDC ) d 8.22-8.20 (d, J = 6.5 Hz, 1H), 6.64 - 6.62 (d, J = 6.5 Hz, 1H), 2.62 (d, J = 0.6 Hz, 3H), 2.51 (d, J = 0.6 Hz, 3H). ES-MS [M+l]+: 210
50%
With polyphosphoric acid; at 85℃; for 23.0h;Sealed tube;
A mixture of 6-chIoro-3-amino-4,5-dimethylpyridazine (550 mg), ethyl 3,3-dietboxypropiQnate (1.02 mL), and poyphosphoric acid (5 mL) was added to a sealed tube. The mixture was stirred at 85 C for 5 hours then cooled to room temperature where additional ethyl 3,3-diethoxypropionate (1.02 L) was added to the reaction mixture. The vial was sealed and the mixture heated to 85 C for 18 hours. The reaction mixture was then slowly added to a stirred solution of 150 L of saturated aqueous NaHCCb and 100 mL of DCM. After complete addition, the mixture was allowed to stir for 20 minutes, the organic layer was separated, and the aqueous layer was further extracted with chioroform/PA (4:1) (x3). The organic layers were pooled, dried over MgSG4, filtered, and concentrated. The crude product was purified using Teledyne ISCO Combi- Flash system (liquid loading with DCM, 40G column, 0 - 70% EtGAc/Hex, 35 min run) to give the title compound as a solid (366 mg, 50% yield). XH MMR (400 MHz, CDC ) d 8.22-8.20 (d, J = 6.5 Hz, 1H), 6.64 - 6.62 (d, J = 6.5 Hz, 1H), 2.62 (d, J = 0.6 Hz, 3H), 2.51 (d, J = 0.6 Hz, 3H). ES-MS [M+l]+: 210






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping