There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
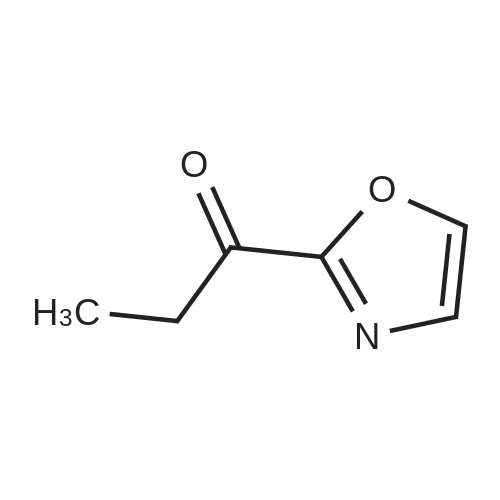
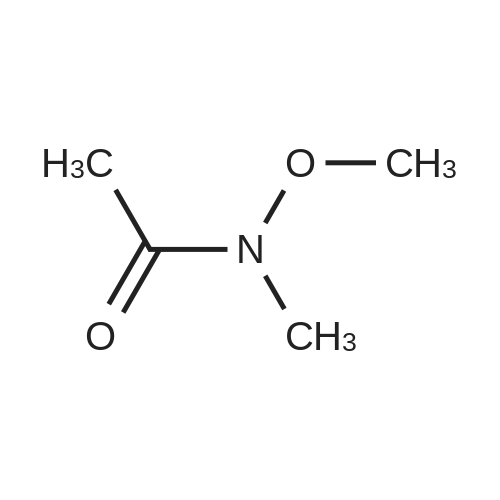
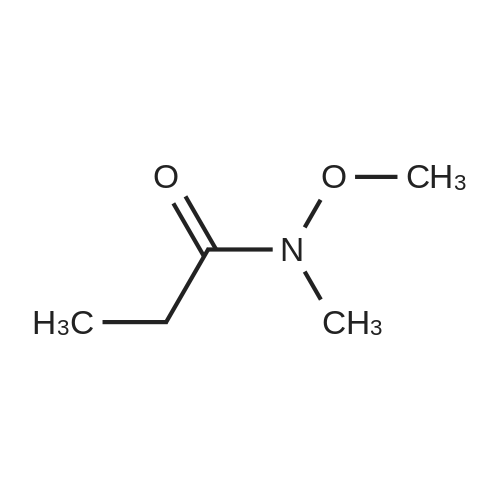
Structure of 104863-65-2 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Angewandte Chemie, International Edition, 2009, vol. 48, p. 4543 - 4545[2] Angewandte Chemie, 2009, vol. 121, p. 4613 - 4615
2
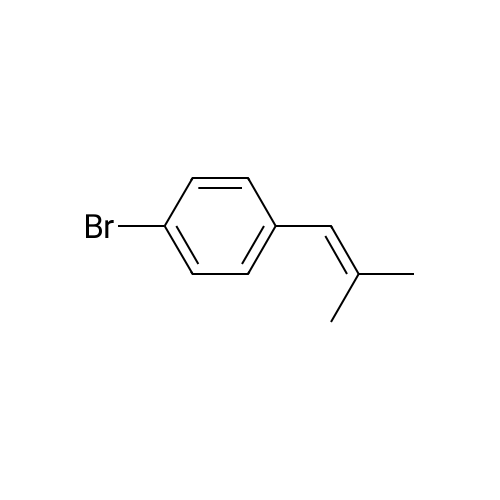
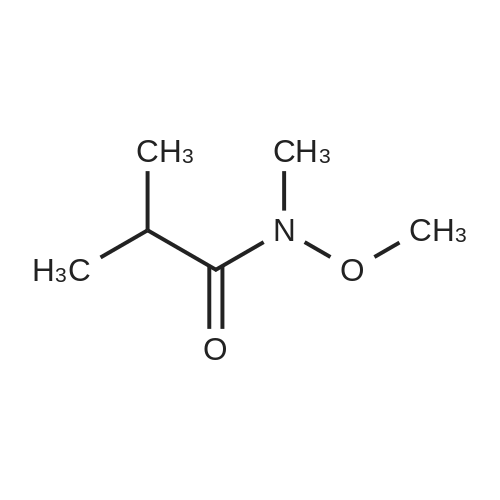
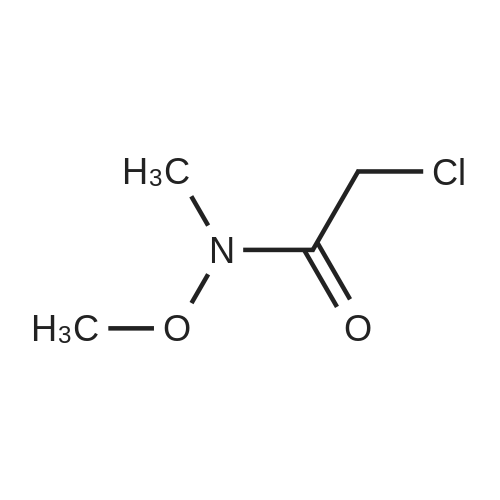
[ 402-43-7 ]
[ 104863-65-2 ]
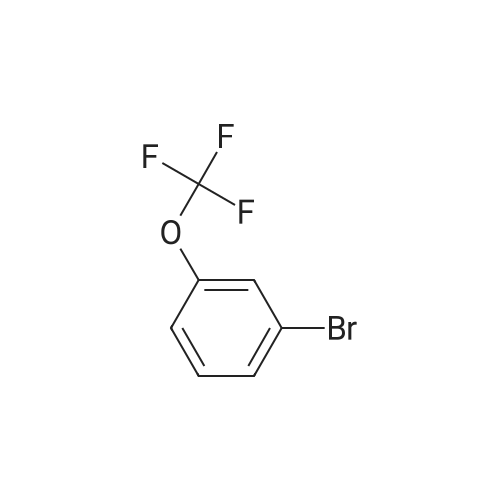
[ 711-33-1 ]
Reference:
[1] Angewandte Chemie, International Edition, 2009, vol. 48, p. 4543 - 4545[2] Angewandte Chemie, 2009, vol. 121, p. 4613 - 4615
3
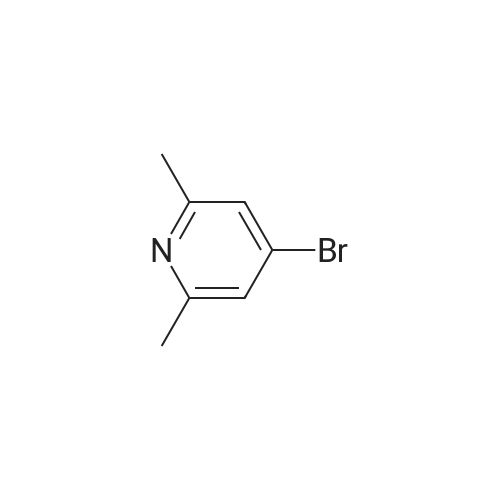
[ 6638-79-5 ]
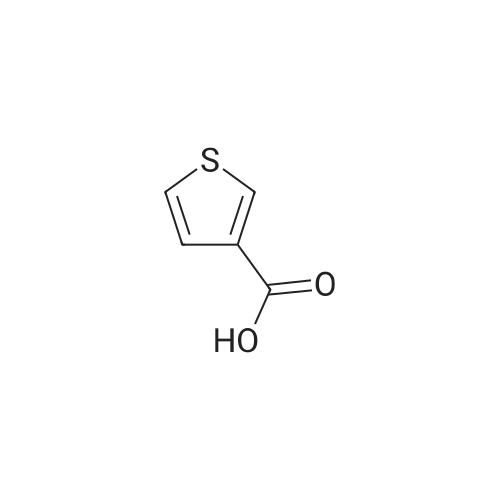
[ 79-03-8 ]
[ 104863-65-2 ]
Yield
Reaction Conditions
Operation in experiment
89%
With pyridine In dichloromethane at 0 - 20℃;
Pyridine (17 mL, 0.4 mol) at 0 0C was added dropwise to a solution of O,Λ/-dimethyl- hydroxylamine hydrochloride (10 g, 0.1 mol) and propionyl chloride (10 g, 0.1 mol) in anhydrous dichloromethane (250 mL). The solution was stirred at room temperature for 24 h, washed with 2x 50 ml of 5percent hydrochloric acid, 100 ml of saturated NaHCO3 and 100 ml brine, dried over MgSO4 and concentrated under reduced pressure to give colorless oil (10 g, 89 percent).
25.6%
With triethylamine In dichloromethane at 20℃;
I. N-Methoxy-N-methylpropanamide. To a solution of N,O-dimethylhydroxylamine hydrochloride (5.61 g, 57.5 mmol) and triethylamine (17.6 mL, 126.5 mmol) in methylene chloride (100 mL) was added propionyl chloride (5 mL, 57.5 mmol) dropwise. The reaction was stirred at room temperature overnight. The reaction was then washed with water (100 mL) and the organic layer was dried over magnesium sulfate. Solvent was removed under reduced pressure and the crude material was purified using column chromatography (SiO2, n-Hexanes to 8:2 n-Hexanes:ethyl acetate) to provide the title compound (1.72 g, 25.6percent). 1H NMR (300 MHz, CD3OD) δ 3.71 (s, 3H), 3.17 (s, 3H), 2.47 (broad m, 2H), 1.09 (t, J=7.42, 3H).
61%
With triethylamine In dichloromethane
Preparation 9 N-Methyl-N-methoxypropionamide A mixture of N,O-dimethyl hydroxylamine hydrochloride (4.43 g, 45.39 mmol) and triethylamine (6.93 mL, 49.71 mmol) in methylene chloride (150 mL) was chilled to 0° C. and propionyl chloride (3.76 mL, 43.23 mmol in 25 mL of methylene chloride with a 25 mL rinse) was added dropwise. The mixture was allowed to warm to ambient temperature and stir over the weekend. The reaction was extracted with water and brine, dried, and concentrated to afford 3.08 g (61percent) of N-methyl-N-methoxypropionamide as a yellow oil which had: NMR δ 3.66 (s, 3 H), 3.16 (s, 3 H), 2.42 (q, J=7.5 Hz, 2 H), 1.12 (t, J=7.5 Hz, 3 H). This material was suitable for use without further purification.
55%
With sodium bicarbonate; sodium chloride; triethylamine In dichloromethane
Reference Example 1 N-Methoxy-N-methylpropionamide (19) To a suspension of 11.8 g (120 mmol) of N,O-dimethylhydroxylamine hydrochloride in 294 ml of dichloromethane were added at 0°C 33.7 ml (242 mmol) of triethylamine and then a solution of 10.0 ml (115 mmol) of propionyl chloride in dichloromethane (6 ml) and the resulting mixture was stirred at room temperature for 21.5 hours. The reaction mixture was washed successively with water, dilute hydrochloric acid, an aqueous solution of sodium hydrogen carbonate, and an aqueous saturated solution of sodium chloride and was concentrated. The residue was distilled under reduced pressure to obtain 7.42 g (55percent) of the objective compound (19). Boiling point: 67°C (21 mmHg) 1H NMR (CDCl3) δ 1.14 (t, J = 7.4 Hz, 3H), 2.45 (q, J = 7.4 Hz, 2H), 3.18 (s, 3H), 3.69 (s, 3H).
61%
With triethylamine In dichloromethane
Preparation 9 N-Methyl-N-methoxypropionamide A mixture of N,O-dimethyl hydroxylamine hydrochloride (4.43 g, 45.39 mmol) and triethylamine (6.93 mL, 49.71 mmol) in methylene chloride (150 mL) was chilled to 0° C. and propionyl chloride (3.76 mL, 43.23 mmol in 25 mL of methylene chloride with a 25 mL rinse) was added dropwise. The mixture was allowed to warm to ambient temperature and stir over the weekend. The reaction was extracted with water and brine, dried, and concentrated to afford 3.08 g (61percent) of N-methyl-N-methoxypropionamide as a yellow oil which had: NMR δ3.66 (s, 3 H), 3.16 (s, 3 H), 2.42 (q, J=7.5 Hz, 2 H), 1.12 (t, J=7.5 Hz, 3 H). This material was suitable for use without further purification.
Reference:
[1] Journal of Medicinal Chemistry, 2017, vol. 60, # 12, p. 4840 - 4860
[2] Organic Letters, 2012, vol. 14, # 9, p. 2250 - 2253
[3] Chemistry - A European Journal, 2001, vol. 7, # 21, p. 4562 - 4571
[4] Patent: WO2010/104488, 2010, A1, . Location in patent: Page/Page column 65
[5] Journal of Organic Chemistry, 2007, vol. 72, # 15, p. 5828 - 5831
[6] Bulletin de la Societe Chimique de France, 1996, vol. 133, # 10, p. 1011 - 1021
[7] ACS Medicinal Chemistry Letters, 2014, vol. 5, # 1, p. 73 - 77
[8] Journal of Chemical Research, Miniprint, 1995, # 9, p. 2111 - 2122
[9] Patent: US2008/242694, 2008, A1, . Location in patent: Page/Page column 65
[10] Patent: US2003/166628, 2003, A1,
[11] Patent: US6046213, 2000, A,
[12] Patent: EP987250, 2000, A1,
[13] Patent: US6258827, 2001, B1,
[14] Patent: US2008/90861, 2008, A1, . Location in patent: Page/Page column 20
4
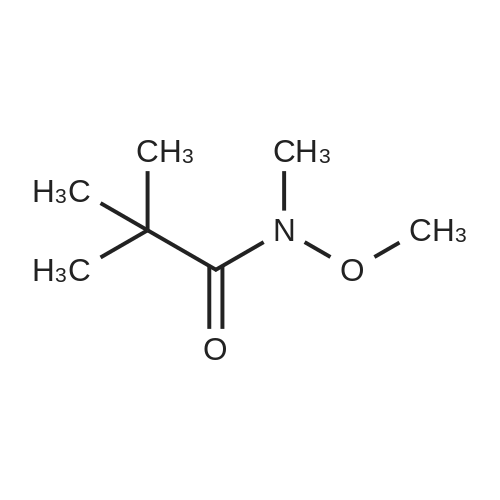
[ 6638-79-5 ]
[ 123-62-6 ]
[ 104863-65-2 ]
Reference:
[1] Angewandte Chemie, International Edition, 2009, vol. 48, p. 4543 - 4545[2] Angewandte Chemie, 2009, vol. 121, p. 4613 - 4615
[3] Patent: WO2015/24905, 2015, A1, . Location in patent: Paragraph 00293-00294
[4] Patent: US2015/80391, 2015, A1, . Location in patent: Paragraph 0570; 0571; 0572
Pyridine (17 mL, 0.4 mol) at 0 0C was added dropwise to a solution of O,Lambda/-dimethyl- hydroxylamine hydrochloride (10 g, 0.1 mol) and propionyl chloride (10 g, 0.1 mol) in anhydrous dichloromethane (250 mL). The solution was stirred at room temperature for 24 h, washed with 2x 50 ml of 5% hydrochloric acid, 100 ml of saturated NaHCO3 and 100 ml brine, dried over MgSO4 and concentrated under reduced pressure to give colorless oil (10 g, 89 %).
25.6%
With triethylamine; In dichloromethane; at 20℃;
I. N-Methoxy-N-methylpropanamide. To a solution of N,O-dimethylhydroxylamine hydrochloride (5.61 g, 57.5 mmol) and triethylamine (17.6 mL, 126.5 mmol) in methylene chloride (100 mL) was added propionyl chloride (5 mL, 57.5 mmol) dropwise. The reaction was stirred at room temperature overnight. The reaction was then washed with water (100 mL) and the organic layer was dried over magnesium sulfate. Solvent was removed under reduced pressure and the crude material was purified using column chromatography (SiO2, n-Hexanes to 8:2 n-Hexanes:ethyl acetate) to provide the title compound (1.72 g, 25.6%). 1H NMR (300 MHz, CD3OD) delta 3.71 (s, 3H), 3.17 (s, 3H), 2.47 (broad m, 2H), 1.09 (t, J=7.42, 3H).
With potassium carbonate; In water; toluene;
Example 6 Potassium carbonate (249.6 g) was added in portions at 0 C. over 7 minutes to a solution of N,O-dimethylhydroxylamine hydrochloride (100 g) in water (1000 ml), then toluene (1000 ml) was added and the mixture was stirred until the internal temperature reached 2 C. Propionyl chloride (86 ml) was added dropwise over 20 minutes, then the mixture was stirred at 0 C. for 30 minutes and at ambient temperature for 2 hours. The organic layer was separated, washed with water (500 ml) and saturated aqueous sodium chloride solution (500 ml), then the combined aqueous phases were shaken with ether (2*300 ml). The combined ether solutions were washed with water (500 ml) and saturated aqueous sodium chloride solution (500 ml), then the ether and toluene extracts were combined, dried (Na2SO4) and the solvents were removed in vacuo to give N-methoxy-N-methylpropionamide (73.0 g) as a pale yellow oil which was used without further purification.
3.08 g (61%)
With triethylamine; In dichloromethane;
Preparation 9 N-Methyl-N-methoxypropionamide A mixture of N,O-dimethyl hydroxylamine hydrochloride (4.43 g, 45.39 mmol) and triethylamine (6.93 mL, 49.71 mmol) in methylene chloride (150 mL) was chilled to 0 C. and propionyl chloride (3.76 mL, 43.23 mmol in 25 mL of methylene chloride with a 25 mL rinse) was added dropwise. The mixture was allowed to warm to ambient temperature and stir over the weekend. The reaction was extracted with water and brine, dried, and concentrated to afford 3.08 g (61%) of N-methyl-N-methoxypropionamide as a yellow oil which had: NMR delta 3.66 (s, 3 H), 3.16 (s, 3 H), 2.42 (q, J=7.5 Hz, 2 H), 1.12 (t, J=7.5 Hz, 3 H). This material was suitable for use without further purification.
7.42 g (55%)
With sodium bicarbonate; sodium chloride; triethylamine; In dichloromethane;
Reference Example 1 N-Methoxy-N-methylpropionamide (19) To a suspension of 11.8 g (120 mmol) of N,O-dimethylhydroxylamine hydrochloride in 294 ml of dichloromethane were added at 0C 33.7 ml (242 mmol) of triethylamine and then a solution of 10.0 ml (115 mmol) of propionyl chloride in dichloromethane (6 ml) and the resulting mixture was stirred at room temperature for 21.5 hours. The reaction mixture was washed successively with water, dilute hydrochloric acid, an aqueous solution of sodium hydrogen carbonate, and an aqueous saturated solution of sodium chloride and was concentrated. The residue was distilled under reduced pressure to obtain 7.42 g (55%) of the objective compound (19). Boiling point: 67C (21 mmHg) 1H NMR (CDCl3) delta 1.14 (t, J = 7.4 Hz, 3H), 2.45 (q, J = 7.4 Hz, 2H), 3.18 (s, 3H), 3.69 (s, 3H).
3.08 g (61%)
With triethylamine; In dichloromethane;
Preparation 9 N-Methyl-N-methoxypropionamide A mixture of N,O-dimethyl hydroxylamine hydrochloride (4.43 g, 45.39 mmol) and triethylamine (6.93 mL, 49.71 mmol) in methylene chloride (150 mL) was chilled to 0 C. and propionyl chloride (3.76 mL, 43.23 mmol in 25 mL of methylene chloride with a 25 mL rinse) was added dropwise. The mixture was allowed to warm to ambient temperature and stir over the weekend. The reaction was extracted with water and brine, dried, and concentrated to afford 3.08 g (61%) of N-methyl-N-methoxypropionamide as a yellow oil which had: NMR delta3.66 (s, 3 H), 3.16 (s, 3 H), 2.42 (q, J=7.5 Hz, 2 H), 1.12 (t, J=7.5 Hz, 3 H). This material was suitable for use without further purification.
With pyridine; In dichloromethane; at 0 - 20℃;
To a stirring mixture of O,N-dimethyl-hydroxylamine hydrochloride (11.1 g) and propionyl chloride (9.4 mL) in dichloromethane (300 mL) at 0 C. under a nitrogen atmosphere was added pyridine (18.2 mL). The cold bath was removed and the mixture was allowed to warm gradually to room temperature, at which temperature it stirred over the weekend. A white precipitate had formed in the stirring mixture. The mixture was treated with 10% aqueous hydrochloric acid and was stirred until the precipitate had dissolved in the biphasic solution. The layers were separated and the dichloromethane phase was washed with saturated aqueous sodium bicarbonate (100 mL) and brine solution (100 mL), was dried over anhydrous magnesium sulfate, and was concentrated under reduced pressure to afford the title compound as a clear colorless oil (12.05 g); 1H-NMR (400 MHz; CDCl3) delta 3.62 (s, 3H), 3.11 (s, 3H), 2.38 (quartet, J=7.6 Hz, 2H), 1.07 (t, J=7.6 Hz, 3H); 13C-NMR (100 MHz; CDCl3) delta 175.5, 61.3, 32.5, 25.4, 8.9; MS (APCI+) m/z 118 (MH+)
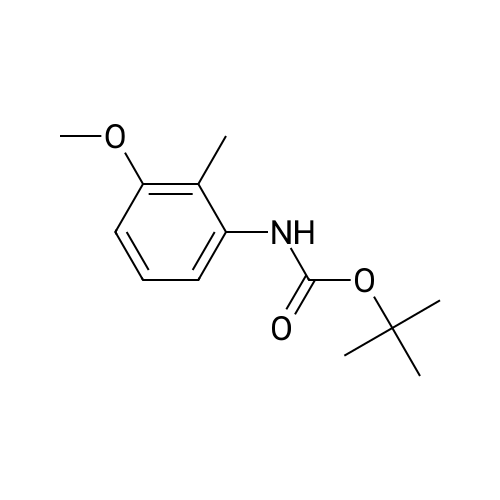
A solution of 140 mL (0.18 mol) of 1.3M sec-butyl lithium in cyclohexane was added slowly to N-tert-butoxycarbonyl-3-methoxy-2-methylaniline (21.3g, 0.09 mol) in 250 mL of THF keeping the temperature below -40C with a dry ice-ethanol bath. The bath was removed and the temperature allowed to rise to 0C and then the bath replaced. After the temperature had cooled to -60C, 18.5g (0.18 mol) of <strong>[104863-65-2]N-methoxy-N-methylpropanamide</strong> in an equal volume of THF was added dropwise. The reaction mixture was stirred 5 minutes, the cooling bath removed and stirred an additional 18 hours. It was then poured into a mixture of 300 mL of ether and 400 mL of 0.5N HCl. The organic layer was separated, washed with water, brine, dried over MgSO4, and concentrated at reduced pressure to give 25.5g of a crude of 1-[2-(tert-butoxycarbonylamino)-6-methoxyphenyl]-2-butanone. This material was dissolved in 250 mL of methylene chloride and 50 mL of trifluoroacetic acid and stirred for a total of 17 hours. The mixture was concentrated at reduced pressure and ethyl acetate and water added to the remaining oil. The ethyl acetate was separated, washed with brine, dried (MgSO4) and concentrated. The residue was chromatographed three times on silica eluting with 20% EtOAc/hexane to give 13.9g of 2-ethyl-4-methoxy-1H-indole.
A solution of 140 mL (0.18 mol) of 1.3M sec-butyl lithium in cyclohexane was added slowly to N-tert-butoxycarbonyl-3-methoxy-2-methylaniline (21.3 g, 0.09 mol) in 250 mL of THF keeping the temperature below -40 C. with a dry ice-ethanol bath. The bath was removed and the temperature allowed to warm to 0 C. and then the bath replaced. After the temperature had cooled to -60 C., 18.5 g (0.18 mol) of <strong>[104863-65-2]N-methoxy-N-methylpropanamide</strong> in an equal volume of THF was added dropwise. The reaction mixture was stirred 5 minutes, the cooling bath removed and stirred an additional 18 hours. It was then poured into a mixture of 300 mL of ether and 400 mL of 0.5N HCl. The organic layer was separated, washed with water, brine, dried over MgSO4, and concentrated at reduced pressure to give 25.5 g of a crude of 1-[2-(tert-butoxycarbonylamino)-6-methoxyphenyl]-2-butanone. This material was dissolved in 250 mL of methylene chloride and 50 mL of trifluoroacetic acid and stirred for a total of 17 hours. The mixture was concentrated at reduced pressure and ethyl acetate and water added to the remaining oil. The ethyl acetate was separated, washed with brine, dried (MgSO4) and concentrated. The residue was chromatographed three times on silica eluting with 20% EtOAc/hexane to give 13.9 g of 2-ethyl-4-methoxy-1H-indole.
With triethylamine; In dichloromethane; at 0 - 20℃; for 5h;
To a solution of propanoyl chloride (15ml) in DCM (250ML) under nitrogen was added N, O-DIMETHYLHYDROXYLAMINE (17g) and triethylamine (72ML) at 0C with stirring. The resulting mixture was allowed to warm to room temperature over 5h then filtered, the filtrate evaporated under reduced pressure and then triturated with diethyl ether. The resulting filtrate was evaporated under reduced pressure to give the sub-title compound AS AN OIL (17. 7g) 8'HCDCL3 1. 14 (3H, t), 2.43 (2H, q), 3.08 (3H, s), 3.67 (3H, s)
A tetrahydrofuran solution (200 ml) of 2-bromo-imidazo[5,1-b]thiazole (16.5 g, 81 mmol) was cooled to -30C under an argon atmosphere. Ethyl magnesium bromide (0.89 M, 100 ml) was added thereto, and the mixture was stirred for 40 min. A tetrahydrofuran solution (100 ml) of <strong>[104863-65-2]N-methyl-N-methoxypropionamide</strong> (10.5 g, 90 mmol) was added thereto, the temperature of the mixture was raised to 15C, followed by stirring for 3.5 hr. A saturated aqueous ammonium chloride solution was added thereto to stop the reaction. The mixture was extracted with ethyl acetate, was dried over anhydrous magnesium sulfate, and was then concentrated under the reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate ? ethyl acetate/methanol = 10/1) to give 2-propionylimidazo[5,1-b]thiazole (11.8 g, 80%). 1H-NMR (400 MHz, CDCl3): delta (ppm) 1.26 (3H, t), 2.89 (2H, q), 7.11 (1H, s), 8.06 (1 H, s), 8.09 (1 H, s); FABMS m/z 181 (M + H)+
With n-butyllithium; In tetrahydrofuran; hexanes; at -78 - 20℃; for 14h;Cooling with acetonitrile-dry ice;Product distribution / selectivity;
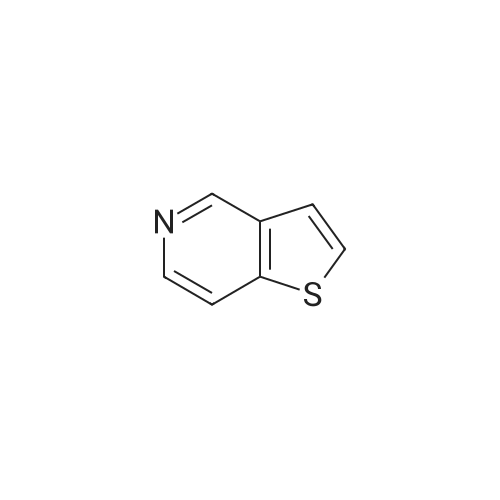
To a stirring mixture of thieno[3,2-c]pyridine (1.0 g) in anhydrous tetrahydrofuran (50 mL) at -40 C. (acetonitrile-dry ice bath) was added a solution of n-butyllithium (1.6 M, 4.7 mL) in hexanes. The mixture was cooled to -78 C. followed by the subsequent addition of a solution of <strong>[104863-65-2]N-methoxy-N-methyl-propionamide</strong> (0.97 g) in anhydrous tetrahydrofuran (10 mL). The cold bath was removed and the reaction mixture was allowed to warm gradually to room temperature. The mixture was stirred overnight (14 hours). The mixture was treated with saturated aqueous ammonium chloride (150 mL). The mixture was then extracted with ethyl acetate (500 mL), and the extract was washed with brine solution (100 mL). The organic phase was dried over anhydrous potassium carbonate and was concentrated under reduced pressure, reconstituted in chloroform, and concentrated again under reduced pressure to afford the orange-brown semisolid (1.17 g). The semisolid was purified by flash silica chromatography using an Analogix silica column (115 g silica), eluding with 0-30% ethyl acetate/heptane, then 30-60% ethyl acetate/heptane. Concentration of fractions containing product gave 0.49 g of product. Starting material (0.45 g) was also isolated by evaporation of appropriate fractions
Example 13: 2-Propionyl-7-(pyridin-3-yl)triethylsilyl oxymethylimidazo[5,1-b]thiazole; A solution of 5.14 g of 2-bromo-7-(pyridin-3-yl)triethylsilyloxymethylimidazo[5,1-b]thiazole in 36 ml of tetrahydrofuran under an argon atmosphere was cooled to - 60C, 13.3 ml of a 0.89 M tetrahydrofuran solution of ethylmagnesium bromide was added thereto, and the mixture was stirred for one hr. N-Methyl-N-methoxypropionamide (1.71 g) was added thereto at -30C, and the mixture was stirred at room temperature for 12 hr. A 20% aqueous ammonium chloride solution was added to stop the reaction, and the reaction mixture was extracted with ethyl acetate. The extract was washed with a saturated aqueous sodium bicarbonate solution and saturated brine in that order and was then dried over anhydrous magnesium sulfate. The solvent was removed by evaporation, and the residue was purified by column chromatography on silica gel (3 to 6% methanol/methylene chloride) to give 3.88 g of 2-propionyl-7-(pyridin-3-yl)triethylsilyloxymethylimidazo[5,1-b]thiazole. 1H-NMR (CDCl3) delta: 0.56 - 0.82 (6H, m), 0.88 - 0.93 (9H, m), 1.25 (3H, t, J = 7.28), 2.86 (2H, d, J = 7.28), 5.98 (1H, s), 7.23 - 7.27 (1H, m), 7.77 - 7.81 (1H, m), 7.97 (1H, s), 7.98 (1H, s), 8.51 (1H, dd, J = 1.9, 4.7 Hz), 8.73 (1H, d, J = 1.9 Hz)
Example 14: 2-Propionyl-7-(pyridin-3-yl)dimethylhydrazonoyl imidazo[5,1-b]thiazole; A solution of 1.75 g of 2-bromo-7-(pyridin-3-yl)dimethylhydrazonoylimidazo[5,1-b]thiazole in 10 ml of tetrahydrofuran was cooled to -50C under an argon atmosphere, 15.4 ml of a 0.89 M tetrahydrofuran solution of ethylmagnesium bromide was added thereto, and the mixture was stirred for one hr. N-Methyl-N-methoxypropionamide (2.2 ml) was added thereto at -20C, and the mixture was stirred at room temperature for one hr. A 20% aqueous ammonium chloride solution was added to stop the reaction, and the reaction mixture was extracted with ethyl acetate. The extract was washed with saturated aqueous sodium bicarbonate solution and saturated brine in that order and was then dried over anhydrous magnesium sulfate. The solvent was removed by evaporation, and the solid was washed with an ethyl acetate : hexane = 2 : 1 solution to give 0.94 g of 2-propionyl-7-(pyridin-3-yl)dimethylhydrazonoylimidazo[5,1-b]thiazole. 1H-NMR (CDCl3) delta: 1.29 (3H, t, J = 7.1 Hz), 2.92 (2H, q, J = 7.1 Hz), 7.31 - 7.35 (1H, m), 7.98 (1H, ddd, J = 1.6, 1.6, 8.2 Hz), 8.87 - 8.88 (1H, m)
Example 15: 7-Dimethoxy(pyridin-3-yl)methyl-2-propionyl imidazo[5,1-b]thiazole; A solution of 10.63 g of 2-bromo-7-dimethoxy(pyridin-3-yl)methylimidazo[5,1-b]thiazole in 100 ml of tetrahydrofuran under an argon atmosphere was cooled to -50C, 40 ml of a 0.89 M tetrahydrofuran solution of ethylmagnesium bromide was added thereto, and the mixture was stirred for one hr. N-Methyl-N-methoxypropionamide (5.3 ml) was added thereto at - 20C, and the mixture was stirred at room temperature for 6 hr. A saturated aqueous ammonium chloride solution was added to stop the reaction, and the reaction mixture was extracted with ethyl acetate. The organic layer was washed with a saturated aqueous sodium bicarbonate solution and saturated brine in that order and was then dried over anhydrous magnesium sulfate. The solvent was removed by evaporation, and the residue was purified by column chromatography on silica gel (5 to 15% methanol/ethyl acetate) to give a crude product of 2-propionyl-7-(pyridin-3-yl)dimethoxymethylimidazo[5,1-b] thiazole. A solution of this crude product in 200 ml of ethyl acetate was washed four times with 100 ml of an aqueous 0.02 N hydrochloric acid solution and was further washed with a 5% aqueous sodium bicarbonate solution and a 20% aqueous sodium chloride solution in that order to give 4.79 g of 7-dimethoxy(pyridin-3-yl)methyl-2-propionylimidazo[5,1-b]thiazole. 1H-NMR (CDCl3) delta: 1.26 (3H, t, J = 7.1 Hz), 2.88 (2H, q, J = 7.1 Hz), 3.22 (6H, s), 7.24 - 7.29 (1H, m), 7.88 - 7.92 (1H, m), 7.90 (1H, ddd, J = 1.9, 1.9, 8.2 Hz), 7.97 (1H, s), 8.02 (1H, s), 8.52 (1H, dd, J = 1.6, 4.9 Hz), 8.74 - 8.75 (1H, m)
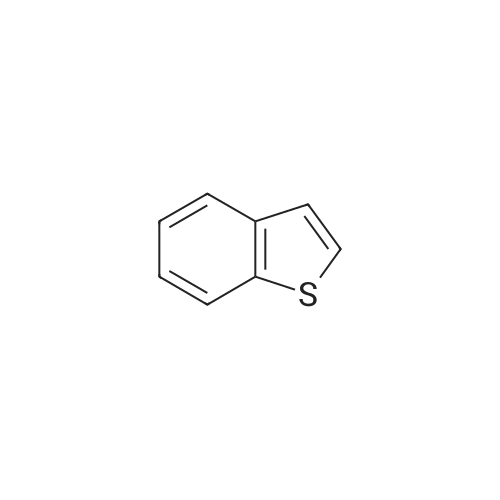
n-Butyllithium (1.6 M solution in hexanes; 28.9 ml) was added dropwise under nitrogen at 0 C. to a stirred solution of benzo[b]thiophene (6.21 g) in ether (90 ml), then the mixture was stirred 0 C. for 15 minutes and cooled to -70 C. A solution of <strong>[104863-65-2]N-methoxy-N-methylpropionamide</strong> (5.42 g) in ether (40 ml) was added over 10 minutes, then the mixture was allowed to warm to ambient temperature and was stirred at ambient temperature for 3.5 hours. The mixture was poured into saturated aqueous ammonium chloride solution (200 ml) and ethyl acetate (100 ml) and water (50 ml) were added. The organic phase was separated, washed with saturated aqueous sodium chloride solution (150 ml), dried (Na2SO4) and the solvents were removed in vacua. The residue was triturated with petroleum ether (b.p. 40-60 QC) (25 ml) and the resulting solid was collected by filtration and dried in vacuo to give 1-(benzo[b]thiophen-2-yl)propan-1-one (7.05 g) as a colourless solid, m.p. 79-81 C.
Example 16 A solution of 3-bromo-4-methylbenzo[b]thiophene (1.69 g) in tetrahydrofuran (10 ml) was added dropwise under nitrogen at -70 C. over 10 minutes to a stirred solution of n-butyllithium (2.5M solution in hexanes; 3.87 ml) in ether (30 ml), then the mixture was stirred at -70 C. for 30 minutes. A solution of <strong>[104863-65-2]N-methoxy-N-methylpropionamide</strong> (1.31 g) in tetrahydrofuran (10 ml) was added dropwise at -70 IC over 10 minutes, the mixture was stirred at -70 C. for 3 hours, then it was allowed to warm to ambient temperature and quenched by the addition of saturated aqueous ammonium chloride solution (30 ml). The organic layer was separated, washed with saturated aqueous sodium chloride solution (30 ml), and dried (MgSO4). The solvents were removed in vacuao to give 1-(4-methyl-benzo[b]thiophen-2-yl)propan-1-one (0.53 g) as an orange solid which was used without further purification.
With n-butyllithium; In tetrahydrofuran; Petroleum ether;
Example 7 A solution of 3-bromo-4-fluorobenzo[b]thiophene (3.0 g) in tetrahydrofuran (10 ml) was added dropwise under nitrogen at -60--70 C. over 10 minutes to a stirred solution of n-butyllithium (2.5M solution in hexanes; 6.75 ml) in ether (30 ml), then the mixture was stirred at -70 C. for 30 minutes. A solution of <strong>[104863-65-2]N-methoxy-N-methylpropionamide</strong> (1.31 g) in tetrahydrofuran (10 ml) was added dropwise at -70 C. over 10 minutes, then the mixture was stirred at -70 C. for 5 hours and allowed to stand at ambient temperature for 12 hours. The mixture was quenched by the addition of saturated aqueous ammonium chloride solution (40 ml), then the organic layer was separated, washed with saturated aqueous sodium chloride solution (40 ml), dried (MgSO4) and the solvents were removed in vacuo. The residue was purified by flash chromatography over silica using a 10:1 mixture of petroleum ether (b.p. 60-80 C.) and ethyl acetate as eluant. Appropriate fractions were combined and the solvents removed in vacuo to give 1-(4-fluorobenzo[b]thiophen-2-yl)propan-1-one (0.54 g) as a colourless solid which was used without further purification.
With hydrogenchloride; iodine; magnesium; In tetrahydrofuran; Petroleum ether;
Example 8 Approximately 3 ml of a solution of 4-bromobenzo[b]thiophene (1.85 g; prepared in a manner similar to that described in Bull. Soc. Chim. Fr., 1966, 111, 3667-3674) in tetrahydrofuran (20 ml) was added under nitrogen to a mixture of magnesium turnings (0.22 g) and tetrahydrofuran (2 ml). Two crystals of iodine were added and heat was applied to initiate the reaction. The remainder of the 4-bromobenzo[b]thiophene solution was added at reflux temperature over 20 minutes, the mixture was heated under reflux for 10 minutes, then it was cooled to ambient temperature. A solution of <strong>[104863-65-2]N-methoxy-N-methylpropionamide</strong> (1.01 g) in tetrahydrofuran (10 ml) was added, the mixture was heated under reflux for 30 minutes, then it was cooled to ambient temperature and quenched by the addition of 2M hydrochloric acid (25 ml). The mixture was allowed to stand at ambient temperature for 18 hours, then the tetrahydrofuran was removed in vacuo, the residue was diluted with water (50 ml) and the product was extracted into ethyl acetate (50 ml). The extract was washed with saturated aqueous sodium chloride solution (2*50 ml), dried (Na2SO4), and the solvents were removed in vacuo. The residue was purified by flash chromatography over silica using a 97:3 mixture of petroleum ether (b.p. 40-60 C.) and ethyl acetate as eluant. Appropriate fractions were combined and the solvents removed in vacuo to give 1-(benzo[b]thiophen-4-yl)propan-1-one (0.615 g) as a colourless oil which was used without further purification.
With hydrogenchloride; iodine; magnesium; In tetrahydrofuran; Petroleum ether;
Approximately 5 ml of a solution of 4-bromobenzo[b]thiophene (9.55 g) in tetrahydrofuran (100 ml) was added under nitrogen to magnesium turnings (1.5 g). Two crystals of iodine were added and heat was applied to initiate the reaction. The remainder of the 4-bromobenzo[b]thiophene solution was added at reflux temperature over 20 minutes, then the mixture was heated under reflux for 15 minutes. A solution of <strong>[104863-65-2]N-methoxy-N-methylpropionamide</strong> (6.0 g) in tetrahydrofuran (50 ml) was added, the mixture was heated under reflux for 40 minutes, then it was allowed to cool to ambient temperature and quenched by the addition of 2M hydrochloric acid (125 ml). The mixture was stirred at ambient temperature for 1 hour, then the product was extracted into ethyl acetate (300 ml). The extract was washed with water (3*200 ml) and saturated aqueous sodium chloride solution (200 ml), dried (Na2SO4), and the solvents were removed in vacuo. The residue was purified by flash chromatography over silica using petroleum ether (b.p. 60-80 C.) followed by a 97:3 mixture of petroleum ether (b.p. 60-80 C.) and ethyl acetate as eluant. Appropriate fractions were combined and the solvents removed in vacuo to give 1-(benzo[b]thiophen-4-yl)propan-1-one (5.5 g) as a colourless oil which was used without further purification.
With n-butyllithium; In tetrahydrofuran; hexane; dichloromethane; water; ethyl acetate;
Preparation 8 4-Propionyl-2-fluorophenol A mixture of 4-bromo-2-fluorophenol (1.0 g, 5.24 mmol) in tetrahydrofuran (15 mL) was chilled to -78 C. and butyllithium (4.6 mL, 11.5 mmol, 2.5 M solution) was added rapidly, dropwise. The reaction was stirred 12 min and <strong>[104863-65-2]N-methyl-N-methoxypropionamide</strong> (the compound of Preparation 9, 0.735 g, 6.28 mmol in 1 mL of tetrahydrofuran with a 1 mL rinse) was added. The reaction was allowed to stir 5 min at -78 C. and then it was warmed to ambient temperature. A few drops of water were added; then the solvent was removed at reduced pressure. The residue was taken up in methylene chloride and washed with aqueous ammonium chloride and brine. The organic layer was dried and concentrated. The residue was flash chromatographed on silica gel (1*2.5 inches packed in hexane) with elution proceeding as follows: 10% ethyl acetate/hexane (250 mL), discarded forerun; 20% ethyl acetate/hexane (250 mL), 0.186 g of a yellow crystalline solid which had NMR identical to that of preparation 7.
N-tert-butoxycarbonyl-4-cyclopentoxy-2-methylaniline[ No CAS ]
[ 598-30-1 ]
[ 104863-65-2 ]
1-[2-(tert-Butoxycarbonylamino)-5-cyclopentoxyphenyl]-2-butanone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
48%
With hydrogenchloride; In tetrahydrofuran; cyclohexane;
D. 1-[2-(tert-Butoxycarbonylamino)-5-cyclopentoxyphenyl]-2-butanone. A solution of 1.3M sec-butyl lithium/cyclohexane (33.3 mL, 0.0433 mol) was added slowly to 6.3 g (0.0216 mol) of N-tert-butoxycarbonyl-4-cyclopentoxy-2-methylaniline in 80 mL of THF while keeping the temperature below -40 C. with a dry ice-ethanol bath. The bath was removed and the temperature allowed to rise to -20 C. and then the bath was replaced. After the temperature had cooled to -60 C., 2.57 g (0.022 mol) of <strong>[104863-65-2]N-methoxy-N-methylpropanamide</strong> in an equal volume of THF was added dropwise. The reaction mixture was stirred 1 hour, the cooling bath removed and stirred an additional 1 hour. It was then poured into a mixture of 200 mL of ether and 200 mL of 1N HCl. The organic layer was separated, washed with water and dried over Na2 SO4. After removing the solvent the residue was crystallized from hexane to give 3.58 g (48% yield) of 1-(2-(tert-butoxycarbonylamino)-5-cyclopentoxyphenyl]-2-butanone, melting at 71-73 C. Analyses: Calc'd for C20 H29 NO4: C, 69.14; H, 8.41; N, 4.03. Found: C, 69.17; H, 8.42; N, 4.14.
B. 2-Ethyl-5-methyl-1H-indole. Using methods described in Example 1, Part D, 11.05 g (0.05 mol) of N-tert-butoxycarbonyl-2,4-dimethylaniline was reacted with 81 mL of 1.3M sec-butyl lithium and 6.1 g (0.05 mol) of <strong>[104863-65-2]N-methoxy-N-methylpropanamide</strong> to give the crude 1-[2-(tert-butoxycarbonylamino)-5-methylphenyl)-2-pentanone. Treatment of this material with trifluoroacetic and crystallization from EtOAc/hexane gave 1.82 g (13% yield) of 2-ethyl-5-methyl-1H-indole, mp, 77-78 C. Analyses: Calc'd for C11 H13 N: C, 82.97; H, 8.23; N, 8.80. Found: C, 83.19; H, 8.35; N, 8.89.
52a. 3-bromo-4-(1-oxopropyl)thiophene A solution of 3,4-dibromothiophene (6.0 g, 24.8 mmol) in 10 mL of ether was cooled to -78 C. and trated with n-butyllithium (9.92 mL, 2.5M solution in hexanes, 24.8 mmol). While holding the temperature at -78 C., the reaction was stirred for 15 minutes, then 2.9 g (24.8 mmol) of <strong>[104863-65-2]N-methyl-N-methoxypropionamide</strong> was added and the reaction was stirred for 1 hour. The reaction was quenched with satd NH4 Cl, and the mixture was diluted with ether. The ether layer was separated, washed with water and brine, dried over MgSO4 and concentrated. The residue was triturated with hexanes, and the mixture was filtered. The solvent was removed to give 2.08 g of a tan solid. MS 236, 238 (M+NH4)+. NMR (CDCl3) delta:7.99 (d, J=3 Hz, 1H), 7.32 (d, J=3 Hz, 1H), 2.98 (q, J=7 Hz, 2H), 1.21 (t, J=7 Hz, 3H).
With hydrogenchloride; In tetrahydrofuran; cyclohexane;
Part B. Preparation of 1-[2-(tert-Butoxycarbonylamino)-5-methoxyphenyl]-2-butanone. A solution of 1.3M sec-butyl lithium/cyclohexane (81 mL, 0.105 mol) was added slowly to 11.85 g (0.05 mol) of N-tert-butoxycarbonyl-4-methoxy-2-methylaniline in 80 mL of THF while keeping the temperature below -40 C. with a dry ice-ethanol bath. The bath was removed and the temperature allowed to rise to -20 C. and then the bath was replaced. After the temperature had cooled to -60 C., 6.1 g (0.052 mol) of <strong>[104863-65-2]N-methoxy-N-methylpropanamide</strong> in an equal volulme of THF was added dropwise. The reaction mixture was stirred 1 hour, the cooling bath removed and stirred an additional 1 hour. It was then poured into a mixture of 200 mL of ether and 200 mL of 1N HCl. The organic layer was separated, washed with water, dried over Na2 SO4 and concentrated at reduced pressure to give 10.9 g (74% yield) of 1-(2-(tert-butoxycarbonylamino)-5-methoxyphenyl]-2-butanone, melting at 80-81 C., after chromatography on silica eluding with 5% EtOAc/toluene. Analyses for C16 H23 NO4: Calculated: C, 65.51; H, 7.90; N, 4.77 Found: C, 65.69; H, 7.89; N, 4.90.
74%
With hydrogenchloride; In tetrahydrofuran; cyclohexane;
B. 1-[2-(tert-Butoxycarbonylamino)-5-methoxyphenyl]-2-butanone. A solution of 1.3M sec-butyl lithium/cyclohexane(81 mL, 0.105 mol) was added slowly to 11.85 g (0.05 mol) of N-tert-butoxycarbonyl-4-methoxy-2-methylaniline in 80 mL of THF while keeping the temperature below -40 C. with a dry ice-ethanol bath. The bath was removed and the temperature allowed to rise to -20 C. and then the bath was replaced. After the temperature had cooled to -60 C., 6.1 g (0.052 mol) of <strong>[104863-65-2]N-methoxy-N-methylpropanamide</strong> in an equal volume of THF was added dropwise. The reaction mixture was stirred 1 hour, the cooling bath removed and stirred an additional 1 hour. It was then poured into a mixture of 200 mL of ether and 200 mL of 1N HCl. The organic layer was separated, washed with water, dried over Na2 SO4 and concentrated at reduced pressure to give 10.9 g (74% yield) of 1-(2-(tert-butoxycarbonylamino)-5-methoxyphenyl)-2-butanone, melting at 80-81 C., after chromatography on silica eluding with 5% EtOAc/toluene. Analyses: Calc'd for C16 H23 NO4: C, 65.51; H, 7.90; N, 4.77. Found: C, 65.69; H, 7.89; N, 4.90.
With lithium diisopropyl amide; In tetrahydrofuran; n-heptane; ethylbenzene; at -78℃; for 2h;
H. 4-Fluoro-3-propanoylbenzenecarbonitrile. 4-Fluorobenzonitrile (1.14 g, 9.44 mmol) and <strong>[104863-65-2]N-methoxy-N-methylpropanamide</strong> (1.32 g, 11.32 mmol) were dissolved in THF (200 mL). The mixture was cooled to -78 C. and LDA (1.8 M in THF/heptane/ethylbenzene, 10.48 mL, 18.88 mmol) was added dropwise. The reaction was stirred at -78 C. for 2 hours and quenched by the addition of water. The reaction was extracted with ethyl acetate (3×100 mL), the organic layers combined and dried over magnesium sulfate. Solvent was removed under reduced pressure and the crude material was purified using column chromatography (SiO2, 8:2 n-Hexanes:ethyl acetate) to provide the title compound (0.268 g, 16%). 1H NMR (300 MHz, DMSO-d6) delta 8.30 (dd, J=6.59, 2.20, 1H), 8.16 (m, 1H), 7.61 (dd, J=10.71, 8.79, 1H), 3.02 (dddd, J=7.14, 7.14, 7.14, 2.47, 2H), 1.07 (t, J=7.14, 3H).
With triethylamine; In dichloromethane; at 0 - 20℃; for 5h;Inert atmosphere;
Intermediate Gen-7-a: To a suspension of Intermediate Gen-9-c (9.53 g, 98 mmol, 1 eq.) in DCM (100 mL) at 0C under stirring and nitrogen are slowly added TEA (28.7 mL, 205 mol, 2.1 eq.) and. Intermediate Gen- 15- g (12.5 mL, 98 mmol, 1 eq). Then the reaction mixture is allowed to warm up to room temperature and stirred at room temperature for 5 h. The reaction is quenched by slowly adding an aqueous NH4C1 solution, the layers are separated and the aqueous layer is extracted again with EtOAc. The combined organic layers are dried over Na2S04, filtered and evaporated to dryness to afford Intermediate Gen-7-a. The crude product is used directly in the next step without purification. lU NMR (300 MHz, CDC -d) delta ppm 3.69 (3 H, s), 3.19 (3 H, s), 2.46 (2 H, d), 1.15 (3 H, t).
With triethylamine; In dichloromethane; at 0 - 20℃; for 5h;Inert atmosphere;
To a suspension of Intermediate Gen-9-c (9.53 g, 98 mmol, 1 eq.) in DCM (100 mL) at 0 C. under stirring and nitrogen are slowly added TEA (28.7 mL, 205 mol, 2.1 eq.) and. Intermediate Gen-15-g (12.5 mL, 98 mmol, 1 eq). Then the reaction mixture is allowed to warm up to room temperature and stirred at room temperature for 5 h. The reaction is quenched by slowly adding an aqueous NH4Cl solution, the layers are separated and the aqueous layer is extracted again with EtOAc. The combined organic layers are dried over Na2SO4, filtered and evaporated to dryness to afford Intermediate Gen-7-a. The crude product is used directly in the next step without purification. 1H NMR (300 MHz, CDCl3-d) delta ppm 3.69 (3H, s), 3.19 (3H, s), 2.46 (2H, d), 1.15 (3H, t).
g.) Preparation of intermediate 9; BuLi (0.080 mol; 50 ml, 1.6 M in hexane) was added drop wise to a mixture of intermediate 3 (0.020 mol) in THF anhydrous (24 ml) at -78C under a nitrogen flow. The mixture was stirred at -78C for 45 minutes. A solution of JV-methoxy-iV-methyl- propanamide (0.100 mol) in THF anhydrous (1 ml) was added dropwise and the resulting reaction mixture stirred at -700C for 2 hours and then allowed to warm up to room temperature. The mixture was stirred at room temperature overnight. Water was added and the mixture was extracted with EtOAc. The organic layer was dried (MgSO4), filtered and evaporated to dryness. The residue was purified by column chromatography (eluent: Petroleum ether / EtOAc = 4:1) The product fractions were collected and the solvent was evaporated, yielding 1.1 g of (23%) of intermediate 9.
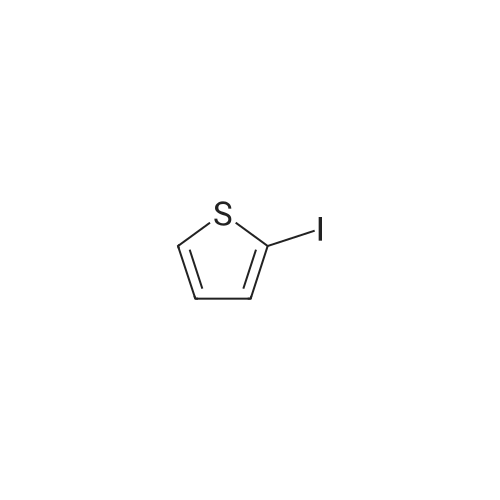
38.4 ml of a 2M solution of lithium diisopropylamide (8.2 g, 76.8 mmol) in THF- heptane-ethylbenzene was added to THF (50 ml) taken in a 500 three neck RB flask equipped with a nitrogen inlet. The mixture was cooled to -400C and then 2- iodothiophene (16.1 g, 76.8 mmol) taken in 50 ml of THF was added with vigorous stirring. After 10 minutes the mixture was warmed to -100C and stirred for 30 min. The reaction mixture was re-cooled to -400C and <strong>[104863-65-2]N-methoxy-N-methylpropionamide</strong> (9 g, 76.8 mmol) taken in 50 ml of THF was added in one portion. The reaction mixture was allowed to warm slowly to O0C and then the reaction was quenched with saturated 50 ml of NH4CI solution. The contents were extracted with 3 x 100 ml of DCM, dried over MgSO4 and concentrated to give an oily residue. This was purified by flash chromatography to give the title compound (10 g, 49%). 1H NMR (500 MHz, CDCI3-d) 7.32 (d, 1 H), 7.27 - 7.30 (m, 1H), 2.86 (q, J = 7.32 Hz, 2H), 1.21 (t, J = 7.32 Hz, 3H).
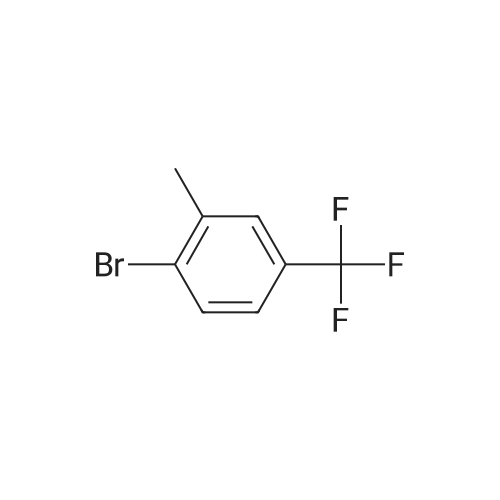
Cool a solution of 1-bromo-2-methyl-4-(trifluoromethyl)benzene (2.00 g, 8.367 mmol) and THF (17 mL) to -71 C., and then add n-butyl lithium (2.5 M in hexanes, 9.204 mmol, 3.681 mL) over 5 minutes. Stir the mixture for 15 minutes at -71 C. Add <strong>[104863-65-2]N-methoxy-N-methylpropanamide</strong> (0.980 g, 8.367 mmol) to the mixture drop-wise over 3-4 minutes keeping temperature below -65 C. Continue to stir the solution at -71 C. for 15-20 minutes; then warm the solution to room temperature. Stir at room temperature for 40 minutes. Quench the reaction with a saturated ammonium chloride aqueous solution. Extract the aqueous layer with Et2O. Wash the combined organic extracts with water and brine. Dry the mixture over sodium sulfate; filter; collect the filtrate; and concentrate the filtrate under reduced pressure. Purify the residue by flash chromatography (40 g RediSep column) eluting with a gradient of 0-40% DCM/pentane to give the title compound as a colorless oil (1.490 g, 82% yield). 1H NMR (399.80 MHz, d6-DMSO) delta 7.84 (d, J=7.8 Hz, 1H), 7.64-7.61 (m, 2H), 2.93 (q, J=7.1 Hz, 2H), 2.39 (s, 3H), 1.03 (t, J=7.2 Hz, 3H).
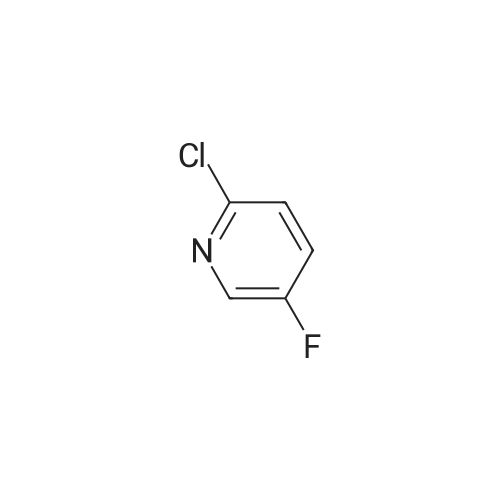
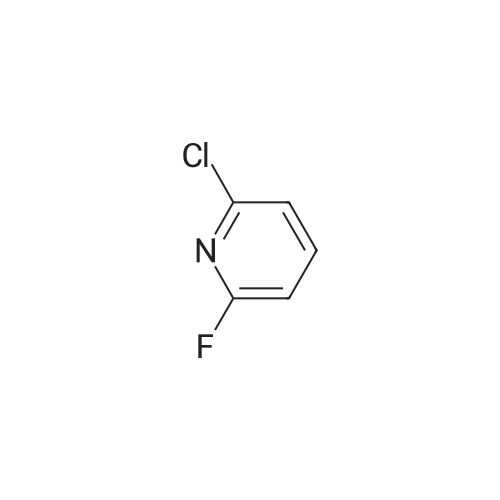
a) Synthesis of: l-(2-chloro-5-fiuoropyridin-4-yl)propan-l-one (1 1)[0258] A solution of 2-chloro-5-fiuoropyridine (1.5 g, 11.4 mmol) in THF (20 mL) was dropped into a solution of n-BuLi (5.5 mL, 2.5M in n-hexane, 13.68 mmol) in THF (20 mL) at about -78C and the solution was stirred at this temperature for about 30 min. Then <strong>[104863-65-2]N-methoxy-N-methylpropionamide</strong> (1.74 g, 14.8 mmol) was added. The resulting mixture was stirred at about -78C for about one hour, then quenched with saturated NH4C1 (20 mL) and diluted with ethyl acetate (20 mL). The organic layer was separated out and the aqueous layer was extracted by ethyl acetate (20 mL) once more. The organic layers were combined, dried and concentrated to get the crude product (910 mg, 42%) as colorless oil, which was used for the next step.
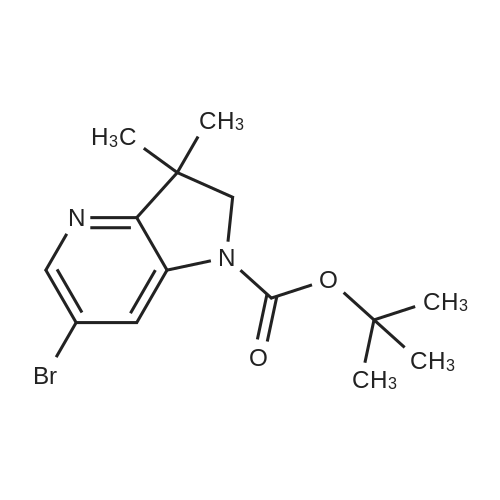
Preparation 39: 3,3-Di methyl-6-propionyl-2,3-dihydro-pyrrolo[3,2-b]pyridine-1 -carboxyl ic acid tert-butyl ester6-Bromo-3, 3-dimethyl-2 , 3-dihydro-pyrrolo[3,2-b]pyridine- 1 -carboxylic acid tert-butyl ester (1.33g, 4.07 mmol) in THF (20.3 mL) was cooled to -78 C under nitrogen and butyllithium (2.5 M in hexanes, 3.75 mL, 9.4 mmol) added. The reaction was stirred at this temperature for 30 minutes. To this was added <strong>[104863-65-2]N-methoxy-N-methyl-propionamide</strong> (0.71 g, 6.1 mmol) and the reaction was stirred for 1 h. Water and EtOAc were added and the organic layer separated, washed with brine (3x) and dried with sodium sulfate, filtered and concentrated.Chromatography (silica gel, gradient elution, 0 - 60%, EtOAc in petrol 40-60) gave the title compound (0.823 g), MS: [M+H] = 305, as a 2:1 mixture with 3,3-dimethyl-2,3-dihydro- pyrrolo[3,2-b]pyridine- 1 -carboxylic acid tert-butyl ester.
3,3-dimethyl-2,3-dihydro-pyrrolo[3,2-b]pyridine-1-carboxylic acid tert-butyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
0.823 g
Preparation 35: 3,3-Dimethyl-6-propionyl-2,3-dihydro-pyrrolo[3,2-b]pyridine-I -carboxylic acid tert-butyl ester Preparation 35: 3,3-Dimethyl-6-propionyl-2,3-dihydro-pyrrolo[3,2-b]pyridine-I -carboxylic acid tert-butyl ester6-Bromo-3, 3-dimethyl-2 , 3-dihydro-pyrrolo[3,2-b]pyridine- 1 -carboxylic acid tert-butyl ester(1.33 g, 4.07 mmol) in THF (20.3 mL) was cooled to -78 C under nitrogen and butyllithium(2.5 M in hexanes, 3.75 mL, 9.4 mmol) added. The reaction was stirred at this temperaturefor 30 minutes. To this was added <strong>[104863-65-2]N-methoxy-N-methyl-propionamide</strong> (0.71 g, 6.1 mmol) andthe reaction was stirred for 1 h. Water and EtOAc were added and the organic layer separated, washed with brine (3x) and dried with sodium sulfate, filtered and concentrated. Chromatography (silica gel, gradient elution, 0 - 60%, EtOAc in petrol 40-60) gave the title compound (0.823 g), MS: [M+H] = 305, as a 2:1 mixture with 3,3-dimethyl-2,3-dihydro- pyrrolo[3,2-b]pyridine- 1 -carboxylic acid tert-butyl ester.
1-(6-chloro-2-fluoro-pyridin-3-yl)-propan-1-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: 1.4. General method E: Synthesis of Intermediate Gen-8 Gen-7 Gen-8 Gen General method A solution of LDA is prepared by adding dropwise a 2.5 M butyllithium solution in hexane (1.1 to 1.2 eq.) to a solution of DIPA (1.07 to 1.2 eq.) in THF under nitrogen at a temperature comprised between -78C and -5C. The reaction mixture is stirred 15 min to 30 min at the same temperature. Then Intermediate Gen-6-a (1 to 1.2 eq.) in THF is added dropwise between -78C and -60C, and the reaction is stirred under nitrogen at -78C for 1 h to 1.33 h. Then Intermediate Gen-7 (1.1 to 1.2 eq.) is added dropwise or portionwise by monitoring the temperature. The mixture is stirred 1.5 h to 3 h between -78C and -70C and quenched with a saturated aqueous NH4C1 solution. EtOAc is added, then the organic layer is separated, dried over Na2S04, filtered and evaporated to dryness. The residue is purified by chromatography of silica gel to give Intermediate Gen-8.
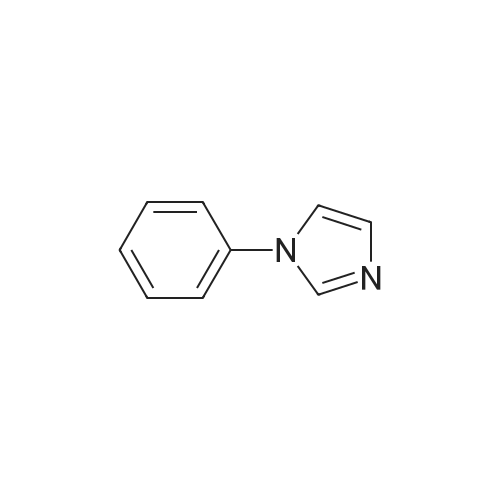
General procedure: General procedure for method A. To a solution of 1-(o-tolyl)-1H-imidazole (1.1 eq.) in THF at78 C was added n-BuLi (1.1 eq.) dropwise. The reaction was stirred at 78 C for 30 min, thenstirred at room temperature for 30 min. The corresponding Weinreb amide (1.0 eq. in THF) wasadded dropwise to the flask after the reaction was cooled back down to 78 C. The overallconcentration of Weinreib amide was 0.4 M. The reaction was allowed to warm to roomtemperature slowly (over a period of 3-4 h) and stirred overnight. The reaction was quenched withAcOH (6.0 eq.) at room temperature and extracted with EtOAc. The organic layer was washed withaqueous saturated NaHCO3 and brine. The combined organic layers were dried over anhydrousNa2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flashchromatography on silica gel (EtOAc/hexane = 1:3) to produce 1a-g.The experimental data of 1a-d, 1f-g are shown below. The other 2-acyl imidazoles (1a?4, 1e6) havebeen reported previously.
General procedure: 1a-1n, 4a, 4b 4d and 4e were synthesized according to the reported literature.2 Accordingly, to a solution of Imidazoles, Benzimidazoles, thiazoles (3.0 eq) in anhydrous THF (0.6 M) at -78 C was added n-BuLi (2.8 eq, 2.4 M in hexanes) dropwise. The reaction mixture was stirred at -78 C for 30 min, then the corresponding Weinreb amide in THF (2.0 M) was added dropwise. The solution was warmed to room temperature and stirred at the temperature overnight. The reaction was quenched with AcOH (6.0 eq) in one portion at -78 C, then allowed to warm up to room temperature over 1 h, diluted with EtOAc (100 mL). The organic phase was washed with aqueous saturated NaHCO3 (1 x 100 mL), brine (1 x 50 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was subjected to a silica gel flash chromatography (eluent: EtOAc/n-hexane = 1:5 to 1:3) to afford the pure substrates 1a-n, 4a, 4b, 4d, 4e. All spectroscopic data were in agreement with the literature3,4.6,7
1-(5-bromo-2-fluoropyridin-3-yl)propan-1-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
43.47%
Preparative Example 8 Step 1: l-(5-bromo-2-fluoropyridin-3-yl)propan-l-one To a solution of diisopropylamine (48.7 g, 482.1 mmol) in tetrahydrofuran (lOOOmL) was added butyllithium (185.7 mL, 464.23 mmol, 2.5 M) at -78 C under nitrogen. After addition, the reaction mixture was stirred for 30 min at -78 C. 5-bromo-2-fluoropyridine (70 g, 357.1 mmol) was added (keeping the temperature under -65 C). After addition, the mixture was stirred for 1 h. N-methoxy- N-methylpropionamide (45.95 g, 392.8 mmol) was added and stirred at -78 C for 1 h. The reaction mixture was quenched with water (lOOOmL), extracted with ethyl acetate (3 x 500 mL), washed with brine, dried over sodium sulfate and concentrated to dryness in vacuo. The resulting residue was purified by column chromatography (silica gel, 100-200 mesh, 0.5% ethyl acetate in petroleum ether) affording l-(5-bromo-2-fluoropyridin-3-yl)propan-l-one (36 g, 43.47%): H NMR (400 MHz, Chloroform-d): delta 8.42 (s, 2H), 3.04 (m, 2H), 1.23 (m. 3H).
1-(3-(trifluoromethoxy)phenyl)propan-1-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
To a solution of 1-bromo-3-(trifluoromethyl)benzene (2.00 g, 8.30 mmol) in THF (20 mL) was dropped a solution of n-BuLi in n-hexane (2.5 M, 3.3 mL, 8.30 mmol) at - 78oC under nitrogen and the mixture was stirred at -78oC for 15 minutes. To the solution at - 78oC was dropped a solution of Compound 4A (1.17 g, 9.93 mmol) in THF (4 mL) and the mixture was stirred at the same temperature for 20 minutes. The reaction mixture was quenched with saturated NH4Cl solution (50 mL) at -20oC, and extracted with ethyl acetate (160 mL). The organic layer was separated, washed with water (200 mL) and brine (200 mL), dried over anhydrous sodium sulfate, filtered, and concentrated to give a crude product, which was purified with flash column chromatography on silica gel (ethyl acetate in petroleum ether, from 0% to 8% v/v) to give Compound 4B. LC-MS (ESI) m/z: 219 [M+H]+.
A 2M solution of LDA in THF/heptane/benzene (7.6 mL) was added dropwiseto a stirred solution of thiophene-3-carboxylic acid (6.8 mmol) in anhydrousTHF (30 mL) at 0 C under nitrogen atmosphere. The resulting mixture wasstirred for 15 min and then, after cooling to 0 C, a solution of the <strong>[104863-65-2]N-methoxy-N-methylpropanamide</strong> (8.89 mmol) was dropwise added. The reaction mixture was allowed to warm to rtand stirred for 4 h. The resulting suspension was diluted with iced water (40mL) and washed with Et2O (20 mL). The aqueous layer was acidifiedwith 3M HCl and then extracted with EtOAc (70 mL x 3), anhydrified (Na2SO4)and evaporated under reduced pressure to yield a solid which was used for thenext step without further purification.70% yield. 1H NMR (DMSO-d6) delta: 1.07 (t, 3H, CH3,J = 7.2 Hz), 2.95 (q, 2H, CH2, J = 7.2 Hz), 7.37 (d, 1H, Ar, J = 5.0Hz), 7.86 (d, 1H, Ar, J = 5.0 Hz),13.38 (br s, 1H, OH).
With magnesium; In tetrahydrofuran; at 20℃;Inert atmosphere; Heating;
(b) under the protection of N2,Add magnesium chips (20 mmol) to a 100 mL round bottom flask.Add 2 mL of dry tetrahydrofuran (THF),(I)-1a (2 mmol) was dissolved in 5 mL of THF with stirring and then injected with a syringe.Heated with a hair dryer,After the mixed solution is kept in a slightly boiling state for 2 minutes,Then put the remaining (I)-1a (18mmol)And N-methoxy-N-methylacetamide (20 mmol)The solution in 10 mL of THF was slowly added dropwise to the reaction system.Keep it in a slightly boiling state,After the addition, add 10 mL of THF.Stirring the reaction at room temperature,After TLC monitors the reaction completely,Add to the reaction solution10mL saturated ammonium chloride solution,After stirring for 10 min, it was extracted three times with ethyl acetate.The organic phase was separated and dried over anhydrous sodium sulfate.After evaporating the solvent, the title product (I)-1b was obtained by silica gel column chromatography. Yield: 80%.
With magnesium; In tetrahydrofuran; at 20℃;Inert atmosphere; Heating;
(b) under the protection of N2,Add magnesium chips (20 mmol) to a 100 mL round bottom flask.Add 2 mL of dry tetrahydrofuran (THF),(I)-25a (2mmol) with stirringDissolve in 5mL THF and inject with a needle.Heated with a hair dryer,After the mixed solution is kept in a slightly boiling state for 2 minutes,Then put the remaining (I)-25a (18mmol)And N-methoxy-N-methylacetamide (20 mmol)The solution in 10 mL of THF was slowly added dropwise to the reaction system.Keep it in a slightly boiling state,After the addition, add 10 mL of THF.Stirring the reaction at room temperature,After TLC monitors the reaction completely,10 mL of a saturated ammonium chloride solution was added to the reaction solution.After stirring for 10 min, it was extracted three times with ethyl acetate.The organic phase was separated and dried over anhydrous sodium sulfate.After evaporating the solvent, silica gel column chromatography gave the objective product (I)-25b. Yield: 77%.
1-(4-bromo-6-methylpyridin-2-yl)butan-2-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85%
With lithium diisopropyl amide;
Step 1: 1-(4-bromo-6-methylpyridin-2-yl)butan-2-one (2B) To a solution of 4-bromo-2,6-dimethylpyridine (2A) (25.5 g, 137.0 mmol) in anhydrous tetrahydrofuran (500 mL) was added lithium diisopropylamide (2M in tetrahydrofuran, 102.4 mL, 204.9 mmol) dropwise over 30 minutes via a dropping addition funnel at -78 C. under nitrogen. The mixture was stirred for 2 h at -78 C. and <strong>[104863-65-2]N-methoxy-N-methylpropionamide</strong> (2A-1) (8.0 g, 68.3 mmol, dissolved in 20 mL THF) was added dropwise over 15 minutes. Then the reaction mixture was stirred for 1 h at -78 C. and then quenched with addition of water. The mixture was warmed to rt, and then concentrated in vacuo. The residue was diluted with EtOAc and H2O. The organic layer was separated and the aqueous layer extracted with EtOAc. The combined organic layers were then washed with water and brine, dried over Na2SO4, concentrated in vacuo. The residue was purified by flash chromatography to afford 2B (14 g, 85%). 1H NMR (400 MHz, CDCl3) delta 7.26-7.23 (m, 1H), 7.23-7.19 (m, 1H), 3.84 (s, 2H), 2.56 (q, J=7.3 Hz, 2H), 2.50 (s, 3H), 1.06 (t, J=7.3 Hz, 3H). LC-MS (ESI): m/z =244.1 [M+H]+.
1-((5-phenylpent-1-en-2-yl)oxy)pyridin-1-ium bis((trifluoromethyl)sulfonyl)amide[ No CAS ]
[ 104863-65-2 ]
2-oxo-5-phenylpentyl propionate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
76%
With water; In dichloromethane; at 60℃; for 3h;
The reaction formula of this embodiment is as follows:In a 25 ml reaction flask, add compound 1a (0.3 mmol, 0.1561 g), compound 2i (15 mmol, 1.7573 g), water (30 muL), DCM (5 mL) to dissolve the above materials, and stir the reaction at 60 C. The reaction time is 3 h. After the reaction was completed, the solvent was removed by spin-drying, and the residue was subjected to flash column chromatography (PE: EA = 20: 1) to obtain pure product 3ai (0.0534 g) with a yield of 76%.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping