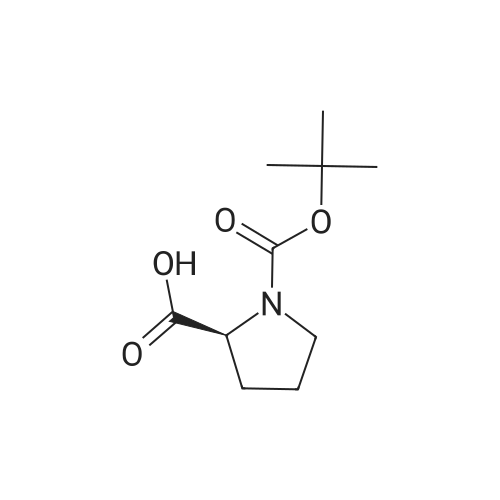
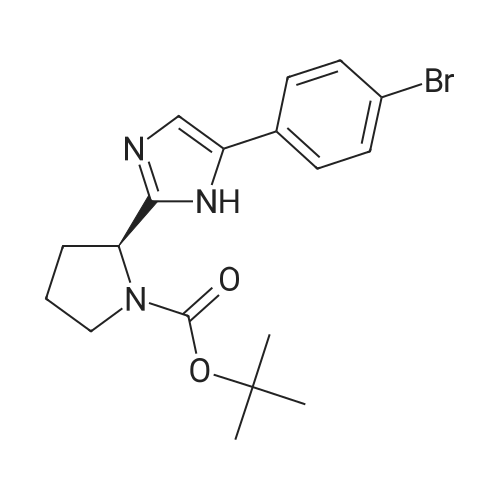
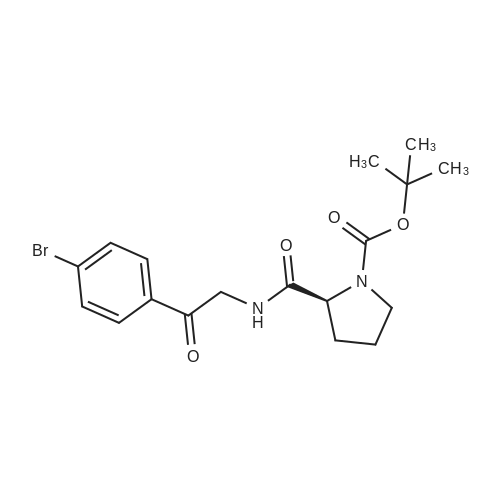
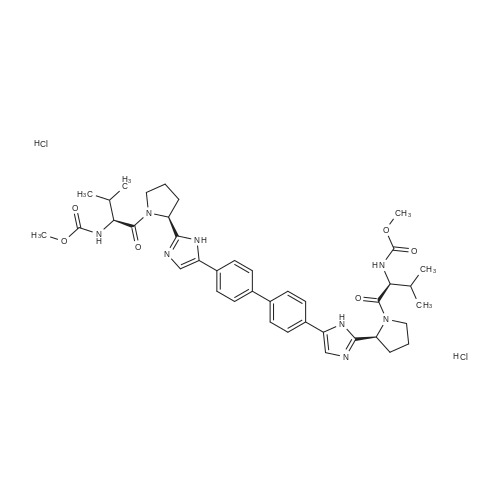
| 77% |
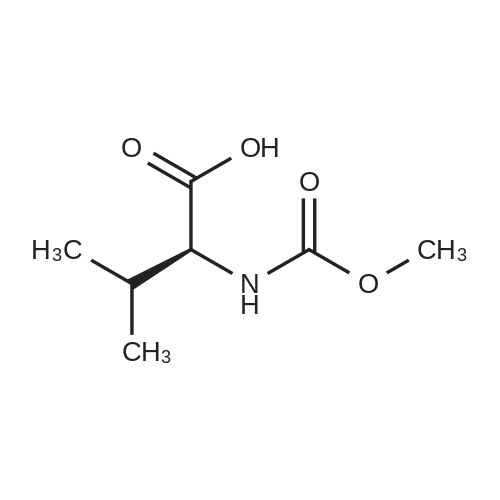
Stage #1: N-methoxycarbonyl-L-valine With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride In acetonitrile at 20℃; for 1h;
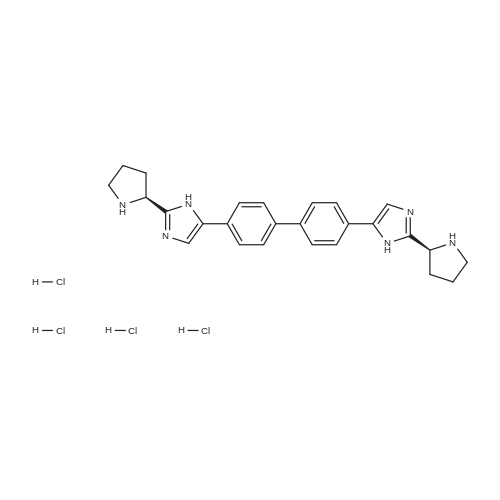
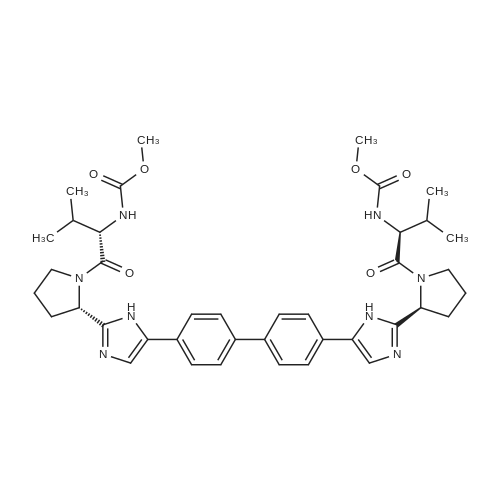
Stage #2: methyl N-[(2S)-1-[(2S)-2-[5-[4-[4-[2-[(2S)-1-[(2S)-2-(methoxycarbonylamino)-3-methylbutanoyl]pyrrolidin-2-yl]-1H-imidazol-5-yl]phenyl]phenyl]-1H-imidazol-2-yl]pyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl]carbamate hydrochloride With N-ethyl-N,N-diisopropylamine In acetonitrile at 0 - 20℃; for 22.75h;
Stage #3: With hydrogenchloride; sodium chloride more than 3 stages; |
24.23
A 50 mL flask equipped with a stir bar was sequentially charged with 2.5 mL acetonitrile, 0.344 g (2.25 mmol, 2.5 equiv) hydroxy benzotriazole hydrate, 0.374 g (2.13 mmol, 2.4 equiv) N-(methoxycarbonyl)-L-valine, 0.400 g (2.09 mmol, 2.4 equiv) 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride and an additional 2.5 mL acetonitrile. The resulting solution was agitated at 20° C. for 1 hour and charged with 0.501 g (0.88 mmol, 1 equiv) Example A-1e-4. The slurry was cooled to about 0° C. and 0.45 g (3.48 mmol, 4 equiv) diisopropylethylamine was added over 30 minutes while maintaining a temperature below 10° C. The solution was slowly heated to 15° C. over 3 hours and held at 15° C. for 16 hours. The temperature was increased to 20° C. and stirred for 3.25 hours. The resulting solution was charged with 3.3 g of 13 wt % aqueous NaCl and heated to 50° C. for 1 hour. After cooling to 20° C., 2.5 mL of isopropyl acetate was added. The rich organic phase was washed with 2×6.9 g of a 0.5 N NaOH solution containing 13 wt % NaCl followed by 3.3 g of 13 wt % aqueous NaCl. The mixture was then solvent exchanged into isopropyl acetate by vacuum distillation to a target volume of 10 mL. The resulting hazy solution was cooled to 20° C. and filtered through a 0.45 μm filter. The clear solution was then solvent exchanged into ethanol by vacuum distillation with a target volume of 3 mL. 1.67 mL (2.02 mmol, 2.3 equiv) of 1.21 M HCl in ethanol was added. The mixture was then stirred at 25° C. for 15 hours. The resulting slurry was filtered and the wet cake was washed with 2.5 mL of 2:1 acetone:ethanol. The solids were dried in a vacuum oven at 50° C. to give 0.550 g (0.68 mmol, 77%) of the desired product.Recrystallization of Example 24-23A solution of Example 24-23 prepared above was prepared by dissolving 0.520 g of the above product in 3.65 mL methanol. The solution was then charged with 0.078 g of type 3 Cuno Zeta loose carbon and allowed to stir for 0.25 hours. The mixture was then filtered and washed with 6 ml of methanol. The product rich solution was concentrated down to 2.6 mL by vacuum distillation. 7.8 mL acetone was added and allowed to stir at 25° C. for 15 h. The solids were filtered, washed with 2.5 mL 2:1 acetone:ethanol and dried in a vacuum oven at 70° C. to give 0.406 g (57.0%) of the desired product as white crystals: 1H NMR (400 MHz, DMSO-d6, 80° C.): 8.02 (d, J=8.34 Hz, 4H), 7.97 (s, 2H), 7.86 (d, J=8.34 Hz, 4H), 6.75 (s, 2H), 5.27 (t, J=6.44 Hz, 2H), 4.17 (t, J=6.95 Hz, 2H), 3.97-4.11 (m, 2H), 3.74-3.90 (m, 2H), 3.57 (s, 6H), 2.32-2.46 (m, 2H), 2.09-2.31 (m, 6H), 1.91-2.07 (m, 2H), 0.88 (d, J=6.57 Hz, 6H), 0.79 (d, J=6.32 Hz, 6H); 13C NMR (75 MHz, DMSO-d6): δ 170.9, 156.9, 149.3, 139.1, 131.7, 127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7; IR (neat, cm-1): 3385, 2971, 2873, 2669, 1731, 1650. Anal. Calcd for C40H52N8O6Cl2: C, 59.18; H, 6.45; N, 13.80; Cl, 8.73. Found C, 59.98; H, 6.80; N, 13.68; Cl, 8.77. mp 267° C. (decomposed). Characteristic diffraction peak positions (degrees 2θ+/-0.1) (at) RT, based on a high quality pattern collected with a diffractometer (CuKα) with a spinning capillary with 2θ calibrated with a NIST other suitable standard are as follows: 10.3, 12.4, 12.8, 13.3, 13.6, 15.5, 20.3, 21.2, 22.4, 22.7, 23.7. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping